

Abstract
Interaction of a mitochondria-specific anionic phospholipid, cardiolipin (CL), with an intermembrane protein, cytochrome c (cyt c), yields a peroxidase complex. During apoptosis, the complex induces accumulation of CL oxidation products that are essential for detachment of cyt c from the mitochondrial membrane, induction of permeability transition and release of proapoptotic factors into the cytosol. Therefore, suppression of the peroxidase activity and prevention of CL oxidation may lead to discovery of new anti-apoptotic drugs. Here, we report a new approach to regulate the cyt c peroxidase activity by using modified CL with oxidizable and fluorescent 7-nitro-2-1,3-benzoxadiazol (NBD) moiety (NBD-CL). We demonstrate that NBD-CL forms high affinity complexes with cyt c and blocks cyt c-catalyzed oxidation of several peroxidase substrates, cyt c self-oxidation, and, most importantly, inhibits cyt c-dependent oxidation of polyunsaturated tetra-linoleoyl CL (TLCL) and accumulation of TLCL hydroperoxides. Electrospray-ionization mass spectrometry and fluorescence analysis revealed that oxidation and cleavage of NBD moiety of NBD-CL underlies the inhibition mechanism. We conclude that modified CL combining a non-oxidizable monounsaturated tri-oleoyl CL with a C12-NBD fragment undergoes a regio-specific oxidation thereby representing a novel inhibitor of cyt c peroxidase activity.
Keywords: cytochrome c; cardiolipin; apoptosis; 7-nitro-2-1,3-benzoxadiazol; phospholipid hydroperoxides; peroxidase
The major physiological function of apoptosis is safe elimination of unwanted or irreparably damaged cells. Excessive apoptosis is usually associated with tissue degeneration during acute injury (e.g., ischemia, stroke, γ-irradiation) and chronic disease conditions (e.g., diabetes, cardio-vascular and neurodegenerative diseases) [1-8]. Stringent regulation of apoptosis is important to avoid massive cell loss. Therefore, significant efforts have been directed towards the development of new regulators acting at early stages of apoptosis – upstream of the point-of-no-return, i.e. release of pro-apoptotic factors and caspase activation [9-16]. In early 90th Radi et al. have been demonstrated that cyt c can play a critical role in hydrogen-peroxide induced lipid peroxidation in isolated mitochondria [17, 18]. We have recently documented that oxidation of a mitochondria-specific phospholipid, cardiolipin (CL), is a required step in the execution of the mitochondrial stage of apoptosis [19]. This is achieved via the formation of a complex of cyt c with polyunsaturated molecular species of CL that confers peroxidase function on the former. The complex can oxidize small reducing substrates, protein tyrosines and most importantly CL [19-22]. Cyt c-driven accumulation of CL hydroperoxides is critical for the detachment of cyt c from the inner mitochondrial membrane, Bax-induced mitochondrial outer membrane permeabilization and release of proapoptotic proteins, including cyt c into the cytosol [23-26]. Therefore suppression of the peroxidase activity and prevention of CL oxidation may lead to discovery of new anti-apoptotic drugs [15, 27]. This can be achieved via direct inhibition of the enzymatic activity, removal of co-factors feeding the peroxidase cycle (e.g., H2O2), or decreasing susceptibility of CL to oxidation (e.g., by biosynthesis of non-oxidizable species of monounsaturated/saturated CLs). Indeed, we demonstrated that mitochondria-targeted conjugates of nitroxides with hemigramicidin S were able to scavenge electrons from damaged carriers, suppress CL peroxidation and protect cells against apoptosis induced by chemical pro-apoptotic agents or irradiation in vitro and in vivo [15, 27]. We further found that enrichment of CL pool of HL-60 cells with highly oxidizable docosahexaenoic acid (C22:6) increased their sensitivity to apoptosis; in contrast, enrichment of cells with oleic acid (C18:1)-containing species resulted in their decreased sensitivity to apoptotsis [19, 20]. Here, we report a new approach to regulate the cyt c peroxidase activity by using modified CL containing an oxidizable moiety re-routing the high oxidizing potential of the complex and “distracting” it from peroxidation of polyunsaturated CLs. We designed such an inhibitor on the basis of two components – non-oxidizable tri-oleoyl-CL and oxidizable and fluorescent 7-nitro-2-1,3-benzoxadiazol (NBD).
MATERIALS AND METHODS
Reagents
Horse heart cytochrome c (cyt c, type C-7752, >95%), horse radish peroxidase (HRP, type VI), etoposide (demethylepipodophyllotoxin-ethyledieneglucopyranoside), 2’,7’-dichlorofluorescein, 4-chloro-7-nitrobenz-2-oxa-1,3-diazole (NBD), diethylenetriaminepentaacetic acid (DTPA), taurine, glycine, hydrogen peroxide (H2O2), tert-butyl hydroperoxide (tBOOH), digitonin and SDS were purchased from Sigma-Aldrich (St. Louis, MO). Amplex Red (N-acetyl-3,7-dihydroxyphenoxazine) reagent was obtained from Molecular Probes (Eugene, OR). 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), 1,1’,2,2’-tetralinoleoyl-cardiolipin (TLCL), and 1,1’,2,2’-tetraoleoyl-cardiolipin (TOCL), 1-oleoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl]-sn-glycero-3-phosphocholine (C6-NBD-PC), 1-oleoyl-2-[12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-sn-glycero-3-phosphocholine (C12-NBD-PC), 1,1’, 2,2’-tetramyristoyl-cardiolipin (TMCL), 1,1’,2,2’-tetralinoleoyl-cardiolipin (TLCL), 1,1’,2,2’-tetramyristoyl-cardiolipin (TMCL) were obtained from Avanti Polar Lipids, Inc. (Alabaster, AL). Mouse anti–cyt c antibody (clone 7H8.2C12) was obtained from BD Biosciences (Franklin Lakes, NJ), goat anti-mouse HRP conjugated antiserum West Femto (ThermoFisher Scienific). 1,1’,2-trioleoyl-2’-[12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-cardiolipin (NBD-CL) was custom-synthesized by Avanti Polar Lipids, Inc. (Alabaster, AL).
Small unilamellar liposomes
Individual phospholipids, stored in chloroform, were mixed and dried under nitrogen. Then lipids were mixed in vortex in HEPES buffer (20 mM, pH 7.4) and sonicated three times for 30 s on ice. Liposomes were used immediately after preparation.
Distribution of NBD-CL between the inner and the outer leaflets of DOPC/TOCL liposomes was estimated by using a reducing agent, sodium dithionite, which can reduce NBD and quench its fluorescence in the outer leaflet only [28]. DOPC/TOCL/NBD-CL liposomes (50:49:1) were treated with dithionite (200 μM) for 1 min. NBD-CL fluorescence was analyzed before and after treatment by using a Shimadzu F5301-PC spectrofluorimeter with the excitation and emission wavelength of 470 and 537 nm, respectively.
Isolation of mitochondria
Mitochondria were isolated from freshly obtained livers of adult male mice according to [29]. The preparation was carried out using MSH buffer (210 mM mannitol, 70 mM sucrose, 5 mM HEPES, 1 mM EDTA pH 7.5). Mitoplasts were immediately prepared from freshly isolated mitochondria by a digitonin method according to Krebs et al. [30] and then depleted of cytochrome c as described in [31].
Incorporation of NBD-CL in mitochondria
Mitochondria were incubated (35 min) with different concentrations of NBD-CL, washed from the excess of the phospholipid and mitochondrial lipids were extracted according to Folch [32]. Amounts of NBD-CL in the extracts were determined by measuring NBD-CL fluorescence in chloroform and using a standard calibration curve.
Native gel electrophoresis
Agarose gel electrophoresis was run by using horizontal gel system “Mupid-21” (Cosmo Bio) at 50 V in nondenaturing HEPES buffer (35 mM, pH 7.4) containing imidazole (43 mM). Samples of cyt c and liposomes were applied onto 0.8% agarose gel (120 pmol protein per sample). Gels were stained for protein with 0.25% Coomassie Brilliant Blue R-250 in 45% methanol and 10% acetic acid.
PAGE and Western Blotting Analysis
Cyt c and its aggregates were separated by 12.5% SDS-PAGE in Tris-glycine buffer. The separated proteins were electro-transferred to nitrocellulose membrane. After blocking with 5% non-fat milk dissolved in phosphate buffered saline Tween-20 (0.05%) (PBS-T) for 1h, membrane was incubated overnight at 4°C with primary antibodies (anti-cyt c). The membranes were washed several times and incubated with HRP-conjugated goat anti-mouse antiserum for 1 h at RT. The protein bands were visualized by using a SuperSignal West Pico Chemiluminescent Substrate (ThermoFisher Scientific). The density of bands was determined by scanning with an Epi Chemi II Darkroom (UVP BioImaging Systems, Upland, CA).
Fluorescence spectrometry
PC1 spectrofluorimeter (ISS Inc., Champaign, IL) equipped with xenon arc lamp as a light source was employed for fluorescence measurements of NBD-CL incorporated in DOPC/TOCL (1:1) liposomes in the amount of 1 mol%. Fluorescence emission spectra of NBD-labeled CL were detected by using an excitation wavelength of 470 nm, excitation and emission slits of 8 nm; fluorescence excitation spectra – by using emission wavelength of 536 nm. Excited state lifetime of NBD-CL was estimated by using a frequency domain method. An excitation wavelength of 470 nm, cutoff emission filter of 515 nm, and modulation frequency in the range of 1-150 MHz were employed to determine the phase delay and the modulation ratio. Data were analyzed by using 2’,7’-dichlorofluorescein as a reference standard and a “Vinci” software.
Assessment of peroxidase activity with Amplex Red reagent was performed by measuring the fluorescence of resorufin, an oxidation product of Amplex Red. Cyt c (0.5 μM) was incubated with liposomes for 10 min. Peroxidase reaction was started by addition of Amplex Red (5-100 μM) and H2O2 (100 μM) and was carried out for 30 min (reaction rate was linear in the entire time interval). Fluorescence was detected by employing an “Fusion α”universal microplate analyzer and by using an excitation wavelength of 535 nm and an emission wavelength of 585 nm.
Peroxidase activity of mitochondria was determined in the presence of Amplex Red (50 μM) and tBOOH (2 mM) after 10 min incubation. Fluorescence was measured using a Shimadzu RF5301-PC spectrofluorimeter (excitation and emission wavelengths of 560 nm and 582 nm, respectively).
EPR Spectroscopy
EPR measurements were performed on a JEOL-RE1X spectrometer at 25°C in gas-permeable Teflon tubings (0.8 mm i.d., 0.013 mm thickness obtained from Alpha Wire Corp. (Elizabeth, NJ). Teflon tube was filled with 50 μl of sample and placed in EPR quartz tube for the measurements. Etoposide phenoxyl radical spectra were recorded under the following conditions: 335.3 mT, center field; 2 mT, sweep width; 0.04 mT, field modulation; 10 mW, microwave power; 0.1 s, time constant; 2 min, time scan. The time course of etoposide radical EPR signal was obtained by repeated scanning of the field (0.15 mT, sweep width; 335.3 mT, center field) corresponding to a part of the EPR signal. Other instrumental conditions were: 0.04 mT, field modulation; 10 mW, microwave power; 0.1 s, time constant; 20 s, time scan; internal mode of recording.
Mass Spectrometry
Electrospray ionization mass spectrometry (ESI-MS) of phospholipids and their oxidation products were analyzed by direct infusion into a linear ion-trap mass spectrometer LXQ (Thermo Electron, San Jose, CA). Samples in chloroform/methanol 2:1 v/v (20 pmol/μl) were utilized directly for acquisition of ESI mass spectra at a flow rate of 5 μl/min. The electrospray probe was operated at 5.0 kV in the negative ion mode. Source temperature was maintained at 150°C. MSn analysis was carried out with relative collision energy ranging from 20 to 40% and with an activation q value at 0.25 for collision-induced dissociation (CID) and a q value at 0.7 for the pulsed-Q dissociation technique. TMCL was used the as internal standard. Isotopic corrections were performed by entering the chemical composition of each species into the Qual browser of Xcalibur (operating system) and using the simulation of the isotopic distribution to make adjustments for the major peaks. Chemical structures of lipid molecular species were confirmed by comparing with the fragmentation patterns presented in Lipid Map Data Base (www.lipidmaps.org).
In our experiments the structure of TLCL oxidized products was confirmed by MSn fragmentation analysis with an additional approach using reduction of hydroperoxy-derivatives by a mild reductant of organic hydroperoxides – triphenylphosphine. These results were reported in our previously published articles [19, 33]. Further, additional quantitative controls to independently confirm the validity of our estimates. Specifically, quantitative analysis of low concentrations of TLCL (5 pmoles/μL) and its oxidation products (less than 0.5 pmoles/μL) was based on a combination of: 1) lipid phosphorus determination by a micro-method [34] and 2) ESI-MS protocol where CL molecular species were quantified by comparing the ratio of the individual peak intensities with that of an internal standard TMCL, which was added to the sample before MS analysis. TMCL was used as internal standard and TLCL was used as a reference standard [35]. Additionally, NBD-CL was used as a reference standard in the presence of TMCL as an internal standard. The linear response and detection limit for CL were established by injection of calibration mixtures with different concentrations of TLCL or NBD-CL at a constant concentration of TMCL. Thus, the concentration of non-oxidized TLCL in the sample was confirmed by two independent methods: by lipid phosphorus determination and ESI-MS protocol. Both methods resulted in quantitatively the same concentration of TLCL. In experiments where TLCL was oxidized by cyt c/H2O2 the total amount of individual molecular species of oxidized TLCL (TLCLox) plus non-oxidized TLCL – which were estimated by MS protocol (using TMCL as internal standard and TLCL as reference standard) – resulted in the same total value of CL that was independently obtained by lipid phosphorus determination. These results indicate that although oxidized TLCL molecular species may have different ionization efficiency than non-oxidized TLCL these differences in MS responses were negligible in the range of low concentrations employed [36].
Statistical Analysis
Data are expressed as means ±S.D. values from at least three experiments. Changes in variables are analyzed by one-way ANOVA for multiple comparisons. Differences are considered significant at p < 0.05.
RESULTS
I. Interactions of cyt c with NBD-CL
Native gel electrophoresis of cyt c/cardiolipin complexes
Interaction of cyt c with various anionic lipids (see Fig. 1) can be assessed by bidirectional gel electrophoresis in native low density gel whereby cationic cyt c and negatively charged liposomes migrate in opposite directions. Binding of cyt c to lipids shifts the protein mobility and can be used for quantitative assessments of the strength of the interactions [19, 37]. We applied this approach to analyze cyt c interactions with CL-, and NBD-CL-containing liposomes (Fig. 2). TOCL and NBD-TOCL were mixed with zwitterionic DOPC at a ratio of 1:1. In the presence of TOCL or NBD-TOCL, the migration profile of cyt c was changed. Zero electrophoretic mobility of cyt c in the presence of TOCL was achieved at a ratio of 5:1 (Fig. 2). CL has a negative charge of 1.3 at pH 7.4 [38], hence, cyt c/CL mixture had a zero net charge at this ratio. Zero electrophoretic mobility of NBD-CL/cyt c was observed at a ratio of 3:1 suggesting that NBD-CL was able to form more stable complexes with cyt c than non-modified TOCL. Moreover, NBD-CL prevented migration of cyt c to the cathode even when the theoretical total charge of the complex was positive. This indicates that NBD group could be involved in the charge distribution in the complex.
Figure 1. Structural formulas of CL, NBD-CL, NBD-PC.
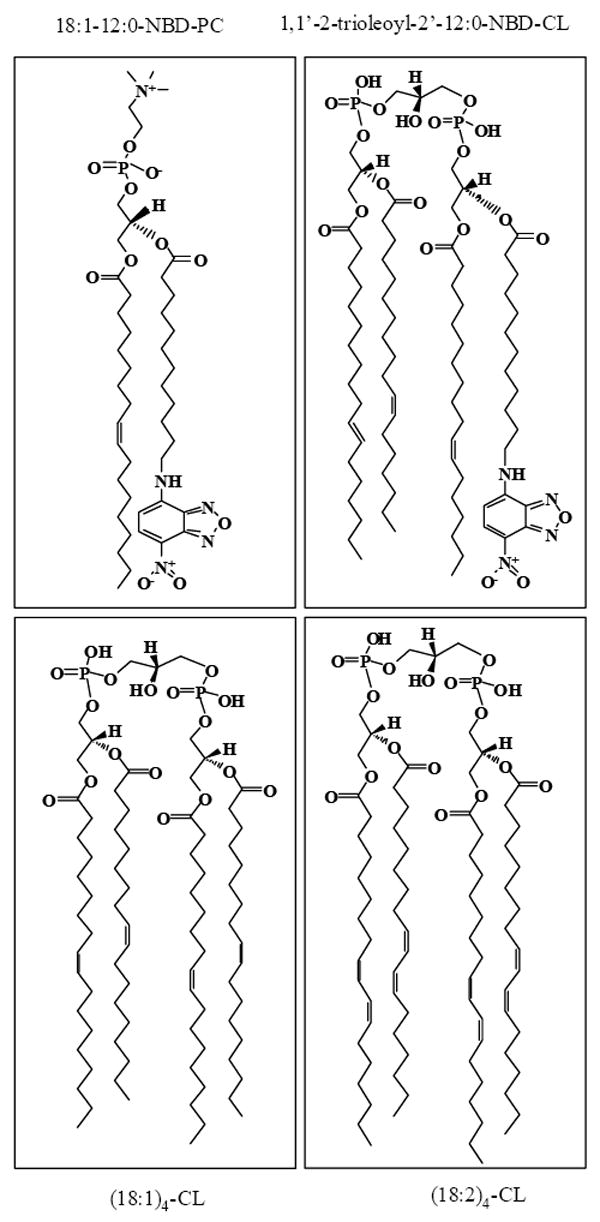
Figure 2. Native agarose gels of cyt c complexes with TOCL and NBD-CL at various anionic phospholipid/cyt c ratios.
Samples of protein mixed with liposomes were loaded onto a 0.8% aragose gel (as indicated on the figure), and the electrophoresis was run in HEPES-imidazole buffer (35 mM HEPES, 43 mM imidazole, pH 7.4). Gels were stained with Coomassie Brilliant Blue R-250. Cyt c, 40 μM; anionic phospholipid/cyt c ratios are indicated. Liposomes were made from PC premixed with equal amounts of TOCL or NBD-CL.
Quenching of NBD-CL fluorescence upon cyt c binding to membranes
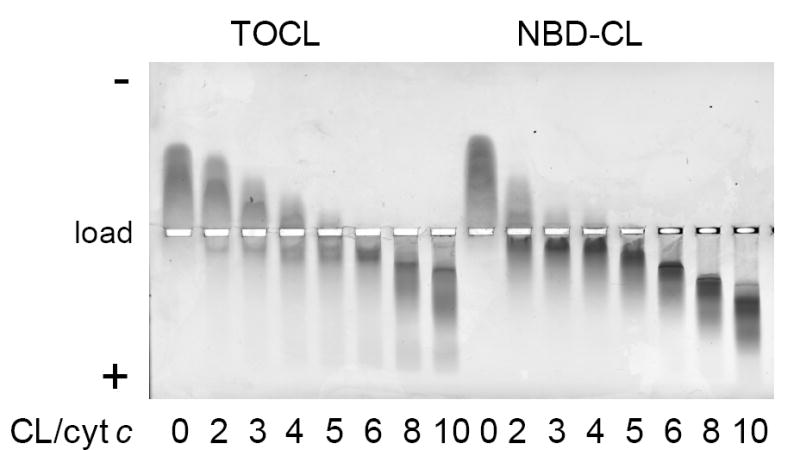
The absorbance spectrum of NBD-labeled CL in TOCL/DOPC liposomes had two characteristic maxima at 338 nm and 468 nm. The fluorescence spectrum had a maximum at 538 nm (Fig. 3A, insert). Fluorescence life time was 5.0 ns as determined by a frequency-domain method [39]. Thus, fluorescence properties of NBD-CL were very close to other NBD-labeled phospholipids [40]. The location of NBD molecule near the phosphoglycerol moiety of lipids in the membrane and the high sensitivity of its fluorescence to the local environment result in fluorescence decay upon protein binding to the membrane [41]. Indeed, instant quenching of NBD-CL fluorescence was observed upon cyt c addition to liposomes (Fig. 3A). To minimize NBD fluorescence self-quenching and direct interaction of NBD-CL with cyt c, only 1 mol% NBD-CL was added to the lipid mixture (compare with 49 mol% of TOCL in CL/PC liposomes). The extent of quenching depended on the cyt c/CL ratio and reached the maximum value of ~80%. NBD-CL was nearly equally distri buted between inner and outer leaflets of the bilayer as it was confirmed by quenching experiments with dithionite [28] (Fig. 3B), therefore a strong quenching effect suggests that cyt c affects NBD fluorescence on both sides of the bilayer. In the outer leaflet fluorescence can be altered due to direct interaction with the protein (via lipid segregation and interaction with amino acid groups). Because NBD fluorescence emission spectrum overlaps with the absorbance spectrum of cyt c heme, an efficient fluorescence resonance energy transfer (FRET) from NBD to heme is anticipated upon protein-phospholipid binding. In the inner leaflet (which is physically inaccessible for cyt c), NBD fluorescence is quenched via FRET mechanism. On the basis of quenching efficiency of NBD-CL in the inner leaflet we approximated the distance of the membrane-bound heme from the center of the bilayer to be in the range of 9-17 A.
Figure 3. Quenching of NBD-CL fluorescence by cyt c (A) and dithionite (B).
A – Dependence of NBD-CL fluorescence on the concentration of cyt c. Incubation time 1 min. Insert – fluorescence emission spectrum of NBD-CL in liposomes.
B – Time course of NBD-CL fluorescence in the presence of dithionite (200 mM). Excitation wavelength was 470 nm. Liposomes were prepared from DOPC/TOCL/NBD-CL at a ratio of 50:49:1 Phospholipid concentration was 20 μM for (A) and 50 μM for (B).
II. Inhibition of cyt c peroxidase activity by NBD-CL
Effect of NBD-CL on etoposide radical generation by cyt c/CL complex
The ability of cyt c to catalyze oxidation of various substrates is markedly augmented upon binding to CL-containing membranes [19-21]. We tested whether NBD-CL could induce the peroxidase activity by analyzing cyt c-dependent production of etoposide phenoxyl radicals, oxidation of Amplex Red, protein aggregation, and generation of CL hydroperoxides.
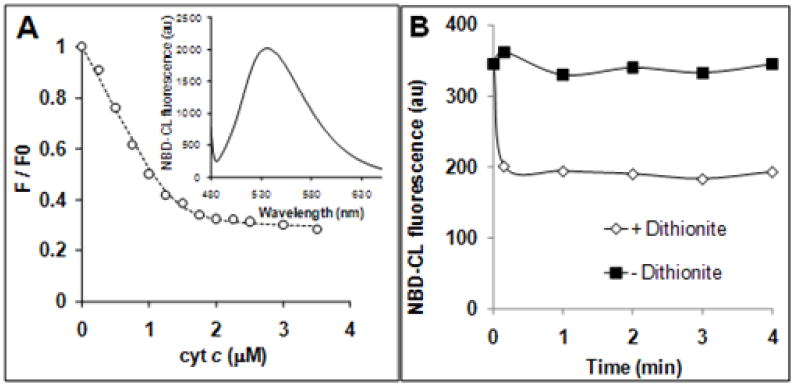
One-electron oxidation of etoposide by peroxidases yields phenoxyl radical intermediates readily detectable by EPR spectroscopy. The radicals were generated by CL-bound (but not by free) cyt c (Fig. 4) – in line with previous demonstrations of the ability of cyt c/CL complex to catalyze H2O2-depended one-electron oxidation of etoposide [21]. Surprisingly, when TOCL was substituted for NBD-CL, etoposide radical production was completely blocked. Similarly, NBD-CL inhibited oxidation and chemiluminescence response from West Femto (ThermoFisher Scientific), a typical substrate of peroxidase reactions catalyzed by cyt c/CL complex (Fig. 4, C).
Figure 4. NBD-CL inhibits generation of etoposide phenoxyl radicals and chemiluminescence substrate oxidation by cyt c.
A – EPR spectrum of etoposide phenoxyl radical generated by cyt c/TOCL complex in the presence of H2O2. B – Relative intensity of EPR signal in the presence of liposomes of different composition: DOPC only, DOPC/TOCL (1:1), DOPC/NBD-CL (1:1). Incubation conditions: phospholipid concentration 1 mM, cyt c 20 μM, etoposide 200 μM, H2O2 100 μM, incubation time 4 min. C – Effect of NBD-CL on oxidation of West Femto sensitive substrate by cyt c/H2O2. Conditions: 5 nM cyt c was mixed with liposomes (DOPC/TOCL 1:1) containing either non-labeled, or labeled TOCL, total volume 100 μL. Reaction was started by addition of substrate solution already containing H2O2.
Effect of NBD-CL on the H2O2-induced oligomerization of cyt c
Formation of protein-centered radicals, likely tyrosyl radicals, has been previously documented in the course of peroxidase reaction of cyt c with H2O2 [42-44], particularly in the complex of cyt c with CL [19, 20]. In the absence of reducing substrates, incubation of cyt c with TOCL-containing liposomes and H2O2 resulted in oligomerization of the protein and decrease of its monomeric form (Fig. 5) due to radical recombination and dityrosine cross-linking [19, 21 22]. In the presence of NBD-CL, the monomeric form of cyt c was preserved (Fig. 5A). The higher molar fraction of NBD-CL was incorporated in the phospholipid vehicles the lower amount of high-molecular weight aggregates was detected by PAGE analysis. Notably, oligomerization of cyt c/TOCL complexes in the presence of H2O2 was inhibited by etoposide proportionally to the amount of this reducing substrate (Fig. 5B). These results suggest that NBD-CL was acting similar to reductants capable of competitive inhibition of oxidative oligomerization of the protein. Of note, NBD-CL was considerably more effective as compared to etoposide. The highest concentration of NBD-CL (25 mol%, 6.25 μM) that caused marked protection against cyt c oligomerization – as evidenced by the amounts of detectable monomeric form of cyt c – was essentially ineffective in the case of etoposide. Even at much higher concentrations – up-to 200 μM – the protective effects of etoposide were less pronounced than those of NBD-CL at 6.25 μM.
Figure 5. NBD-CL and etoposide inhibit H2O2-dependent oligomerization of cyt c.
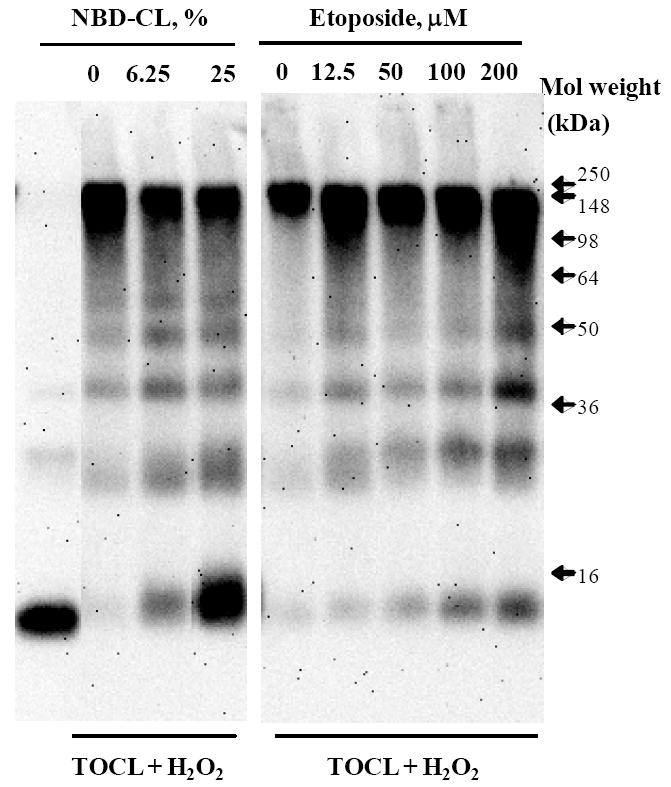
Typical Western blots of cyt c and its oligomeric products after staining with anti-cyt c antibody. Cyt c (1.5 μM) was pre-incubated with liposomes (concentration of phospholipids 25 μM) in HEPES buffer (20 mM, pH 7.4) for 15 min, and then aliquots of H2O2 (at a final concentration of 25 μM) were added every 15 min during 1 h interval at 37°C. Liposomes were prepared from DOPC/TOCL (1:1). Mol % of NBD-CL in liposomes and etoposide concentration were as shown above the gel.
Effect of NBD-CL on the cyt c-catalyzed accumulation of cardiolipin hydroperoxides (CL-OOH)
In the presence of polyunsaturated CL, cyt c catalyzes H2O2-dependent peroxidation of CL with a characteristic pattern of products in ESI mass spectra [19, 33]. As shown on Fig. 6, peroxidation of tetralinoleoyl-CL (TLCL) by cyt c/H2O2 yielded oxidation products, which include mono-, di- and tri-hydroperoxides as well as different hydroxy-derivatives. In the presence of 50 mol % NBD-CL, the production of CL-OOH was inhibited 3-fold; similarly, the content of TLCL oxidation products with several hydroperoxy- and/or hydroxy-groups was markedly decreased (with the exception of CL with mono-hydroxy-group with m/z 731) (Fig. 6). Thus, NBD-CL inhibited cyt c/CL-induced peroxidation of TLCL.
Figure 6. NBD-CL inhibits cyt c-dependent formation of cardiolipin hydroperoxides.
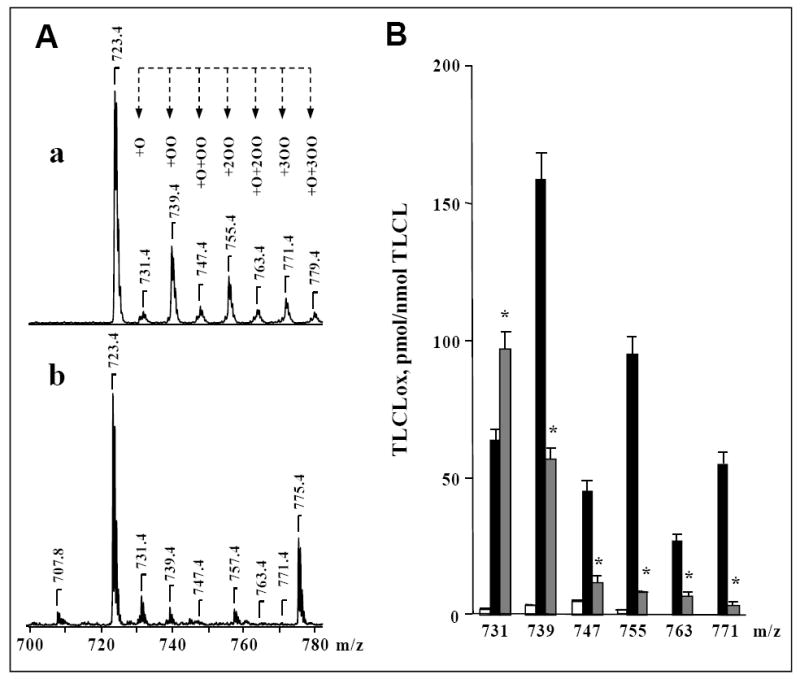
A, a, b – typical mass spectra of TLCL and its H2O2-induced oxidation products formed in the presence of cyt c. Peaks at m/z 723 and 775 correspond to doubly charged TLCL and NBD-CL, respectively. TLCL oxidation products are represented by previously characterized monohydroxy (m/z 731), monohydroperoxy (m/z 739), monohydroxy/monohydroperoxy (m/z 747), dihydroperoxy (m/z 755), monohydroxy/dihydroperoxy (m/z 763), and trihydroperoxy (m/z 771) derivatives of TLCL [33]. Cyt c (5 μM) was incubated with DOPC/TLCL (2:2) liposomes (A, a) or DOPC/TLCL/NBD-CL (2:1:1) liposomes (A, b) (total phospholipids concentration was 500 μM) in the presence of H2O2 (100 μM) for 30 min at 37°C. In the end of incubation, excess of H2O2 was removed by addition of catalase (1.4 U/μL). B – Quantitative analysis of oxygenated molecular species of TLCL. Open bars - control (TLCL/cyt c); filled black bars - after incubation of TLCL/cyt c with H2O2; filled grey bars - after incubation TLCL/cyt c with H2O2 in the presence of NBD-CL. Lipid phosphorus was determined by a micro-method [34]. CL molecular species were quantified by comparing the peak intensities with that of an internal CL standard, which was added to the sample before MS analysis. TMCL was used as an internal standard and TLCL was used as a reference standard [35]. The linear response and detection limit for TLCL were established by injection of calibration mixtures with different concentrations of TLCL and a constant concentration of TMCL. *p<0.05 versus cyt c/H2O2, n=6 independent experiments.
III. Mechanism of cyt c and peroxidase inhibition by NBD-CL
Analysis of the NBD-CL inhibition mechanism of cyt c peroxidase activity by Amplex Red fluorescence assay
We further utilized Amplex Red (N-acetyl-3,7-dihydroxyphenoxazine), as a substrate for the peroxidase activity of cyt c/CL complexes – catalyzing its oxidation to a highly fluorescent resorufin [45] – in the presence and absence of NBD-CL. Expectedly, NBD-CL inhibited oxidation of Amplex Red by cyt c/CL complex. Kinetic studies revealed the decreased maximum reaction rate and apparent affinity of the enzyme for the substrate in the presence of NBD-CL (Fig. 7A,B). The extent of inhibition depended on the molar fraction of NBD-CL in the liposomes; the inhibitory effect was observed even at high Amplex Red concentrations (up to 100 μM). On the basis of Michaelis-Menton formalism, our results can be interpreted as a reversible mixed mechanism of inhibition. Interestingly, C12-NBD-PC (for structure see Fig. 1) also exerted a slight inhibitory effect, which was not observed in the presence of excess Amplex Red (Fig. 7C). As can be seen from a Lineweaver-Burk plot, the affinity for Amplex Red (but not the reaction rate) decreased in the presence of C12-NBD-PC (Fig. 7D). Of note, C6-NBD-PC did not affect the peroxidase activity of cyt c/CL complex (Fig. 7E,F).
Figure 7. Effects of NBD-CL on oxidation of Amplex Red by cyt c/H2O2.
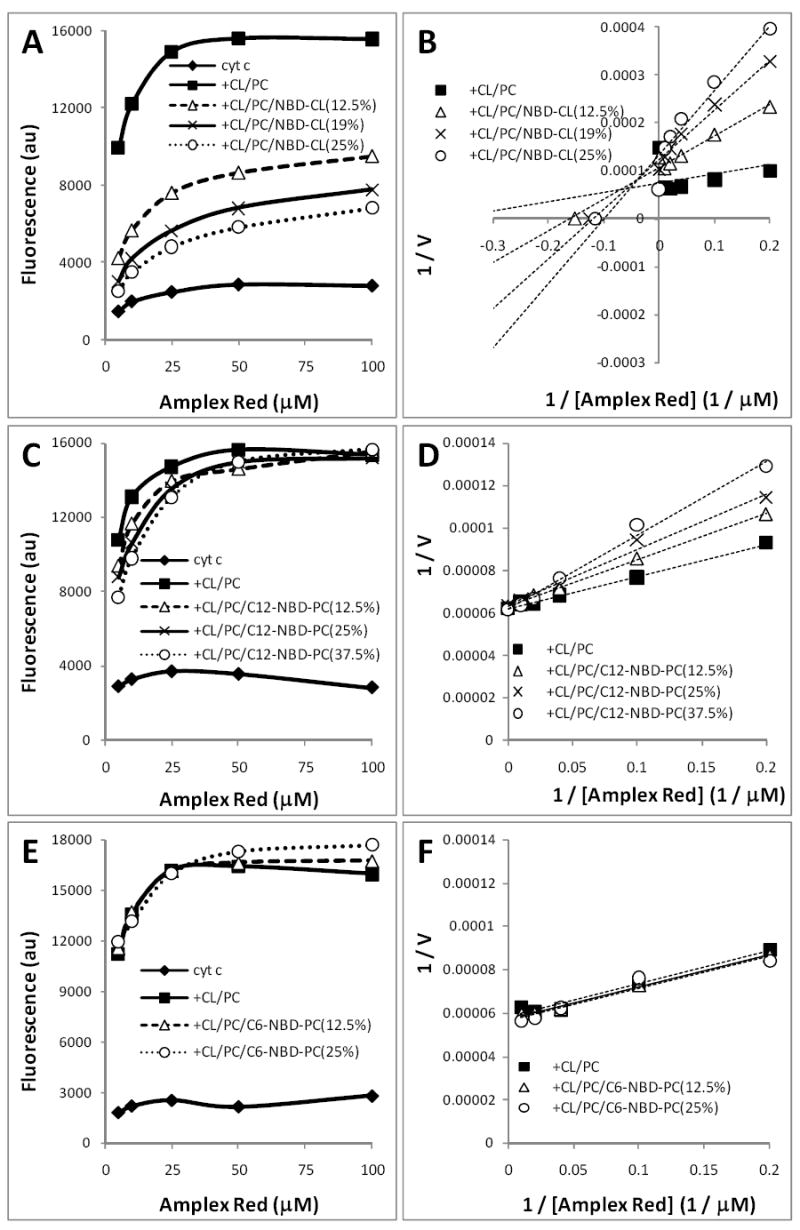
Cyt c (0.5 μM) was pre-incubated for 10 min with DOPC/TOCL liposomes (1:1) at a ratio of 1:25. Mol % of NBD-TOCL, C12-NBD-PC and C6-NBD-PC in liposomes was as indicated. Inhibition of the peroxidase activity of cyt c/CL complexes by NBD-CL (A, B), C12-NBD-PC (C, D) and C6-NBD-PC (E, F) was assessed by fluorescence in 20 mM HEPES buffer (pH 7.4) containing H2O2 (100 μM), DTPA (100 μM), and Amplex Red in the range of 5-100 μM. A, C, E – dependence of the reaction rate on the substrate concentration; B, D, F – Lineweaver-Burk plots for peroxidase activity.
Mass spectrometry of NBD-CL and its products formed by cyt c/TLCL/H2O2
Combined our results suggest that NBD-CL acted as a substrate for the peroxidase activity of cyt c/CL complex. To further confirm this, we performed ESI-MS analysis of NBD-CL incubated with cyt c and TLCL-DOPC (see Fig. 1) liposomes in the presence of H2O2. As shown in Fig. 8 (A) singly charged ions of TLCL and NBD-CL in negative mode were represented by two different molecular clusters with m/z 1447 and 1551 with their Na- adducts at m/z 1469 and 1573, respectively. After the incubation, in addition to a signal corresponding to trioleoyl-C12-NBD-CL (m/z 1551), a prominent signal with m/z 1388 appeared in the spectrum (Fig. 8B, b) consistent with [M – trioleoyl-C12-NH2-CL- H]-. Another peak at m/z 1410 represented its Na-adduct. The identity of an ion at m/z 1388 was unambiguously supported by the structural analysis using CID tandem MS, which revealed fragments of glycerol backbone plus PO4 (m/z 152), oleic acid (m/z 281) and aminomyristoic acid (m/z 214); neither NBD nor NBD-myristoic acid fragments were detected. ESI-MS analysis of NBD-CL before and after its incubation with cyt c/H2O2 confirmed the formation of NBD-derivative with m/z 180 (Fig 8B, a). In addition, we observed trace amounts of an ion with m/z 1405, probably corresponding to a hydroxy-derivative of the compound with m/z 1388 (trioleoyl-C12-NHOH-CL). Quantitative analysis showed a drastic decomposition of molecular ion of NBD-CL at m/z 1551 after incubation of liposomes with cyt c/H2O2 (Fig. 8C, a).
Figure 8. Mass spectrometry and fluorescence analysis of NBD-CL and its oxidation products produced by cyt c/CL complex.
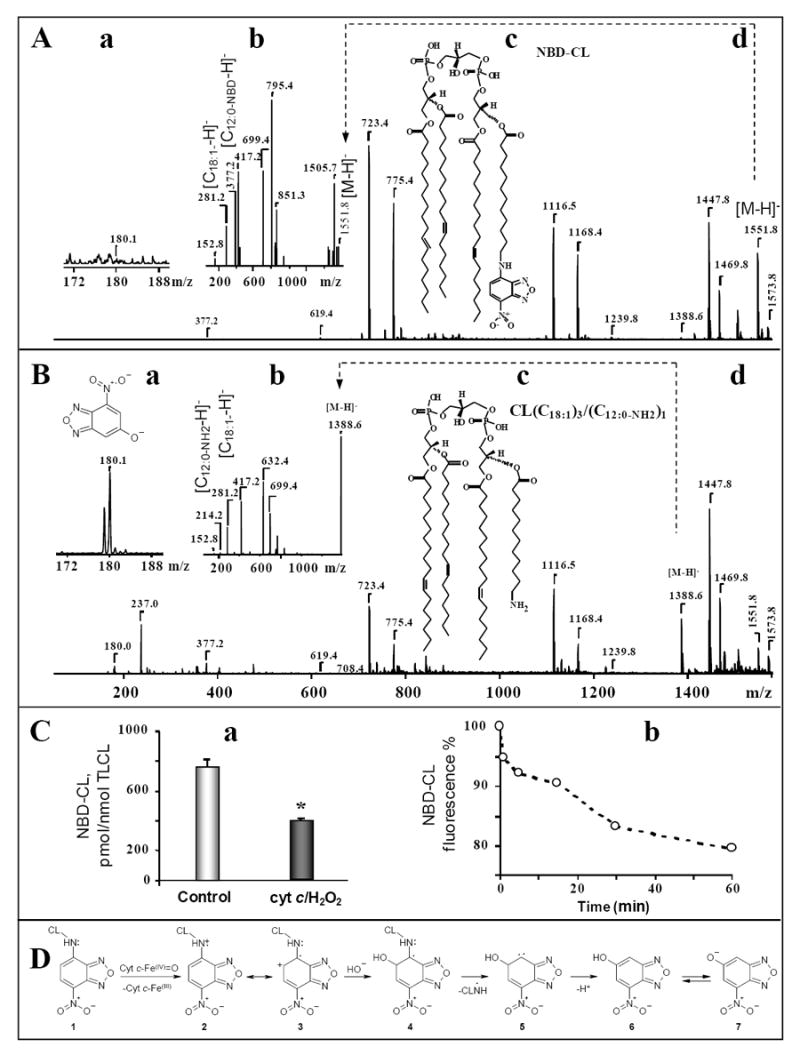
A, d, B, d,– Typical mass spectra of TLCL, NBD-CL, and its H2O2-induced oxidation product in the presence of cyt c were acquired in negative ion mode using full-range zoom (m/z 50-1600) scans. Typical MS/MS fragmentation profiles of singly charged molecular species of NBD-CL before and after its incubation with cyt c/H2O2 are presented on panels A, b, B, b, respectively. Panels A, c and B, c show chemical structures of parent ions that were fragmented. Panels A, a, B, a, show the formation of NBD-CL degradation fragment with m/z 180 (insert) and its structure after incubation of liposomes with cyt c/H2O2.
C, a – Quantitative analysis of NBD-CL decomposition. Cyt c (5 μM) was incubated with DOPC/TLCL/NBD-CL (2:1:1) liposomes (total concentration of phospholipids was 500 μM) in the presence of H2O2 (100 μM) for 30 min. The concentration of NBD-CL in the samples was confirmed in independent experiments which included ESI-MS protocol and lipid phosphorus determination. TMCL was used as internal standard and NBD-CL was used as a reference standard. *p<0.05 versus control, n=6 independent experiments.
C, b – Typical time course of NBD-CL fluorescence decay induced by cyt c via peroxidase mechanism. Cyt c (1 μM) was pre-incubated for 10 min with DOPC/TOCL/NBD-CL liposomes (50:49:1) at the cyt c/CL ratio of 1:33. NBD-CL fluorescence was measured during 1 h incubation with H2O2 (50 μM).
D - Schema of reaction intermediates illustrating the proposed mechanism of NBD-CL degradation by the peroxidase activity of cyt c in the presence of H2O2.
H2O2-dependent decay of NBD-CL fluorescence in the presence of cyt c
In addition to instant fluorescence quenching upon binding of cyt c to NBD-CL/CL-containing liposomes, further fluorescence decay was induced by H2O2 (Fig. 8C, b). The time-course of H2O2-dependent fluorescence decay was characterized by a small and rapid initial drop followed by a more pronounced and slower fluorescence reduction (Fig. 8C, b). The effect of H2O2 was universally observed within a broad range of cyt c concentrations: from a relatively low protein content (cyt c/CL ratio of 1:100, when binding was associated with only 2% of initial fluorescence quenching), to a high protein level (cyt c/CL ratio of 1:4, when cyt c binding sites were completely saturated and caused 75% quenching of initial fluorescence) (data not shown).
IV. Incorporation of NBD-CL in mitochondria and assessment of mitochondrial peroxidase activity
Incubation of NBD-CL with mitochondria was associated with the increase of its fluorescence. Quantitative assessments showed that incorporation of NBD-CL into mitochondria was concentration-dependent (data not shown). At 20 and 200 μM NBD-CL, around 0.5 and 8 nmoles NBD-CL/mg protein (8 and 12% of total, respectively) were integrated. Using these two concentrations of NBD-CL, we assessed its effects on peroxidase activity of mitochondria induced by tert-butyl hydroperoxide (tBOOH). We employed measurements of fluorescence of resorufin produced upon oxidation of Amplex Red. Resorufin formation was only detected in the presence of both tBOOH and mitochondria (Fig. 9A). NBD-CL suppressed peroxidase activity and the inhibitory effect depended on the amount of NBD-CL incorporated in mitochondria (Fig. 9B). Similar results were obtained using mitoplasts (data not shown).
Figure 9. Effect of NBD-CL on peroxidase activity of mouse liver mitochondria.
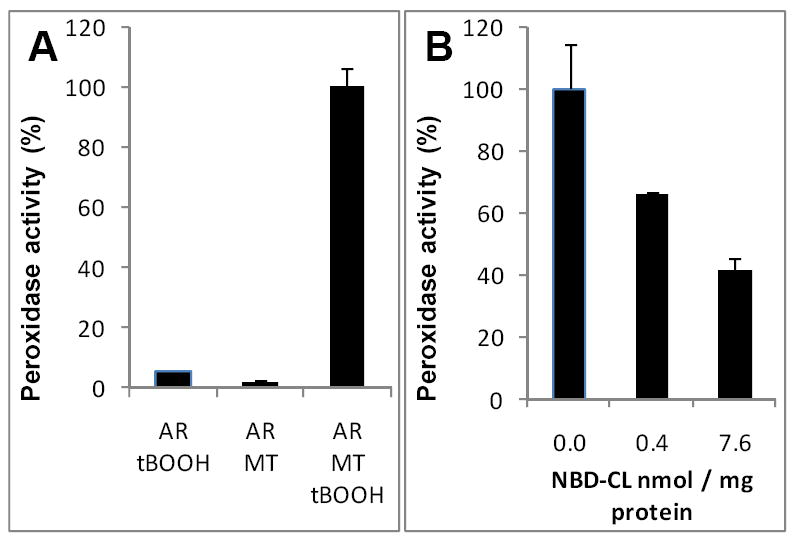
A,B – mitochondrial peroxidase activity assayed with Amplex Red (AR) as a substrate. Incubation conditions: mitochondria (MT) (0.25 mg protein/mL), AR (50 μM), tBOOH (2 mM), HEPES (25 mM, pH 7.4), incubation time 10 min.
DISCUSSION
Mitochondria are central to the execution of intrinsic apoptosis triggered by different damaging stimuli [46]. Mitochondrial generation of ROS is important in this cell death pathway and manipulations of ROS production were shown to cause changes in the sensitivity of cells to apoptosis [10, 47-49]. Recently, we demonstrated that ROS can act via a cyt c–dependent mechanism whereby cyt c binds to mitochondrial CL, adopts a new function of a peroxidase and catalyzes H2O2-dependent oxidation of CL [19]. CL oxidation products were shown essential for the mitochondrial permeability transition and release of pro-apoptotic factors from mitochondria [23, 25, 26]. This implies that inhibition of CL oxidation may be a promising approach to regulate apoptosis. Indeed, we reported that prevention of the production of H2O2 – feeding the peroxidase cycle of cyt c/CL complexes – by mitochondria-targeted conjugates of hemigramicidin S with nitroxides inhibited CL oxidation and protected cells against apoptosis induced by different agents both in cultured cells and in vivo [15, 27]. We further demonstrated that mitochondria-targeted donors of NO• activated by the peroxidase function of cyt c/CL complexes were also capable of preventing CL oxidation and decreasing sensitivity of cells to apoptosis [50]. Here, we report yet another approach to inhibit CL peroxidation based on the utilization of modified CLs with functional moieties capable of “consuming” the high oxidizing potential of the peroxidase complex thus “distracting” it from peroxidation of polyunsaturated CLs. We designed such an inhibitor on the basis of two components – non-oxidizable tri-oleoyl-CL and oxidizable and fluorescent 7-nitro-2-1,3-benzoxadiazol (NBD).
Using tri-oleoyl cardiolipin with C12-NBD as the forth acyl chain (NBD-CL), we assessed the binding of cyt c to the modified CL. Native gel electrophoresis of mixtures of cyt c with various amounts of DOPC/NBD-CL liposomes clearly demonstrated that NBD-labeled CL can successfully compete with unlabeled CL for cyt c. Analysis of cyt c effect on fluorescence of NBD-CL integrated into NBD-CL/TOCL/DOPC liposomes revealed an unusually strong quenching, which plateaued at ~80% upon gradual addition of cyt c. The amount of cyt c required for the maximal fluorescence quenching was 16 times lower than the amount of phospholipid available for the protein binding in the outer leaflet. This again points to very avid cyt c/NBD-CL interactions. Although the complexity of mechanisms of NBD fluorescence quenching by cyt c does not permit unambiguous estimation of cyt c/NBD-CL affinity constant, our data indicate that the lower limit for the binding constant of cyt c/CL is 5×108 M-1.
A rough approximation of Forster radius for NBD-CL and heme yields values in the range from 28 to 36 A° that is shorter than the distance between NBD moieties located in the outer and inner leaflet (>38A°). These R0 values indicate that quenching efficiency of 80% could be achieved only if (1) fluorescence of NBD-CL located in the outer membrane leaflet was completely quenched (by ~100%) at the saturated level of binding, that (2) fluorescence of inner leaflet NBD-CL was quenched by ~60% and that (3) this NBD has to be at least within ~20A° from the heme. Thus, heme has to be located within the membrane between C7 atom of the acyl chain and the glycerol moiety of phospholipids. These data support the view that cyt c undergoes partial unfolding upon binding to the membrane whereby the heme (or at least its edge) is inserted in the lipid bilayer. Moreover, this data indicate that the NBD moiety may physically interact with the protein and, most importantly, with the heme.
Analysis of the peroxidase activity of cyt c/CL complex demonstrated that NBD-CL was able to inhibit oxidation of prototypical substrates - etoposide and Amplex Red - to an extent that no oxidation was observed upon complete substitution of TOCL for NBD-CL. H2O2-induced protein oligomerization via recombination of protein-centered radicals – likely tyrosyl radicals – was also markedly and competitively inhibited by both etoposide and NBD-CL. This suggests that, similar to etoposide, NBD-CL acted as a substrate for the peroxidase activity of cyt c/NBD-CL complexes. Kinetic analysis of Amplex Red oxidation revealed that NBD-CL’s inhibitory effects were due to both competitive and noncompetitive mechanisms.
Finally, NBD-CL was able to block cyt c-dependent peroxidation of TLCL. Thus NBD-CL formed a strong complex with cyt c, which, however, had no oxidative potency towards endogenous polyunsaturated CLs. Importantly, NBD-CL was by far a more potent inhibitor of the peroxidase activity than C12-NBD-PC, thus clearly demonstrating selectivity of NBD-CL. Given that C6-NBD-PC had no effect on the peroxidase activity of cyt c/CL complex at all, it is tempting to speculate that the presence of a long acyl chain is required for the positioning of NBD moiety within the protein environment and for its interaction with active site.
Additional insights into the mechanisms underlying the inhibition of cyt c/CL peroxidase activity by NBD-CL were gained from spectral analysis of NBD moiety. H2O2-induced decomposition of NBD manifested itself by fluorescence decay in the presence of cyt c/CL complex. Moreover, MS data revealed that NBD-CL was predominantly converted into a product lacking NBD moiety thus suggesting that cleavage of NBD occurred at the amine-benzene bond. The amino group participates in the formation of the intramolecular charge transfer state responsible for the fluorescence of NBD. Thus, the loss of amine by NBD should lead to the fluorescence loss as observed in our experiments.
Four major mechanisms of NBD metabolism by cytochromes P450 and peroxidases have been proposed including heteroatom oxidation, carbon hydroxylation, reductive ring opening and reduction of nitro group. Heteroatom oxidation is one of the most widespread reactions catalyzed by oxoferryl species of both cytochromes P450 and peroxidases. Various peroxidases including horseradish peroxidase, cytochrome c peroxidase, lactoperoxidase and prostaglandin synthetase are able to catalyze oxidation of primary, secondary and tertiary aromatic and heterocyclic amines [51-55]. These reactions are usually initiated by electron transfer from a nitrogen atom to reactive heme intermediate and result in N-dealkylation of amines, radical dimerization or in the adduct formation [52, 54, 55]. Generation of nitrogen-centered cation radical, the first intermediate of amine oxidation, has been reported during peroxidase-dependent oxidation of several aromatic amines [52, 53]. Similarly, P450-catalyzed oxidation of nitrogen atoms proceeds via electron abstraction from nitrogen, cation radical formation and leads to N-dealkylation (Reviewed in [56]). Hydrocarbon hydroxylation involves oxygen incorporation at the carbon atoms of the benzene ring or at the adjacent carbon atom to the amino group. This classical for P450 enzymes reaction has been suggested to be catalyzed by oxoferryl species [57, 58]. NBD contains oxadiazole ring, which may undergo a reductive N-O bond cleavage to generate the ring-opened products as has been reported for oxadiazoles and their close analogue, isoxazole [59, 60]. In addition, NBD’s nitro group, which is essential for fluorescence properties of the molecule, could be reduced by P450 enzymes to amine [61]. The latter mechanism is the least likely to occur, since there were no efficient electron donors for nitro-reduction (like NADPH) in our enzymatic system. Hydroxylation of carbon atoms in the benzene ring has not been observed by MS-analysis after cyt c/H2O2-dependent NBD-CL oxidation. Instead, MS data revealed cleavage of the bond between the benzene ring and amine. This suggests that secondary amine that links acyl chain and NBD, is a likely oxidation site. The only difference with a common heteroatom oxidation mechanism is that NBD-CL oxidation resulted in the bond cleavage between nitrogen and the ring system rather than in N-dealkylation. This can be explained, at least in part, by a reported lower probability of N-dealkylation with increase of acyl chain length (i.e., N-demethylation is favored over N-deethylation by factor of 16) [62]; in our case, the acyl moiety was represented by C12 chain. Overall, the proposed mechanism for the regio-specific oxidation of NBD-moiety in the modified CL is illustrated by the schema (Fig. 8, D).
Finally, we demonstrated that NBD-CL is incorporated into isolated mitochondria, where it inhibits tBuOOH-dependent peroxidase activity towards Amplex Red. Most of current approaches aimed at protection of mitochondria against oxidative insults are based on the use of conjugates with mitochondria-targeted moieties (e.g., tri-phenyl-phosphonium) and active cargoes represented by antioxidants – spin traps and nitroxides, quinones, vitamins, peptides [9-11, 13-16]. A combination of non-oxidizable tri-oleoyl moiety with C12-NBD fragment within one CL molecule is a promising novel approach to create inhibitors of peroxidase activity of cyt c/CL complexes by exploiting high affinity binding of NBD-CL to cyt c and regio-specific catabolism of the NBD-moiety. Thus, NBD-CL functions not as a random chain-breaking lipid antioxidant/free radical scavenger, but specifically inhibits enzymatic activity of cyt c involved in the execution of apoptotic program in mitochondria. By effectively preventing peroxidation of endogenous polyunsaturated molecular species of CL, NBD-CL may act as a promising regulator of apoptosis.
Acknowledgments
This work was supported by NIH grants NIAID U19AI068021, HL70755 and R03TW007320, by Pennsylvania Department of Health SAP 4100027294 and by Human Frontier Science Program.
Abbreviations
- cyt c
cytochrome c
- HRP
horse radish peroxidase
- NBD
7-nitro-2-1,3-benzoxadiazol
- CL
cardiolipin
- (CL-OOH)
cardiolipin hydroperoxides
- NBD-CL
1,1’,2-trioleoyl-2’-[12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-cardiolipin
- C12-NBD-PC
1-oleoyl-2-[12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-sn-glycero-3-phosphocholine
- C6-NBD-PC
1-oleoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl]-sn-glycero-3-phosphocholine
- TLCL
1,1’,2,2’-tetralinoleoyl-cardiolipin
- TOCL
1,1’,2,2’-tetraoleoyl-cardiolipin
- TMCL
1,1’,2,2’-tetramyristoyl-cardiolipin
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPS
1,2-dioleoyl-sn-glycero-3-phospho-L-serine
- tBOOH
tert-butyl hydroperoxide
References
- 1.Graham SH, Chen J. Programmed Cell Death in Cerebral Ischemia. J Cereb Blood Flow Metab. 2001;21:99–109. doi: 10.1097/00004647-200102000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Fei P, El-Deiry WS. P53 and radiation responses. Oncogene. 2003;22:5774–5783. doi: 10.1038/sj.onc.1206677. [DOI] [PubMed] [Google Scholar]
- 3.Chowdhry MF, Vohra HA, Galiñanes M. Diabetes increases apoptosis and necrosis in both ischemic and nonischemic human myocardium: role of caspases and poly-adenosine diphosphate-ribose polymerase. J Thorac Cardiovasc Surg. 2007;134:124–131. doi: 10.1016/j.jtcvs.2006.12.059. [DOI] [PubMed] [Google Scholar]
- 4.Liadis N, Murakami K, Eweida M, Elford AR, Sheu L, Gaisano HY, Hakem R, Ohashi PS, Woo M. Caspase-3-dependent beta-cell apoptosis in the initiation of autoimmune diabetes mellitus. Mol Cell Biol. 2005;25:3620–3629. doi: 10.1128/MCB.25.9.3620-3629.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Narula J, Haider N, Arbustini E, Chandrashekhar Y. Mechanisms of Disease: apoptosis in heart failure—seeing hope in death. Nat Clin Pract Cardiovasc Med. 2006;3:681–688. doi: 10.1038/ncpcardio0710. [DOI] [PubMed] [Google Scholar]
- 6.Geng Y-J. Molecular mechanisms for cardiovascular stem cell apoptosis and growth in the hearts with atherosclerotic coronary disease and ischemic heart failure. Ann N Y Acad Sci. 2003;1010:687–697. doi: 10.1196/annals.1299.126. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y, Liu W, McPhie D, Hassinger L, Neve R. APP-BP1 mediates APP-induced apoptosis and DNA synthesis and is increased in Alzheimer’s disease brain. J Cell Biol. 2003;163:27–33. doi: 10.1083/jcb.200304003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pompl P, Yemul S, Xiang Z, Ho L, Haroutunian V, Purohit D, Mohs R, Pasinetti GM. Caspase gene expression in the brain as a function of the clinical progression of Alzheimer disease. Arch Neurol. 2003;60:369–376. doi: 10.1001/archneur.60.3.369. [DOI] [PubMed] [Google Scholar]
- 9.Smith RA, Porteous CM, Coulter CV, Murphy MP. Selective targeting of an antioxidant to mitochondria. Eur J Biochem. 1999;263:709–716. doi: 10.1046/j.1432-1327.1999.00543.x. [DOI] [PubMed] [Google Scholar]
- 10.Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, Smith RA, Murphy MP. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- 11.Murphy MP, Echtay KS, Blaikie FH, Asin-Cayuela J, Cocheme HM, Green K, Buckingham JA, Taylor ER, Hurrell F, Hughes G, Miwa S, Cooper CE, Svistunenko DA, Smith RA, Brand MD. Superoxide activates uncoupling proteins by generating carbon-centered radicals and initiating lipid peroxidation: studies using a mitochondria-targeted spin trap derived from a-phenyl-N-tert-butylnitrone. J Biol Chem. 2003;278:48534–48545. doi: 10.1074/jbc.M308529200. [DOI] [PubMed] [Google Scholar]
- 12.Filipovska A, Kelso GF, Brown SE, Beer SM, Smith RA, Murphy MP. Synthesis and characterization of a triphenylphosphoniumconjugated peroxidase mimetic: insights into the interaction of ebselen with mitochondria. J Biol Chem. 2005;280:24113–24126. doi: 10.1074/jbc.M501148200. [DOI] [PubMed] [Google Scholar]
- 13.Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 14.Sheu SS, Nauduri D, Anders MW. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim Biophys Acta. 2006;1762:256–265. doi: 10.1016/j.bbadis.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 15.Wipf P, Xiao J, Jiang J, Belikova NA, Tyurin VA, Fink MP, Kagan VE. Mitochondrial targeting of selective electron scavengers: synthesis and biological analysis of hemigramicidin-TEMPO conjugates. J Am Chem Soc. 2005;127:12460–12461. doi: 10.1021/ja053679l. [DOI] [PubMed] [Google Scholar]
- 16.Skulachev VP. A biochemical approach to the problem of aging: “megaproject” on membrane-penetrating ions. The first results and prospects. Biochemistry (Mosc) 2007;72:1385–96. doi: 10.1134/s0006297907120139. [DOI] [PubMed] [Google Scholar]
- 17.Radi R, Bush KM, Freeman BA. The role of cytochrome c and mitochondrial catalase in hydroperoxide-induced heart mitochondrial lipid peroxidation. Arch Biochem Biophys. 1993;300:409–15. doi: 10.1006/abbi.1993.1055. [DOI] [PubMed] [Google Scholar]
- 18.Radi R, Sims S, Cassina A, Turrens JF. Roles of catalase and cytochrome c in hydroperoxide-dependent lipid peroxidation and chemiluminescence in rat heart and kidney mitochondria. Free Radic Biol Med. 1993;15:653–9. doi: 10.1016/0891-5849(93)90169-u. [DOI] [PubMed] [Google Scholar]
- 19.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 20.Kagan VE, Borisenko GG, Tyurina YY, Tyurin VA, Jiang J, Potapovich AI, Kini V, Amoscato A. Oxidative lipidomics of apoptosis: redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Rad Biol Med. 2004;37:1963–1985. doi: 10.1016/j.freeradbiomed.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 21.Tyurina YY, Kini V, Tyurin VA, Vlasova II, Jiang J, Kapralov AA, Belikova NA, Yalowich JC, Kurnikov IV, Kagan VE. Mechanisms of Cardiolipin Oxidation by Cytochrome c: Relevance to Pro- and Antiapoptotic Functions of Etoposide. Mol Pharmacol. 2006;70:706–717. doi: 10.1124/mol.106.022731. [DOI] [PubMed] [Google Scholar]
- 22.Kapralov AA, Kurnikov IV, Vlasova II, Belikova NA, Tyurin VA, Basova LV, Zhao Q, Tyurina YY, Jiang J, Bayir H, Vladimirov YA, Kagan VE. The Hierarchy of Structural Transitions Induced in Cytochrome c by Anionic Phospholipids Determines Its Peroxidase Activation and Selective Peroxidation during Apoptosis in Cells. Biochemistry. 2007;46:14232–14244. doi: 10.1021/bi701237b. [DOI] [PubMed] [Google Scholar]
- 23.Kowaltowski AJ, Castilho RF, Grijalba MT, Bechara EJ, Vercesi AE. Effect of Inorganic Phosphate Concentration on the Nature of Inner Mitochondrial Membrane Alterations Mediated by Ca2+ Ions. J Biol Chem. 1996;271:2929–2934. doi: 10.1074/jbc.271.6.2929. [DOI] [PubMed] [Google Scholar]
- 24.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci U S A. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petrosillo G, Ruggiero FM, Pistolese M, Paradies G. Ca2+-induced Reactive Oxygen Species Production Promotes Cytochrome c Release from Rat Liver Mitochondria via Mitochondrial Permeability Transition (MPT)-dependent and MPT-independent Mechanisms. J Biol Chem. 2004;279:53103–53108. doi: 10.1074/jbc.M407500200. [DOI] [PubMed] [Google Scholar]
- 26.Petrosillo G, Casanova G, Matera M, Ruggiero FM, Paradies G. Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: Induction of permeability transition and cytochrome c release. FEBS Lett. 2006;580:6311–6316. doi: 10.1016/j.febslet.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 27.Fink MP, Macias CA, Xiao J, Tyurina YY, Jiang J, Belikova N, Delude RL, Greenberger JS, Kagan VE, Wipf P. Hemigramicidin-TEMPO conjugates: novel mitochondria-targeted anti-oxidants. Biochem Pharmacol. 2007;74:801–9. doi: 10.1016/j.bcp.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 28.McIntyre JC, Sleight RG. Fluorescence assay for phospholipid membrane asymmetry. Biochemistry. 1991;30:11819–27. doi: 10.1021/bi00115a012. [DOI] [PubMed] [Google Scholar]
- 29.Schnaitman C, Greenawalt JW. Enzymatic properties of the inner and outer membranes of rat liver mitochondria. J Cell Biol. 1968;38:158–75. doi: 10.1083/jcb.38.1.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krebs JJ, Hauser H, Carafoli E. Asymmetric distribution of phospholipids in the inner membrane of beef heart mitochondria. J Biol Chem. 1979;254:5308–16. [PubMed] [Google Scholar]
- 31.Gnaiger E, Kuznetsov AV. Mitochondrial respiration at low levels of oxygen and cytochrome c. Biochem Soc Trans. 2002;30:252–8. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 32.Folch J, Lees M, Sloane-Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 33.Tyurina YY, Tyurin VA, Epperly MW, Greenberger JS, Kagan VE. Oxidative lipidomics of gamma-irradiation-induced intestinal injury. Free Radic Biol Med. 2008;44:299–314. doi: 10.1016/j.freeradbiomed.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 34.Böttcher CSF, Van Gent CM, Fries C. A rapid and sensitive sub-micro phosphorus determination. Anal Chim Acta. 1961;24:203–204. [Google Scholar]
- 35.Valianpour F, Wanders RJA, Barth PG, Overmars H, van Gennip AH. Quantitative and Compositional Study of Cardiolipin in Platelets by Electrospray Ionization Mass Spectrometry: Application for the Identification of Barth Syndrome Patients. Clin Chem. 2002;48:1390–1397. [PubMed] [Google Scholar]
- 36.Koivusalo M, Haimi P, Heikinheimo L, Kostiainen R, Somerharju P. Quantitative determination of phospholipid compositions by ESI-MS: effects of acyl chain length, unsaturation, and lipid concentration on instrument response. J Lipid Res. 2001;42:663–672. [PubMed] [Google Scholar]
- 37.Tyurin VA, Tyurina YY, Osipov AN, Belikova NA, Basova LV, Kapralov AA, Bayir H, Kagan VE. Interactions of cardiolipin and lyso-cardiolipins with cytochrome c and tBid: conflict or assistance in apoptosis. cell Death Diff. 2007;14:872–875. doi: 10.1038/sj.cdd.4402068. [DOI] [PubMed] [Google Scholar]
- 38.Haines TH, Dencher NA. Cardiolipin: a proton trap for oxidative phosphorylation. FEBS Lett. 2002;528:35–39. doi: 10.1016/s0014-5793(02)03292-1. [DOI] [PubMed] [Google Scholar]
- 39.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2. Kluwer Academic/Plenum Publishers; New York: 1999. pp. 142–162. [Google Scholar]
- 40.Mazeres S, Schram V, Tocanne J-F, Lopez A. 7-Nitrobenz-2-oxa-1,3-diazole-4-yI-Labeled Phospholipids in Lipid Membranes: Differences in Fluorescence Behavior. Biophys J. 1996;71:327–335. doi: 10.1016/S0006-3495(96)79228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borisenko GG, Iverson SL, Ahlberg S, Kagan VE, Fadeel B. Milk fat globule epidermal growth factor 8 (MFG-E8) binds to oxidized phosphatidylserine: implications for macrophage clearance of apoptotic cells. cell Death Diff. 2004;11:943–945. doi: 10.1038/sj.cdd.4401421. [DOI] [PubMed] [Google Scholar]
- 42.Barr DP, Gunther MR, Deterding LJ, Tomer KB, Mason RP. ESR Spin-trapping of a Protein-derived Tyrosyl Radical from the Reaction of Cytochrome c with Hydrogen Peroxide. J Biol Chem. 1996;271:15498–15503. doi: 10.1074/jbc.271.26.15498. [DOI] [PubMed] [Google Scholar]
- 43.Chen YR, Deterding LJ, Sturgeon BE, Tomer KB, Mason RP. Protein Oxidation of Cytochrome c by Reactive Halogen Species Enhances Its Peroxidase Activity. J Biol Chem. 2002;277:29781–29791. doi: 10.1074/jbc.M200709200. [DOI] [PubMed] [Google Scholar]
- 44.Lawrence A, Jones CM, Wardman P, Burkitt MJ. Evidence for the role of a peroxidase compound I-type intermediate in the oxidation of glutathione, NADH, ascorbate, and dichlorofluorescin by cytochrome c/H2O2. Implications for oxidative stress during apoptosis. J Biol chem. 2003;278:29410–29419. doi: 10.1074/jbc.M300054200. [DOI] [PubMed] [Google Scholar]
- 45.Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem. 1997;253:162–8. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]
- 46.Danial NN, Korsmeyer SJ. Cell Death: Critical Control Points. cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 47.Jacobson MD, Raff MC. Programmed cell death and Bcl-2 protection in very low oxygen. Nature. 1995;374:814–816. doi: 10.1038/374814a0. [DOI] [PubMed] [Google Scholar]
- 48.Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J. 2000;351:183–193. doi: 10.1042/0264-6021:3510183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.James AM, Cocheme HM, Smith RA, Murphy MP. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J Biol Chem. 2005;280:21295–21312. doi: 10.1074/jbc.M501527200. [DOI] [PubMed] [Google Scholar]
- 50.Stoyanovsky DA, Vlasova II, Belikova NA, Kapralov A, Tyurin V, Greenberger JS, Kagan VE. Activation of NO donors in mitochondria: peroxidase metabolism of (2-hydroxyamino-vinyl)-triphenyl-phosphonium by cytochrome c releases NO and protects cells against apoptosis. FEBS Lett. 2008;582:725–728. doi: 10.1016/j.febslet.2008.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sivarajah K, Lasker JM, Eling TE, Abou-Donia MB. Metabolism of N-alkyl compounds during the biosynthesis of prostaglandins. N-Dealkylation during prostaglandin biosynthesis. Mol Pharmacol. 1982;21:133–41. [PubMed] [Google Scholar]
- 52.Van der Zee J, Duling DR, Mason RP, Eling TE. The oxidation of N-substituted aromatic amines by horseradish peroxidase. J Biol Chem. 1989;264:19828–19836. [PubMed] [Google Scholar]
- 53.Roe JA, Goodin DB. Enhanced oxidation of aniline derivatives by two mutants of cytochrome c peroxidase at tryptophan 51. J Biol Chem. 1993;268:20037–20045. [PubMed] [Google Scholar]
- 54.Gorlewska-Roberts KM, Teitel CH, Lay JO, Jr, Roberts DW, Kadlubar FF. Lactoperoxidase-Catalyzed Activation of Carcinogenic Aromatic and Heterocyclic Amines. chem Res Toxicol. 2004;17:1659–1666. doi: 10.1021/tx049787n. [DOI] [PubMed] [Google Scholar]
- 55.Chiavarino B, Cipollini R, Crestoni ME, Fornarini S, Lanucara F, Lapi A. Probing the Compound I-like reactivity of a bare high-valent oxo iron porphyrin complex: the oxidation of tertiary amines. J Am Chem Soc. 2008;130:3208–3217. doi: 10.1021/ja077286t. [DOI] [PubMed] [Google Scholar]
- 56.De Montellano PR, De Voss JJ. Oxidizing species in the mechanism of cytochrome P450. Nat Prod Rep. 2002;19:477–493. doi: 10.1039/b101297p. [DOI] [PubMed] [Google Scholar]
- 57.Ogliaro F, de Visser SP, Cohen S, Kaneti J, Shaik S. The experimentally elusive oxidant of cytochrome P450: a theoretical “trapping” defining more closely the “real” species. chembiochem. 2001;11:848–851. doi: 10.1002/1439-7633(20011105)2:11<848::AID-CBIC848>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 58.De Visser SP, Shaik S. A proton-shuttle mechanism mediated by the porphyrin in benzene hydroxylation by cytochrome p450 enzymes. J Am Chem Soc. 2003;125:7413–24. doi: 10.1021/ja034142f. [DOI] [PubMed] [Google Scholar]
- 59.Dalvie DK, Kalgutkar AS, Khojasteh-Bakht SC, Obach RS, O’Donnell JP. Biotransformation reactions of five-membered aromatic heterocyclic rings. chem Res Toxicol. 2002;15:269–299. doi: 10.1021/tx015574b. [DOI] [PubMed] [Google Scholar]
- 60.Isin EM, Guengerich FP. Complex reactions catalyzed by cytochrome P450 enzymes. Biochim Biophys Acta. 2007;1770:314–329. doi: 10.1016/j.bbagen.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 61.Silvers KJ, Eddy EP, McCoy EC, Rosenkranz HS, Howard PC. Pathways for the Mutagenesis of 1-Nitropyrene and Dinitropyrenes in the Human Hepatoma Cell Line HepG2. Environ Health Perspect. 1994;102:195–200. doi: 10.1289/ehp.94102s6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guengerich FP, Yun C-H, Macdonald TL. Evidence for a 1-electron oxidation mechanism in N-dealkylation of N,N-dialkylanilines by cytochrome P450 2B1. Kinetic hydrogen isotope effects, linear free energy relationships, comparisons with horseradish peroxidase, and studies with oxygen surrogates. J Biol Chem. 1996;271:27321–27329. doi: 10.1074/jbc.271.44.27321. [DOI] [PubMed] [Google Scholar]