

Abstract
Recently, phospholipid peroxidation products gained a reputation as key regulatory molecules and participants in oxidative signaling pathways. During apoptosis, a mitochondria-specific phospholipid, cardiolipin (CL), interacts with cytochrome c (cyt c) to form a peroxidase complex that catalyzes CL oxidation; this process plays a pivotal role in the mitochondrial stage of the execution of the cell death program. This review is focused on redox mechanisms and essential structural features of cyt c's conversion into a CL-specific peroxidase that represent an interesting and maybe still unique example of a functionally significant ligand change in hemoproteins. Furthermore, specific characteristics of CL in mitochondria – its asymmetric trans-membrane distribution and mechanisms of collapse, regulation of its synthesis, remodeling and fatty acid composition – are given significant consideration. Finally, new concepts in drug discovery based on the design of mitochondria-targeted inhibitors of cyt c/CL peroxidase and CL peroxidation with anti-apoptotic effects are presented.
Keywords: cytochrome c, cardiolipin, peroxidase, lipidomics, oxidative stress, apoptosis, autophagy, mitochondrial targeting
Introduction
“Life is pleasant. Death is peaceful. It's the transition that's troublesome.”
Isaac Asimov (1920 - 1992)
As a triplet biradical with two parallel spins, molecular oxygen readily interacts with other radicals – e.g., lipid alkyl radicals, thiyl radicals – but it has a very poor reactivity towards molecules with fully paired electrons (non-radicals). As a kid, everyone was amazed by a famous experiment in chemistry class when the teacher burned a small strip of iron in an atmosphere of oxygen. Radicals generated by the high temperature of the flame and combustion facilitated the oxidation of iron. Remarkably, iron is vital to functions of diverse enzymes where it catalyzes reactions with oxygen; however, the chemistry of life does not burn our body. On the contrary, aerobically living cells have developed a safe and sophisticated machinery to activate oxygen and catalyze slow and well controlled oxidation (but not combustion) processes. Yet, oxygen radicals are continuously produced in our body via a univalent reduction of molecular oxygen. While all one-electron products of oxygen reduction are called “reactive oxygen species”, only one of them – the hydroxyl radical HO˙ (a three electron reduction intermediate of oxygen) – is notorious for its remarkably high and calamitously indiscriminative reactivity towards most biomolecules.
It is a common belief that strict control and elimination of superoxide and hydrogen peroxide (H2O2) are protective mechanisms preventing cell damage and death [1]. Recently, however, superoxide radicals and hydrogen peroxide gained a reputation as regulatory molecules and participants in oxidative signaling pathways. Superoxide dismutases (SODs) – in the mitochondrial matrix and intermembrane space, in the cytosol, and in extracellular compartments – convert superoxide radicals into H2O2. Thus, SODs may be acting as important regulators and sources of H2O2. An important process through which cells utilize H2O2 for signaling purposes is the peroxidase catalytic cycle of hemoproteins1. While activation of H2O2 by peroxidases is usually effectively controlled by the participating protein moieties, it is still a high risk endeavor; changes in the redox environment, protein structure, or genotoxic events may lead to unregulated activation of H2O2 and the production of hydroxyl radicals. In this review, we will focus on cyt c – a well known hemoprotein electron transporter in mitochondria – to illustrate possible mechanisms and consequences stemming from peroxidase activation of this protein by physiologically relevant anionic phospholipids.
Multiple functions of cytochrome c in cells
Over last two decades, we witnessed the collapse of an old dogma of biochemistry: one gene->one protein->one function. Discoveries of new functions of cyt c are one of stunning hallmarks of this paradigm shift. In addition to its well established role as an electron shuttle between respiratory complexes III and IV in mitochondria, the antioxidant role of cyt c has been linked to its propensity to catalyze the oxidation of superoxide radicals to molecular oxygen. Thus, cyt c can act as a superoxide scavenger [2]. In addition to mechanisms associated with cyt c's electron-transporting capacities, it has been identified as a critical cell death factor capable of initiating the caspase cascade via its binding to apoptosis protease activating factor, Apaf-1, and the formation of apoptosomes [3]. Mueller et al. have demonstrated that cyt c catalyzes the amidation of fatty acids and the formation of important physiological regulators – long chain fatty acyl glycines – through yet to be identified pathways [7,8]. All these important biological activities of cyt c are realized in its native structure. Studies from several laboratories documented that unfolding of the cyt c's globule reveals a new function as a peroxidase [9]. The structural destabilization of the protein can be induced by chemical modification (i.e. oxidation, nitration) [10-12] or by its association with hydrophobic anions, including anionic phospholipids [13-19]. Our studies uncovered the mechanisms through which cyt c peroxidase activity propagates the oxidation of a mitochondria-specific phospholipid, cardiolipin, and the pivotal role of this reaction in the execution of apoptosis (via mitochondrial membrane permeabilization and release of pro-apoptotic factors from mitochondria) [20,21]. The mechanisms and essential features of cyt c's conversion into a peroxidase represent an interesting and maybe still unique example for a functionally significant ligand change in hemoproteins. These properties of cyt c may represent an interesting case of the recently developed concept of “intrinsically disordered” or “ill-structured” proteins, whereby anionic membrane phospholipids induce the “controlled chaos” [22,23] required for the emergence of a new peroxidase function. These structural rearrangements of cyt c – induced by its binding with CL, the appearance of the peroxidase function, CL peroxidation and the subsequent “dreadful” consequences for both mitochondria and cells – are the major focus of this review.
Interactions of cyt c with anionic phospholipids lead to peroxidase activation
Binding modes of cyt c to anionic phospholipids
During the last three decades, studies of cyt c revealed several protein binding sites for anionic lipids; at least 30% of the protein surface constitutes so-called A-, C- and L- candidate binding domains believed to participate in interactions with anionic lipids (see schema 1) [24-26]. Both the penetration of phospholipid acyl chains into the protein globule as well as the protein integration into the phospholipid bilayer of the membrane were suggested as possible binding modes. Interactions of cyt c with anionic phospholipids are complex, and multiple factors can contribute to the unfolding capacity of the lipids.
Schema 1.
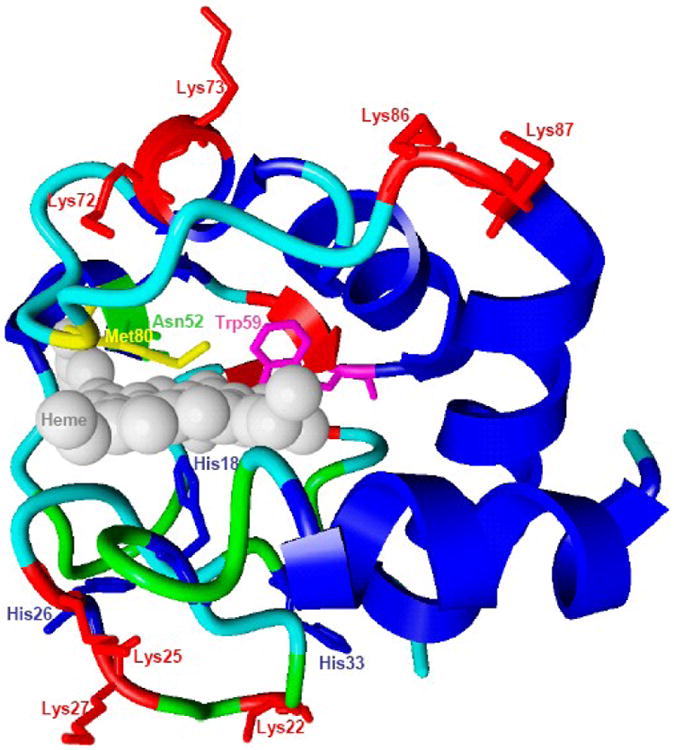
Possible cardiolipin (CL) binding sites on cytochrome c.
Structure of native cyt c (1HRC). Several domains that are likely involved in interactions with CL and acting as heme-iron ligands include the following amino acid residues: Lys72, Lys73, Lys86 and Lys87 (A-site), Asn52 (C-site) and Lys22, Lys25, His26, Lys27 and His33 (L-site). Met80 and His18 form coordination bonds with heme. Intrinsic fluorescence of Trp59 is quenched by the proximity to the heme moiety.
Electrostatic forces are one of the major factors that govern cyt c-lipid interactions. Positively charged cyt c molecules (isoelectric point is near pH 10, net charge is +8e at neutral pH) are strongly attracted to the negatively charged headgroups of anionic lipids [20,24,27,28]. There are no signs of unfolding of cyt c and activation of its peroxidase activity in the absence of electrostatic interactions - with non-charged (zwitterionic) lipids such as phosphatidylcholine (PC) - or with anionic phospholipids in high-ionic strength buffers, as evidenced by electrophoretic measurements of the cyt c/lipid complex [17,19,29]. Site A has been designated as the anion-binding center that likely includes the basic residues Lys72 and Lys73 [24,25]. By studying interactions of the spin-labeled protein with spin-labeled phospholipids, Kostrzewa et al. found that the membrane interface of the protein includes Lys72, Lys86 and Lys87 [30]. Similarly, recent mutation studies of yeast cyt c revealed an involvement of Lys72 and Lys73 in cyt c-CL binding [31]. The Nantes' group reported the existence of an additional electrostatic binding site on cyt c, named L site, which includes Lys-22, Lys-25, His-26, Lys-27, and His-33, and participates in protein-membrane interactions at pH<7.0. Through simultaneous interactions of sites L and A with CL-containing membranes, cyt c can promote vehicle fusion at low pH [26].
Electrostatic interactions between cyt c and lipid membranes, however, are not the only factors that affect unfolding of the protein. Hydrogen bonding between the C site represented by Asn52 and protonated acidic phospholipids was proposed to stabilize the high affinity binding of the protein [24,25]. By detecting tertiary rearrangements, it was demonstrated that cyt c-CL binding is a two step process involving high and low affinity sites, which are believed to be A- and C-sites, respectively [19]. Studies of cyt c binding with fluorescence resonance energy transfer (FRET) from labeled lipid to heme have also confirmed the presence of two interaction modes – an electrostatic low affinity binding to deprotonated CL molecules and a high affinity binding stabilized by electrostatic and H-bonding to partially protonated CL [32]. The partial involvement of these two binding sites depended on pH, ionic strength and the mole fraction of CL; yet both binding modes were operational at physiologically relevant conditions.
Hydrophobic interactions between nonpolar acyl residues of the lipid molecules and nonpolar regions of cyt c (normally buried inside the protein) also effect the formation of the cyt c/lipid complex [17,28,33]. Tuominen et al. suggested that the C-site-mediated interaction of cyt c includes a “lipid anchorage” and provided a direct demonstration of phospholipid acyl chain interaction with the hydrophobic interior of the protein [28]. It has been proposed that a single CL molecule could be involved in both the electrostatic interaction of a head group with Lys72 and in the hydrophobic anchoring of the protein by acyl chain insertion either in the channel surrounded by Asn52, Lys72 and Lys73 or between the non-polar polypeptide strands 67–71 and 82–85 [31,32]. Our recent finding that the reaction rate of both membrane bound and free cyt c with fatty acid hydroperoxides is about three orders of magnitude higher than the rate of H2O2-dependent peroxidase activity strongly argues in favor of the importance of hydrophobic interactions (Belikova et al., unpublished observations).
It has been postulated that the formation of the complex commences with electrostatic interactions that promote the penetration of cyt c into the lipid bilayer, where it interacts hydrophobically with interior lipids [17,19]. This model suggests that the presence of unsaturated lipids is a requirement for effective membrane interfacing of cyt c and formation of the complex. Indeed, alterations in saturation of the lipid acyl chains have a dramatic effect on cyt c binding and unfolding. Mono-unsaturated tetraoleoyl cardiolipin (TOCL) was found to be a much stronger inducer of cyt c conformational changes and activator of its peroxidase activity than saturated tetramyristoyl cardiolipin (TMCL); the structural rearrangements triggered by polyunsaturated tetralinoleoyl cardiolipin (TLCL) appeared to be even stronger than those initiated by TOCL [17]. In support of this hypothesis, interaction of cyt c with CL evaluated by competitive binding with acridine 10-nonenyl bromide (nonenyl acridine orange, NAO) increased in the same order TMCL ≪ TOCL < TLCL < BHCL (bovine heart cardiolipin), i.e. interactions progressed proportional to the number of double bonds in the CL acyl chains.
An interesting mode for cyt c interactions with anionic phosphatidylglycerol (PG) has been proposed by Oellerich et al. on the basis of viscosity and turbidity of lipid/protein mixtures. The binding mechanism depends on the lipid to protein ratio. At low coverage of the membrane surface, the binding is peripheral; at a ratio from 18:1 to 12:1 partial protein penetration into the membrane occurs. At lower ratios, peripheral binding dominates again [16]. In line with this observation, the analysis of FRET from anthroylvinyl-labeled PC to cyt c heme indicated that the high affinity binding involves a partial penetration of the protein into the lipid bilayer to the depth of several proximal carbons of the acyl chains [32]. Approximate estimates of heme location relative to the membrane/aqueous interface by FRET from NBD-labeled CL (1,1′,2-trioleoyl-2′-[12-;-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-cardiolipin) are in agreement with these findings [34].
In summary, the molecular description of cyt c interactions with CL (and other anionic phospholipids) is not complete. There are at least two sites on the cyt c protein surface that can contribute to CL binding. The binding may be realized via different modes whose relative contributions depend on the experimental conditions, such as pH, ionic strength, lipid composition and lipid/protein ratios. However, detailed information on the structure of “foldons” in cyt c/CL complexes and the molecular dynamics of CL-driven protein unfolding is lacking. Further studies to better our understanding of cyt c-membrane interactions are warranted.
Structural rearrangements and peroxidase activity of cyt c upon its interactions with anionic lipids
Interaction of cyt c with negatively charged lipid membranes induces considerable disruption of the native compact structure of the protein and induces intermediate conformations between the native and fully unfolded states, called a “molten globule”. This state, an “alternative folding”, is defined as a compact conformation with a secondary structure comparable to that of the native state and fluctuating tertiary conformation due to a high enhancement of intramolecular motion [13,27,35,36]. In solution, stability and unfolding of cyt c were extensively studied using deuterium exchange [37] and other experimental techniques [38-41]. Stabilities of different regions of the protein were found to be very dissimilar: five distinct domains of cyt c (foldons) with non-equivalent stabilities were identified, which participate in cooperative folding–unfolding of the protein in a stepwise sequential way [37]. These structural domains are folded around the heme of cyt c which is covalently attached to the polypeptide chain by residues Cys14 and Cys17.
Binding of cyt c to membranes is accompanied by changes in the tertiary protein conformation and opening of a heme crevice [13-15]. An early finding of Fe-S(Met80) bond disruption upon binding was further advanced by analysis of the heme configuration using resonance Raman spectroscopy, which revealed the coexistence of a mixture of hexa-coordinated low spin states (His-Fe-Me and His-Fe-His) or hexa-coordinated low spin (His-Fe-His) and high spin states (His-Fe-H20 and His-Fe-) in DOPG bound protein, depending on the lipid to protein ratio [16]. These coordination states of heme are similar to those present in microperoxidases, heme-containing peptides produced by proteolytic digestion of cyt c [42,43].
Changes in the heme environment also affected CD spectra, causing shifts in the 375-425 nm region (heme moiety) as well as changes in the UV-region (250-280 nm) that were observable even at relatively low CL/cyt c ratios (2:1 and 4:1). By detecting changes in 416 nm dichroic signal upon cyt c binding to CL, Sinibaldi et al. described two-step alterations in the heme pocket associated with Fe-S(Met80) bond disruption and rearrangements of the tertiary structure [19]. They found that the peroxidase activation of cyt c also followed a two-step transition profile, thus demonstrating a strong link between conformational and functional properties of the protein. In concordance with this observation, an acidic environment further favored the high affinity binding of the protein to CL and the destabilization of its tertiary structure [32,44] as well as simultaneously enhanced peroxidase activity of CL-bound cyt c (Borisenko, unpublished observations). The appearance of Trp59 fluorescence (quenched by its proximity to heme in native cyt c) is also characteristic of CL binding to cyt c; this effect indicates a substantial conformational shift in the protein, leading to a proportional increase in the peroxidase activity [18].
Because high affinity binding of CL with cyt c – accompanied by unfolding of the protein – is realized largely through an electrostatic interface between negatively charged phosphates on CL and positively charged lysines on cyt c, as well as through hydrophobic interactions of CL's acyl groups with a hydrophobic domain of the protein, other negatively charged phospholipids may also bind and unfold cyt c via analogous mechanisms. Indeed, several anionic phospholipids tested for their ability to change the structure of cyt c – phosphatidic acid (PA), dioleoyl-glycero-3-phosphoinositol 4,5-bisphosphate (PIP2) and dioleoyl-glycero-3- phosphoinositol 3,4,5-trisphosphate (PIP3) – revealed significant binding with cyt c accompanied by structural rearrangements of the protein. CL and PA were most effective as inducers of cyt c unfolding assessed by several criteria such as the levels of Trp59 fluorescence, a full disruption of the Fe-S(Met80) bond and a nitrosylation of the cyt c heme. Similarly, CL and PA were stronger inducers of cyt c's peroxidase activity than other anionic phospholipids [18].
The activation energy of the peroxidase reaction, ΔGperox, was found to be ∼1 kcal/mol less than the free energy needed to unfold the cyt c domain containing Trp59 (ΔGfluor) and lower by 0.3-0.6 kcal/mol than the ΔG for disruption of Fe-S(Met80). This interesting kinetic effect is consistent with the assessment of a 5 to 12 kcal/mol range of stability energies for the five domains of cyt c [45-47] compared to an activation energy for the peroxidase catalytic function of ∼3.8 kcal/mol [48]. ΔGperox appeared to be very close to ΔG of heme nitrosylation, probably because it is very similar to the formation of the complex between heme and hydrogen peroxide – a prerequisite for the catalysis of the hydrogen peroxide reduction [18]. These comparisons of structural parameters and peroxidase activity indicate that the peroxidase function of cyt c is strongly activated under conditions that do not markedly change the protein tertiary structure (assessed by Trp59 fluorescence). In other words, unfolding can favor peroxidase activity, yet this activity does not require complete protein unfolding. Peroxidase activity is upregulated when the dissociation energy of the Fe-S(Met80) bond is lowered due to protein-lipid interaction, yet the bond is still retained, thus suggesting that a relatively small perturbation of the protein structure by electrostatic interactions with membrane components is sufficient for the substitution of H2O2 (or nitric oxide, NO) for Met80 and for peroxidase activation. This alternative protein (un)folding and disruption of the Fe-S(Met80) bond inevitably leads to an enhancement of peroxidase activity, similar to that caused by chemical modifications such as carboxymethylation [49] and interactions with peroxynitrite [50] or hypochlorite [51], or complete unfolding in the presence of guanidine hydrochloride. The augmented availability of heme iron in cyt c/CL complex for small molecules is reminiscent of microperoxidases in which one of hexa-coordinated ligands, H2O, can be readily displaced by a variety of exogenous ligands resulting in high peroxidase activity [52].
In summary, high affinity interactions of cyt c with anionic phospholipids may involve binding at two different sites to the membrane, partial unfolding of the protein globule and partial insertion of the protein into the membrane. Structural transitions of cyt c include: (i) opening of the heme crevice (detected by CD and NMR spectroscopy); (ii) reduction of the volume of hydrophobic core (detected by NMR spectroscopy); (iii) disruption of the 6th ligation of heme Fe with the Met80 residue (detected by UV-VIS and Raman spectroscopy); (iv) emergence of penta-coordinated high spin heme and hexa-coordinated Fe with a new ligand, presumably His33 (shown by Raman and EPR spectroscopy); (v) rearrangement of the protein's tertiary structure with the preservation of its secondary structure (observed by CD spectroscopy); and (vi) emergence of Trp59 fluorescence that is completely quenched by heme in the native protein.
To satisfy these experimental observations, the conformation of the protein in the complex with the membrane has to include electrostatic and hydrophobic protein-lipid interactions, relocation of His33 from the proximal to the distal side of the heme and transition of Trp59 from the plane of the heme to the plane perpendicular to the heme (i.e., a relocation whereby a minimal shift in distance will produce the highest gain in fluorescence quantum yield). We propose the following model for the protein-lipid interactions (see Scheme 2): cyt c binds to an anionic phospholipid via Lys72 (A-site) and then to a second anionic lipid via Lys27 (L-site). As a result, the heme will be located perpendicular to the plane of the membrane. Subsequent concerted rearrangements of the tertiary structure include: (i) a slight shift of a low energy foldon (residues 72-86 according to [37]) along the membrane surface and out of the heme plane, (ii) a substantial movement of a low energy foldon (residues 37-59) out of the heme plane and rearrangement of a high energy foldon (residues 20-36) around the heme, leading to the occupation of a distal heme pocket by His33. According to this structure, the heme edge has to be inserted into the membrane and the hydrophobic residues Ile81 or Phe82 anchor the protein in the membrane. The heme and the heme pocket become readily available for interaction with phospholipid acyl chains. Three other high energy foldons (residues 1-19, 60-71 and 88-104) do not undergo perturbations. Residues 1-19 and 88-104 are located above the heme, while residues 60-71 interact with the protein coils of residues 72-86. This model of cyt c structural rearrangements by CL (anionic lipids) is associated with minimal energy cost.
Schema 2.

Proposed structural model of alternative (un)folding of cyt c bound to anionic membrane surface. The model explains reported physico-chemical properties of cyt c including its peroxidase activity.
A – side view, membrane interface is at the bottom of the protein; B – view from the membrane side. Protein backbone is encoded by color in accord with unfolding energy (in the order from low to high energy: white, red, yellow, green, and blue [37]). Amino acid residues important for binding with the membrane and peroxidase activation are highlighted.
Structure of cyt c catalytic site
Highly specialized peroxidases, like horseradish peroxidase (HRP) or myeloperoxidase (MPO), have particular heme environments, allowing the catalytic sites to effectively bind, and cleave H2O2. The exceptionally high reaction rates – 106-107 M-1s-1 of peroxidases [53] are dependent on the presence of His as a 5th iron ligand in the proximal heme pocket as well as His and Arg residues in the distal heme pocket. The latter two residues participate in the so-called “push-pull” catalysis of H2O2 cleavage [54-56] and the formation of the first reactive intermediate, compound I (schema 3). Analysis of HRP mutants revealed that substitution of distal Arg and His was associated with 103- and 105-fold decreases in the rate of Compound I formation, respectively [55,57]. Mutation of the proximal His slowed this reaction 106 fold [58], thus clearly indicating the hierarchy of these amino acids in catalysis.
Schema 3.

Catalytic mechanism of horseradish peroxidase.
A – ferric enzyme with H2O2 bound as a ligand in the 6th coordination position of iron; B – oxoferryl iron and porphyrin centered radical (Compound I).
Although cyt c possesses some of the pre-requisites of a peroxidase – a heme moiety and a proximal His – it has a very weak peroxidase activity in its native state [59,60]. The catalytic site of cyt c lacks Arg or His in the distal pocket of the heme. Moreover, the distal ligand – Met80 – is located 2.5Å away from the Fe, thus precluding access of the heme to H2O2 and other peroxides in the native protein [20]. The above considerations based on crystallography or NMR studies of native cyt c are hardly applicable to its markedly changed organization in complexes with anionic lipid membranes [17,27,28,35,61]. Unfortunately, neither crystallographic data nor detailed NMR analysis of these complexes with anionic phospholipids are currently available. However, several studies employing less direct techniques are indicative of electronic and structural changes in the protein/CL complexes favoring its peroxidase function. Upon binding and unfolding of cyt c by anionic lipids or surfactants as well as after its chemical modification (for example, by carboxymethylation, oxidation or nitration [49-51]), Met80 moves away from the heme site and thus releases the sixth iron coordination bond. Cyt c bound to CL displays a shift of the Soret band to a shorter-wavelength region suggesting a high-spin state of heme in the new cyt c conformation (Belikova unpublished observations). Raman spectroscopy studies confirmed that a penta-coordinated high spin state is one of the major forms of heme in cyt c both bound to DOPG vesicles and unfolded by detergents [16,62]. This heme coordination state is characteristic of peroxidases and is believed to be important for peroxidase function [63].
Weakening and disruption of the Met80-Fe bond, the distal movement of Trp59 from the heme and changes of the protein tertiary structure facilitate engagement of several essential ligands – His26, His33, Arg38 and Arg91 – in the catalytic peroxidase process in cyt c/CL complexes. Raman spectroscopy analysis of ferrous and ferri cyt c/PG complexes revealed the presence of heme with hexa-coordinated ligands represented by His33 or His26 [16,62], thus suggesting a close location of His to the heme in the distal pocket.
The structural rearrangements in cyt c/CL complexes dramatically affect its redox properties. In complexes with CL, the redox potential for the Fe(II)/Fe(III) couple is ∼400 mV more negative than in intact cyt c [64]. As a result, two important functions of free cyt c – electron shuttling between complexes III and IV of the mitochondrial respiratory chain and scavenging of superoxide radicals – become unfeasible. In peroxidases, the Fe(II)/Fe(III) redox potential is linked to the stability of highly oxidized heme intermediates via electron donation from anionic axial ligands to the heme [65-66]. The stability of these intermediates is essential for peroxidase catalysis. Structural factors contributing to the regulation of the redox potential in hemoproteins include the hydrophobicity of the heme pocket, the nature of the axial ligands on iron and electrostatic interactions at the active site. In cyt c, His-Met ligation of iron and a hydrophobic protein core are two important factors that determine its positive redox potential (+260 mW) [67,68]. Loss of these features upon binding of cyt c with CL is likely accountable for a substantially more negative redox potential, which falls into the range characteristic of peroxidases (f. ex., the redox potentials of cyclooxygenase (COX) and HRP are -160 mV and -250 mV, respectively). The CL-induced shift in the redox potential may be a substantial contributor to the higher peroxidase activity of cyt c/CL complexes.
Heterolytic and homolytic pathways of cyt c peroxidase catalysis
Heme-containing peroxidases can oxidize reducing substrates by utilizing different peroxides (e.g., H2O2, and organic peroxides including lipid hydroperoxides) as a source of oxidizing equivalents. As a heme-protein with a proximal His18, cyt c can function as a peroxidase, albeit at a very low rate of about 1 M-1s-1 [59,60]. Partially unfolded cyt c in its complexes with CL exerts almost a 100-fold higher catalytic activity in the presence of H2O2 than the native protein [20], yet it is still a relatively weak peroxidase [17]. H2O2 has been traditionally recognized as the preferred source of oxidizing equivalents for many peroxidases such as horseradish peroxidase, lactoperoxidase, and myeloperoxidase, which do not display significant peroxidase activity with fatty acid hydroperoxides [53]. In contrast, cyt c/CL peroxidase activity can be supported by other hydroperoxides, particularly those of lipids. In fact, oxidized CL (hydroperoxy-CL, CL-OOH) is evidently a very good source of oxidizing equivalents for the peroxidase activity of cyt c. Low concentrations of CL-OOH – accumulated during its storage or by its initial oxidation in the cyt c catalytic peroxidase cycle – effectively propagated the peroxidation of CL without any additional supplementation with H2O2. Our recent studies demonstrated that peroxidase activity of cyt c/CL complexes can be enhanced up to 1,000-fold when fatty acid hydroperoxides were utilized as a source of oxidizing equivalents instead of H2O2 (Belikova et al., unpublished results). In a way, this is similar to the requirement in arachidonic acid hydroperoxides (Ar-OOH) for the full expression of the peroxidase function of cyclooxygenases (COX-1 and COX-2), which catalyze the reduction of lipid hydroperoxides (i.e. PGG2 derivatives) 2-3 orders of magnitude more effectively compared to H2O2 [69]. Interestingly, both cyclooxygenases lack Arg in the distal pocket, while Gln adjacent to His is not involved in the peroxidase reaction. Instead, COXs have enlarged hydrophobic domains that are likely involved in the binding of bulky fatty acid moieties and the cleavage of the O-O bond by the distal His and heme iron [70-72]. Similarly to COX, cyt c/CL complexes accommodate an esterified fatty acid residue of CL (and probably free fatty acid) in their hydrophobic heme pockets upon interaction with the anionic membrane [28].
Two major catalytic mechanisms potentially involved in the cleavage of hydroperoxides by peroxidases are the homolytic and heterolytic pathways [53]. Homolytic splitting of H2O2 leads to the formation of highly and indiscriminately reactive HO˙. As a consequence, the homolytic mechanism is likely associated with a non-specific oxidation of reducing substrates as well as the oxidative modification of the enzyme itself. Homolytic splitting of hydroperoxies by hemin and several hemoproteins such as HRP mutants, cytochrome c peroxidase (CcP) mutants, myoglobin, and cytochrome P450 has been reported [54,55,73-76]. Based on earlier studies of the Mason group with cyt P450 and lipoxygenase [77,78], Iwahashi et al. [79] proposed a mechanism for a one-electron reductive homolytic decomposition of fatty acid hydroperoxide (FA-OOH) by cyt c, leading to the formation of alkoxyl radicals:
| (1) |
where R is a protein chain, Por is heme, R-Por-Fe(III) is ferri cyt c, R-Por-Fe(IV)=O is the oxyferryl state of heme in cyt c, and LO˙ is an alkoxyl radical of a fatty acid.
In contrast, heterolytic cleavage of the O-O bond – leading to the formation of Compound I – requires significantly higher activation energy than homolysis (370 versus 40 kcal/mol [71]). Several peroxidases, such as HRP, CcP, MPO, and COX utilize this pathway [53]. The employment of either of these alternative peroxidase mechanisms depends on several factors, such as the spin-state of the heme (high or low), the organization of catalytic pocket, and the nature and properties of the oxidant. A “Push-pull” catalysis relies on positively charged Arg and His residues to shift the electron density sufficiently for H2O2 heterolytic splitting. Accordingly, the His42Leu mutant of HRP is characterized by the preference for homolytic cleavage of H2O2 and a significantly lower rate of peroxidase reaction [55,57]. In concurrence with these considerations, the catalytic site of native cyt c lacking Arg or His in close vicinity of the heme in the distal pocket should favor the homolytic peroxidase mechanism.
The heterolytic mechanism of peroxidase action utilizes reactive intermediates leading to oxidation products specific for a particular enzyme (i.e., PGH2 produced by COX, and hypochlorous acid by MPO). In contrast to classic peroxidases, no spectroscopic evidence has been obtained so far for the formation of compounds I and II for chemically modified cyt c or cyt c/CL complexes. Combined with readily detectable protein-centered radicals [80,81], this is indicative of the generation of highly unstable heme intermediates. Stereospecific oxidation by cyt c/CL complexes can be achieved via interactions with relatively stable protein-centered radical intermediates generated during the cleavage of the O-O bond. The specificity of cyt c as a peroxidase may be due to the proper orientation of strongly-bound reducing substrates such as polyunsaturated cardiolipins.
Mass spectrometric studies identified several species of CL-OOH and CL-OH as major oxidation products formed by complexes of cyt c with polyunsaturated CL both in vitro and in vivo [20,21,82,83]. This indicates that not only H2O2 but also CL-OOHs are utilized as sources of oxidizing equivalents in this reaction. It is possible that both mechanisms – homolytic and heterolytic – may be involved at different stages of cyt c peroxidase reactions. Initiation of the reaction by H2O2 and subsequent switching to CL-OOH or FA-OOH (see below) may be also associated with different contributions of each of the two mechanisms. Interestingly, employment of both catalytic mechanisms has been demonstrated for mammalian cyclooxygenases [84]. A highly specific COX-1 isoform catalyzes primarily a two-electron reduction of FFA-OOH via its heterolytic cleavage. In contrast, COX-2 catalyzes both one- and two-electron reductions [84].
Protein-derived radicals and oligomerization of cyt c by the peroxidase activity of its complexes with anionic phospholipids
Characteristic reactive intermediates of peroxidase-catalyzed reactions are protein-immobilized radicals [85-88]. The formation of these radicals is markedly enhanced in enzymes that have relatively unstable Compounds I and II. Particularly, the highly reactive Compound I in COX-2 (half-life ∼100 ms) produces a tyrosyl radical that is presumably involved in the oxidation of other protein groups, and enzyme inactivation occurs within seconds [89]. Cyt c contains several potentially oxidizable aminoacid residues: four tyrosines, some of which (Tyr67, Tyr48) are within 5.0 Å of the heme porphyrin ring, and one tryptophan residue [90]. The radical intermediates from these residues can be detected by low temperature EPR spectroscopy [85] as well as by an immuno-spin trapping technique in which the immunoreactive protein-immobilized spin adducts formed during interaction of radicals with a spin trap, DMPO, can be detected by an anti-DMPO antibody [86,88,91]. EPR spectroscopy experiments demonstrated that anionic phospholipids facilitate the H2O2-dependent production of protein immobilized radicals on cyt c. The effectiveness of dioleoyl (DO) phospholipids in inducing protein-derived radicals increased in the order DOPA > TOCL > DOPS > DOPC (DOPS, dioleoylphosphatidylserine). In the absence of reducing substrates or spin-traps, recombination of protein-derived tyrosyl radicals results in oligomerization of the protein via dityrosine cross-links (which are non-dissociable by S-S reducing reagents) and the disappearance of its monomeric form [51,92]. The strengths of phospholipids in inducing peroxidase dependent oligomerization of cyt c ranked similarly to their effects on peroxidase activity and the formation of protein-derived radicals: TOCL ∼ DOPA > PIP3 > PIP2 > DOPS. The importance of cyt c/CL oligomerization is that it may facilitate not only the accumulation of homo-oligomers but also cause a hetero-oligomerization by co-oxidation of other proteins. This may be particularly relevant to proteins that have anionic lipid binding sites, such as alpha-synuclein, resulting in an accumulation of poorly digestible cross-linked aggregates characteristic of neurodegenerative diseases. Interestingly, both cyt c and alpha-synuclein are abundant components of Lewy bodies which accumulate in the brain of patients with Parkinson's disease.
Peroxidase function of cyt c/CL complexes in apoptosis
Multiple functions of cyt c – in mitochondrial electron transport, peroxidase oxidation of CL, interactions with Apaf-1 in the cytosol – raise a question about regulation and switching mechanisms involved in its diverse pathways. One of these mechanisms is a marked negative shift of cyt c's redox potential upon its interaction with CL, thus precluding its operation as an electron acceptor form mitochondrial complex III or from superoxide radicals (see above). Another important regulatory mechanism is availability of oxidizing equivalents - H2O2 or lipid hydroperoxides - feeding the peroxidase cycle of cyt c/CL complexes. Disrupted electron transport, particularly at Complexes I and III, are considered the major sources of superoxide production. Enzymatic mechanisms – by MnSOD in the matrix or Cu,Zn-SD in the inrtermembrane space – and spontaneous (non-enzymatic) pathway convert superoxide radicals into H2O2 [93,94]. A mitochondrial outer membrane enzyme, monoamine oxidase, catalyzes deamination of biogenic amines and directly generates H2O2. In addition, 12/15-lipoxygenase is a potent cytosolic source of lipid hydroperoxides. Both monoamine oxidase and 12/15-lipoxygenase have been implicated in the development of cell death [95,96]. Finally, strict compartmentalization of CL, that prevents its random non-specific binding with cyt c, likely represents one of the most effective regulators of cyt c's peroxidase activity – as detailed below.
Collapse of CL asymmmetry in mitochondria during apoptosis
Peroxidase function of cyt c requires its direct physical interaction with CL. Normally, however, CL is confined almost exclusively (>80%) to the inner mitochondrial membrane (IMM) [20,97-101] whereby it is distributed between the inner and the outer leaflets at a ratio of 60:40 [102,103]. Thus, binding of cyt c to CL depends on the availability of the latter in the outer leaflet of the IMM. Moreover, significant demand for high-affinity CL binding by other mitochondrial proteins such as mitochondrial respiratory Complexes I, III and IV as well as other mitochondrial proteins also limits access of cyt c to CL [104,105]. However, within mitochondrial contact sites – zones of close apposition of the inner and outer membranes – the content of CL may be high and comparable to that of PC and PE – up to 24% of total lipids [101].
During apoptosis, the asymmetric distribution of CL collapses and the level of CL in the outer mitochondrial membrane increases to 40% of its total content, while 60% of it still resides in the inner membrane [20]. About 30-40% of the CL present in the IMM of apoptotic cells, is confined to its inner leaflet [20]. The decrease of the CL level in the inner membrane is accompanied by changes in the inter-membrane distribution of CL. Only 30-40% of CL is found in the inner leaflet of the inner membrane in mitochondria of apoptotic cells [20].
tBid and Scramblase-3: involvement in CL trans-membrane redistribution in apoptosis?
The mechanisms of CL mitochondrial translocation are not well understood. It has been shown that CL inter- and intra-membrane changes in mitochondria occur at early stages of apoptosis [20], before dissipation of membrane potential and prior to PS externalization on the cell surface [106]. Activities of two proteins have been associated with apoptotic trans-migration of CL: tBid and phospholipid scramblase-3 (PLS-3) [103,107,108]. Early work has established that tBid has a CL binding domain [109,110]. While tBid is bound to the outer mitochondrial membrane at both contact and non-contact sites [111], it preferentially inserts into the negative lipids of the mitochondrial contact sites between the inner and outer membranes [112], possibly in proximity to the polyunsaturated CL species [101]. A significant trans-membrane distribution of CL from the inner to the outer leaflets of the inner mitochondrial membrane and subsequent appearance of CL in the outer mitochondrial membrane was observed in tBid-treated mitochondria [100,103].
PLS-3 is another mitochondrial protein believed to play a role in CL translocation during apoptosis [107]. PLS-3 is a member of the PLS family responsible for the bi-directional movement of phospholipids [113]. PLS-3 contains 295 amino acids and its gene is located in chromosome 17 in humans [114]. PLS-3 is activated by calcium and disruption of its calcium binding motif results in its inactivation [107]. Cells transfected with this inactive PLS3 mutant contained fewer mitochondria that were larger than those in control cells [107]. These cells were also less sensitive to UV- and tBid-induced apoptosis. In contrast, cells overexpressing PLS-3 displayed increased sensitivity to UV-induced apoptosis and enhanced CL translocation [107]. PLS-3 undergoes post-translational modification by phosphorylation of a specific Thr residue (Thr21) by protein kinase C delta (PKCδ) [115]. Using pyrene-PC labeled liposomes, He et al., [116] assessed the lipid flip-flop activity of PLS-3 and showed that the phosphomimetic mutant of PLS-3 (T21D) was more effective than wild-type protein or phosphoinhibitory mutant (T21A) [116]. However, the exact mechanisms of how phosphorylation of PLS-3 leads to CL translocation are not known. It has been reported that phosphomimetic form of PLS-3 (T21D) facilitates the mitochondrial targeting of tBid [116]. Thus, apoptosis-associated phosphorylation and activation of PLS-3 may be involved – possibly via tBid-dependent pathways – in trans-membrane re-distribution of CL that is critical to the execution of the mitochondrial stage of the apoptotic program.
Biosynthesis and remodeling of CL
Compartmentalization/topography and sufficiency of CL via its binding to cyt c is an important regulatory mechanism of apoptosis. Therefore, the supply of CL via its de novo biosynthesis and remodeling may affect the sensitivity of cells to apoptosis. As a unique mitochondrial phospholipid, CL may be represented predominantly by one or only few molecular species in some tissues (e.g., heart, skeletal muscle, liver, kidney or intestines) [83,117-119] or by hundreds of individual species in other tissues (e.g., brain) [120-122]. There are two major metabolic pathways for CL biosynthesis and turnover. De novo synthesis of CL takes place in mitochondria. Several enzymes catalyzing the multi-stage CL synthesis – those responsible for the formation of phosphatidic acid from acyl-CoA fatty acid and glycerol-3-phosphate, production of activated phosphatidyl (phosphatidyl-CMP), its transfer to another glycerol-3-phosphate and consequent hydrolysis of the latter yielding phosphatidylglycerol – are localized in mitochondria [123]. The final and rate-limiting synthetic step whereby PG is combined with CDP-diacylglycerol to yield CL, is catalyzed by CL synthase, the active site of which is exposed to the mitochondrial matrix [124-126]. Knocking down CL synthase by using RNA interference results in a significant decrease of CL content [127,128]; however, the CL molecular species in CL-deficient cells remain unchanged [128]. Importantly, CL-deficiency was associated with the increased resistance to apoptosis induced by actinomycin D, X-ray irradiation and rotenone in HeLa cells, likely due to decreased amounts of productive cyt c/CL complexes participating in the peroxidation of CL [128]. Recently, Gonzalves et al. demonstrated that in the type II apoptotic response, CL is important for the anchoring, translocation and embedding of caspase 8 in the mitochondrial membrane. This event is vital for caspase 8 oligomerization and further release of apoptotic factors from mitochondria to cytosol [129].
The second mechanism of CL transformation includes re-acylation or remodeling of CL acyl chains [130]. Remodeling has been recognized as an important step of CL post-synthetic maturation. It is believed that the de novo synthesis predominantly contributes to the diversity of CL molecular species, while acyl-specific remodeling yields a limited number of CL molecular species [131]. Neuronal CL remodeling occurs shortly after birth in mammalian species and causes alterations in the physical properties of the mitochondrial membrane [120]. CL remodeling can occur both in mitochondria [131-133] as well as in the endoplasmic reticulum [134]. The re-acylation process requires a hydrolysis of CL by phospholipase A2 [131] to monolyso-CL (MLCL) or dilyso-CL (DLCL) and its coenzyme A dependent [134] or independent re-acylation [135]. Coenzyme A dependent re-acylation of MLCL is orchestrated by acyl-CoA:lysocardiolipin acyltransferase 1 (ALCAT1). ALCAT1 is localized in the endoplasmic reticulum and recognizes both MLCL and DLCL as substrates with a preference for linoleoyl-CoA and oleoyl-CoA as acyl donors [134]. CoA-independent phospholipid trans-acylase (or so-called “tafazzin”) can catalyze the acylation of both lyso-phosphatidylcholine with CL-derived acyl groups and the acylation of MLCL with phosphatidylcholine-derived acyl groups [135]. It has been reported that defects in CL remodeling associated with Barth syndrome lead to accumulation of CL derivatives with abnormal fatty acid composition (reviewed in [136]). Remodeling of CL species may be important as a mechanism controlling the level of CL oxidation by cyt c. For example, substitution of abundant and highly polyunsaturated C22:6, C22:5, and C20:4 species of CL in the brain for less oxidizable C18:2 or non-oxidizable mono-unsaturated C18:1 should inevitably result in lower susceptibility to pro-apoptotic agents. Conversely, targeted manipulation of CL to more oxidizable molecular species may lead to a desirable decreased resistance to apoptosis in tumor cells.
Peroxidation of cardiolipins by cyt c
The distinction of CLs as substrates of peroxidase activity of cyt c is mostly associated with their participation in the execution of the mitochondrial stage of apoptosis. Polyunsaturated phospholipids are known precursors of many important signaling molecules through their hydrolytic or oxidative metabolism; this is particularly relevant to eicosanoid and docosanoid pathways [137-144]. Phospholipids containing polyunsaturated acyl groups are the major substrates of non-enzymatic free radical oxidation that can be initiated and propagated as a chain reaction. Interestingly, cyt c catalyzed peroxidation of CLs also utilizes polyunsaturated molecular species, whereas saturated and monounsaturated CL molecules do not undergo peroxidation [92]. However, cyt c catalyzed peroxidation reactions display significant specificity: anionic phospholipids, particularly CL, PS and PI – are the preferred oxidation substrates. This is most likely due to the proximity of these cyt c-bound phospholipids to the sites where reactive peroxidase intermediates are generated [83,145,146]. In different types of cultured cells triggered to apoptosis (e.g., by gamma-irradiation, or staurosporine or actinomycin D exposure) as well as in animal tissues with a significant number of apoptotic cells (e.g. induced by traumatic brain injury or gamma-irradiation), accumulation of phospholipid hydroperoxides decreases in the order CL≫PS≫PI⋙PE>PC [21,82,83,120,128,147,148]. A notable example is the selective and robust oxidation of two anionic phospholipids – CL in mitochondria and PS outside of mitochondria – in the small intestine of gamma-irradiated mice (Schema 4) [83]. Two molecular species of CL containing C18:2 as potentially oxidizable species indeed underwent oxidation after exposure of mice to total body irradiation. In line with this argument, several hydroperoxy-derivatives – (C18:2)3/(C18:2–OOH)1; (C18:2)2/(C18:2–OOH)2; (C18:2)1/(C18:2–OOH)3; (C18:2–OOH)4 – were detectable in the MS of CLs from irradiated animals. Notably, more abundant phospholipids with higher contents of more polyunsaturated acyl chains – PC, PE, and PI – remained non-oxidized [83]. This underscores the role of cyt c/CL interactions as major factors in determining the substrate specificity of CL oxidation.
Schema 4.
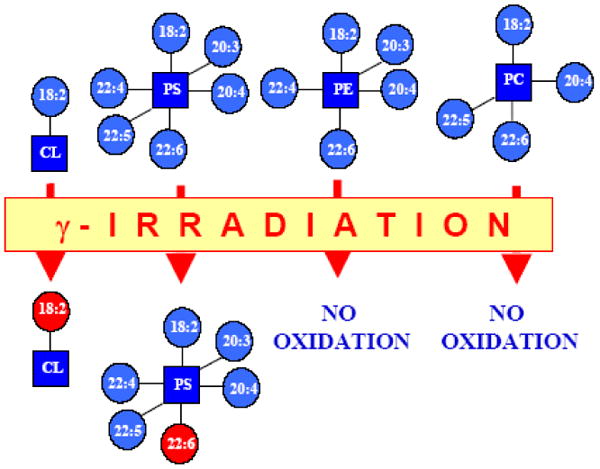
Oxidative lipidomics “Hit-map” of small intestine from mice subjected to total body irradiation.
A non-random, cyt c-driven mechanism is involved in the catalysis of γ-irradiation induced peroxidation of intestinal phospholipids. Selective oxidation of CL followed by oxidation of PS takes place after γ-irradiation and is a part of the intestinal apoptosis in vivo.
Selective oxidation of CL was also found in the lungs of mice exposed to hyperoxia. A 7.5-fold increase of pulmonary CL-hydroperoxide content(to 33.8 ± 8.0 pmol/nmol CL) was detected after hyperoxia (72 h, 99.9% of oxygen) compared to the normal lungs of C57BL/6 mice. The MS analysis of CL oxidation products identified CL molecular species containing hydroperoxy-linoleic acid (C18:2-OOH) along with palmitic C16:0, linoleic C18:2, and stearic C18:0 fatty acids [149]. Accumulation of CL hydroperoxides was also characteristic of the lung of C57BL/6 mice exposed through inhalation (for 4 consecutive days, 5 h/day) to single walled carbon nanotubes. Up to 87.1± 9.7 pmol of CL hydroperoxides per nmol of CL were detected in lungs at 1 and 7 days after inhalation associated with a robust inflammatory response.
The major products of cyt c-catalyzed peroxidation include different hydroperoxy- and hydroxy-derivatives [17,18,20,82,83]. The latter are formed by the peroxidase activity of cyt c whereby phospholipid-hydroperoxides (PL-OOH) are utilized as a source of oxidizing equivalents and reduce PL-OOH to PL-OH at the expense of oxidation of new CL molecules [82,83]. Our recent results indicate that alternatively to the reductive metabolism, CL-OOH may be involved in hydrolytic reactions catalyzed by cyt c [150]. This phospholipase A2-like activity of cyt c yields two major products – non-oxidized monolyso-CL and hydroperoxy-free fatty acids [150]. It is possible that this pathway is involved in CL remodeling as well as the production of oxygenated fatty acids with potentially important physiological functions.
Possible role of CL in interactions between autophagy and apoptosis
In addition to apoptosis, autophagy represents an alternative pathway of programmed cell death (the so called autophagic (type II) cell death). It is well accepted that these two types of cell death mechanisms are interconnected, while the link between autophagy and apoptosis is highly ambiguous [151]. Autophagy is an evolutionary conserved mechanism that provides cells with a mechanism for the continuous turnover of damaged and obsolete macromolecules and organelles (e.g. mitophagy and reticulophagy) [152], and may serve as a mechanism of adaptation to stress (and hence suppress apoptosis), whereas in many other circumstances, it constitutes an alternative cell-death pathway (type II) [153]. For instance, Bax/Bak double knockout mouse embryonic fibroblast cells failed to undergo apoptosis when challenged by DNA damaging reagents such as etoposide. Instead, massive autophagy and delayed cell death was observed [154]. Amaravadi et al. found that autophagy inhibition with either chloroquine or ATG5 shRNA enhanced the ability of either p53 activation or alkylating drug therapy-induced apoptosis in a Myc-induced model of lymphoma [155]. The cytoprotection via autophagy was mainly attributed to its ability of removing protein aggregates and injured organelles, and regulating the cell cycle. In a different experimental setting, perturbations of the apoptotic machinery in lipopolysaccharide treated U937 monocytoid cells and macrophages by the pan-caspase inhibitor Z-VAD-FMK resulted in autophagic cell death, which could be attenuated by RNAi-mediated knockdown of beclin [156]. The cytotoxicity of autophagy could be explained by the destructive potential of uncontrolled massive autophagy. Autophagic removal of mitochondria has been shown to be triggered following a process of induction/blockade of apoptosis. While the detailed molecular mechanisms of autophagy cargo recognition remain unclear, the existence of a selective autophagy of mitochondria (mitophagy) [157,158] indicates that specific mitochondrial signal(s) are involved in triggering the autophagy signaling pathway and “tag” the damaged mitochondria. Indeed, UTH1, which encodes a mitochondrial protein in yeast, has been demonstrated to be required for effective targeting of mitochondria for autophagic degradation [159]. We hypothesized that – in analogy to phosphatidylserine externalization on plasma membrane during apoptosis, and subsequent uptake and digestion of apoptotic cells by professional phagocytes - externalized CL might serve as a mitochondrial version of an “eat-me” signal in an autophagy signaling pathway. Moreover, accumulation of peroxidized CL may act as a molecular switch that initiates the development of pro-apoptotic events when autophageal mechanisms fail to effectively eliminate damaged mitochondria as depicted on Schema 5. Thus CL-mediated signaling may be a key-point in regulation of both autophagy and apoptosis. As a matter of fact, Kissova et al. previously reported that the inhibition of the oxidation of mitochondrial lipids slowed down mitochondria autophagy [160]. Dadakhujaev et al. showed that TrkA overexpression causes ROS accumulation via reduced catalase expression, ultimately leading to autophagic cell death [161]. Further experimental testing of this hypothesis is warranted to define the possible roles of CL externalization/oxidation in these fundamental mechanisms of programmed cell death.
Schema 5.

Cross-roads of mitophagy and apoptosis.
Autophagy and apoptosis are two processes that may be mutually inhibitory; autophagy usually precedes apoptosis while triggering of apoptosis is associated with blocked autophagy. It is likely that these two pathways are intrinsically interconnected via molecular switches that turn on the autophagy process (with still inhibited apoptosis) followed by activation of apoptosis (with turned off autophagy). Both processes function as parts of an essential combined mechanism of elimination of irreparably damaged cells. Phospholipid signaling, particularly deregulation of characteristic for normal cells asymmetry of phospholipids, has been discovered as one of important factors in both autophagy and apoptosis. Collapse of cardiolipin asymmetry in mitochondria and covalent association of phosphatidylethanolamine (or phosphatidylserine) with LC31 are the two major events in signaling, culminating in apoptotic cell death and mitophagy, respectively. It is possible that changes of cardiolipin asymmetry in mitochondria are at the center of the chain of events leading to cell death. This chain includes several consecutive levels of regulation:
1. Synthesis of cardiolipin (CL) and its molecular speciation with a balance of poly- and mono-unsaturated molecular forms as well as saturated CLs).
2. Scramblase-3 (SCR-3) is inactive; maintenance of asymmetry of CL between the inner and outer mitochondrial membranes; CL and cyt c are spatially separated.
3. Regulation of cyt c/CL interactions via cyt c phosphorylation hindering binding of negatively charged CL to cyt c.
4. Low levels of H2O2 production leading to insufficiency of oxidizing equivalents.
5. Cyt c can undergo phosphorylation of its Tyr 97 (in the heart) [168] Tyr 48 (in the liver) [169] likely via a cAMP dependent pathway. Phosphorylation of Y97 is associated with changes of the absorbance at 695 nm which suggests subtle structural changes in the heme environment [168]. Peroxidase activity of cyt c/CL complex involves formation of Tyr radicals [20]. It is possible that phosphorylation of Y97 and Y48 affects binding of cyt c with CL as well as its peroxidase activity.
6. During initiation of autophagy, SCR phosphorylation (resulting in its activation) moves CL to the outer mitochondrial membrane and stimulates (turns on) mitophagy. There is no CL oxidation at this time, because cyt c may be phosphorylated and H2O2 is still unavailable. As damage develops, cyt c can be dephosphorylated and more avidly binds with CL. This disrupts electron transport and stimulates H2O2 production. As a result, CL gets oxidized, thus initiating apoptosis and the end of autophagy (mitophagy).
Inhibition of CL peroxidation as a new approach to anti-apoptotic drug discovery
The discovery of the specific oxygenase activity of cyt c towards CL peroxidation and its essential role in the execution of the apoptotic program indicates possible directions for an effective regulation of apoptosis. Prevention of CL peroxidation may be important because it can be accomplished in mitochondria before the release of pro-apoptotic factors into the cytosol, i.e. prior to “the point-of-no-return” associated with the activation of the caspase cascades [20]. To achieve a substantial effectiveness of the anti-apoptotic action, our recent efforts have been focused on designing and developing several mitochondria-targeted inhibitors of CL peroxidation (Schema 6).
Schema 6.

Inhibition of CL-activated peroxidase activity of cyt c and prevention of CL oxidation in mitochondria leading to suppression of apoptosis.
Peroxidase activity of cyt c/CL complexes leads to CL oxidation and accumulation of products required for the release of pro-apoptotic factors from mitochondria. Consequently, agents and factors that inhibit the peroxidase activity and prevent CL oxidation may act as anti-apoptotic agents. A new approach to regulate the cyt c peroxidase activity is based on the use of modified CL with an oxidizable and fluorescent 7-nitro-2,1,3-benzoxadiazole (NBD) moiety (NBD-CL). NBD-CL forms high-affinity complexes with cyt c and blocks cyt c-catalyzed oxidation of several peroxidase substrates, cyt c self-oxidation, and, most importantly, inhibits cyt c-dependent oxidation of polyunsaturated CL and accumulation of CL hydroperoxides. Mitochondrial targeting of such agents may lead to discovery of new potent drugs. Several options shown on the schema include mitochondria-targeted conjugates of nitroxide radicals (TEMPO) with hemi-gramicidin S (GS) or triphenyl-phosphonium. Specifically, GS-TEMPO is selectively accumulated in mitochondria where it acts as an electron scavenger capable of preventing superoxide formation and its dismutation into H2O2 that is necessary for CL oxidation. GS-TEMPO is also an effective anti-apoptotic agent. Mitochondria-targeted donors of nitric oxide (NO˙) – such as 2-(hydroxyamino-vinyl)-triphenyl-phosphonium (HVTP) - activatable by peroxidase activity of cyt c owe their anti-apoptotic potency to the NO˙-dependent reduction of reactive intermediates of the peroxidase cycle.
The peroxidase reaction of cyt c/CL complexes requires a source of oxidizing equivalents – such as H2O2 – to feed and maintain the peroxidation cycle. It is likely that apoptotic disruption of electron transport and diversion of electron flow to molecular oxygen acts as the major supplier of superoxide radicals and its dismutation product, H2O2. This implies that effective electron scavengers might be potent inhibitors of CL, provided their levels in mitochondria can be increased sufficiently. We took advantage of the well known high effectiveness of stable nitroxide radicals as acceptors of electrons from respiratory carriers [162] and conjugated the electron-scavenging cargo to mitochondria-targeted vehicles. Two different types of vehicles have been employed: (i) fragments of the known antibiotic gramicidin S with high affinity to the mitochondrial inner membrane and (ii) a positively-charged organic cation, triphenylphosphonium, that is readily “electrophorized” into mitochondria due to their membrane potential [163]. In both cases, we found that targeted delivery of nitroxides achieved its goal – re-routing of electron flow from oxygen and preventing superoxide production [164]. As a result, CL peroxidation was blocked in a number of different cell types triggered to apoptosis by different pro-apoptotic stimuli – actinomycin D, staurosporine, or gamma-irradiation [164]. This strategy proved to be successful and resulted in effective prevention of cyt c release into the cytosol, hence protection against apoptosis. Most importantly, in vivo utilization of nitroxide conjugates showed significant protection against hemorrhagic shock induced in rats [165,166] as well as against irradiation of mice (Greenberger et al. unpublished observations). Optimization of this strategy may turn out to bear promise in a number of disease conditions where massive apoptosis represents the major contributor to the mechanisms of pathogenesis.
An alternative strategy to block CL peroxidation may be based on inhibition of the peroxidase function of cyt c/CL complexes. We tested two different pathways with the intent to achieve selectivity of the inhibitory effect. First, we chose to utilize precursors of NO donors which could be activated by the peroxidase function emerging in cyt c during apoptosis upon its interaction with CL. We designed and synthesized several oximes, whose oxidation converts them into NONOates; the latter are known to readily release NO˙ [167]. Indeed, we were able to demonstrate that the peroxidase function of cyt c/CL complexes caused the production of NO˙ from (2-hydroxyamino-vinyl)-triphenylphosphonium (HVTP) [167]. As expected, NO˙ acted as a potent reductant for the reactive peroxidase intermediates and prevented CL peroxidation in model systems and in cells. Experiments with mouse embryonic cells demonstrated that HVTP displayed significant protective potency against apoptosis induced in cells by either actinomycin D or irradiation [167].
Another attractive opportunity to prevent accumulation of CL peroxidation products is to utilize an alternative substrate capable of competing with endogenous CL substrates. If successful, this may re-direct the oxidizing power of cyt c as a peroxidase to non-essential CL-like substrates that will be ineffective in inducing the mitochondrial permeability transition and the release of pro-apoptotic factors from mitochondria. To experimentally explore this concept, we chose to use chemically modified CL, NBD-CL, and demonstrated that this conjugate formed high affinity complexes with cyt c and blocked cyt c-catalyzed oxidation of peroxidase substrates, oxidation of polyunsaturated TLCL and accumulation of TLCL hydroperoxides [34]. Upon incorporation in mitochondria, NBD-CL inhibited peroxidase activity in these organelles and, hence, may act as a promising regulator of apoptosis.
The above examples are only a few of many feasible approaches that may control and regulate peroxidase activity of cyt c towards CL peroxidation. Currently, experiments are underway to utilize effective ligands of cyt c heme to occupy its sixth coordination bond (Met80-Fe) to strongly and irreversibly “knock-out” the peroxidase potential of cyt c/CL complexes.
Creating CL deficiency is another good strategy to achieve increased resistance of cells to apoptosis. As mentioned above, we were successful in creating clones of CL-deficient HeLa cells in which CL content was decreased to 40mol% of its levels in parental cells. This success was associated with a markedly increased resistance to apoptosis induced by gamma-irradiation, rotenone and actinomycin D [128].
Not only inhibition but also stimulation of cyt c/CL peroxidase activity and CL peroxidation may be important for elaborating new therapeutic strategies. As an example, it may be very significant to enhance CL peroxidation in tumor cells, hence trigger their apoptotic machinery towards initiation of programmed cell death. One of potentially important approaches may be based on changing the molecular speciation of cardiolipins to enrich them with highly oxidizable individual chains readily interacting with cyt c. We examined this hypothesis by enriching cells with docosahexaenoic acid and noted that this led to increased sensitivity to proapototic stimulation [20]. Our further attempts are directed towards a selective delivery of the desired precursors of fatty acids into mitochondria using different targeting vehicles.
Future experiments will also elucidate a possible direction of work focused on studies of the CL remodeling pathway – the tafazzine gene – as a potentially important way to regulate CL speciations in targeted tissues, hence manipulating their sensitivity to apoptosis.
Concluding Remarks
The organization of native cyt c favors its most common function as an electron shuttle between complexes III and IV of mitochondria. Hexa-coordination of heme-iron, utilization of not readily oxidizable Met(80) as the distal ligand, lack of Arg and His residues in close proximity to heme, remote location of electron-accepting Trp or Tyr residues – all of these features decrease the occurrence of peroxidase functions in native cyt c. However, the binding of cyt c to anionic phospholipids unfolds the protein and converts it from an electron shuttle into a potent peroxidase. A removal of a relatively weak ligand, Met(80), changes of spin-state and structural rearrangements pave the way for opening of the heme catalytic site to small molecules (including H2O2, FA-OOH) to bolster its catalytic activity to levels comparable to those of genuine peroxidases. In mitochondria, this peroxidase activity displays remarkable specificity towards cardiolipin, causing oxidation as well as hydrolysis of CL-OOH but not other more abundant phospholipids. In cells, this specificity is utilized during the execution of the apoptotic program realized via accumulation of CL-OOH. Normally, the regulation of cyt c as a peroxidase is achieved through the very low availability of CL that prevents the formation of productive cyt c/CL complexes. Upon pro-apoptotic stimulation, phosphorylation and activation of scramblase-3 likely triggers the trans-membrane redistribution of CL facilitating its interactions with cyt c. Phosphorylation of cyt c is another regulatory factor that can fine-tune its interactions with CL. The newly discovered role of cyt c in apoptosis allows the exploitation of this knowledge for drug discovery purposes. Indeed several types of new, mitochondria-targeted compounds have been designed and successfully tested as anti- and/or pro-apoptotic agents based on their ability to manipulate CL peroxidation in cyt c/CL complexes.
Acknowledgments
This work was supported by NIH Grants NIAID U19AI068021, HL70755, HD057587, NS061817, DAMD 17-01-2-637, and R03TW007320, by the Pennsylvania Department of Health SAP 4100027294, and by the Human Frontier Science Program.
List of Abbreviations
- cyt c
Cytochrome c
- CL
cardiolipin
- CL-OOH
hydroperoxy-CL
- MLCL
monolyso-CL
- DLCL
dilyso-CL
- TOCL
tetraoleoyl cardiolipin
- TMCL
tetramyristoyl cardiolipin
- TLCL
tetralinoleoyl CL
- BHCL
bovine heart cardiolipin
- NBD-CL
1,1′,2-trioleoyl-2′-[12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-cardiolipin
- PC
phosphatidylcholine
- PA
phosphatidic acid
- PIP2
dioleoyl-glycero-3-phosphoinositol 4,5-bisphosphate
- PIP3
dioleoyl-glycero-3- phosphoinositol 3,4,5-trisphosphate
- PS
phosphatidylserine
- PG
phosphatidylglycerol
- FRET
fluorescence resonance energy transfer
- SOD
superoxide dismutase
- NO
nitric oxide
- H2O2
hydrogen peroxide
- HRP
horseradish peroxidase
- MPO
myeloperoxidase
- COX
cyclooxygenase
- CcP
cytochrome c peroxidase
- FA-OOH
fatty acid hydroperoxide
- ALCAT1
acyl-CoA:lysocardiolipin acyltransferase 1
- PLS-3
phospholipid scramblase-3
- PKCδ
protein kinase C delta
- IMM
inner mitochondrial membrane
- HVTP
(2-hydroxyamino-vinyl)-triphenyl-phosphonium
Footnotes
Peroxidases are diverse and widespread enzymes capable of two-electron reduction of peroxides at the expense of various oxidizable substrates [4]. There are non-heme peroxidases (thiol peroxidase, NADH peroxidase, etc.) and heme-containing peroxidases. Among the latter are plant enzymes – ascorbate peroxidase, cytochrome c peroxidase etc. – and animal peroxidases - cyclooxygenase superfamily, which includes prostaglandin H synthase (cyclooxygenase), myeloperoxidase, lactoperoxidase and others. Most of heme peroxidases can subtract electrons from specific and nonspecific substrates and generate corresponding free radicals and protein-centered radicals (A– → A•). In addition, some peroxidases (i.e., cyclooxygenase-2) may catalyze only one-electron reduction of peroxides, thus producing O-centered radicals. A similar catalytic mechanism is utilized by various heme proteins including hemoglobin, myoglobin and cytochrome c. Lipids represent one class of physiologically important reducing substrates for peroxidases. Prostraglandin H synthase is capable of specific oxidation of arachidonic acid. Myeloperoxidase is less discriminative towards lipid substrates and catalyzes peroxidation of different lipids [5,6]. Catalytic properties of cyt c/CL complexes and their specificity towards peroxidation of anionic phospholipids – cardiolipin, phosphatidylserine, phsphatidylinositol - are considered in this review.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Halliwell B, Gutteridge J. Free Radicals in Biology and Medicine. Oxford, U.K.: Clarendon Press; 1999. [Google Scholar]
- 2.Skulachev VP. Cytochrome c in the apoptotic and antioxidant cascades. FEBS Lett. 1998;423:275–280. doi: 10.1016/s0014-5793(98)00061-1. [DOI] [PubMed] [Google Scholar]
- 3.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 4.Passardi F, Theiler G, Zamocky M, Cosio C, Rouhier N, Teixera F, Margis-Pinheiro M, Ioannidis V, Penel C, Falquet L, Dunand C. PeroxiBase: the peroxidase database. Phytochemistry. 2007;68:1605–11. doi: 10.1016/j.phytochem.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Rouzer CA, Marnett LJ. Mechanism of free radical oxygenation of polyunsaturated fatty acids by cyclooxygenases. Chem Rev. 2003;103:2239–304. doi: 10.1021/cr000068x. [DOI] [PubMed] [Google Scholar]
- 6.Malle E, Marsche G, Arnhold J, Davies MJ. Modification of low-density lipoprotein by myeloperoxidase-derived oxidants and reagent hypochlorous acid. Biochim Biophys Acta. 2006;1761:392–415. doi: 10.1016/j.bbalip.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 7.Mueller GP, Driscoll WJ. In vitro synthesis of oleoylglycine by cytochrome c points to a novel pathway for the production of lipid signaling molecules. J Biol Chem. 2007;282:22364–22369. doi: 10.1074/jbc.M701801200. [DOI] [PubMed] [Google Scholar]
- 8.Driscoll WJ, Chaturvedi S, Mueller GP. Oleamide synthesizing activity from rat kidney: identification as cytochrome c. J Biol Chem. 2007;282:22353–22363. doi: 10.1074/jbc.M610070200. [DOI] [PubMed] [Google Scholar]
- 9.Gebicka L. Peroxidase-like activity of cytochrome c in the presence of anionic surfactants. Res Chem Intermed. 2001;27:717–723. [Google Scholar]
- 10.Santucci R, Brunori M, Ascoli F. Unfolding and flexibility in hemoproteins shown in the case of carboxymethylated cytochrome c. Biochim Biophys Acta. 1987;914:185–9. doi: 10.1016/0167-4838(87)90062-8. [DOI] [PubMed] [Google Scholar]
- 11.Chen YR, Chen CL, Liu X, Li H, Zweier JL, Mason RP. Involvement of protein radical, protein aggregation, and effects on NO metabolism in the hypochlorite-mediated oxidation of mitochondrial cytochrome c. Free Radic Biol Med. 2004;37:1591–1603. doi: 10.1016/j.freeradbiomed.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 12.Abriata LA, Cassina A, Tórtora V, Marín M, Souza JM, Castro L, Vila AJ, Radi R. Nitration of solvent-exposed tyrosine-74 on cytochrome c triggers heme iron-methionine-80 bond disruption: Nuclear magnetic resonance and optical spectroscopy studies. J Biol Chem. 2008 doi: 10.1074/jbc.M807203200. In press. http://www.jbc.org/cgi/reprint/M807203200v1. [DOI] [PMC free article] [PubMed]
- 13.Pinheiro TJ, Watts A. Lipid specificity in the interaction of cytochrome c with anionic phospholipid bilayers revealed by solid-state 31P NMR. Biochemistry. 1994;33:2451–2458. doi: 10.1021/bi00175a013. [DOI] [PubMed] [Google Scholar]
- 14.de Jongh HH, Ritsema T, Killian JA. Lipid specificity for membrane mediated partial unfolding of cytochrome c. FEBS Lett. 1995;360:255–260. doi: 10.1016/0014-5793(95)00115-p. [DOI] [PubMed] [Google Scholar]
- 15.Sanghera N, Pinheiro TJ. Unfolding and refolding of cytochrome c driven by the interaction with lipid micelles. Protein Sci. 2000;9:1194–1202. doi: 10.1110/ps.9.6.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oellerich S, Lecomte S, Paternostre M, Heimburg T, Hildebrandt P. Peripheral and Integral Binding of Cytochrome c to Phospholipids Vesicles. J Phys Chem B. 2004;108:3871–3878. [Google Scholar]
- 17.Belikova NA, Vladimirov YA, Osipov AN, Kapralov AA, Tyurin VA, Potapovich MV, Basova LV, Peterson J, Kurnikov IV, Kagan VE. Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin-containing membranes. Biochemistry. 2006;45:4998–5009. doi: 10.1021/bi0525573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kapralov AA, Kurnikov IV, Vlasova II, Belikova NA, Tyurin VA, Basova LV, Zhao Q, Tyurina YY, Jiang J, Bayir H, Vladimirov YA, Kagan VE. The Hierarchy of Structural Transitions Induced in Cytochrome c by Anionic Phospholipids Determines Its Peroxidase Activation and Selective Peroxidation during Apoptosis in Cells. Biochemistry. 2007;46:14232–14244. doi: 10.1021/bi701237b. [DOI] [PubMed] [Google Scholar]
- 19.Sinibaldi F, Fiorucci L, Patriarca A, Lauceri R, Ferri T, Coletta M, Santucci R. Insights into cytochrome c-cardiolipin interaction. Role played by ionic strength. Biochemistry. 2008;47:6928–6935. doi: 10.1021/bi800048v. [DOI] [PubMed] [Google Scholar]
- 20.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 21.Belikova NA, Jiang J, Tyurina YY, Zhao Q, Epperly MW, Greenberger J, Kagan VE. Cardiolipin-specific peroxidase reactions of cytochrome C in mitochondria during irradiation-induced apoptosis. Int J Radiat Oncol Biol Phys. 2007;69:176–186. doi: 10.1016/j.ijrobp.2007.03.043. [DOI] [PubMed] [Google Scholar]
- 22.Uversky VN, Dunker AK. Biochemistry. Controlled chaos. Science. 2008;322:1340–1341. doi: 10.1126/science.1167453. [DOI] [PubMed] [Google Scholar]
- 23.Gsponer J, Futschik ME, Teichmann SA, Babu MM. Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science. 2008;322:1365–1368. doi: 10.1126/science.1163581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rytomaa M, Kinnunen PK. Evidence for two distinct acidic phospholipid-binding sites in cytochrome c. J Biol Chem. 1994;269:1770–1774. [PubMed] [Google Scholar]
- 25.Rytomaa M, Kinnunen PK. Reversibility of the binding of cytochrome c to liposomes. Implications for lipid-protein interactions. J Biol Chem. 1995;270:3197–3202. doi: 10.1074/jbc.270.7.3197. [DOI] [PubMed] [Google Scholar]
- 26.Kawai C, Prado FM, Nunes GL, Di Mascio P, Carmona-Ribeiro AM, Nantes IL. pH-Dependent interaction of cytochrome c with mitochondrial mimetic membranes: the role of an array of positively charged amino acids. J Biol Chem. 2005;280:34709–17. doi: 10.1074/jbc.M412532200. [DOI] [PubMed] [Google Scholar]
- 27.Pinheiro TJ, Elove GA, Watts A, Roder H. Structural and kinetic description of cytochrome c unfolding induced by the interaction with lipid vesicles. Biochemistry. 1997;36:13122–13132. doi: 10.1021/bi971235z. [DOI] [PubMed] [Google Scholar]
- 28.Tuominen EK, Wallace CJ, Kinnunen PK. Phospholipid-cytochrome c interaction: evidence for the extended lipid anchorage. J Biol Chem. 2002;277:8822–8826. doi: 10.1074/jbc.M200056200. [DOI] [PubMed] [Google Scholar]
- 29.Nicholls P. Cytochrome c binding to enzymes and membranes. Biochim Biophys Acta. 1974;346:261–310. doi: 10.1016/0304-4173(74)90003-2. [DOI] [PubMed] [Google Scholar]
- 30.Kostrzewa A, Páli T, Froncisz W, Marsh D. Membrane location of spin-labeled cytochrome c determined by paramagnetic relaxation agents. Biochemistry. 2000;39:6066–6074. doi: 10.1021/bi992559l. [DOI] [PubMed] [Google Scholar]
- 31.Kalanxhi E, Wallace CJ. Cytochrome c impaled: investigation of the extended lipid anchorage of a soluble protein to mitochondrial membrane models. Biochem J. 2007;407:179–187. doi: 10.1042/BJ20070459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gorbenko GP, Molotkovsky JG, Kinnunen PK. Cytochrome C interaction with cardiolipin/phosphatidylcholine model membranes: effect of cardiolipin protonation. Biophys J. 2006;90:4093–4103. doi: 10.1529/biophysj.105.080150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chattopadhyay K, Mazumdar S. Stabilization of partially folded states of cytochrome c in aqueous surfactant: effects of ionic and hydrophobic interactions. Biochemistry. 2003;42:14606–14613. doi: 10.1021/bi0351662. [DOI] [PubMed] [Google Scholar]
- 34.Borisenko G, Kapralov A, Tyurin V, Maeda A, Stoyanovsky D, Kagan V. Molecular design of new inhibitors of peroxidase activity of cyt c/cardiolipin complexes: Fluorescent oxadiazol-derivatized cardiolipin. Biochemistry. 2009 doi: 10.1021/bi801507s. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muga A, Mantsch HH, Surewicz WK. Membrane binding induces destabilization of cytochrome c structure. Biochemistry. 1991;30:7219–7224. doi: 10.1021/bi00243a025. [DOI] [PubMed] [Google Scholar]
- 36.Spooner PJ, Watts A. Cytochrome c interactions with cardiolipin in bilayers: a multinuclear magic-angle spinning NMR study. Biochemistry. 1992;31:10129–10138. doi: 10.1021/bi00156a037. [DOI] [PubMed] [Google Scholar]
- 37.Maity H, Maity M, Englander SW. How cytochrome c folds, and why: submolecular foldon units and their stepwise sequential stabilization. J Mol Biol. 2004;343:223–233. doi: 10.1016/j.jmb.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 38.Berners-Price SJ, Bertini I, Gray HB, Spyroulias GA, Turano P. The stability of the cytochrome c scaffold as revealed by NMR spectroscopy. J Inorg Biochem. 2004;98:814–823. doi: 10.1016/j.jinorgbio.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 39.Naeem A, Khan RH. Characterization of molten globule state of cytochrome c at alkaline, native and acidic pH induced by butanol and SDS. Int J Biochem Cell Biol. 2004;36:2281–2292. doi: 10.1016/j.biocel.2004.04.023. [DOI] [PubMed] [Google Scholar]
- 40.Varhac R, Antalik M, Bano M. Effect of temperature and guanidine hydrochloride on ferrocytochrome c at neutral pH. J Biol Inorg Chem. 2004;9:12–22. doi: 10.1007/s00775-003-0492-1. [DOI] [PubMed] [Google Scholar]
- 41.Wen X, Patel KM, Russell BS, Bren KL. Effects of heme pocket structure and mobility on cytochrome c stability. Biochemistry. 2007;46:2537–2544. doi: 10.1021/bi602380v. [DOI] [PubMed] [Google Scholar]
- 42.Tsou CL. Cytochrome c modified by digestion with proteolytic enzymes. 1. Digestion. Biochem J. 1951;49:362–7. doi: 10.1042/bj0490362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marques HM, Perry CB. Hemepeptide models for hemoproteins: the behavior of N-acetylmicroperoxidase-11 in aqueous solution. J Inorg Biochem. 1999;75:281–91. doi: 10.1016/s0162-0134(99)00100-2. [DOI] [PubMed] [Google Scholar]
- 44.Rytomaa M, Mustonen P, Kinnunen PKJ. Reversible, nonionic, and pH-dependent association of cytochrome c with cardiolipin-phosphatidylcholine liposomes. J Biol Chem. 1992;267:22243–22248. [PubMed] [Google Scholar]
- 45.Maity H, Maity M, Krishna MM, Mayne L, Englander SW. Protein folding: the stepwise assembly of foldon units. Proc Natl Acad Sci U S A. 2005;102:4741–4746. doi: 10.1073/pnas.0501043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maity H, Rumbley JN, Englander SW. Functional role of a protein foldon--an Omega-loop foldon controls the alkaline transition in ferricytochrome c. Proteins. 2006;63:349–355. doi: 10.1002/prot.20757. [DOI] [PubMed] [Google Scholar]
- 47.Krishna MM, Maity H, Rumbley JN, Lin Y, Englander SW. Order of steps in the cytochrome C folding pathway: evidence for a sequential stabilization mechanism. J Mol Biol. 2006;359:1410–1419. doi: 10.1016/j.jmb.2006.04.035. [DOI] [PubMed] [Google Scholar]
- 48.Diederix RE, Ubbink M, Canters GW. Peroxidase activity as a tool for studying the folding of c-type cytochromes. Biochemistry. 2002;41:13067–13077. doi: 10.1021/bi0260841. [DOI] [PubMed] [Google Scholar]
- 49.Prasad S, Maiti NC, Mazumdar S, Mitra S. Reaction of hydrogen peroxide and peroxidase activity in carboxymethylated cytochrome c: spectroscopic and kinetic studies. Biochim Biophys Acta. 2002;1596:63–75. doi: 10.1016/s0167-4838(02)00205-4. [DOI] [PubMed] [Google Scholar]
- 50.Cassina AM, Hodara R, Souza JM, Thomson L, Castro L, Ischiropoulos H, Freeman BA, Radi R. Cytochrome c nitration by peroxynitrite. J Biol Chem. 2000;275:21409–21415. doi: 10.1074/jbc.M909978199. [DOI] [PubMed] [Google Scholar]
- 51.Chen YR, Deterding LJ, Sturgeon BE, Tomer KB, Mason RP. Protein oxidation of cytochrome C by reactive halogen species enhances its peroxidase activity. J Biol Chem. 2002;277:29781–29791. doi: 10.1074/jbc.M200709200. [DOI] [PubMed] [Google Scholar]
- 52.Marques HM. Insights into porphyrin chemistry provided by the microperoxidases, the haempeptides derived from cytochrome c. Dalton Trans. 2007;39:4371–85. doi: 10.1039/b710940g. [DOI] [PubMed] [Google Scholar]
- 53.Everse J, Everse KE, Grisham MB. Peroxidases in Chemistry and Biology. II. Boca Raton: CRC Press; 1991. [Google Scholar]
- 54.Poulos TL, Kraut J. A hypothetical model of the cytochrome c peroxidase.cytochrome c electron transfer complex. J Biol Chem. 1980;255:10322–10330. [PubMed] [Google Scholar]
- 55.Rodriguez-Lopez JN, Smith AT, Thorneley RN. Role of arginine 38 in horseradish peroxidase. A critical residue for substrate binding and catalysis. J Biol Chem. 1996;271:4023–4030. doi: 10.1074/jbc.271.8.4023. [DOI] [PubMed] [Google Scholar]
- 56.Hiner AN, Raven EL, Thorneley RN, Garcia-Canovas F, Rodriguez-Lopez JN. Mechanisms of compound I formation in heme peroxidases. J Inorg Biochem. 2002;91:27–34. doi: 10.1016/s0162-0134(02)00390-2. [DOI] [PubMed] [Google Scholar]
- 57.Rodriguez-Lopez JN, Lowe DJ, Hernandez-Ruiz J, Hiner AN, Garcia-Canovas F, Thorneley RN. Mechanism of Reaction of Hydrogen Peroxide with Horseradish Peroxidase: Identification of Intermediates in the Catalytic Cycle. J Am Chem Soc. 2001;123:11838–11847. doi: 10.1021/ja011853+. [DOI] [PubMed] [Google Scholar]
- 58.Newmyer SL, Sun J, Loehr TM, OrtizdeMontellano PR. Rescue of the horseradish peroxidase His-170-->Ala mutant activity by imidazole: importance of proximal ligand tethering. Biochemistry. 1996;35:12788–12795. doi: 10.1021/bi9609331. [DOI] [PubMed] [Google Scholar]
- 59.Florence TM. The degradation of cytochrome c by hydrogen peroxide. J Inorg Biochem. 1985;23:131–141. doi: 10.1016/0162-0134(85)83017-8. [DOI] [PubMed] [Google Scholar]
- 60.Radi R, Thomson L, Rubbo H, Prodanov E. Cytochrome c-catalyzed oxidation of organic molecules by hydrogen peroxide. Arch Biochem Biophys. 1991;288:112–117. doi: 10.1016/0003-9861(91)90171-e. [DOI] [PubMed] [Google Scholar]
- 61.Bernad S, Oellerich S, Soulimane T, Noinville S, Baron MH, Paternostre M, Lecomte S. Interaction of horse heart and thermus thermophilus type c cytochromes with phospholipid vesicles and hydrophobic surfaces. Biophys J. 2004;86:3863–3872. doi: 10.1529/biophysj.103.025114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Droghetti E, Oellerich S, Hildebrandt P, Smulevich G. Heme coordination states of unfolded ferrous cytochrome C. Biophys J. 2006;91:3022–3031. doi: 10.1529/biophysj.105.079749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dunford HB, Stillman JS. On the function and mechanism of action of peroxidases. Coord Chem Reviews. 1976;19:187–251. [Google Scholar]
- 64.Basova LV, Kurnikov IV, Wang L, Ritov VB, Belikova NA, Vlasova II, Pacheco AA, Winnica DE, Peterson J, Bayir H, Waldeck DH, Kagan VE. Cardiolipin switch in mitochondria: shutting off the reduction of cytochrome c and turning on the peroxidase activity. Biochemistry. 2007;46:3423–3434. doi: 10.1021/bi061854k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gunsalus IC, Meeks JR, Lipscomb JD, Debrunner P, Munk E. Molecular Mechanism of Oxygen Activation. New York: Academic Press; 1974. [Google Scholar]
- 66.Adachi S, Nagano S, Ishimori K, Watanabe Y, Morishima I. Roles of Proximal Ligand in Heme Proteins: Replacement of Proximal Histidine of Human Myoglobin with Cysteine and Tyrosine by Site-Directed Mutagenesis as Models for P-450, Chloroperoxidase, and Catalase. Biochemistry. 1993;32:241–252. doi: 10.1021/bi00052a031. [DOI] [PubMed] [Google Scholar]
- 67.Di Marino M, Marassi R, Santucci R, Brunori M, Ascoli F. A Spectroelectrochemical Study Of Carboxymethylated Cytochrome-c. J Electroanal Chem. 1987;17:27–34. [Google Scholar]
- 68.Barker PD, Nerou EP, Cheesman MR, Thomson AJ, de Oliveira P, Hill HA. Bis-Methionine Ligation to Heme Iron in Mutants of Cytochrome b562. 1. Spectroscopic and Electrochemical Characterization of the Electronic Properties. Biochemistry. 1996;35:13618–13626. doi: 10.1021/bi961127x. [DOI] [PubMed] [Google Scholar]
- 69.Bakovic M, Dunford HB. Reactions of prostaglandin endoperoxide synthase and its compound I with hydroperoxides. J Biol Chem. 1996;271:2048–2056. doi: 10.1074/jbc.271.4.2048. [DOI] [PubMed] [Google Scholar]
- 70.Chubb AJ, Fitzgerald DJ, Nolan KB, Moman E. The productive conformation of prostaglandin G2 at the peroxidase site of prostaglandin endoperoxide H synthase: docking.; molecular dynamics.; and site-directed mutagenesis studies. Biochemistry. 2006;45:811–820. doi: 10.1021/bi051973k. [DOI] [PubMed] [Google Scholar]
- 71.Liu J, Seibold SA, Rieke CJ, Song I, Cukier RI, Smith WL. Prostaglandin endoperoxide H synthases: peroxidase hydroperoxide specificity and cyclooxygenase activation. J Biol Chem. 2007;282:18233–18244. doi: 10.1074/jbc.M701235200. [DOI] [PubMed] [Google Scholar]
- 72.Ichimura S, Uchida T, Taniguchi S, Hira S, Tosha T, Morishima I, Kitagawa T, Ishimori K. Unique peroxidase reaction mechanism in prostaglandin endoperoxide H synthase-2: compound I in prostaglandin endoperoxide H synthase-2 can be formed without assistance by distal glutamine residue. J Biol Chem. 2007;282:16681–16690. doi: 10.1074/jbc.M610785200. [DOI] [PubMed] [Google Scholar]
- 73.Shimizu T, Murakami Y, Hatano M. Glu318 and Thr319 mutations of cytochrome P450 1A2 remarkably enhance homolytic O-O cleavage of alkyl hydroperoxides. An optical absorption spectral study. J Biol Chem. 1994;269:13296–304. [PubMed] [Google Scholar]
- 74.Van der Zee J, Barr DP, Mason RP. ESR spin trapping investigation of radical formation from the reaction between hematin and tert-Butyl hydroperoxide. Free Radic Biol Med. 1996;20:199–206. doi: 10.1016/0891-5849(95)02031-4. [DOI] [PubMed] [Google Scholar]
- 75.Allentoff A, Bolton J, Wilks A, Thompson J, Ortiz de Montellano P. Heterolytic versus homolytic peroxide bond cleavage by sperm whale myoglobin and myoglobin mutants. J Am Chem Soc. 1992;114:9744–9749. [Google Scholar]
- 76.Egawa T, Yoshioka S, Takahashi S, Hori H, Nagano S, Shimada H, Ishimori K, Morishima I, Suematsu M, Ishimura Y. Kinetic and spectroscopic characterization of a hydroperoxy compound in the reaction of native myoglobin with hydrogen peroxide. J Biol Chem. 2003;278:41597–606. doi: 10.1074/jbc.M210383200. [DOI] [PubMed] [Google Scholar]
- 77.Chamulitrat W, Hughes MF, Eling TE, Mason RP. Superoxide and peroxyl radical generation from the reduction of polyunsaturated fatty acid hydroperoxides by soybean lipoxygenase. Arch Biochem Biophys. 1991;290:153–159. doi: 10.1016/0003-9861(91)90601-e. [DOI] [PubMed] [Google Scholar]
- 78.Rota C, Barr DP, Martin MV, Guengerich FP, Tomasi A, Mason RP. Detection of free radicals produced from the reaction of cytochrome P-450 with linoleic acid hydroperoxide. Biochem J. 1997;328(Pt 2):565–571. doi: 10.1042/bj3280565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Iwahashi H, Nishizaki K, Takagi I. Cytochrome c catalyses the formation of pentyl radical and octanoic acid radical from linoleic acid hydroperoxide. Biochem J. 2002;361:57–66. doi: 10.1042/0264-6021:3610057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barr DP, Gunther MR, Deterding LJ, Tomer KB, Mason RP. ESR spin-trapping of a protein-derived tyrosyl radical from the reaction of cytochrome c with hydrogen peroxide. J Biol Chem. 1996;271:15498–15503. doi: 10.1074/jbc.271.26.15498. [DOI] [PubMed] [Google Scholar]
- 81.Kagan VE, Borisenko GG, Tyurina YY, Tyurin VA, Jiang J, Potapovich AI, Kini V, Amoscato A. Oxidative lipidomics of apoptosis: redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic Biol Med. 2004;37:1963–1985. doi: 10.1016/j.freeradbiomed.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 82.Tyurin VA, Tyurina YY, Kochanek PM, Hamilton R, DeKosky ST, Greenberger JS, Bayir H, Kagan VE. Oxidative lipidomics of programmed cell death. Methods Enzymol. 2008;442:375–393. doi: 10.1016/S0076-6879(08)01419-5. [DOI] [PubMed] [Google Scholar]
- 83.Tyurina YY, Tyurin VA, Epperly MW, Greenberger JS, Kagan VE. Oxidative lipidomics of gamma-irradiation-induced intestinal injury. Free Radic Biol Med. 2008;44:299–314. doi: 10.1016/j.freeradbiomed.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 84.Landino LM, Crews BC, Gierse JK, Hauser SD, Marnett LJ. Mutational analysis of the role of the distal histidine and glutamine residues of prostaglandin-endoperoxide synthase-2 in peroxidase catalysis, hydroperoxide reduction, and cyclooxygenase activation. J Biol Chem. 1997;272:21565–21574. doi: 10.1074/jbc.272.34.21565. [DOI] [PubMed] [Google Scholar]
- 85.Qian SY, Chen YR, Deterding LJ, Fann YC, Chignell CF, Tomer KB, Mason RP. Identification of protein-derived tyrosyl radical in the reaction of cytochrome c and hydrogen peroxide: characterization by ESR spin-trapping, HPLC and MS. Biochem J. 2002;363:281–288. doi: 10.1042/0264-6021:3630281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Detweiler CD, Lardinois OM, Deterding LJ, de Montellano PR, Tomer KB, Mason RP. Identification of the myoglobin tyrosyl radical by immuno-spin trapping and its dimerization. Free Radic Biol Med. 2005;38:969–976. doi: 10.1016/j.freeradbiomed.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 87.Svistunenko DA. Reaction of haem containing proteins and enzymes with hydroperoxides: the radical view. Biochim Biophys Acta. 2005;1707:127–155. doi: 10.1016/j.bbabio.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 88.Ehrenshaft M, Mason RP. Protein radical formation on thyroid peroxidase during turnover as detected by immuno-spin trapping. Free Radic Biol Med. 2006;41:422–430. doi: 10.1016/j.freeradbiomed.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 89.Smith WL, Song I. The enzymology of prostaglandin endoperoxide H synthases-1 and -2. Prostaglandins Other Lipid Mediat. 2002;68-69:115–128. doi: 10.1016/s0090-6980(02)00025-4. [DOI] [PubMed] [Google Scholar]
- 90.Tsong TY. Ferricytochrome c chain folding measured by the energy transfer of tryptophan 59 to the heme group. Biochemistry. 1976;15:5467–5473. doi: 10.1021/bi00670a007. [DOI] [PubMed] [Google Scholar]
- 91.Keszler A, Mason RP, Hogg N. Immuno-spin trapping of hemoglobin and myoglobin radicals derived from nitrite-mediated oxidation. Free Radic Biol Med. 2006;40:507–515. doi: 10.1016/j.freeradbiomed.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 92.Tyurina YY, Kini V, Tyurin VA, Vlasova II, Jiang J, Kapralov AA, Belikova NA, Yalowich JC, Kurnikov IV, Kagan VE. Mechanisms of cardiolipin oxidation by cytochrome c: relevance to pro- and antiapoptotic functions of etoposide. Mol Pharmacol. 2006;70:706–717. doi: 10.1124/mol.106.022731. [DOI] [PubMed] [Google Scholar]
- 93.Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J Bioenerg Biomembr. 1999;31:347–66. doi: 10.1023/a:1005427919188. [DOI] [PubMed] [Google Scholar]
- 94.Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 2004;279:49064–73. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 95.Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Rådmark O, Wurst W, Bornkamm GW, Schweizer U, Conrad M. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–48. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 96.Toninello A, Pietrangeli P, De Marchi U, Salvi M, Mondovì B. Amine oxidases in apoptosis and cancer. Biochim Biophys Acta. 2006;1765:1–13. doi: 10.1016/j.bbcan.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 97.Nilsson OS, Dallner G. Transverse asymmetry of phospholipids in subcellular membranes of rat liver. Biochim Biophys Acta. 1977;464:453–458. doi: 10.1016/0005-2736(77)90019-0. [DOI] [PubMed] [Google Scholar]
- 98.Sperka-Gottlieb CD, Hermetter A, Paltauf F, Daum G. Lipid topology and physical properties of the outer mitochondrial membrane of the yeast, Saccharomyces cerevisiae. Biochim Biophys Acta. 1988;946:227–234. doi: 10.1016/0005-2736(88)90397-5. [DOI] [PubMed] [Google Scholar]
- 99.Gallet PF, Petit JM, Maftah A, Zachowski A, Julien R. Asymmetrical distribution of cardiolipin in yeast inner mitochondrial membrane triggered by carbon catabolite repression. Biochem J. 1997;324(Pt 2):627–634. doi: 10.1042/bj3240627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Daum G. Lipids of mitochondria. Biochim Biophys Acta. 1985;822:1–42. doi: 10.1016/0304-4157(85)90002-4. [DOI] [PubMed] [Google Scholar]
- 101.Ardail D, Privat JP, Egret-Charlier M, Levrat C, Lerme F, Louisot P. Mitochondrial contact sites. Lipid composition and dynamics. J Biol Chem. 1990;265:18797–18802. [PubMed] [Google Scholar]
- 102.Hovius R, Thijssen J, van der Linden P, Nicolay K, de Kruijff B. Phospholipid asymmetry of the outer membrane of rat liver mitochondria. Evidence for the presence of cardiolipin on the outside of the outer membrane. FEBS Lett. 1993;330:71–76. doi: 10.1016/0014-5793(93)80922-h. [DOI] [PubMed] [Google Scholar]
- 103.Tyurin VA, Tyurina YY, Osipov AN, Belikova NA, Basova LV, Kapralov AA, Bayir H, Kagan VE. Interactions of cardiolipin and lyso-cardiolipins with cytochrome c and tBid: conflict or assistance in apoptosis. Cell Death Differ. 2007;14:872–875. doi: 10.1038/sj.cdd.4402068. [DOI] [PubMed] [Google Scholar]
- 104.Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg M, Schagger H. Cardiolipin stabilizes respiratory chain supercomlexes. J Biol Chem. 2003;278:52873–52880. doi: 10.1074/jbc.M308366200. [DOI] [PubMed] [Google Scholar]
- 105.Sharpley MS, Shannon RJ, Draghi F, Hirst J. Interactions between phospholipids and nadh:ubiquinone oxidoreductase (complex i) from bovine mitochondria. Biochemistry. 2006;45:241–248. doi: 10.1021/bi051809x. [DOI] [PubMed] [Google Scholar]
- 106.Garcia Fernandez M, Troiano L, Moretti L, Nasi M, Pinti M, Salvioli S, Dobrucki J, Cossarizza A. Early changes in intramitochondrial cardiolipin distribution during apoptosis. Cell Growth Differ. 2002;13:449–455. [PubMed] [Google Scholar]
- 107.Liu J, Dai Q, Chen J, Durrant D, Freeman A, Liu T, Grossman D, Lee RM. Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol Cancer Res. 2003;1:892–902. [PubMed] [Google Scholar]
- 108.Gonzalvez F, Pariselli F, Dupaigne P, Budihardjo I, Lutter M, Antonsson B, Diolez P, Manon S, Martinou JC, Goubern M, Wang X, Bernard S, Petit PX. tBid interaction with cardiolipin primarily orchestrates mitochondrial dysfunctions and subsequently activates Bax and Bak. Cell Death Differ. 2005;12:614–626. doi: 10.1038/sj.cdd.4401571. [DOI] [PubMed] [Google Scholar]
- 109.Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol. 2000;2:754–761. doi: 10.1038/35036395. [DOI] [PubMed] [Google Scholar]
- 110.Esposti MD, Cristea IM, Gaskell SJ, Nakao Y, Dive C. Proapoptotic Bid binds to monolysocardiolipin, a new molecular connection between mitochondrial membranes and cell death. Cell Death Differ. 2003;10:1300–1309. doi: 10.1038/sj.cdd.4401306. [DOI] [PubMed] [Google Scholar]
- 111.Kim TH, Zhao Y, Ding WX, Shin JN, He X, Seo YW, Chen J, Rabinowich H, Amoscato AA, Yin XM. Bid-cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome C release. Mol Biol Cell. 2004;15:3061–3072. doi: 10.1091/mbc.E03-12-0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Van Mau N, Kajava AV, Bonfils C, Martinou JC, Harricane MC. Interactions of Bax and tBid with lipid monolayers. J Membr Biol. 2005;207:1–9. doi: 10.1007/s00232-005-0799-7. [DOI] [PubMed] [Google Scholar]
- 113.Van Q, Liu J, Lu B, Feingold KR, Shi Y, Lee RM, Hatch GM. Phospholipid scramblase-3 regulates cardiolipin de novo biosynthesis and its resynthesis in growing HeLa cells. Biochem J. 2007;401:103–109. doi: 10.1042/BJ20060373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wiedmer T, Zhou Q, Kwoh DY, Sims PJ. Identification of three new members of the phospholipid scramblase gene family. Biochim Biophys Acta. 2000;1467:244–53. doi: 10.1016/s0005-2736(00)00236-4. [DOI] [PubMed] [Google Scholar]
- 115.Liu J, Chen J, Dai Q, Lee RM. Phospholipid scramblase 3 is the mitochondrial target of protein kinase C delta-induced apoptosis. Cancer Res. 2003;63:1153–1156. [PubMed] [Google Scholar]
- 116.He Y, Liu J, Grossman D, Durrant D, Sweatman T, Lothstein L, Epand RF, Epand RM, Lee RM. Phosphorylation of mitochondrial phospholipid scramblase 3 by protein kinase C-delta induces its activation and facilitates mitochondrial targeting of tBid. J Cell Biochem. 2007;101:1210–12121. doi: 10.1002/jcb.21243. [DOI] [PubMed] [Google Scholar]
- 117.Schlame M, Ren M, Xu Y, Greenberg ML, Haller I. Molecular symmetry in mitochondrial cardiolipins. Chem Phys Lipids. 2005;138:38–49. doi: 10.1016/j.chemphyslip.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 118.Han X, Yang K, Yang J, Cheng H, Gross RW. Shotgun lipidomics of cardiolipin molecular species in lipid extracts of biological samples. J Lipid Res. 2006;47:864–879. doi: 10.1194/jlr.D500044-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang HY, Jackson SN, Woods AS. Direct MALDI-MS analysis of cardiolipin from rat organs sections. J Am Soc Mass Spectrom. 2007;18:567–577. doi: 10.1016/j.jasms.2006.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bayir H, Tyurin VA, Tyurina YY, Viner R, Ritov V, Amoscato AA, Zhao Q, Zhang XJ, Janesko-Feldman KL, Alexander H, Basova LV, Clark RS, Kochanek PM, Kagan VE. Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis. Ann Neurol. 2007;62:154–169. doi: 10.1002/ana.21168. [DOI] [PubMed] [Google Scholar]
- 121.Cheng H, Mancuso DJ, Jiang X, Guan S, Yang J, Yang K, Sun G, Gross RW, Han X. Shotgun lipidomics reveals the temporally dependent, highly diversified cardiolipin profile in the mammalian brain: temporally coordinated postnatal diversification of cardiolipin molecular species with neuronal remodeling. Biochemistry. 2008;47:5869–5880. doi: 10.1021/bi7023282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kiebish MA, Han X, Cheng H, Lunceford A, Clarke CF, Moon H, Chuang JH, Seyfried TN. Lipidomic analysis and electron transport chain activities in C57BL/6J mouse brain mitochondria. J Neurochem. 2008;106:299–312. doi: 10.1111/j.1471-4159.2008.05383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schlame M, Rua D, Greenberg ML. The biosynthesis and functional role of cardiolipin. Prog Lipid Res. 2000;39:257–288. doi: 10.1016/s0163-7827(00)00005-9. [DOI] [PubMed] [Google Scholar]
- 124.Schlame M, Haldar D. Cardiolipin is synthesized on the matrix side of the inner membrane in rat liver mitochondria. J Biol Chem. 1993;268:74–79. [PubMed] [Google Scholar]
- 125.Zhao M, Schlame M, Rua D, Greenberg ML. Cardiolipin synthase is associated with a large complex in yeast mitochondria. J Biol Chem. 1998;273:2402–2408. doi: 10.1074/jbc.273.4.2402. [DOI] [PubMed] [Google Scholar]
- 126.Chen D, Zhang XY, Shi Y. Identification and functional characterization of hCLS1, a human cardiolipin synthase localized in mitochondria. Biochem J. 2006;398:169–176. doi: 10.1042/BJ20060303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Choi SY, Gonzalvez F, Jenkins GM, Slomianny C, Chretien D, Arnoult D, Petit PX, Frohman MA. Cardiolipin deficiency releases cytochrome c from the inner mitochondrial membrane and accelerates stimuli-elicited apoptosis. Cell Death Differ. 2007;14:597–606. doi: 10.1038/sj.cdd.4402020. [DOI] [PubMed] [Google Scholar]
- 128.Huang Z, Jiang J, Tyurin VA, Zhao Q, Mnuskin A, Ren J, Belikova NA, Feng W, Kurnikov IV, Kagan VE. Cardiolipin deficiency leads to decreased cardiolipin peroxidation and increased resistance of cells to apoptosis. Free Radic Biol Med. 2008;44:1935–1944. doi: 10.1016/j.freeradbiomed.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gonzalvez F, Schug ZT, Houtkooper RH, MacKenzie ED, Brooks DG, Wanders RJ, Petit PX, Vaz FM, Gottlieb E. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol. 2008;183:681–696. doi: 10.1083/jcb.200803129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ma BJ, Taylor WA, Dolinsky VW, Hatch GM. Acylation of monolysocardiolipin in rat heart. J Lipid Res. 1999;40:1837–1845. [PubMed] [Google Scholar]
- 131.Danos M, Taylor WA, Hatch GM. Mitochondrial monolysocardiolipin acyltransferase is elevated in the surviving population of H9c2 cardiac myoblast cells exposed to 2-deoxyglucose-induced apoptosis. Biochem Cell Biol. 2008;86:11–20. doi: 10.1139/o07-156. [DOI] [PubMed] [Google Scholar]
- 132.Taylor WA, Xu FY, Ma BJ, Mutter TC, Dolinsky VW, Hatch GM. Expression of monolysocardiolipin acyltransferase activity is regulated in concert with the level of cardiolipin and cardiolipin biosynthesis in the mammalian heart. BMC Biochem. 2002;3:9. doi: 10.1186/1471-2091-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Taylor WA, Hatch GM. Purification and characterization of monolysocardiolipin acyltransferase from pig liver mitochondria. J Biol Chem. 2003;278:12716–12721. doi: 10.1074/jbc.M210329200. [DOI] [PubMed] [Google Scholar]
- 134.Cao J, Liu Y, Lockwood J, Burn P, Shi Y. A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. J Biol Chem. 2004;279:31727–31734. doi: 10.1074/jbc.M402930200. [DOI] [PubMed] [Google Scholar]
- 135.Xu Y, Malhotra A, Ren M, Schlame M. The enzymatic function of tafazzin. J Biol Chem. 2006;281:39217–39224. doi: 10.1074/jbc.M606100200. [DOI] [PubMed] [Google Scholar]
- 136.Schlame M, Ren M. Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 2006;580:5450–5455. doi: 10.1016/j.febslet.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 137.Nigam S, Schewe T. Phospholipase A(2)s and lipid peroxidation. Biochim Biophys Acta. 2000;1488:167–181. doi: 10.1016/s1388-1981(00)00119-0. [DOI] [PubMed] [Google Scholar]
- 138.McGinley CM, van der Donk WA. Enzymatic hydrogen atom abstraction from polyunsaturated fatty acids. Chem Commun (Camb) 2003;(23):2843–2846. doi: 10.1039/b311008g. [DOI] [PubMed] [Google Scholar]
- 139.Bazan NG. Neuroprotectin D1 (NPD1): a DHA-derived mediator that protects brain and retina against cell injury-induced oxidative stress. Brain Pathol. 2005;15:159–166. doi: 10.1111/j.1750-3639.2005.tb00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bazan NG. Omega-3 fatty acids, pro-inflammatory signaling and neuroprotection. Curr Opin Clin Nutr Metab Care. 2007;10:136–141. doi: 10.1097/MCO.0b013e32802b7030. [DOI] [PubMed] [Google Scholar]
- 141.Bazan NG. Neurotrophins induce neuroprotective signaling in the retinal pigment epithelial cell by activating the synthesis of the anti-inflammatory and anti-apoptotic neuroprotectin D1. Adv Exp Med Biol. 2008;613:39–44. doi: 10.1007/978-0-387-74904-4_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lambert IH, Pedersen SF, Poulsen KA. Activation of PLA2 isoforms by cell swelling and ischaemia/hypoxia. Acta Physiol (Oxf) 2006;187:75–85. doi: 10.1111/j.1748-1716.2006.01557.x. [DOI] [PubMed] [Google Scholar]
- 143.Schneider C, Pratt DA, Porter NA, Brash AR. Control of oxygenation in lipoxygenase and cyclooxygenase catalysis. Chem Biol. 2007;14:473–488. doi: 10.1016/j.chembiol.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tyurina YY, Kawai K, Tyurin VA, Liu SX, Kagan VE, Fabisiak JP. The plasma membrane is the site of selective phosphatidylserine oxidation during apoptosis: role of cytochrome c. Antioxid Redox Signal. 2004;6:209–225. doi: 10.1089/152308604322899288. [DOI] [PubMed] [Google Scholar]
- 146.Tyurina YY, Serinkan FB, Tyurin VA, Kini V, Yalowich JC, Schroit AJ, Fadeel B, Kagan VE. Lipid antioxidant, etoposide, inhibits phosphatidylserine externalization and macrophage clearance of apoptotic cells by preventing phosphatidylserine oxidation. J Biol Chem. 2004;279:6056–6064. doi: 10.1074/jbc.M309929200. [DOI] [PubMed] [Google Scholar]
- 147.Tyurin VA, Tyurina YY, Feng W, Mnuskin A, Jiang J, Tang M, Zhang X, Zhao Q, Kochanek PM, Clark RS, Bayir H, Kagan VE. Mass-spectrometric characterization of phospholipids and their primary peroxidation products in rat cortical neurons during staurosporine-induced apoptosis. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05728.x. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jiang J, Huang Z, Zhao Q, Feng W, Belikova NA, Kagan VE. Interplay between bax, reactive oxygen species production, and cardiolipin oxidation during apoptosis. Biochem Biophys Res Commun. 2008;368:145–50. doi: 10.1016/j.bbrc.2008.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tyurina YY, Wasserloos KJ, Stewart RC, Stitt MS, Kaynar A, Pitt BR, Kagan VE. Oxidative lipidomics of hyperoxic lung injury. 47th Annual Meeting, Society of Toxicology; Seattle, WA. The Toxicologist; 2008. p. 1213. [Google Scholar]
- 150.Tyurin VA, Jung MY, Tyurina YY, Prieto T, Zhao Q, Kapralov AA, Belikova NA, Nantes IL, Kagan VE. Cardiolipin oxidation, hydrolysis and accumulation of monolysocardiolipins and oxidized free fatty acids during apoptosis: role of cytochrome c. 48th Annual Meeting, Society of Toxicology; Baltimore, Maryland. The Toxicologist; 2009. [Google Scholar]
- 151.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 152.Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–312. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 153.Kim KW, Mutter RW, Cao C, Albert JM, Freeman M, Hallahan DE, Lu B. Autophagy for cancer therapy through inhibition of pro-apoptotic proteins and mammalian target of rapamycin signaling. J Biol Chem. 2006;281:36883–36890. doi: 10.1074/jbc.M607094200. [DOI] [PubMed] [Google Scholar]
- 154.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a nonapoptotic programmed cell death dependent on autophagy genes. Nature Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 155.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Xu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage cell death. J Biol Chem. 2006;281:19179–19187. doi: 10.1074/jbc.M513377200. [DOI] [PubMed] [Google Scholar]
- 157.Xue L, Fletcher GC, Tolkovsky AM. Autophagy is activated by apoptotic signalling in sympathetic neurons: an alternative mechanism of death execution. Mol Cell Neurosci. 1999;14:180–198. doi: 10.1006/mcne.1999.0780. [DOI] [PubMed] [Google Scholar]
- 158.Xue L, Fletcher GC, Tolkovsky AM. Mitochondria are selectively eliminated from eukaryotic cells after blockade of caspases during apoptosis. Curr Biol. 2001;11:361–365. doi: 10.1016/s0960-9822(01)00100-2. [DOI] [PubMed] [Google Scholar]
- 159.Kissová I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem. 2004;279:39068–39074. doi: 10.1074/jbc.M406960200. [DOI] [PubMed] [Google Scholar]
- 160.Kissová I, Deffieu M, Samokhvalov V, Velours G, Bessoule JJ, Manon S, Camougrand N. Lipid oxidation and autophagy in yeast. Free Radic Biol Med. 2006;41:1655–1661. doi: 10.1016/j.freeradbiomed.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 161.Dadakhujaev S, Noh HS, Jung EJ, Hah YS, Kim CJ, Kim DR. The reduced catalase expression in TrkA-induced cells leads to autophagic cell death via ROS accumulation. Exp Cell Res. 2008 In press. [Google Scholar]
- 162.Chapman DA, Killian GJ, Gelerinter E, Jarrett MT. Reduction of the spin-label TEMPONE by ubiquinol in the electron transport chain of intact rabbit spermatozoa. Biol Reprod. 1985;32:884–893. doi: 10.1095/biolreprod32.4.884. [DOI] [PubMed] [Google Scholar]
- 163.Murphy MP, Smith RAJ. Targeting Antioxidants to Mitochondria by Conjugation to Lipophilic Cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 164.Jiang J, Belikova NA, Hoye AT, Zhao Q, Epperly MW, Greenberger JS, Wipf P, Kagan VE. A mitochondria-targeted nitroxide/hemigramicidin S conjugate protects mouse embryonic cells against gamma irradiation. Int J Radiat Oncol Biol Phys. 2008;70:816–825. doi: 10.1016/j.ijrobp.2007.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fink MP, Macias CA, Xiao J, Tyurina YY, Delude RL, Greenberger JS, Kagan VE, Wipf P. Hemigramicidin-TEMPO conjugates: novel mitochondria-targeted antioxidants. Crit Care Med. 2007;35(9 Suppl):S461–467. doi: 10.1097/01.CCM.0000279192.96303.E7. [DOI] [PubMed] [Google Scholar]
- 166.Macias CA, Chiao JW, Xiao J, Arora DS, Tyurina YY, Delude RL, Wipf P, Kagan VE, Fink MP. Treatment with a novel hemigramicidin-TEMPO conjugate prolongs survival in a rat model of lethal hemorrhagic shock. Ann Surg. 2007;245:305–314. doi: 10.1097/01.sla.0000236626.57752.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Stoyanovsky DA, Vlasova II, Belikova NA, Kapralov A, Tyurin V, Greenberger JS, Kagan VE. Activation of NO donors in mitochondria: peroxidase metabolism of (2-hydroxyamino-vinyl)-triphenyl-phosphonium by cytochrome c releases NO and protects cells against apoptosis. FEBS Lett. 2008;582:725–728. doi: 10.1016/j.febslet.2008.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lee I, Salomon AR, Yu K, Doan JW, Grossman LI, Hüttemann M. New prospects for an old enzyme: mammalian cytochrome c is tyrosine-phosphorylated in vivo. Biochemistry. 2006;45:9121–8. doi: 10.1021/bi060585v. [DOI] [PubMed] [Google Scholar]
- 169.Yu H, Lee I, Salomon AR, Yu K, Hüttemann M. Mammalian liver cytochrome c is tyrosine-48 phosphorylated in vivo, inhibiting mitochondrial respiration. Biochim Biophys Acta. 2008;1777:1066–71. doi: 10.1016/j.bbabio.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]