

Abstract
Allosteric regulation provides highly specific ligand recognition and signaling by transmembrane protein receptors. Unlike functions of protein molecular machines that rely on large-scale conformational transitions, signal transduction in receptors appears to be mediated by more subtle structural motions that are difficult to identify. We describe a theoretical model for allosteric regulation in receptors that addresses a fundamental riddle of signaling: What are the structural origins of the receptor agonism (specific signaling response to ligand binding)? The model suggests that different signaling pathways in bovine rhodopsin or human β2-adrenergic receptor can be mediated by specific structural motions in the receptors. We discuss implications for understanding the receptor agonism, particularly the recently observed “biased agonism” (selected activation of specific signaling pathways), and for developing rational structure-based drug-design strategies.
Keywords: allostery, signal transduction, agonism, GPCR
Allostery [Greek for “other site” (1, 2)] indicates a mechanism in which a stimulus acting at a regulatory protein site (often called the orthosteric site or the effector site) causes a response at a spatially distant functional site (the allosteric site) (3). Allosteric regulatory mechanisms (i.e., regulation of protein functions by binding an effector molecule at an allosteric site) are ubiquitous throughout biology; they control vital biomolecular and cellular functions, including enzymatic catalysis and biosynthesis, molecular recognition and response to messengers, cell signaling, energy transduction, cell division and cancer, and many others (4–7). Signaling is perhaps the most fascinating and significant set of processes in which allosteric communication plays a central role (8, 9); biochemical signaling pathways lie at the core of most biological processes in cells, most notably signaling responses to binding endogenous ligands and drugs (10–12). Therefore, an understanding of allosteric signal transduction mechanisms in protein receptors is critical for describing cell regulation.
The investigation of allosteric regulatory mechanisms in signaling is difficult because of the complexity of receptor structure, the presence of thermal fluctuations, the range of relevant time scales, and, often, the subtlety of the underlying structural changes (13–15). Although experimental techniques such as FRET (16), time-resolved X-ray crystallography (17), time-resolved spectroscopy (18), cryo-EM (19), and biochemical methods (20, 21) provide valuable insights into specific aspects of allosteric interactions, a comprehensive atomic-level description of the connection between receptor structure and signaling is still beyond reach. As such, advanced computational methods could be instrumental for revealing the structural origins of allosteric regulation in signal transduction.
Computational approaches have been used recently in attempts to explore signal transduction in receptors. Although all-atom molecular dynamics (MD) simulations of various membrane proteins revealed nanosecond-long structural motions (22–27), it is unclear how exactly these motions relate to signal transduction. Normal mode analysis (NMA) based on coarse-grained (CG) protein models successfully described allostery in a variety of molecular machines that exhibit large-scale conformational transitions (28–35), but these methods are less suited for studying subtle structural changes that mediate signaling (13–15). Although bioinformatics methods based on the analysis of conserved amino acids in the receptor sequence (36–38) identify some allosteric sites, these methods do not provide a direct mechanistic link between structure and allostery. Therefore, developing a theoretical description of allosteric regulation in signaling ultimately requires new strategies.
We describe a model for allosteric regulation in protein receptors that addresses a key puzzle of signal transduction: the structural origins of the receptor agonism, i.e., the specific signaling response to ligand binding. How can minor differences in ligand structure translate into major differences in receptor response? Can a single receptor relay multiple signaling pathways, and if so, how are different signals “encoded”? Can ligand binding selectively activate one signaling pathway and suppress another, and how could such ligands be identified (or designed) for a specific receptor? To address these questions, we explore allosteric interactions between different protein sites in bovine rhodopsin (39, 40) and human β2-adrenoreceptor (B2AR) (41, 42), 2 members of the G-protein coupled receptor (GPCR) superfamily that comprises over 50% of the marketed drug targets (43). Our analysis suggests that different signaling pathways can be mediated by specific structural motions within the receptor, which may be triggered by the binding of different ligands. Although investigating specific signaling responses will require modeling on the atomic scale, structural origins of the receptor agonism can be inferred at the CG level.
Model
Our model is largely motivated by the recent approach developed by Zheng, Brooks and Thirumalai (32, 33) and by Ming and Wall (34), who used first-order perturbation theory combined with NMA to study the coupling between ligand binding and collective protein motions. We extend this approach to describe the allosteric coupling between specific protein sites. We model the primary signaling event (such as ligand binding or photoexcitation) and the response (a structural change at the distant allosteric site) as local structural perturbations that are coupled by collective structural motions in the receptor. These motions are described by protein normal modes, assuming that signal transduction in receptors is mediated by relatively small structural motions around equilibrium (13–15). Fig. 1 explains the connection between thermal motion along a normal mode k and the corresponding structural distortion in the environment of the CG atom m (characterized by a region Sm, hereafter taken as a sphere of radius Rs centered at the CG atom m). The motion of atoms within Sm in the original CG structure (blue) along normal mode k by the mode unit vector (solid arrows) generates a new structure (magenta), resulting in a structural perturbation within Sm as well as a translation and rotation of Sm as a whole. To characterize the structural perturbation alone, the translation and rotation effects are removed by root mean square aligning the region Sm in the new structure to the same region in the original structure (dashed arrows) (44). The mean square deviation between the CG atoms within Sm in the aligned structure (red) and the original structure quantifies the local structural perturbation within Sm as follows:
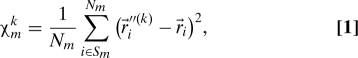 |
where are Cartesian coordinates of CG atom i in the original structure, are the coordinates of CG atom i in the aligned structure, and Nm is the number of CG atoms within Sm. Notably, χmk is a purely geometric characteristic of the CG structure, and it does not depend on temperature or the NMA potential spring constant. Although its magnitude depends on the size of Sm, the model predictions were found to be robust to variations of Rs from 7 to 11 Å (at larger Rs values, the model no longer resolves individual amino acids). Similar definitions of χmk, e.g., those based on a local coordinate system instead of the structural alignment, are also possible.
Fig. 1.

The coupling between a local structural perturbation and a collective normal mode (Eq. 1). A fragment of the original CG structure is shown with the blue open circles (atoms) and the blue curve (protein backbone). The gray sphere marks the region Sm around the CG atom m (coordinates ). The CG structure obtained by moving each CG atom along the corresponding component of the normalized eigenvector for normal mode k (solid arrows) is shown in magenta (coordinates ). The CG structure aligned to the region Sm of the original structure (dashed arrows) (44) is shown in red (coordinates ).
The allosteric coupling between local structural perturbations within regions Sm and Sn mediated by a single normal mode k is then defined as
where γ is the NMA spring constant (45), and λk is the eigenvalue of the kth normal mode. The prefactor in Eq. 2 describes the mean square magnitude of thermal fluctuations along the kth normal mode (see Eqs. 6 and 7 in the Appendix). Finally, assuming that structural perturbations arising from different normal modes are independent, we obtain the net allosteric coupling between regions Sm and Sn by adding all normal mode contributions:
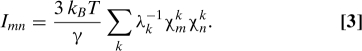 |
Importantly, the definition of the allosteric coupling matrix I is formally similar to the definition of the covariance matrix C (Eq. 7 in the Appendix). This similarity indicates that either quantity describes a connection between 2 distant localized events (atomic motion for C, structural distortion for I) that is mediated by collective protein motions described by normal modes.
Similar forms of coupling between a local structural perturbation and a collective protein motion have been recently used to describe allosteric effects in biomolecular machines (see, e.g., ref. 46 for a review). For example, Miyashita et al. (28), Maragakis et al. (29), Hyeon et al. (30), and Whitford et al. (31) used local structural distortion to characterize mechanical strain arising from conformational transitions in the enzyme adenylate kinase and the molecular motor kinesin. Their analysis suggested that local protein unfolding (“cracking”) in regions where elastic stress becomes sufficiently high may lower the transition free-energy barrier and thereby facilitate the transition (28–31). Zheng et al. investigated structural motions in several molecular machines including the chaperonin GroEL (32) and the enzyme hepatitis C virus NS3 helicase (33). They identified functionally important protein sites by modeling effects of point mutations as local perturbations of the NMA spring constants at each site (one at a time) and analyzing the influence of these perturbations on protein normal modes (32, 33). Chennubhotla et al. (35) combined NMA with probabilistic modeling of interactions among amino acids in GroEL. The success of these and similar studies depends largely on the fact that function of the molecular machines is coupled to well-characterized large-scale conformational transitions that are often dominated by one or a few normal modes (46).
Functional motions in protein receptors are, however, essentially different from those in molecular machines. Receptors do not exhibit well-manifested large-scale transitions between distinct conformations; instead, they undergo small-scale motions among multiple metastable states that are hard to characterize experimentally (13–15). The magnitude of these motions is comparable with that of thermal fluctuations, making it difficult to detect signal transduction events in the “sea” of thermal noise (13–15). As such, conventional NMA methods that rely on well-manifested transitions are not well suited for studying protein receptors. Conversely, our model, defined by Eqs. 1–3, does not depend on specific transitions, and it is capable of identifying allosteric sites and functional motions in receptors, as described below.
Results
Allosteric Coupling Analysis Vs. Conventional NMA.
A comparison of our analysis with conventional NMA for bovine rhodopsin [PDB code 1U19 (39)] and human B2AR [PDB code 2RH1 (41)] is illustrated in Fig. 2 The covariance matrices calculated by using Eq. 8 in the Appendix for rhodopsin (Fig. 2A) and B2AR (Fig. 2B) are similar, suggesting that they reflect the common 7-transmembrane helix (TMH) fold, rather than distinguishing features of the individual structures. Although the maps show the nearest neighbor and higher-order interactions among the TMHs, the orthosteric site (ligand environment) and the main allosteric site [the G-protein binding site located at the E(D)RY motif (40)] are not identified. Neither does conventional NMA identify what normal modes are most relevant for signal transduction. These results clearly indicate that NMA alone can not properly describe allosteric interactions in protein receptors.
Fig. 2.

Covariance maps (A and B), allosteric coupling maps (C and D), and the identified allosteric sites (E and F) in bovine rhodopsin (Left) and human B2AR (Right), respectively. For clarity, amino acids resolved in the B2AR crystal structure were reenumerated continuously starting with 1. Ligands are shown as green spheres, and the E(D)RY motif is shown as orange spheres. The covariance maps reflect the common 7TMH structure of the receptors, whereas the allosteric coupling maps identify pairs of distance sites involved in allosteric communication. The identified allosteric sites are marked with red arrows in A–D and with red ribbons in E and F. (E) In rhodopsin, residues 113–115, 182–184, and 299–301 are located in the ligand environment, residues 135–137 nearly coincide with the ERY motif (residues 134–136), residues 66–68, 225–227, 143–147, 235–245, and 312–323 are known to participate in G-protein binding (40), and residues 18–20 and 278–283 may influence signaling by interacting with ligands (47, 48). The C-terminal residue 348 could interact with allosteric modulators. The sites predicted by Ranganathan et al. (36) are shown with magenta ribbons, the sites predicted by Nussinov et al. (38) are shown with blue ribbons, and experimentally identified sites are shown with yellow ribbons (21). (F) In B2AR, the ligand binding sites inferred from the receptor structure are shown with blue ribbons, and the cholesterol binding sites are shown with magenta ribbons (49). Residues 196–200 and 295–305 are in contact with the ligand environment, residues 136–140 are in close spatial proximity to the DRY motif (residues 130–132), and residues 226–230 are located near the β-arrestin binding site (40). Residues 62–66, 148–152, and 335–340 may influence cholesterol binding, and residues 95–99 and 178–183, as well as the N-terminal residues 29–34, could interact with allosteric modulators (13, 43).
The allosteric coupling maps calculated by using Eq. 3 for rhodopsin (Fig. 2C) and B2AR (Fig. 2D) exhibit distinct peaks. The diagonal elements of I are strongly correlated with those of C: The correlation coefficient is 0.85 for rhodopsin and 0.90 for B2AR. This correlation indicates that both metrics provide similar (although not identical) measures of local protein flexibility at each amino acid. The strongest off-diagonal peaks of I form distinct grid-like patterns with the rows and columns indicating protein allosteric sites. Most of these sites are involved in multiple allosteric interactions; these sites are marked with arrows in Fig. 2. In both receptors, the allosteric maps identify the orthosteric site as well as the main allosteric site (40). Notably, the identified allosteric sites in the 2 receptors are similar, albeit not identical, indicating that the allosteric coupling I reflects the common overall fold as well as the structural differences between the receptors.
Allosteric Sites.
The allosteric sites predicted from our model, from bioinformatics analysis of conserved residues (36, 38), and obtained in experiments (21) are shown in Fig. 2 E and F. In rhodopsin (Fig. 2E), our model identifies residues 113–115, 182–184, and 299–301 (located in the immediate environment of the retinal cofactor), residues 135–137, which nearly coincide with the ERY motif [residues 134–136 (40)], and residues 66–68, 225–227, 143–147, 235–245, and 312–323 (the eighth α-helix), which are also known to participate in G-protein binding (40). Our model also suggests that loop residues 278–283 at the periplasmic side may be involved in allosteric regulation by influencing ligand binding (47, 48).
The results of the allosteric coupling analysis described here are consistent with experimental data (21). The allosteric map indicates the importance of the interactions of TMH3, TMH5, and TMH7 with the retinal cofactor and identifies most of the residues previously implicated in G-protein binding. The more rigid protein core residues are not identified in the allosteric maps at this level of coarse-graining, indicating that multiscale modeling may be needed to identify those residues. The theoretically identified allosteric sites are also consistent with the sequence-based analysis of Ranganathan et al. (36) [residues 113, 293–295, 299, and 302 (301 in our analysis)] and with the topology-based analysis of Nussinov et al. (38) (residues 67, 301, and 312). Although the predictions of the different models are not identical, they are consistent and successful in capturing the key allosteric sites, including the ligand environment and the G-protein binding site.
In B2AR (Fig. 2F), our model identifies residues 196–200 and 295–305 which are located near residues 203, 204, 207, 293, 308, and 312, which in turn are known to be involved in ligand binding (41, 42, 49). Residues 136–140 are in close spatial proximity to the DRY motif (residues 130–132), and residues 226–230 are located near the assumed β-arrestin binding site (40). Interestingly, the allosteric coupling map also identifies residues 62–66, 148–152, and 335–340, which are located near residues 70, 147–151, 154, 158, and 341 which may be involved in cholesterol binding (49). Residues 95–99 and 178–183, which form solvated loops, may interact with allosteric modulators. Indeed, the significance of the extracellular loop conformations for the receptor function has been observed in experiments (13, 43), and simulations showed that loop motions may occur on time scales much slower than submicroseconds (27).
Our model suggests that, in both rhodopsin and B2AR, the protein N- and C-termini may be involved in multiple allosteric interactions. This suggestion needs to be examined further, because the higher flexibility of the protein termini [the “tip effect” (50)] might influence the allosteric analysis to some extent. Experiments indicate that the protein termini may indeed engage in interactions with signaling and scaffold proteins (42) or lipids (49). As such, the role of the protein termini in signal transduction requires additional investigation.
Allosteric Interactions.
Mapping the strongest allosteric couplings onto the structures of rhodopsin and B2AR yields the allosteric interaction networks shown in Fig. 3. The 2 networks are similar on the cytoplasmic side, indicating similar mechanisms of activating the G-protein in the 2 receptors. On the periplasmic side, however, the networks are different, in part because of the different solvated loop conformations in the 2 receptors that reflect the different activation mechanisms: photoexcitation of retinal in rhodopsin vs. agonist binding-induced activation in B2AR.
Fig. 3.

The network of the strongest allosteric interactions in bovine rhodopsin (A) and human B2AR (B). Amino acids shown in green indicate the ligand binding site, those shown in orange indicate the E(D)RY motif, and those shown in yellow indicate the assumed β-arrestin binding site. The allosteric sites are explained in Fig. 2. In rhodopsin, normal mode 25 is indicated in red, normal mode 31 in purple, normal mode 52 in pink, and normal mode 65 in blue. In B2AR, normal mode 25 is shown in red, normal mode 26 in purple, normal mode 28 in pink, and normal mode 29 in blue. The allosteric interactions are represented as cylinders with radii proportional to the interaction strengths. For clarity, only long-range interactions are shown. The cylinders are color coded by the dominant normal mode for each interaction (see Eq. 3 and Fig. 5). Strong allosteric interactions controlled by 2 or more normal modes are shown in silver.
What kinds of structural motion underlie the strongest allosteric interactions? To address this question, we examined the contributions of each normal mode to the allosteric couplings between different receptor sites (Eq. 3). Fig. 4 shows the normal mode “spectra” for a strong allosteric interaction R29–P138 and a randomly chosen weak interaction G162–L230 in B2AR. Remarkably, >50% of the allosteric coupling R29–P138 is provided by 1 normal mode (number 26). Other strong allosteric interactions are also dominated by 1 or, less often, 2 normal modes. In contrast, the weak interaction G162–L230, as well as other nonfunctional interactions, is influenced by a large number of normal modes. Fig. 3 shows that, in both receptors, the allosteric network is dominated by only a few normal modes, and interactions between different receptor sites are mediated by different modes. In particular, the coupling to the β-arrestin binding site in B2AR is dominated by normal mode 25, and the coupling to the G-protein binding site (the DRY motif) is dominated by normal mode 26. The structural motions associated with these modes are heavily localized at the allosteric sites, and the rest of the receptor (particularly the more rigid receptor core) exhibits small-scale motion (see Movies S1 and S2).
Fig. 4.

Dissection of the allosteric coupling strength Imn into contributions from individual normal modes, Xmnk (see Eq. 3) for a strong allosteric interaction between the extracellular side and the G-protein binding site (R29–P138, red) and a randomly picked weak nonfunctional interaction (G162–L230, blue) in human B2AR. More than 50% of the allosteric interaction strength is provided by normal mode 26 alone. In contrast, the weak nonallosteric interaction is influenced by a large number of normal modes.
It is truly intriguing that only the allosteric couplings between protein functional sites are strong and dominated by one or a few normal modes, whereas couplings between non-functional sites are weak and mediated by a large number of modes. This fact indicates that positions of allosteric sites in receptors, as well as structural motions that couple these sites, are defined by the receptor structure and can be identified with our analysis. Although many other normal modes exhibit similar types of motion, only a few are important for allostery (see Movies S1–S4). Although allostery dominated by a few normal modes has been observed in protein molecular machines that undergo well-manifested large-scale motions (46), it is surprising that the same occurs in signaling receptors that do not exhibit such motions.
Discussion
Our model provides a framework for understanding the structural origins of receptor agonism. In a simplified view, a ligand binding event triggers a ligand-specific structural change at the receptor orthosteric site. How does this local structural change translate into signaling responses at the remote allosteric sites, and if it does not, why not? The answer depends on whether this structural change facilitates any of the structural motions that mediate signal transduction to the allosteric sites. As shown, in the Results section, our model identifies these motions, and the couplings between these motions and the ligand-induced structural change can be calculated by using Eqs. 1–2. If none of these couplings is significant, the structural perturbation triggered by ligand binding will not be relayed to any of the receptor allosteric sites, and no signaling response will follows (antagonist ligand). If one or more of these couplings are sufficiently large, ligand binding will cause structural changes at the corresponding allosteric sites that can either activate the associated signaling pathways (agonist ligand) or suppress them (inverse agonist ligand). The specific response will then depend on atomic-scale details of the structural changes at the allosteric sites (that can be identified by using, e.g., all-atom MD simulations). If the receptor mediates 2 or more signaling pathways, ligand binding could activate one pathway but suppress another. Indeed, selected activation of the “G protein-mediated” pathway or the “β-arrestin-mediated” pathway in B2AR (termed “biased agonism” or “functional selectivity”) has been recently discovered in experiments (47, 48).
Understanding the signal transduction mechanisms revealed by our model may facilitate computational structure-based drug design for protein receptors. After the collective structural motions that dominate allosteric communications in the receptor are identified, further analysis can focus on the couplings between these motions and the ligand-induced structural changes at the receptor orthosteric site. The goal then will be to identify (or to design) ligands that maximize the couplings to specific structural motions and minimize the couplings to the other motions. Although that analysis needs to be performed at the atomic level, it may be limited to the orthosteric site environment, which is much smaller than the entire receptor and has much shorter relaxation times. As such, the proposed strategy could facilitate the design of receptor-targeting drugs with higher selectivity and reduced side effects (51–53). Although a comprehensive atomic-scale theory of the receptor signaling response lies ahead, our results indicate that key features of the allosteric regulation mechanisms in receptors are revealed at a CG level.
Supplementary Material
Acknowledgments.
I.A.B. thanks Nagrajan Vaidehi (City of Hope Medical Center, Duarte, CA) for providing early B2AR structural models and Vadim Cherezov, Mikhail Dobrikov, and Ian Dance for extremely helpful discussions. This research was supported by National Institutes of Health Grants GM-048043 and GM-061870.
Appendix: Normal Mode Analysis
The NMA was performed by using an anisotropic elastic network model (45, 54, 55). The protein CG models were based on the crystallographic structures of rhodopsin [PDB code 1U19 (39)] and B2AR [PDB code 2RH1 (41, 42)], coarse-grained to Cα atoms. Calculations were performed by using several CG models of retinal (rhodopsin) and carazolol (B2AR) ligands that included from 1 to 7 CG atoms. The contact matrix Γ included N × N blocks of 3 × 3 elements, where N is the number of the CG atoms in the model. The stepwise distance dependence of the spring constant used in ref. 45 was replaced with a smooth sigmoidal distance dependence, improving the robustness of the NMA eigenvectors to the protein conformation. The off-diagonal contact matrix elements were
where indices i and j indicate the interacting CG atoms, is the vector that begins at CG atom i and ends at CG atom j, indices α and β indicate coordinate axes x, y, or z, is a unit vector on axis α (α = x, y, z), Rc is the cutoff distance, and q is the width of the sigmoid transition. The diagonal elements of the contact matrix were defined as (45):
The cutoff distance Rc was set to 11 Å, and the transition width q was set to 2 Å. Simulations with the cutoff distance varying from 7–20 Å and different transition widths confirmed that the chosen values of Rc and q are well in the range of the model's robustness.
The covariance matrix that describes the correlation of atomic fluctuations is (45):
where γ is the spring constant (45). Since the first 6 eigenvalues of the contact matrix Γ are zero (they correspond to translations and rotations of the molecule as a whole), C was computed by using pseudoinversion of Γ (45):
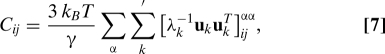 |
where uk is the kth eigenvector of Γ, and the summation over k starts with the seventh normal mode. In our analysis, γ was chosen such that the average B-factor (which is proportional to the diagonal elements of C) is equal to the average B-factor for the Cα atoms obtained from the crystallographic data. For clarity, visualizations of correlations in protein motion (Fig. 2) are based on the normalized covariance matrix
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0901811106/DCSupplemental.
References
- 1.Monod J, Wyman J, Changeux JP. On nature of allosteric transitions: A plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 2.Koshland DEJ, Némethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–368. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 3.Berg J, Tymoczko J, Stryer L. Biochemistry. 6th Ed. New York: W. H. Freeman; 2006. [Google Scholar]
- 4.Goodey NM, Bencovic SJ. Allosteric regulation and catalysis emerge via a common route. Nat Chem Biol. 2008;4:474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 5.Kuriyan J, Eisenberg D. The origin of protein interactions and allostery in colocalization. Nature. 2007;450:983–990. doi: 10.1038/nature06524. [DOI] [PubMed] [Google Scholar]
- 6.Gunasekaran B, Ma B, Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins Struct Funct Genet. 2004;57:443–453. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 7.Kern D, Zuiderweg ERP. The role of dynamics in allosteric regulation. Curr Opin Struct Biol. 2003;13:748–757. doi: 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 8.Lefkowitz RJ, Shehoy SK. Transduction of receptor signals by β-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 9.Christopoulos A, May LT, Avlani VA, Sexton PM. G-protein coupled receptor allosterism: The promise and the problems. Biochem Soc Trans. 2004;32:873–877. doi: 10.1042/BST0320873. [DOI] [PubMed] [Google Scholar]
- 10.Krauss G. Biochemistry of Signal Transduction and Regulation. 3rd Ed. Weinheim, Germany: Wiley; 2003. [Google Scholar]
- 11.Okazaki K, Takada S. Dynamic energy landscape view of coupled binding and protein conformational change: Induced-fit versus population-shift mechanisms. Proc Natl Acad Sci USA. 2008;105:11182–11187. doi: 10.1073/pnas.0802524105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boehr DD, Wright P. How do proteins interact? Science. 2008;320:1429–1430. doi: 10.1126/science.1158818. [DOI] [PubMed] [Google Scholar]
- 13.Vauquelin G, Van Liefde I. G protein-coupled receptors: a count of 1001 conformations. Fundam Clin Pharmacol. 2005;19:45–56. doi: 10.1111/j.1472-8206.2005.00319.x. [DOI] [PubMed] [Google Scholar]
- 14.Popovych N, Sun S, Elbright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cui Q, Karplus M. Allostery and cooperativity revisited. Protein Sci. 2008;17:1–13. doi: 10.1110/ps.03259908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rueda D, et al. Single-molecule enzymology of RNA: Essential functional groups impact catalysis from a distance. Proc Natl Acad Sci USA. 2004;101:10066–10071. doi: 10.1073/pnas.0403575101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knapp JE, Pahl R, Srajer V, Royer WE. Allosteric action in real time: Time-resolved crystallographic studies of a cooperative dimeric hemoglobin. Proc Natl Acad Sci USA. 2006;103:7649–7654. doi: 10.1073/pnas.0509411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato A, Gao Y, Kitagawa T, Mizutani Y. Primary protein response after ligand photodissociation in carbonmonoxy myoglobin. Proc Natl Acad Sci USA. 2007;104:9627–9632. doi: 10.1073/pnas.0611560104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu GZ, Liu RT, Zak O, Aisen P, Chance MR. Structural allostery and binding of the transferrin center dot receptor complex. Mol Cell Proteomics. 2005;4:1959–1967. doi: 10.1074/mcp.M500095-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Johnson SJ, Beese LS. Structures of mismatch replication errors observed in a DNA polymerase. Cell. 2004;116:803–816. doi: 10.1016/s0092-8674(04)00252-1. [DOI] [PubMed] [Google Scholar]
- 21.Madabushi S, Gross AK, Philippi A, Meng EC, Lichtarge O. Evolutionary trace of G protein-coupled receptors reveals clusters of residues that determine global and class-specific functions. J Biol Chem. 2004;279:8126–8132. doi: 10.1074/jbc.M312671200. [DOI] [PubMed] [Google Scholar]
- 22.Dror RO, et al. Identification of two distinct inactive conformations of the β2-adrenergic receptor reconciles structural and biochemical observations. Proc Natl Acad Sci USA. 2009;106:4689–4694. doi: 10.1073/pnas.0811065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huber T, Menon S, Sakmar TP. Structural basis for ligand binding and speci-ficity in adrenergic receptors: Implications for GPCR-targeted drug discovery. Biochem. 2008;47:11013–11023. doi: 10.1021/bi800891r. [DOI] [PubMed] [Google Scholar]
- 24.Abankwa D, et al. A novel switch region regulates H-ras membrane orientation and signal output. EMBO J. 2008;27:727–735. doi: 10.1038/emboj.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Filizola M, Wang SX, Weinstein H. Dynamic models of G-protein coupled receptor dimers: Indications of asymmetry in the rhodopsin dimer from molecular dynamics simulations in a POPC bilayer. J Comput Aided Mol Des. 2006;20:405–416. doi: 10.1007/s10822-006-9053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spijker P, Vaidehi N, Freddolino PL, Hilbers PAJ, Goddard WAI. Dynamic behavior of fully solvated β2-adrenergic receptor, embedded in the membrane with bound agonist or antagonist. Proc Natl Acad Sci USA. 2006;103:4882–4887. doi: 10.1073/pnas.0511329103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faraldo-Comez JD, et al. Conformational sampling and dynamics of membrane proteins from 10-nanosecond computer simulations. Proteins Struct Funct Genet. 2004;57:783–791. doi: 10.1002/prot.20257. [DOI] [PubMed] [Google Scholar]
- 28.Miyashita O, Onuchic JN, Wolynes PG. Nonlinear elasticity, proteinquakes, and the energy landscapes of functional transitions in proteins. Proc Natl Acad Sci USA. 2003;100:12570–12575. doi: 10.1073/pnas.2135471100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maragakis P, Karplus M. Large amplitude conformational change in proteins explored with a plastic network model: Adenylate kinase. J Mol Biol. 2005;352:807–822. doi: 10.1016/j.jmb.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 30.Hyeon C, Onuchic JN. Internal strain regulates the nucleotide binding site of the kinesin leading head. Proc Natl Acad Sci USA. 2007;104:2175–2180. doi: 10.1073/pnas.0610939104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitford PC, Gosavi S, Onuchic JN. Conformational transitions in adenylate kinase: Allosteric communication reduces misligation. J Biol Chem. 2008;283:2042–2048. doi: 10.1074/jbc.M707632200. [DOI] [PubMed] [Google Scholar]
- 32.Zhen gW, Brooks BR, Thirumalai D. Allosteric transitions in the chaperonin GroEL are captured by a dominant normal mode that is most robust to sequence variations. Biophys J. 2007;93:2289–2299. doi: 10.1529/biophysj.107.105270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng W, Liao JC, Brooks BR, Doniach S. Toward the mechanism of dynamical couplings and translocation in hepatitis C virus NS3 helicase using elastic network model. Proteins Struct Funct Genet. 2007;67:886–896. doi: 10.1002/prot.21326. [DOI] [PubMed] [Google Scholar]
- 34.Ming D, Cohn JD, Wall ME. Fast dynamics perturbation analysis for prediction of protein functional sites. BMC Struct Biol. 2008;8:5. doi: 10.1186/1472-6807-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chennubhotla C, Yang Z, Bahar I. Coupling between global dynamics and signal transduction pathways: A mechanism of allostery for chaperonin GroEL. Mol Biosystems. 2008;4:287–292. doi: 10.1039/b717819k. [DOI] [PubMed] [Google Scholar]
- 36.Süel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 37.Hardy JA, Wells JA. Searching for new allosteric sites in enzymes. Curr Opin Struct Biol. 2004;14:706–715. doi: 10.1016/j.sbi.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 38.del Sol A, Fujihashi H, Amoros D, Nussinov R. Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Molecular Syst Biol. 2006;2:1–12. doi: 10.1038/msb4100063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okada T, et al. The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 40.Menon ST, Han M, Sakmar TP. Rhodopsin: Structural basis of molecular physiology. Physiol Rev. 2001;81:1659–1688. doi: 10.1152/physrev.2001.81.4.1659. [DOI] [PubMed] [Google Scholar]
- 41.Cherezov V, et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosenbaum DM, et al. GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 43.Avlani VA, et al. Critical role for the second extracellular loop in the binding of both orthosteric and allosteric G protein-coupled receptor ligands. J Biol Chem. 2007;282:25677–25686. doi: 10.1074/jbc.M702311200. [DOI] [PubMed] [Google Scholar]
- 44.Kabsch W. Discussion of solution for best rotation to relate two sets of vectors. Acta Crystallogr Sect A. 1978;34:827–828. [Google Scholar]
- 45.Atilgan AR, et al. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys J. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Onuchic JN, Kobayashi C, Miyashita O, Jennings P, Baldridge KK. Exploring biomolecular machines: Energy landscape control of biological reactions. Philos Trans R Soc London B. 2006;361:1439–1443. doi: 10.1098/rstb.2006.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shukla AK, et al. Distinct conformational changes in β-arrestin report biased agonism at seven-transmembrane receptors. Proc Natl Acad Sci USA. 2008;105:9988–9993. doi: 10.1073/pnas.0804246105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kenakin T. Collateral efficacy in drug discovery: taking advantage of the good (allosteric) nature of 7TM receptors. Trends in Pharm Sci. 2007;28:407–415. doi: 10.1016/j.tips.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 49.Hanson MA, et al. A specific cholesterol binding site is established by the 2.8 Å structure of the human β2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma J. New advances in normal mode analysis of supermolecular complexes and applications to structural refinement. Curr Protein Pept Sci. 2004;5:119–123. doi: 10.2174/1389203043486892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drake MT, et al. β-arrestin-biased agonism at the β2-adrenergic receptor. J Biol Chem. 2008;283:5669–5676. doi: 10.1074/jbc.M708118200. [DOI] [PubMed] [Google Scholar]
- 52.Gilchrist A. Modulating G-protein-coupled receptors: from traditional pharmacology to allosterics. Trends in Pharm Sci. 2007;28:431–437. doi: 10.1016/j.tips.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 53.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 54.Tirion MM. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys Rev Lett. 1997;77:1905–1908. doi: 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- 55.Bahar I, Jernigan RL. Inter-residue potentials in globular proteins and the dominance of highly specific hydrophilic interactions at close separation. J Mol Biol. 1997;266:195–214. doi: 10.1006/jmbi.1996.0758. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.