

Abstract
AIMS
Omalizumab, a subcutaneously administered anti-IgE antibody, is effective for moderate-to-severe persistent allergic asthma. The aims were to (i) describe the population pharmacodynamics of free IgE with a mechanism-based, nonlinear, omalizumab–IgE binding model; (ii) deduce a target-free IgE suppression level by correlation with clinical outcomes; and (iii) check the adequacy of current approved dosing tables and explore potential doses and regimens beyond.
METHODS
Concentration data (omalizumab, free and total IgE) were obtained from 1781 patients aged 12–79 years, in four sparsely sampled randomized, placebo-controlled studies and 152 subjects in a richly sampled single-dose study. NONMEM predictive performance across the range of bodyweights (39–150 kg) and baseline IgE (19–1055 IU ml−1) was checked by simulation. Predicted free IgE levels were correlated with time-averaged patient diary clinical outcomes.
RESULTS
The model accurately predicted observed omalizumab, free and total IgE concentrations. Free IgE concentrations correlated well with clinical signs and symptoms, allowing a target concentration of 14 ng ml−1, at the midpoint of 4-week clinical observation periods, to be set for determining the dose and regimen for omalizumab.
CONCLUSIONS
The omalizumab–IgE binding model is predictive for free IgE and demonstrates a nonlinear time-dependent relationship between free IgE suppression and clinical outcomes in asthma. Although currently approved dosing tables are close to optimal, it should be possible to treat patients with higher levels of baseline IgE if higher doses can be administered.
Keywords: asthma, binding, IgE, NONMEM, omalizumab
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
It had been hypothesized that there must be a relationship between free IgE concentrations and the signs and symptoms of asthma – after all, this is what drove the development of omalizumab.
However, although many statistical analyses of free IgE and spirometry data for patients equilibrated on omalizumab had shown a difference between placebo and treatment, no consistent time- and IgE-dependent relationship had been shown due to the narrow range of free IgE being studied and the sparse nature of the sampling and clinical measurements in Phase III trials.
WHAT THIS STUDY ADDS
The above problem was solved using a pharmacokinetic–pharmacodynamic (PK–PD) model to estimate free IgE concentrations for all time points throughout and after treatment with omalizumab, together with patient daily diary data.
This allowed for the first time the direct correlation between free IgE and signs and symptoms of asthma to be observed.
Doses and regimens for omalizumab could then be derived, through PK–PD model simulation, for suppressing free IgE to a point correlated with an improvement in clinical symptoms.
Introduction
Asthma is a cause of substantial mortality and morbidity and has a considerable economic impact [1–4]. An estimated two-thirds of patients with asthma have allergic asthma [5] and the causal role of immunoglobulin E (IgE) is well established [6–9]. The allergic cascade is initiated when allergen binds to and cross-links IgE bound to high-affinity IgE receptors (FcεRI) on the surface of basophils and mast cells, triggering the release of mediators such as histamine, prostaglandin D2, leukotriene C4 and tumour necrosis factor-α[5, 10]. Subsequent release of cytokines and chemokines promotes adhesion and infiltration of circulating inflammatory cells into the tissues and results in the characteristic symptoms of asthma [5, 10].
Omalizumab is a recombinant DNA-derived humanized IgE monoclonal antibody that selectively binds human IgE at the epitope that would otherwise engage the FcεR. Omalizumab is an IgG1κ with a human framework and complementarity-determining region (CDR) from a humanized anti-IgE murine antibody [11, 12]. Omalizumab binds all forms of circulating IgE, whatever its allergen specificity, and prevents subsequent IgE-mediated responses [11, 12]. By reducing IgE, omalizumab also leads to the downregulation of FcεRI on mast cells/basophils [9]. The clinical utility of targeting IgE in patients with allergic (IgE-mediated) asthma has been demonstrated in numerous trials of omalizumab involving patients with moderate-to-severe and severe persistent allergic asthma [13–15].
Following subcutaneous administration, omalizumab is absorbed slowly, reaching peak serum concentrations after an average of 7–8 days [16]. Omalizumab binds IgE in a reversible reaction to form small, biologically inert, noncomplement-fixing complexes with a molecular mass and stoichiometry which varies from 1 : 2, through 1 : 1 (actually 3 : 3), to 2 : 1 depending upon the molar ratio of total IgE to total omalizumab [17, 18]. The complexes are cleared via interactions with Fcγ receptors of the hepatic sinusoidal and other endothelial cells of the reticuloendothelial system [19, 20]. Overall, the clearance of the monoclonal antibody is slow (mean 2.4 ± 1.1 ml kg−1 day−1), with a terminal half-life (t1/2) of 26 days [16], although the clearance of the IgG–IgE complex is faster than that of the free IgG [21, 22]. By pushing the binding equilibrium away from free IgE towards the complex, the major effects of omalizumab are to (i) increase total IgE (since the clearance of the IgG–IgE complex is slower than that of free IgE, total IgE, which is mainly the complex, builds up); (ii) reduce free unbound IgE; and subsequently (iii) reduce the clinical signs and symptoms of allergic asthma.
Pharmacokinetic–pharmacodynamic (PK–PD) models based upon this binding reaction have been published [21, 22]. In this study, we built upon a previously published PK–PD model [22] and applied it to sparse omalizumab and IgE concentration data collected from four randomized, double-blind, placebo-controlled trials [13, 23–25]. We have extended the investigations into the relationship between the model-derived free IgE concentrations and changes in clinical outcomes in patients with severe persistent allergic asthma [26] in order to define a target level for the suppression of free IgE. Given a clear relationship between the biomarker, free IgE, and several clinical end-points important in asthma, it then became straightforward to deduce a posology by using the model in an automated iterative dose-escalating Monte-Carlo simulation process. We thus explored doses and regimens for omalizumab beyond the current licensed dosing tables.
Materials and methods
Study design and conduct
The analysis included data from five clinical studies, four of which were Phase III: (i) INNOVATE, a 28-week treatment, 16-week follow-up, randomized, double-blind, placebo-controlled study in patients with inadequately controlled severe persistent allergic asthma [13]; (ii) and (iii) two 7-month, randomized, double-blind, parallel-group, placebo-controlled, multicentre studies with 5-month blinded extension periods in adolescents and adults with moderate-to-severe allergic asthma requiring daily treatment with inhaled corticosteroids (ICS) [23, 24]; (iv) a 32-week, randomized, double-blind, parallel-group, placebo-controlled, multicentre pilot trial to assess corticosteroid reduction in adolescents and adults with severe allergic asthma requiring daily treatment with high-dose ICS, with or without oral corticosteroids [25]; and (v) a single-dose bioequivalence study [Novartis, unpublished data].
In the Phase III studies, omalizumab was administered by subcutaneous (s.c.) injection every 2 or 4 weeks according to patients' pretreatment bodyweight and baseline IgE levels using either the earlier US dosing table or the later and more individualized EU dosing table (Table 1). Further details of the patient populations and study designs have been published previously for the multiple-dose studies [13, 23–25]. The bioequivalence study was a single-dose, parallel-group investigation of omalizumab (150 and 300 mg s.c.) in healthy but atopic volunteers with total IgE above normal levels (30–300 IU ml−1) at the screening visit, conducted at four centres in the USA.
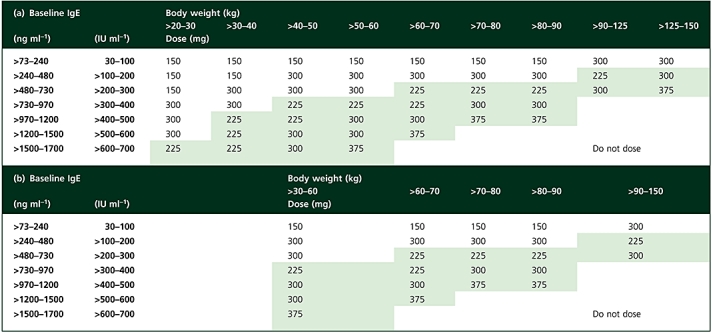
Table 1.
Omalizumab dosing table used in (a) the INNOVATE study (EU dosing table) [13] and (b) the other three Phase III studies (US dosing table) [23–25]; the omalizumab dose was administered by subcutaneous injection every 4 weeks (white cells) or 2 weeks (green-shaded cells)
 |
All studies were approved by Institutional Review Boards and all patients gave informed written consent. The studies were conducted in accordance with the declaration of Helsinki and Good Clinical Practice guidelines.
Data for model development
Samples (n= 2182) were obtained from 440 patients in the INNOVATE study [13] and analysed for total omalizumab and free and total IgE at weeks 1, 2, 12, 28 and 44. The other three Phase III studies provided an additional 3129 samples from 525 patients [23], 2632 samples from 539 patients [24] and 885 samples from 277 patients [25].
Given the sparseness of the concentration data available from the Phase III studies, data from 152 subjects in a richly sampled single-dose bioequivalence study were also added to the dataset for the development of the PK–PD model; this study provided 3925 additional blood samples for analysis. Blood samples in the bioequivalence study were collected just prior to dosing then at 6 and 12 h postdose, then on days 1, 2, 3, 5, 7, 10, 14, 21, 28, and every 14 days thereafter through to 84 days.
The model was developed using data from both omalizumab-treated and placebo patients. The full dataset contained 23 488 observations (5938 omalizumab, 11 034 total IgE, and 6156 free IgE) from 1928 patients and volunteers. Subjects with missing covariate data were excluded (Table 2). The numbers of patients included in the analysis and their baseline demographic data are summarized in Table 2.
Table 2.
Patient numbers and baseline demographic data for studies contributing to the population PK–PD model analysis
| Patient numbers | Demographic data Subjects used for analysis, mean ± SD (range) | |||||
|---|---|---|---|---|---|---|
| Study | Treated | Used in the analysis | Age (years) | Bodyweight (kg) | Baseline IgE (ng ml−1) | |
| Bioequivalence | 155 | 152 | 35 ± 12 (18–64) | 71 ± 12 (48–91) | 186 ± 124 (47–620) | |
| INNOVATE [13] | 482 core study 420 in follow-up (211 active, 209 placebo) | 440 226 active 214 placebo | Active | 42 ± 14 (12–79) | 79 ± 20 (45–143) | 509 ± 375 (51–1692) |
| Placebo | 43 ± 13 (14–74) | 77 ± 17 (39–146) | 479 ± 387 (53–2173) | |||
| [23] | 268 active | 268 active | Active | 39 ± 13 (12–73) | 80 ± 20 (39–150) | 417 ± 341 (48–2081) |
| 257 placebo | 257 placebo | Placebo | 39 ± 14 (12–74) | 78 ± 19 (39–136) | 451 ± 345 (51–1699) | |
| [24] | 274 active | 271 active | Active | 40 ± 15 (12–76) | 77 ± 17 (46–136) | 541 ± 411 (51–1900) |
| 272 placebo | 268 placebo | Placebo | 39 ± 14 (12–72) | 78 ± 18 (40–148) | 498 ± 391 (53–1970) | |
| [25] | 176 active | 133 active | Active | 44 ± 14 (12–73) | 76 ± 18 (41–135) | 578 ± 461 (63–2553) |
| 165 placebo | 144 placebo | Placebo | 43 ± 14 (12–74) | 74 ± 14 (41–115) | 605 ± 448 (46–1902) | |
Analysis of omalizumab, total and free IgE
The enzyme-linked immunosorbent assay (ELISA) for total omalizumab used human myeloma-derived IgE from the U266B1 cell line as the capture antibody (American Type Culture Collection number TIB-196266; American Type Culture Collection, Manassas, VA, USA). Plate-bound omalizumab was detected with a monoclonal antibody developed against the CDR of omalizumab and conjugated with horseradish peroxidase. Colourimetric detection utilized the o-phenylenediamine reaction with hydrogen peroxide. The assay range was 0.16–10 ng ml−1, but the dilution used in this case gave a lower limit of quantification (LLOQ) of 16 ng ml−1. The coefficient of variation was 4.9% at 0.156 ng ml−1. Only 199 (3%) of the omalizumab samples were below the LLOQ. Free IgE in serum was determined using a previously reported ELISA [27]. The assay had an upper limit of quantification (ULOQ) of 150 ng ml−1, a LLOQ of 0.78 ng ml−1, and coefficients of variation of 10.1% at 1.51 ng ml−1 and 5.4% at 21.8 ng ml−1. Of the free IgE samples, 355 (6%) were above the ULOQ and therefore excluded. A commercially available microparticle enzyme immunoassay test kit (Abbott Inc., Abbott Park, IL, USA) was used to measure pretreatment total serum IgE for selection of dose/frequency and subsequent measurements of total IgE levels. The LLOQ of total IgE was 2.4 ng ml−1, with a coefficient of variation of 13.6% at 3.6 ng ml−1. Measurements for total IgE in IU ml−1 were converted to ng ml−1 (1 IU ml−1 total IgE is 2.42 ng ml−1 total IgE). No total IgE samples were out of range of the assays. Since in the absence of omalizumab there can be no formation of omalizumab–IgE complexes, free and total IgE should be the same; therefore, for placebo patients, the few samples from the lower baseline IgE patients where free IgE was quantifiable were excluded (<150 ng ml−1 or 62 IU ml−1) in order to prevent any bias by including more data on a small subset of patients.
Omalizumab–IgE model
The nonlinear mixed-effect model of omalizumab–IgE turnover and binding has been published previously [22]. The same model was used in this analysis for estimating the concentrations of free IgE at time points when the clinical measures were assessed. Briefly, the binding of omalizumab with IgE is written as a system of three differential equations, one for the s.c. administration site, one for total omalizumab (free plus complex) and another for total IgE (free plus complex). The differential equations, in terms of molar masses of omalizumab, IgE and the complex, were:
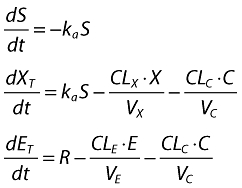 |
(1) |
where C (complex) is the solution to the quadratic for the equilibrium binding equation:
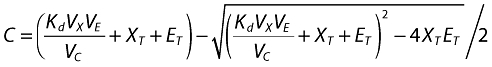 |
where
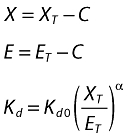 |
(2) |
and S is the subcutaneous site; XT and ET are molar masses of total omalizumab and IgE; X and E are free omalizumab and IgE; ka is the absorption rate constant; R is the rate of production (or expression) of IgE; CLn and Vn are the clearances and volumes of free omalizumab, free IgE and the complex; Kd is the equilibrium binding constant, and α is the change in the affinity of binding between omalizumab and IgE as a function of the molar ratio of total omalizumab to total IgE. Weight-based doses were converted to molar amounts in the program code, then weight-based concentrations were calculated from the molar masses by dividing by the respective volumes, then correcting for the molecular mass for each component (free and complex). The free and complex concentrations were them summed to attain total omalizumab and IgE concentrations.
The generic population parameter, P, relationship for the ith individual was modelled as
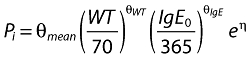 |
(3) |
where θmean represents the population mean of the parameter, θWT and θIgE the covariate relationships to be estimated and η a Gaussian distribution with mean zero.
Although both bodyweight (WT) and baseline IgE (IgE0) are shown in the previous equation, they were not applied to all structural parameters. Bodyweight centred on 70 kg was specified in an allometric (power) relationship on all clearances and volumes, and on IgE production rate (R). These were estimated rather than fixed at a default of 0.75 and 1 [28–30], as the objective of the exercise was to provide the best description of the data from the population studied in order to compare the results of a model-based dosage regimen with the original algorithm. Baseline IgE is a function of rate of production and clearance. The observed distribution of baseline IgE in the trials was not normal, so could not be represented with a Gaussian distribution. Therefore baseline IgE, which was used to determine the dose, was used as a covariate to help predict IgE clearance and production rate for any individual patient. Any remaining variances in the parameters were considered residual interpatient variance and specified as log-normal distributions. Correlation between parameters was investigated in preliminary models but, finally, only the covariance between clearance of free omalizumab and its volume, and between the clearance of the complex and IgE production rate, were estimated.
Natural logarithm-transformed data for omalizumab, free and total IgE concentrations were used with corresponding logarithm transforms of the output–concentration functions. The residual error model specified in the code was therefore additive, rather than the more frequently used exponential notation. All concentrations outside the quantification range were excluded. The first-order estimation method was used. First-order conditional estimation with interaction (FOCEI) was attempted with the final model, but it terminated when the maximum number of iterations (9999) was exceeded. Given that this run took 94.79 days to reach this point, it was not restarted. Nonetheless, there was no visible difference in the diagnostic plots (pred and ipred vs. DV) between the FO estimation and that from the termination point with FOCEI (results not shown). As a matter of note, the FO estimation took 1.23 days to complete, 0.79 for initial convergence, 0.44 days for the $COV procedure, on a 1.8-GHz IBM computer running NONMEM version VI.
The differences between the model used here and that previously published by Hayashi et al.[22] were: (i) Kd was allowed to vary between patients to take account of the possibility that differing levels of Fcε-expressing factors may compete for omalizumab binding with IgE; (ii) baseline IgE was introduced as a covariate on Kd; and (iii) given the larger population available for parameter estimation, bodyweight covariates were introduced on the IgE production and clearance parameters in addition to the covariates previously included [22]. Beyond bodyweight and baseline IgE, one further potential population covariate, that of body mass index (BMI) (weight in kg divided by height in m2), which was hypothesised as potentially important given the different fluid volume properties of lean and adipose tissue, was explored graphically and is presented in this analysis.
Once the parameters had been successfully estimated, a predictive check was performed. The 1933-patient dataset, with dosing regimens, sampling times and patient covariates (bodyweight and baseline IgE) from each patient, was replicated 10 times, then used with the final model to simulate free IgE concentrations at steady state, approximately 6 months after dosing. The patients were then divided into nine subsets, based on the minimum to lower third, lower to middle third, and middle third to maximum baseline IgE values (21, 146.5, 308.9, 860 IU/ml) and bodyweights (39, 67.5, 91.6, 150 kg). For each subset, a histogram of the observed steady-state free IgE levels was overlaid with that predicted from the simulation, to test the ability of the model to predict the free IgE across the dosing table.
Clinical outcomes
For the Phase III studies, based on patients' diary card data, mean changes from baseline were determined for total asthma symptoms score [sum of the daytime (range 0–4), night-time (range 0–4) and morning (range 0–1) score], mean morning peak expiratory flow and rescue medication use (mean number of daily puffs). For each patient, the changes from baseline in clinical outcomes were summarized into 28-day arithmetic means. The midpoint of each of the averaged periods was then used to correlate clinical outcomes with model-derived free IgE concentrations.
Correlations between free IgE and the clinical variables were constructed by producing, for each patient, post hoc predictions of their free IgE at the midpoint of each 28-day clinical observation period, i.e. at 14 days, 42 days and every 28 days thereafter. The time-matched pairs of values for each observation period were then summarized across patients, using arithmetic means for the change in clinical variables (which were normally distributed) and geometric means for free IgE (which were log-normally distributed); the pairs were then plotted against each other. From these correlations, a target level of free IgE suppression, corresponding to the maximum observed clinical effect, could be determined.
Deduction of posology through simulation
Using the model, the optimal dose and regimen for each cell of the dosing table – i.e. that which suppressed the free IgE in the patients to a mean level equal to or below the target level – was deduced through simulation. In an iterative fashion, the following dosing regimens were tested, sequentially, on each simulated patient population until the target IgE suppression was obtained: 150, 300, 450 and 600 mg once every 4 weeks (q4w), then 150, 300, 450 and 600 mg every 4 weeks (q2w). This loop was repeated for each cell in the dosing table, defined by a range of bodyweights and a range of baseline IgE values.
The simulation methodology was as follows. Using S-Plus®, 1000 values were randomly selected from a uniform distribution ranging from the lower to upper bodyweight in the cell; similarly, 1000 values of baseline IgE were sampled. These covariates were paired to create 1000 virtual patients. The selected dosing regimen (e.g. 150 mg q4w), along with the patient covariates, was then incorporated into a template NONMEM dataset in which patients were dosed from time zero until steady state; omalizumab and free IgE concentrations were ‘measured’ daily from 182 to 210 days, a 28-day dosing interval. The NONMEM simulated data were then imported into S-Plus. The free IgE at the midpoint of the dosing interval was recorded for each subject and the geometric mean calculated and compared against the target free IgE; if the mean was less than or equal to the target, the simulation loop terminated and the current dosing regimen was recorded. Otherwise, the simulation proceeded to the next dosing regimen on the list. The iterations continued until the target was reached or the last dosing regimen (600 mg q2w) was tested without reaching the target.
Software and settings
Estimation of population PK–PD parameters and their variances, followed by calculation of individual patient omalizumab, free and total IgE concentration–time predictions, was carried out using NONMEM (Version V level 1.1 or Version VI level 1.0) with the ADVAN6 subroutine. This utilised the Runga–Kutta integrator, for which a tolerance of 5 was specified. NONMEM was run both in a DOS environment (IBM and Dell Pentium 4 with Compaq Visual Fortran 6.6) and under UNIX (XL Fortran compiler 8.1.1). Both NONMEM versions V and VI, and DOS and UNIX gave the same result, differing only occasionally at the third significant figure and, more noticeably, in the slopes of the partial differentials during optimization, perhaps reflecting slightly different floating point calculations with the different Fortran compilers or the optimization of NONMEM with version VI.
The above-described versions of NONMEM were also used to perform the simulations for the predictive check as well as the iterative simulations to determine the optimal dosing regimen for the cells of the dosing table. S-Plus® 6.2 for Windows (Insightful Corp., Seattle, WA, USA) was used to automate the iterative simulations and to format the data and create the graphics for the predictive check and the correlation between the free IgE and the clinical signs and symptoms. Datasets and descriptive tables were computed with SAS 8.2 (SAS Institute Inc., Cary, NC, USA) on the Unix platform. NONMEM output datasets were read by SAS 8.2. Graphics were created using R 2.4.1 and S-Plus 6.2.
Results
Model parameter estimation and predictive check
Unusually, compared with some Phase III studies, there was a very high level, nearly 98%, of PK and PD sampling from the patients, providing confidence that the model was representative for the moderate to severe allergic asthma population. Furthermore, few of these samples were out of range of the assays, with <3% of omalizumab being below the LLOQ and only 6% of the free IgE being above the ULOQ. No total IgE assays were out of range. Although there are recent reports on likelihood techniques to utilize information from samples that were measured but were out of range [31], these were not used in the current work as they require conditional estimation methods with the Laplacian option. As mentioned earlier, FOCE was attempted but took >90 days before terminating with rounding errors. Nonetheless, all three markers were accurately described by the model and each contributed information to the others through the molecular binding equation, so even when data were missing for one measurement (e.g. free IgE) it was supported by the others. Of the 6% free IgE samples out of range, 261 (4.3%) were correctly predicted to be above the limit of quantification, whereas only 1.5% were measured >LOQ and predicted <LOQ, and 0.9% measured <LOQ and predicted to be above. Furthermore, there were no notable deviations of the model curve from the observed data, as shown by the diagnostic plots of residuals vs. both concentration and time (Figure 1). The majority of measured samples were within 0.5 loge units, i.e. within +65% or −39%, of the individual patients' predicted curves. Therefore it was judged that the model fitted well both single-dose data from healthy but atopic volunteers and longer-term multiple-dose data from patients with severe persistent allergic asthma. Examples of individual predicted curves from the omalizumab–IgE model applied to data from the INNOVATE study [13] patients are presented in Figure 2. The fits to the data from the other studies were comparable (plots not shown). The addition of the rich data from the bioequivalence study confirmed that the model captured the rapid suppression of free IgE and the return to baseline following treatment cessation. Furthermore, the single-dose data enabled better characterization of the s.c. absorption kinetics, which could not be accurately estimated from the limited samples available from the Phase III studies. After a single dose, omalizumab concentrations rose above 10,000 ng ml−1, whereas free IgE was suppressed to approximately 10 ng ml−1. Free IgE was further suppressed with multiple dosing as the drug accumulated. At the same time, total IgE increased, owing to the formation of omalizumab–IgE complexes, which have a longer half-life than free IgE [21, 22].
Figure 1.
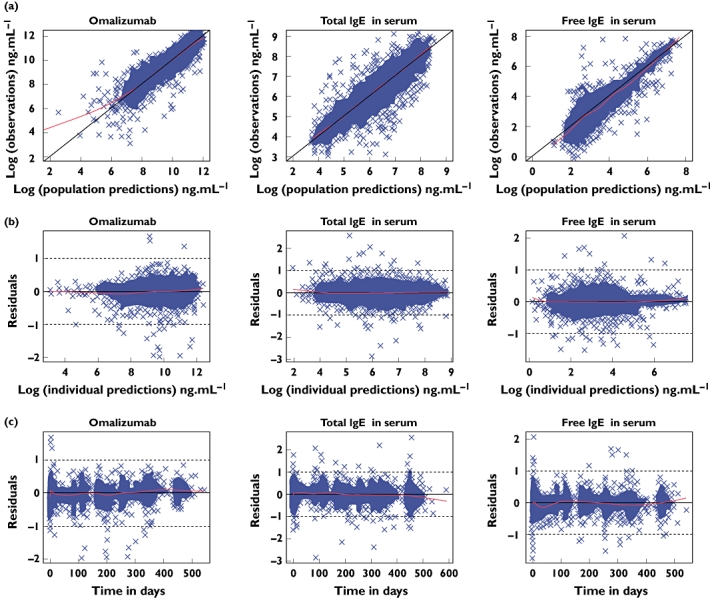
Diagnostic plots for assessment of model fit to the data: (a) observations vs. population prediction, (b) residuals vs. concentration and (c) residuals vs. time. The line through the centre is a local regression with a span of 0.25 generated using the LOESS function in R. Given that an additive error model was used on log-transformed data, the units for the residuals are loge of the concentrations in ng ml−1; 0.5 loge unit is 1.6 fold, or, on the linear scale, +65%, −39%
Figure 2.
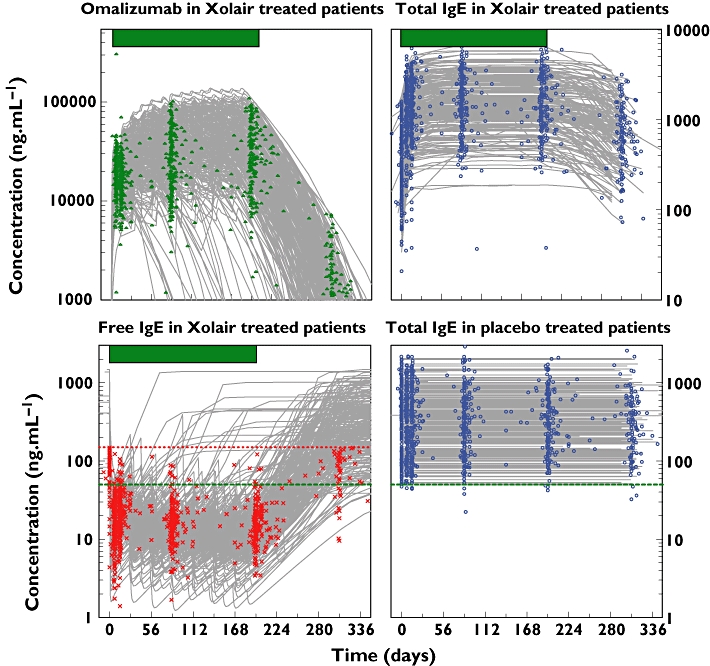
Individual predicted curves from the omalizumab–IgE model applied to data from INNOVATE study patients [13]. For the omalizumab and free IgE, those individual predictions that decrease or increase before the end of the treatment period (indicated by the bar above the plots) represent patients who discontinued the study early. The upper limit of quantification for free IgE is marked with a red dotted line. The 50 ng ml−1 target level of free IgE at trough is shown by the green dashed line
The estimated parameter values are shown in Table 3. Clearance (CL/F) and volume of distribution (V/f) of free omalizumab varied approximately proportionally with bodyweight (exponents of 1.0 and 0.83, respectively); for the clearance of free IgE (0.5) and clearance and volume of the omalizumab–IgE complex (0.67, 0.55), the bodyweight variation was less than proportional. Unexplained interindividual random variability ranged from 22% for the clearance of IgE to a rather high 141% for the drug absorption rate. The residual variability of 24–26% was, unsurprisingly, greater than the 5% (omalizumab) to 14% (total IgE) analytical specifications for the assays. The parameters were in general precisely estimated, with standard errors of the parameter estimates ranging from 1.6% (CLX and VX) through 14% (ka) of the means for the structural parameters; from 2.6% (baseline IgE on IgE production rate) to 24% (bodyweight on IgE production rate) for the covariates; and from 12% (ηCLX) to 47% (ηka) for the interpatient variances. The least precise parameter estimate was the covariance between ηCLC/F and ηRE/F with a standard error of 68%; however, the extent of the covariance was quite small. Unlike previously reported work [22], a between-patient variance in Kd was included rather than assuming a fixed value for the entire population. The NONMEM log-likelihood objective function was −26 625.602 for a fixed Kd, and −28 106.608 when the variance on Kd was included. The decrease of 1553.13 is highly significant. Further, including baseline IgE as a covariate on Kd also improved the model, generating a further decrease in the log-likelihood objective function of 72.124 (P < 0.001 for one degree of freedom).
Table 3.
Estimated model parameters for the population from the Phase III studies [13, 23–25] and the bioequivalence study
| Omalizumab or IgE parameter | Population mean [θ± SEM] | Interindividual variance [(%CV) ω± SEM] | Shrinkage, post hocs†† |
|---|---|---|---|
| Clearance omalizumab CLX/F [l day−1]* | 0.208 ± 0.00338 | (40%) 0.162 ± 0.0196 | 12% |
| Clearance, IgE, CLE/F [l day−1]† | 3.85 ± 0.155 | (23%) 0.0479 ± 0.0208 | 37%, 34%¶ |
| Clearance, complex CLC/F [l day−1]* | 0.832 ± 0.0344 | (26%) 0.0649 ± 0.00921 | 22% |
| Volume, omalizumab and IgE, VX/f & VE/f [l]* | 9.33 ± 0.147 | (22%) 0.0901 ± 0.00762 | 42%, 25%¶ |
| Volume, complex, VC/f[l]* | 6.31 ± 0.196 | (26%) 0.0519 ± 0.00829 | 63% |
| Rate IgE production, RE/f[µg day−1]† | 1220 ± 49.9 | (30%) 0.0701 ± 0.0186 | 23%, 18%¶ |
| Absorption rate, ka[day−1] | 0.458 ± 0.0626 | (141%) 2.01 ± 0.940 | 60% |
| Binding dissociation constant, Kd[nmol l−1] | 1.81 ± 0.0808 | (31%) 0.0991 ± 0.00722 | 18% |
| Kd change with total omalizumab to total IgE ratio α | 0.0902 ± 0.0108 |
| Covariates (exponents) | |||
|---|---|---|---|
| Bodyweight on CLX/F | 1.00 ± 0.0662 | Baseline IgE on RE/f | 0.594 ± 0.0156 |
| Bodyweight on CLE/F | 0.499 ± 0.114 | Baseline IgE on CLE/F | 0.372 ± 0.0158 |
| Bodyweight on CLC/F | 0.671 ± 0.108 | Baseline IgE on Kd | 0.115 ± 0.0142 |
| Bodyweight on VX/f | 0.828 ± 0.0635 | Covariance ηCLX/F:ηVX/F | 0.103 ± 0.0183 |
| Bodyweight on VC/f | 0.549 ± 0.0936 | Covariance ηCLC/F:ηRE/F | −0.0101 ± 0.00687 |
| Bodyweight on RE/f | 0.491 ± 0.116 | ||
| Residual variance (%CV), σ | Shrinkage‡‡ | |
|---|---|---|
| Omalizumab | (24%) 0.0568 ± 0.00429 | 13% |
| Total IgE | (26%) 0.0671 ± 0.00324 | 12%, 21%¶ |
| Free IgE | (24%) 0.0600 ± 0.00352 | 13% |
| Objective function | −28 178.732‡,§ | |
For convenience, inter- and intra-individual variances are shown as %CV as well as the original values determined by NONMEM.
Value at 70 kg bodyweight.
Value at 70 kg and 365 ng ml−1 of baseline IgE.
Value without covariate of baseline IgE on Kd was −28,106.608, an increase of 72.124.
Value without interindividual variance or the covariate of baseline IgE on Kd was −26,625.602, an increase of 1481.006 from (3), or the 1553.13 from the final model.
Value for the treated population only.
Shrinkage in the post hoc η was calculated from ηsh= 1−SD(ηph)/√ω[32].
Shrinkage in the ε calculated from εsh= 1−SD(residual)/√ε.
Shrinkage in the empirical Bayes estimates of the individual patients' parameters, calculated according to Karlsson and Savic [32], varied from 12% and 18% for CLX and Kd to 60% and 63% for ka and VC, respectively (Table 3). The shrinkage in ka was probably due to the paucity of early postdose sampling times in the sparsely sampled Phase III studies, such that post hoc estimates shrank to the values estimated from the richly sampled bioequivalence study. Similar to the absorption rate constant, the period where there is maximum information to support the estimation of the volume for the complex is in the first 2 weeks postdose of the total IgE data. Samples for this were only present, to any great extent, in the bioequivalence study. Shrinkage in the residual error was small, being only 13% for the omalizumab pharmacokinetics and the free IgE, the key prediction required for correlation with the clinical data, assuring that the individual patient predictions were close to each patients' data.
To ensure that the model not only fits the data as assessed by conventional analysis of the residuals and standard errors of the parameter estimates, but could also simulate correctly the suppression of free IgE across the dosing table, a predictive check was carried out. Figure 3 shows the results of a prediction from the model on to the combined data from the four Phase III studies, subsetted for low, medium and high bodyweight and baseline IgE. Both the medians and the shapes of the distributions were well predicted.
Figure 3.
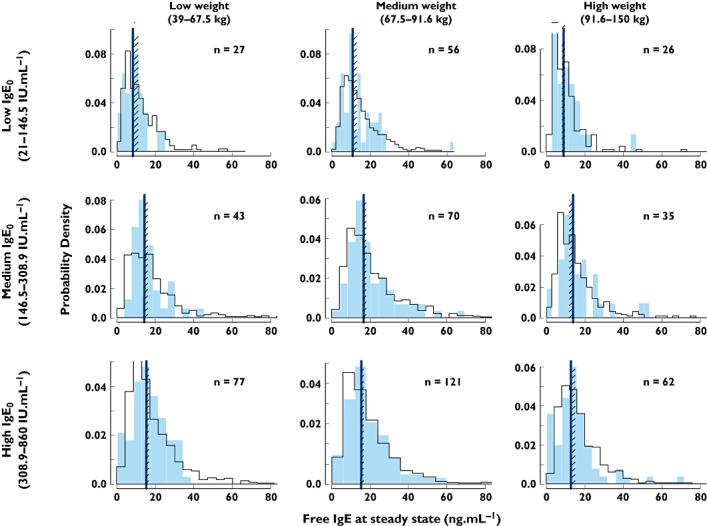
Predictive check of the omalizumab–IgE model on to the distribution of free IgE at steady state for different regions of the dosing table, using the combined data from Phase III studies [13, 23–25]. The solid histograms are the observed data from the Phase III studies for the numbers of patients indicated. The open histograms are from a model simulation of a total of 19 330 patients, split into the nine subsets. The lines are the medians of the observed data, the cross-hatched areas the 95% confidence interval for the median from the simulations. observed ( ); predicted (
); predicted ( )
)
In addition to bodyweight and baseline IgE, BMI was also graphically explored as a potential covariate. Plots of the unexplained interpatient variability in the eight parameters of the model failed to show any notable trends with BMI (Figure 4). It was therefore concluded that there was no need to consider the BMI, in addition to bodyweight, as a basis for adjusting the dose of omalizumab. Furthermore, the lack of correlation with BMI suggests that the disposition of the omalizumab–IgG, or the IgG–IgE complex, is unlikely to be different for adipose than for lean tissue.
Figure 4.
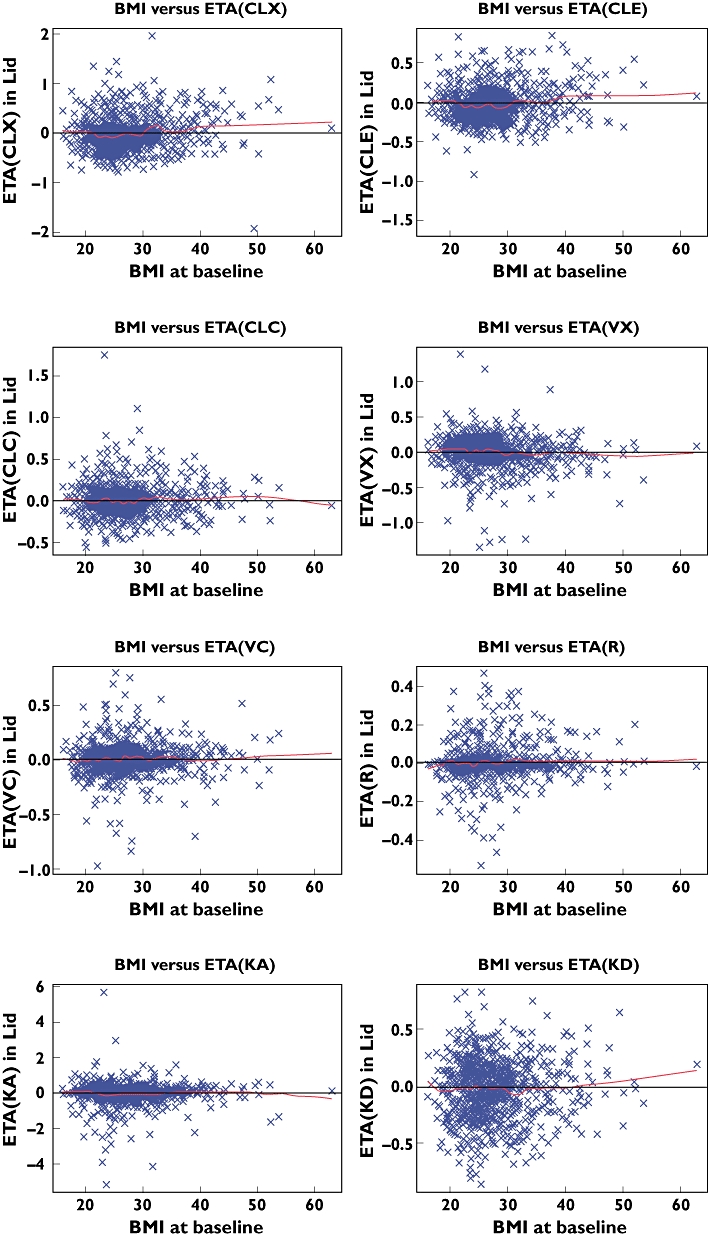
Relationship between body mass index and parameters for omalizumab and IgE, across the five studies. ETA for the parameters is the random variability between patients that is not explained by the bodyweight and baseline IgE covariates included in the model. The line through the centre is a local regression calculated in R with the LOESS function, with a span of 0.25
Since the model fitted and predicted the observed data well, the entire time courses of omalizumab, free and total IgE for patients in the Phase III studies could be reconstructed, even though only sparse samples were collected. In particular, the model enabled patients to be included even when they dropped out of the study prematurely, albeit with reduced numbers of clinical measurements; the technique of last observation carried forward thus being unnecessary. Given their dosing histories and at least two or three samples for drug and IgE, they could still be well fitted by the model and provide IgE and clinical symptom information up to each patient's last observation, individual estimates of free IgE being matched, time point for time point, with clinical measurements of asthma.
Correlation of free IgE concentrations with clinical measures
Although free IgE is extensively and rapidly suppressed after commencing treatment with omalizumab (Figure 2), it takes up to 3 months before clinical symptoms achieve their new equilibrium values [26]. Furthermore, following cessation of treatment, it takes some time for the symptoms of asthma to re-emerge. Nevertheless, once the changes in the clinical responses were summarized into blocks of time, in this case periods of 4 weeks, time could be removed and model-derived concentrations of free IgE at the midpoint of each of the blocks of time plotted against the mean clinical measurements. Figure 5 shows that, as free IgE was suppressed by binding with omalizumab, the mean total asthma symptoms score and rescue medication use decreased and the morning peak expiratory flow increased. Although follow-up data were not collected in two of the studies [23, 24], it can be seen from the INNOVATE study [13] that, after omalizumab treatment, clinical symptoms of asthma return as free IgE returns towards baseline concentrations. The same can be seen, albeit partially, in the fourth study [25], although the results on the change in total symptom score were confounded by the parallel reduction in steroid usage. When the data from the four studies were combined, the full significance of these results became very apparent from the small size of the standard errors (Figure 5, lower portion).
Figure 5.
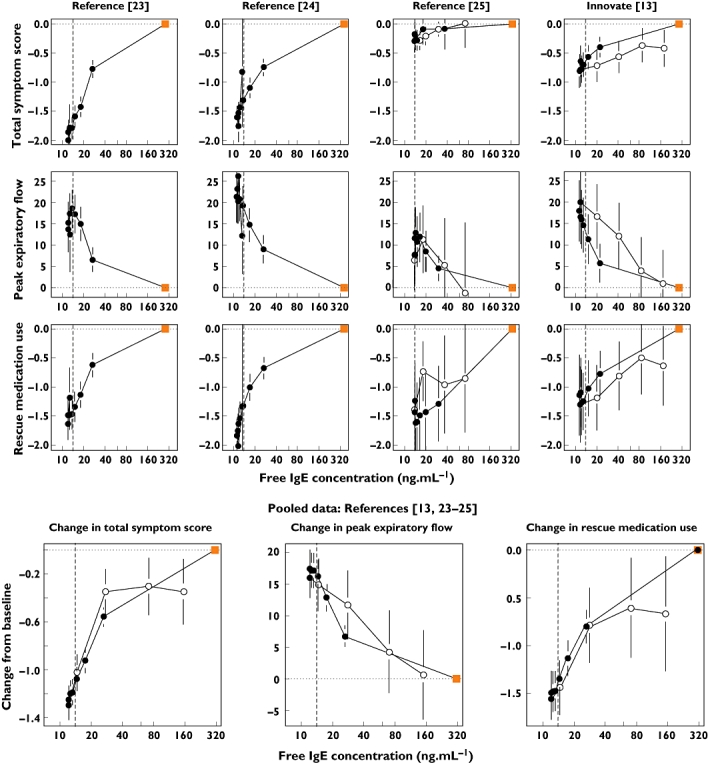
Correlation between the change from the baseline (time zero) value of the total symptom score, morning peak expiratory flow and rescue medication use with free IgE across four Phase III studies [13, 23–25]. The symptoms are the mean values for 28-day observation periods; the free IgE value is that predicted for the midpoint of each 28-day period. The square symbol represents the baseline IgE value and zero point for changes in symptoms, with the closed circles being the values whilst on treatment, the open circles during follow-up after treatment cessation. The lower figures show the correlations for the integrated dataset of four studies. All plots show a vertical indicator line at 14 ng ml−1. The error bars are the standard errors for the means. The units for peak expiratory flow are l min−1, those for rescue medication use are puffs per day
Deduction of dose and regimen through simulation from the model
From the correlation with the signs and symptoms of asthma, a 14 ng ml−1 target level of free IgE suppression was selected, this being the point below which the three clinical responses stabilize whilst on treatment (Figure 5, lower portion). A dose and regimen were deduced using Monte-Carlo simulations for 1000 patients for each subset of bodyweight and baseline IgE in the omalizumab dosing table. The dose was increased, in 150-mg increments, changing from q4w to q2w when the monthly dose went above 600 mg, until the geometric mean free IgE at the midpoint of steady-state 28-day observation periods was ≤14 ng ml−1. The results are shown in Table 4. In the model-derived table, the algorithm successfully selected doses and regimens such that the geometric mean free IgE concentrations were at or below the target level. Comparison of the model-derived table with the reference simulation for the current EU licensed table reveals some apparent inconsistencies due to differing dosing frequencies. For example, the 30–40 kg, 600–700 IU ml−1 baseline IgE cell was predicted to achieve 14.4 ng ml−1 free IgE, yet the equivalent cell in the model-derived table was 12.8 ng ml−1 for the same q4w dose. This is explained by the fact that the model-derived regimen is 450 mg q4w, whereas in the EU table this is split into a 225-mg q2w regimen. The midpoint of the 4-week clinical assessment period was a midpoint for a q4w regimen, but a trough for q2w. The differing dosing frequencies reflect the objective of the exercise, to explore new doses beyond the current EU table with the possibility of administering up to 600 mg at each visit.
Table 4.
Monthly doses for omalizumab and corresponding predicted free IgE concentrations: (a) is that deduced by dose escalation simulations from the omalizumab–IgE model. The target for escalation was the suppression of geometric mean free IgE, at the midpoint of a steady-state 28-day dosing interval, to be ≤14 ng ml−1. For each dosing cell, Monte-Carlo simulations of 1000 patients with a uniform distribution of bodyweight and baseline IgE were performed, including variability in all PK and IgE parameters. The doses in the cells filled in dark grey are increases from the current EU dosing table, those in light grey, decreases. Doses up to 600 mg are given q4w; above this the doses are split and given in a q2w regimen; (c) is the licensed EU table. Doses >300 mg are split into two q2w doses; (b) is the predicted geometric mean free IgE for the model-derived table, (d) free IgE for the EU table; in (d) mean free IgE values that did not meet the target criteria of 14 ng ml−1 are in bold
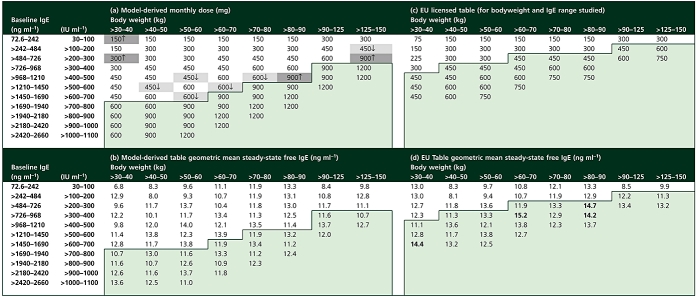 |
In addition to deriving doses and regimens for the combinations of bodyweight and baseline IgE included in the EU dosing table, the model was used to explore doses and regimens for patients outside the licensed table. The ranges of baseline IgE levels and bodyweights explored were those included in the analysis dataset (Table 2). The predictions for these patients are shown in the lightly shaded areas of Table 4a for the monthly dose, and Table 4b for the predicted free IgE levels. In all cases free IgE is predicted to be suppressed to <14 ng ml−1 without exceeding 1200 mg per 4-week period. Doses >600 mg were given as q2w regimens.
Discussion
We have extended a prior mechanism-based population PK–PD model of the binding of the anti-IgE monoclonal antibody, omalizumab, [22, 26] to calculate individual patient concentrations of drug and free IgE from limited samples throughout and after a treatment in four Phase III clinical studies in patients with severe persistent allergic asthma [13, 23–25]. In developing this model, data from a richly sampled bioequivalence study supplemented that of the Phase III studies in order to provide estimates of drug absorption parameters and to demonstrate that the model accurately describes the return of free IgE to baseline. Estimated population PK and IgE model parameters compared well with and, owing to the increased amount of data, extend previously published analyses [21, 22, 26]. Although calculating a half-life for a nonlinear system is somewhat illogical, the theoretical half-lives, calculated from the clearances and volumes of free omalizumab, the omalizumab–IgE complex and free IgE, assuming they were to exist in isolation, were 31, 5.3 and 1.7 days, respectively. However, it must be remembered that what is observed in vivo is total omalizumab, the sum of free and complex, the proportion of which varies over time and with the production or expression level of IgE. Any observed ‘half-life’ will, similarly, vary over time and with IgE production. The results of the present study confirm the earlier findings of Hayashi and colleagues [22] that such models can predict both the central tendency and population distribution of free IgE suppression for any combination of dose, regimen and the major covariates, these being bodyweight and baseline IgE.
BMI, although hypothesized by many colleagues to be important, did not show any notable relationships with any of the parameters. However, it is interesting that the volumes of distribution had exponents significantly less than unity. The volume of distribution of a monoclonal antibody is, however, not a straightforward actual physiological volume. In the current model, a single compartment is specified, even though IgG is known to distribute to the interstitial fluid of tissues. Therefore the volumes estimated in the current model will reflect both the systemic blood volume, plus a proportion of the tissue interstitium. The distribution of IgG to the tissue interstitium is a function of its permeation through holes, fenestrae, in the vascular endothelium of some tissues such as liver and spleen, plus endocytosis–exocytosis, or transcytosis, across blood vessel walls. Transcytosis is an energy-requiring process, therefore more likely to scale with an exponent less than unity according to classical theory [28, 29]. Further, the movement of proteins through fenestrae will be dependent upon solvent drag by fluid flowing into the tissues, later returned to the blood circulation via the lymphatics. Together, this makes it unsurprising that the bodyweight exponents are less than unity. Nevertheless, the lack of any notable explanatory relationship with BMI suggests that adipose is not notably different from lean tissue in terms of immunoglobulin distribution, clearance or binding of IgE.
In addition, the model described here is the first to be applied to data from patients with inadequately controlled, severe persistent allergic asthma despite receiving high-dose ICS and a long-acting β-agonist and additional controller medication in approximately 60% of cases [13].
Interestingly, using the rather larger database of patient data, in this case a total of 1928 asthma patients (55% treated), the model could be extended and improved by including random interpatient variability in the omalizumab–IgE dissociation constant, Kd, plus a covariate of baseline IgE on Kd. The biological explanation for this observation is not fully known, but a plausible hypothesis is that omalizumab competes for binding IgE with Fcε receptors, both high (RI) and low affinity (RII). The high-affinity Fcε receptor has a Kd in the same region as for omalizumab [33]; therefore, if there were to be greater amounts of this receptor in the body, it would make it appear, due to the competition, that the affinity of omalizumab binding were lower (higher Kd). Increased expression of Fcε receptor may then occur in tandem with increased production of IgE, which increases the baseline IgE, heightening sensitivity to IgE. IgE has been shown to induce B-cell Fcε expression [34] and, following treatment with omalizumab, a decrease in the expression of the type 1 high-affinity Fcε receptor has been observed [35–37]. The end result is that, with the covariates bodyweight and baseline IgE being predictive of IgE production and clearance (i.e. IgE turnover), baseline IgE being predictive of the binding affinity, and bodyweight predicting omalizumab clearance and volume, the omalizumab–IgE binding model has the ability to predict the suppression of free IgE across the range of the dosing table, even though the entire system is inherently nonlinear.
Given that the model fits and describes the data well, both for rich and sparse sample collection, individual patient IgE suppression time profiles could be calculated even though only three to seven samples were taken over the course of each clinical trial. Concentrations of free IgE were estimated for times matching those of the measurement of clinical signs and symptoms. When free IgE was correlated with peak expiratory flow, rescue medication use and a total symptom score, the classical curvilinear or logarithmic sigmoidal S-shaped relationship typical of ligand–receptor binding and pharmacological responses became apparent. The correlation was not confined to a single study but was observed, consistently, in four independently conducted Phase III trials. This correlation allowed us to set a target for free IgE suppression of 14 ng ml−1, corresponding to the maximum clinical effects. It then became straightforward to carry out repetitive Monte-Carlo simulations for sets of patients with defined ranges of bodyweight and baseline IgE, with increasing doses of omalizumab, until the criterion for suppression of free IgE was met. This resulted in a PK–PD model-deduced dosing table that ensures more even and balanced suppression than the current EU dosing table. Although the original algorithm that was used to create the dosing table (0.016 mg per kg bodyweight, per IU ml−1 baseline IgE, per 4 weeks), performs very well, there were a few cells that were predicted to have less than optimal suppression, plus some where the regimen could be changed to a more convenient 4-weekly frequency.
In summary, this study has described a robust PK–PD model that allows the calculation of free IgE, omalizumab and total IgE concentrations at any time point during treatment with omalizumab, given information on the patient's bodyweight, baseline (total) IgE concentration, dosing history and limited concentration data for omalizumab, free and total IgE during treatment. In patients treated with omalizumab dosed in accordance with the dosing table, model-derived omalizumab and free IgE concentrations correlated well with changes in clinical measures of asthma, further detailing the relationship between free IgE and clinical symptoms. Finally, given the clear clinical end-point-linked biomarker criterion, iterative dose-escalating Monte-Carlo simulations allowed us, in an automatic process, to deduce doses and regimens for narrow subsets of patients. With increments of 10 kg for bodyweight and 100 IU ml−1 for baseline IgE, the dosing table is close to individualized treatment. The PK–PD-deduced table (Table 4a) revealed that the currently licensed omalizumab dosing table is close to optimal, although perhaps a few cells could be adjusted. The objective, to deduce potential doses and regimens that would be required to treat patients who are currently not covered by the EU dosing table, was met by exploring the range of bodyweights and baseline IgE levels present in the integrated dataset of four Phase III studies. Although the model is mechanistic in nature and therefore should be able to extrapolate beyond the range of bodyweights and baseline IgE values present in the dataset, further work is necessary to confirm that the model can extrapolate and be used to deduce posologies for patients with high baseline IgE and bodyweights.
Competing interests
All authors work for and own shares in Novartis Pharma AG, manufacturers of omalizumab (XOLAIR®).
The authors wish to thank professional medical writer Tom McMurray (ACUMED) and editorial assistant Helen Venables (ACUMED) for editorial assistance with the manuscript. This support was funded by Novartis Pharma AG.
REFERENCES
- 1.World Health Organization. The European Health Report. Geneva: World Health Organization; 2002. [Google Scholar]
- 2.Masoli M, Fabian D, Holt S, Beasley R. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy. 2004;59:469–78. doi: 10.1111/j.1398-9995.2004.00526.x. [DOI] [PubMed] [Google Scholar]
- 3.Beasley R. The burden of asthma with specific reference to the United States. J Allergy Clin Immunol. 2002;109:S482, 9. doi: 10.1067/mai.2002.122716. [DOI] [PubMed] [Google Scholar]
- 4.European Respiratory Society and the European Lung Foundation. European Lung White Book. Brussels, Belgium: European Respiratory Society and the European Lung Foundation; 2003. [Google Scholar]
- 5.Novak N, Bieber T. Allergic and nonallergic forms of atopic diseases. J Allergy Clin Immunol. 2003;112:252–62. doi: 10.1067/mai.2003.1595. [DOI] [PubMed] [Google Scholar]
- 6.Ishizaka K, Ishizaka T. Identification of gamma-E-antibodies as a carrier of reaginic activity. J Immunol. 1967;99:1187–98. [PubMed] [Google Scholar]
- 7.Johansson SGO, Bennich H. Immunological studies of an atypical (myeloma) immunoglobulin. Immunology. 1967;13:381–94. [PMC free article] [PubMed] [Google Scholar]
- 8.Burrows B, Martinez FD, Halonen M, Barbee RA, Cline MG. Association of asthma with serum IgE levels and skin-test reactivity to allergens. N Engl J Med. 1989;320:271–7. doi: 10.1056/NEJM198902023200502. [DOI] [PubMed] [Google Scholar]
- 9.Holgate S, Casale T, Wenzel S, Bousquet J, Deniz Y, Reisner C. The anti-inflammatory effects of omalizumab confirm the central role of IgE in allergic inflammation. J Allergy Clin Immunol. 2005;115:459–65. doi: 10.1016/j.jaci.2004.11.053. [DOI] [PubMed] [Google Scholar]
- 10.Prussin C, Metcalfe DD. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol. 2003;111:S486–94. doi: 10.1067/mai.2003.120. [DOI] [PubMed] [Google Scholar]
- 11.Presta L, Shields R, O'Connell L, Lahr S, Porter J, Gorman C, Jardieu P. The binding site on human immunoglobulin E for its high affinity receptor. J Biol Chem. 1994;269:26368–73. [PubMed] [Google Scholar]
- 12.Presta LG, Lahr SJ, Shields RL, Porter JP, Gorman CM, Fendly BM, Jardieu PM. Humanization of an antibody directed against IgE. J Immunol. 1993;151:2623–32. [PubMed] [Google Scholar]
- 13.Humbert M, Beasley R, Ayres J, Slavin R, Hébert J, Bousquet J, Beeh KM, Ramo S, Canonica GW, Hedgecock S, Fox H, Blogg M, Surrey K. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy. 2005;60:309–16. doi: 10.1111/j.1398-9995.2004.00772.x. [DOI] [PubMed] [Google Scholar]
- 14.Bousquet J, Cabrera P, Berkman N, Buhl R, Holgate S, Wenzel S, Fox H, Hedgecock S, Blogg M, Cioppa GD. The effect of treatment with omalizumab, an anti-IgE antibody, on asthma exacerbations and emergency medical visits in patients with severe persistent asthma. Allergy. 2005;60:302–8. doi: 10.1111/j.1398-9995.2004.00770.x. [DOI] [PubMed] [Google Scholar]
- 15.Corren J, Casale T, Lanier BQ, Blogg M, Reisner C, Gupta N. Omalizumab is well tolerated in adolescent/adult patients (≥12 years) with moderate-to-severe persistent asthma. J Allergy Clin Immunol. 2005:115–S75. abstract. [Google Scholar]
- 16.Omalizumab. Xolair, label information. http://www.fda.gov/cder/foi/label/2003/omalgen062003LB.pdf.
- 17.Fox JA, Hotaling TE, Struble C, Ruppel J, Bates DJ, Schoenhoff MB. Tissue distribution and complex formation with IgE of an anti-IgE antibody after intravenous administration in cynomolgus monkeys. J Pharmacol Exp Ther. 1996;279:1000–8. [PubMed] [Google Scholar]
- 18.Liu J, Lester P, Builder S, Shire SJ. Characterization of complex formation by humanized anti-IgE monoclonal antibody and monoclonal human IgE. Biochemistry. 1995;34:10474–82. doi: 10.1021/bi00033a020. [DOI] [PubMed] [Google Scholar]
- 19.Ghetie V, Hubbard JG, Kim JK, Tsen MF, Lee Y, Ward ES. Abnormally short serum half-lives of IgG in beta 2-microglobulin-deficient mice. Eur J Immunol. 1996;26:690–6. doi: 10.1002/eji.1830260327. [DOI] [PubMed] [Google Scholar]
- 20.Mariani G, Strober W. Immunoglobulin metabolism. In: Metzger H, editor. Receptors and the Action of Antibodies. Washington DC: American Society of Microbiology; 1990. pp. 94–177. [Google Scholar]
- 21.Meno-Tetang GML, Lowe PJ. On the prediction of the human response: a recycled mechanistic PK/PD approach. Basic Clin Pharmacol Toxicol. 2005;96:182–92. doi: 10.1111/j.1742-7843.2005.pto960307.x. [DOI] [PubMed] [Google Scholar]
- 22.Hayashi N, Tsukamoto Y, Sallas WM, Lowe PJ. A mechanism-based binding model for the population pharmacokinetics and pharmacodynamics of omalizumab. Br J Clin Pharmacol. 2007;63:548–61. doi: 10.1111/j.1365-2125.2006.02803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Busse W, Corren J, Lanier BQ, McAlary M, Fowler-Taylor A, Cioppa GD, van As A, Gupta N. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol. 2001;108:184–90. doi: 10.1067/mai.2001.117880. [DOI] [PubMed] [Google Scholar]
- 24.Solèr M, Matz J, Townley R, Buhl R, O'Brien J, Fox H, Thirlwell J, Gupta N, Della Cioppa G. The anti-IgE antibody omalizumab reduces exacerbations and steroid requirement in allergic asthmatics. Eur Respir J. 2001;18:254–61. doi: 10.1183/09031936.01.00092101. Erratum in Eur Respir J 2001; 18: 739–40. [DOI] [PubMed] [Google Scholar]
- 25.Holgate ST, Chuchalin AG, Hébert J, Lötvall J, Persson GB, Chung KF, Bousquet J, Kerstjens HA, Fox H, Thirlwell J, Cioppa GD, Omalizumab 011 International Study Group Efficacy and safety of a recombinant anti-immunoglobulin E antibody (omalizumab) in severe allergic asthma. Clin Exp Allergy. 2004;34:632–8. doi: 10.1111/j.1365-2222.2004.1916.x. [DOI] [PubMed] [Google Scholar]
- 26.Slavin RG, Ferioli C, Tannenbaum SJ, Martin C, Blogg M, Lowe PJ. Asthma symptom re-emergence after omalizumab withdrawal correlates well with increasing IgE and decreasing pharmacokinetic concentrations. J Allergy Clin Immunol. 2009;123:107–13. doi: 10.1016/j.jaci.2008.09.050. [DOI] [PubMed] [Google Scholar]
- 27.Ädelroth E, Rak S, Haahtela T, Aasand G, Rosenhall L. Recombinant humanized mAb-E25, an anti-IgE mAb, in birch pollen-induced seasonal allergic rhinitis. J Allergy Clin Immunol. 2000;106:253–9. doi: 10.1067/mai.2000.108310. [DOI] [PubMed] [Google Scholar]
- 28.West GB, Brown JH, Enquist BJ. A general model for the origin of allometric scaling laws for biology. Science. 1997;276:122–6. doi: 10.1126/science.276.5309.122. [DOI] [PubMed] [Google Scholar]
- 29.West GB, Brown JH, Enquist BJ. The fourth dimension of life: fractal geometry and allometric scaling of organisms. Science. 1999;284:1677–9. doi: 10.1126/science.284.5420.1677. [DOI] [PubMed] [Google Scholar]
- 30.Bouwmeester NJ, Anderson BJ, Tibboel D, Holford NHG. Developmental pharmacokinetics of morphine and its metabolites in neonates, infants and young children. Br J Anaesth. 2004;92:208–17. doi: 10.1093/bja/aeh042. [DOI] [PubMed] [Google Scholar]
- 31.Ahn JE, Karlsson MO, Dunne A, Ludden TM. Likelihood based approaches to handling data below the quantitation limit using NONMEM VI. J Pharmacokinet Pharmacodyn. 2008;35:401–21. doi: 10.1007/s10928-008-9094-4. [DOI] [PubMed] [Google Scholar]
- 32.Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther. 2007;82:17–20. doi: 10.1038/sj.clpt.6100241. [DOI] [PubMed] [Google Scholar]
- 33.Riske F, Hakimi J, Mallmaci M, Griffin M, Pilson R, Tobkes N, Lin P, Danho W, Kochan J, Chizzonite R. High affinity human IgE receptor (FcεRI) analysis of functional domains of the α-subunit with monoclonal antibodies. J Biol Chem. 1991;266:11245–51. [PubMed] [Google Scholar]
- 34.Marcelletti JF, Katz DH. FcR epsilon+ lymphocytes and regulation of the IgE antibody system. I. A new class of molecules, termed IgE-induced regulants (EIR), which modulate FcR epsilon expression by lymphocytes. J Immunol. 1984;133:2821–8. [PubMed] [Google Scholar]
- 35.Beck LA, Marcotte GV, MacGlashan D, Togias A, Saini S. Omalizumab-induced reductions in mast cell Fc epsilon RI expression and function. J Allergy Clin Immunol. 2004;114:527–30. doi: 10.1016/j.jaci.2004.06.032. [DOI] [PubMed] [Google Scholar]
- 36.Lin H, Boesel KM, Griffith DT, Prussin C, Foster B, Romero FA, Townley R, Casale TB. Omalizumab rapidly decreases nasal allergic response and FcepsilonRI on basophils. J Allergy Clin Immunol. 2004;113:297–302. doi: 10.1016/j.jaci.2003.11.044. [DOI] [PubMed] [Google Scholar]
- 37.MacGlashan DW, Jr, Bochner BS, Adelman DC, Jardieu PM, Togias A, McKenzie-White J, Sterbinsky SA, Hamilton RG, Lichtenstein LM. Down-regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients with anti-IgE antibody. J Immunol. 1997;158:1438–45. [PubMed] [Google Scholar]