

Abstract
AIMS
To investigate the safety, pharmacokinetics and pharmacodynamics of rivaroxaban, an oral, direct Factor Xa (FXa) inhibitor, in healthy, male Chinese subjects.
METHODS
Two randomized, single-blind, placebo-controlled, dose-escalation studies were conducted in healthy Chinese men aged 18–45 years. In the single-dose study, subjects received single, oral doses of rivaroxaban 2.5, 5, 10, 20 and 40 mg. In the multiple-dose study, oral rivaroxaban was administered in doses of 5, 10, 20 and 30 mg twice daily for 6 days.
RESULTS
Rivaroxaban, in single and multiple doses up to 60 mg, was well tolerated. Rapid absorption was observed in both studies (time to Cmax 1.25–2.5 h). In the multiple-dose study, rivaroxaban exposure increased dose-proportionally after the first dose and at steady state (for the 5–20-mg doses). The half-life of rivaroxaban was up to 7.9 h in the single-dose study. Maximal inhibition of FXa activity was achieved within 1–3 h of dosing in the single-dose study [at 20 mg FXa inhibition as a median percentage change from baseline, 45.92; 95% confidence interval (CI) 44.64, 50.70] and 2–3 h after administration at steady state in the multiple-dose study (at 20 mg median FXa inhibition as a median percentage change from baseline, 60.25; 95% CI 56.16, 63.05), in line with maximum rivaroxaban plasma concentrations.
CONCLUSIONS
Rivaroxaban demonstrated predictable pharmacokinetics and pharmacodynamics in healthy Chinese subjects, in line with findings observed previously in White subjects. This suggests that fixed doses of rivaroxaban may be administered to all patients, regardless of their ethnic origin.
Keywords: anticoagulant, Chinese subjects, Factor Xa inhibitor, rivaroxaban
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Rivaroxaban is an oral, direct Factor Xa inhibitor in advanced clinical development for the prevention and treatment of thromboembolic disorders.
In single- and multiple-dose Phase I studies in White subjects, rivaroxaban was safe and demonstrated predictable, dose-dependent pharmacokinetics and pharmacodynamics.
WHAT THIS STUDY ADDS
The Phase III programme with rivaroxaban is being conducted worldwide.
Therefore, it is necessary to determine whether the pharmacokinetics, pharmacodynamics and tolerability of rivaroxaban are altered in patients of different ethnic origins.
Dose-escalation studies were conducted to determine the safety, pharmacokinetics and pharmacodynamics of single and multiple doses of rivaroxaban in healthy Chinese subjects.
Introduction
Anticoagulants are recommended for numerous medical conditions, including the prevention and treatment of venous thromboembolism (VTE) [1, 2], stroke prevention in patients with atrial fibrillation [3], and secondary prevention in acute coronary syndromes [4]. These conditions are predominant in the elderly, and because of the growing ageing population worldwide [5] it is likely that their prevalence will increase over the coming years. For example, the prevalence of atrial fibrillation is expected to reach 12.1 million in the USA by 2050 [6]. Therefore, anticoagulants are likely to be used more frequently, increasing the need for greater choice.
Currently available anticoagulants include vitamin K antagonists (VKAs), unfractionated heparin and the low-molecular-weight heparins (LMWHs). Each of these classes has limitations, resulting in several unmet needs that must be addressed by new therapies [7, 8]. The VKAs are administered orally, but have unpredictable pharmacokinetics (PK) and pharmacodynamics (PD), resulting in a narrow therapeutic window. In addition, VKAs are associated with numerous food and drug interactions, limiting their convenience and ease of use for patients [9–12]. The pharmacological profile of the VKAs results in a requirement for frequent monitoring to ensure anticoagulant effects remain within the appropriate range [9–12]. Unfractionated heparin also requires coagulation monitoring, and is administered parenterally [7]. The LMWHs are effective and do not require coagulation monitoring, but they too must be administered parenterally, making the heparins inconvenient for long-term use [8].
Several specific targets in the coagulation cascade are being further investigated as therapeutic strategies for anticoagulation [13, 14]. Factor Xa (FXa) is an attractive target for novel anticoagulants, because it acts at the convergence point of the intrinsic and extrinsic coagulation pathways [15]. By catalysing the conversion of prothrombin to thrombin through the prothrombinase complex, one molecule of FXa results in the generation of >1000 thrombin molecules [16]. Inhibition of FXa activity may be expected to block the amplification of thrombin generation, thereby limiting thrombin-mediated activation of coagulation and platelets, without affecting existing thrombin levels [14].
Rivaroxaban (Xarelto®) is an oral, direct FXa inhibitor [17] in advanced clinical development for the prevention and treatment of thromboembolic disorders. In single- and multiple-dose Phase I studies in White subjects, rivaroxaban has demonstrated predictable, dose-dependent PK and PD across the wide dose ranges tested (single dose 1.25–80 mg; multiple dose total daily doses of 5–60 mg for 7 days) [18, 19]. Rivaroxaban was absorbed rapidly after oral administration, reaching Cmax within 2–4 h. The half-life of rivaroxaban was up to 9 h in healthy subjects and up to 12 h in elderly subjects [18, 20]. The rapid absorption of rivaroxaban was mirrored by pharmacodynamic findings, with maximum inhibition of FXa activity achieved within 1–4 h of rivaroxaban dosing in the single-dose study [18]. Further studies have shown that the PK and PD of rivaroxaban are not significantly affected by age, weight or gender, suggesting that fixed dosing is likely to be possible in all patients [20, 21].
A large Phase II clinical study programme has been conducted with rivaroxaban, investigating its efficacy and safety for the prevention of VTE after major orthopaedic surgery, and for the treatment of VTE [22–26]. These studies led to the initiation of an extensive, worldwide Phase III study programme. The Phase II studies were conducted mainly in Europe and North America, and the vast majority (>90%) of patients were White. Because the Phase III programme with rivaroxaban is being conducted worldwide, it is necessary to determine whether the PK, PD and tolerability of rivaroxaban are altered in patients of different ethnic origins.
Body mass index (BMI) has been reported to be lower in the Chinese population when compared with Whites [27], and such differences can affect drug pharmacokinetic behaviour. Earlier Phase I studies have been conducted in Japan, Germany and the USA since 2001 [18], where tolerability has been confirmed with single doses up to 80 mg and multiple doses of 30 mg twice daily (b.i.d.) administered for 5 days. The results from foreign single-dose studies under fasting conditions were compared with those from a Japanese single-dose study, demonstrating the plasma concentration profile of rivaroxaban to be similar at each dose compared with White and Japanese subjects. Accordingly, to evaluate further and determine the safety, pharmacokinetic and pharmacodynamic properties of single- and multiple-dose rivaroxaban in the Asian population, two dose-escalation studies were conducted in healthy Chinese men.
Methods
Both studies were conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. The protocols were approved by the Independent Ethics Committee and the Institutional Review Board of Peking University First Hospital and the State Food and Drug Administration of China. The approval number of the State Food and Drug Administration was 2005 L00796. Prior to the beginning of the study, all subjects provided written informed consent.
Subjects
Healthy, male Chinese subjects aged 18–45 years, with a BMI of 19–27 kg m−2, were enrolled. Subjects with any known coagulation disorder (e.g. von Willebrand's disease, haemophilias) were excluded, as were those with conditions that have an increased bleeding risk (e.g. haemorrhoids, acute gastritis and peptic ulcer). Other exclusion criteria included recent blood donation, known hypersensitivity to the study medication, and relevant pathological changes in the electrocardiogram (ECG), such as a second- or third-degree arteriovenous block, prolongation of the QRS complex >120 ms, or of the QTc-interval >450 ms.
Study design
These were randomized, single-blind, placebo-controlled, parallel-group, dose-escalation studies. Both studies were conducted at a single site. In the single-dose study, subjects received rivaroxaban doses of 2.5, 5, 10, 20 or 40 mg. In the multiple-dose study, rivaroxaban was administered for 6 days: subjects received rivaroxaban 5, 10, 20 or 30 mg once daily (o.d.) on days 1 and 6, and b.i.d. on days 2–5. In both studies, eight subjects were randomized to receive rivaroxaban and two to receive placebo at each dose level.
In the single-dose study, subjects entered the study unit the day before drug administration, single doses of rivaroxaban or placebo were administered with water, the next morning after an overnight fast (≥10 h). Subjects were discharged 48 h after drug administration. Subjects were requested to return to the study unit 7 days after drug administration for a follow-up visit.
In the multiple-dose study, subjects entered the study unit the day before drug administration, received a single dose of rivaroxaban or placebo on the morning of day 1, rivaroxaban or placebo doses every 12 h (after breakfast and dinner) on days 2–5, and a single dose on the morning of day 6. Both rivaroxaban and placebo were administered with water within 5 min of standardized meals. No concomitant medication was permitted during the study period. Subjects were discharged on day 8, and returned for follow-up visits on days 9, 10 and 12.
Safety and tolerability assessments
Subjective tolerability was assessed by questioning subjects about any adverse events, and by spontaneous reporting. Objective tolerability was assessed by monitoring blood pressure, heart rate, ECG parameters, clinical laboratory parameters, and bleeding time (using the Template Bleeding Time method in both studies [28]; in addition, the Rumpel–Leede test [29] was used in the single-dose study). Adverse event severity was classified according to Medical Dictionary for Regulatory Activities criteria.
Sample collection
For the pharmacokinetic analysis, blood samples (6 ml and 4 ml in the single- and multiple-dose studies, respectively) were collected into tubes containing ammonium heparinate and centrifuged immediately to obtain plasma samples. These samples were then transferred to polypropylene vials and kept frozen below −20°C until analysis. In the single-dose study, samples were collected at dosing (time 0) and 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 15, 24 and 36 h after dosing. In the multiple-dose study, samples were collected at these same times on days 1 and 6, before the morning dose on day 3, and 36, 48 and 60 h after the last dose. Urine was collected predose (10 ml), and also pooled in the time ranges 0–4, 4–8, 8–12, 12–24 and 24–36 h after dosing. Urine from the same time ranges was also pooled in the multiple-dose study on days 1 and 6; additional samples were collected for up to 60 h after the last dose. After the total volume of urine for each time range was measured, 10 ml was taken from it and kept frozen at −20°C until analysis.
For the pharmacodynamic analysis, blood samples (8 ml and 4 ml in the single- and multiple-dose studies, respectively) were collected in tubes containing sodium citrate at dosing (time 0) and 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 15 and 24 h after dosing. These samples were also centrifuged immediately to obtain plasma samples, which were transferred to polypropylene vials and kept frozen below −20°C until analysis.
Analytical assay
All frozen samples of plasma and urine were sent to a central laboratory at Bayer HealthCare AG (Wuppertal, Germany) for analysis. All samples were analysed within 8 days after sampling. Rivaroxaban concentrations were determined using a fully validated high-performance liquid chromatography method, coupled to tandem mass spectrometry (plasma) or ultraviolet detection (urine). As described previously [21], samples were analysed after solid-phase extraction of rivaroxaban and the internal standard from the matrix using C18 cartridges. A close chemical analogue of rivaroxaban was used as an internal standard [30]. Monitored ions were 436→145 (rivaroxaban) and 464→145 (internal standard). The concentrations were validated by assaying quality control samples of blank plasma spiked with known concentrations of rivaroxaban. Rivaroxaban samples above the lower limit of quantification (0.5 µg l−1) were determined with an accuracy of 99.5–107.4% and a precision of 3.5–5.6%.
Pharmacokinetic parameters
In the single-dose study, the pharmacokinetic parameters assessed included: area under the plasma concentration–time curve (AUC), AUC divided by dose (AUC/D), maximum plasma concentration (Cmax), Cmax divided by dose (Cmax/D), time to Cmax (tmax), terminal half-life (t1/2), apparent oral clearance (CL/f), apparent volume of distribution during the terminal phase (Vz/F) and amount excreted in the urine (Aeur). The pharmacokinetic parameters determined in the multiple-dose study included the AUC during the dosage interval τ (AUCτ), AUCτ divided by dose per kg body weight (AUCτ(norm)), Cmax, Cmax divided by dose per kg body weight (Cmax,norm), tmax, t1/2, Aeur and accumulation ratio of RA4 (AUCτ at steady state/AUCτ after the first dose). The PK parameters of rivaroxaban were calculated from the plasma concentration–time data by noncompartmental methods.
Pharmacodynamic parameters
FXa activity was determined by a two-step photometric assay. FXa in plasma was completely activated with Russell's viper venom (RVV; Haemochrom Diagnostica, Essen, Germany). The concentration of RVV in the final assay volume (150 µl) was 14.5 µg ml−1, added as 50 µl of a 1 : 1 mix of one part 87 µg ml−1 RVV solution and one part 0.1 M CaCl2. Thus, the final concentration of Ca ions added was 16.7 mM. There was no dedicated pre-incubation step with the RVV before adding the substrate. The chromogenic substrate Z-D-Arg-Gly-Arg-pNA (S-2765™; Chromogenix, Milan, Italy) was then hydrolysed by FXa, releasing pNA, which was quantified by spectrophotometry at 405 nm. Standards and controls were prepared from the Third International Standard Coagulation Factors II and X Concentrate, Human 98/590 (National Institute for Biological Standards and Control, Potters Bar, UK). Prothrombin time (PT) was assessed using freeze-dried thromboplastin from rabbit brain (Neoplastin Plus®; Roche Diagnostics, Mannheim, Germany), and activated partial thromboplastin time (aPTT) was assessed using a kaolin-activated test (Roche Diagnostics). PT, aPTT and HepTest (Haemachem, St Louis, MO, USA) were measured using a ball coagulometer KC 10 (Amelung, Lemgo, Germany) according to the manufacturer's instructions.
In the single-dose study, thrombin and antithrombin (AT) activity were also assessed. The activity of thrombin was determined after ecarin-mediated activation (ecarin incubation time was 3 min) of prothrombin (ecarin 0.6 IU ml−1; Haemochrom Diagnostica, Essen, Germany) by use of the chromogenic substrate HD-Phe-Pip-Arg-pNA (S-2238; Haemochrom Diagnostica). All standards and controls were prepared from the 3rd International Standard Coagulation Factors II and X Concentrate, Human, 98/590 (National Institute for Biological Standards and Control). To determine AT activity, plasma was incubated with an excess of FXa and heparin, and the quantity of unbound FXa was then determined by cleavage of S-2765 and release of pNA, measured by photometer at 405 nm. The pNA level is inversely proportional to the AT activity of the plasma sample. All reagents used were obtained from the COAMATIC Antithrombin III kit (Chromogenix). All standards were prepared from Calibration Plasma (Haemochrom Diagnostica). All controls were prepared from the Second International Standard Antithrombin Plasma, Human, 93/768 (National Institute for Biological Standards and Controls).
Statistical analyses
The pharmacokinetic parameters AUC/D and Cmax/D in the single-dose study, and AUCτ(norm) and Cmax,norm in the multiple-dose study, were analysed assuming log-normally distributed data. To investigate whether these parameters increased dose proportionally, an exploratory analysis of variance was performed on the log-transformed values. Point estimates [least squares (LS)-means ratios] with 95% confidence intervals (CI) for these parameters were calculated by retransformation of the logarithmic data. In order to investigate the relationship between PK and PD, scatter plot and correlation analyses were performed.
Results
Single-dose study
Subjects
A total of 50 subjects were enrolled between May and July 2005; all subjects completed the study and, because there were no protocol violations, all subjects were included in the safety, pharmacokinetic and pharmcaodynamic analyses. The groups were well matched with respect to demographic characteristics (Table 1). The mean age of subjects was 34.7 years. Minor between-group differences in BMI were not statistically significant.
Table 1.
Demographic characteristics of subjects enrolled in single- and multiple-dose studies
| Rivaroxaban dose | |||||||
|---|---|---|---|---|---|---|---|
| Placebo | 2.5 mg | 5 mg | 10 mg | 20 mg | 40 mg | Total | |
| Single-dose study | (n= 10) | (n= 8) | (n= 8) | (n= 8) | (n= 8) | (n= 8) | (n= 50) |
| Age (range; years) | 33.2 | 34.6 | 34.4 | 35.1 | 35.8 | 35.8 | 34.7 |
| (30–38) | (30–39) | (30–39) | (30–39) | (32–39) | (32–39) | (30–39) | |
| Weight (kg) | 63.8 ± 7.5 | 62.4 ± 7.4 | 66.0 ± 9.4 | 58.4 ± 4.9 | 59.1 ± 5.4 | 62.8 ± 4.7 | 62.1 ± 6.9 |
| BMI (kg m−2) | 22.3 ± 1.7 | 21.9 ± 1.8 | 22.5 ± 1.9 | 20.9 ± 1.6 | 21.6 ± 1.1 | 22.9 ± 1.2 | 22.0 ± 1.7 |
| Rivaroxaban dose | |||||||
| Placebo | 5 mg b.i.d. | 10 mg b.i.d. | 20 mg b.i.d. | 30 mg b.i.d. | Total | ||
| Multiple-dose study | (n= 8) | (n= 8) | (n= 8) | (n= 9) | (n= 8) | (n= 41) | |
| Age (range; years) | 34.5 | 34.6 | 33.8 | 35.3 | 35.0 | – | 34.7 |
| (31–39) | (30–39) | (31–37) | (32–39) | (32–39) | (30–39) | ||
| Weight (kg) | 66.3 ± 8.8 | 65.4 ± 7.6 | 64.0 ± 6.9 | 68.3 ± 5.7 | 65.6 ± 7.0 | – | 66.0 ± 7.0 |
| BMI (kg m−2) | 22.9 ± 1.9 | 23.2 ± 1.9 | 22.5 ± 2.0 | 24.4 ± 1.5 | 22.6 ± 1.7 | – | 23.2 ± 1.8 |
Values are shown as mean ± SD, except for age, which is shown as mean (range). BMI, body mass index.
Safety and tolerability
Ten adverse events were reported; of these, two occurred before study drug administration. The eight treatment-emergent adverse events were reported by seven of the 50 subjects. No adverse events were reported in the rivaroxaban 40 mg group, one was reported in each of the rivaroxaban 2.5, 10 and 20 mg groups, two were reported in the rivaroxaban 5 mg group and three were reported in the placebo group. Three of the eight treatment-emergent adverse events were considered to be related to rivaroxaban. These were separate incidences of increased serum amylase and increased lipase in one subject receiving rivaroxaban 5 mg, and a transient prolongation of bleeding time (to 3.3 times baseline) in a subject receiving rivaroxaban 10 mg, which was not associated with a bleeding event. All adverse events were mild in intensity and resolved without treatment.
No changes in vital signs or ECG parameters, including the QTc interval, were observed. Median bleeding times were not substantially prolonged compared with baseline with any rivaroxaban dose, except for a transient prolongation in one patient receiving rivaroxaban 10 mg (mentioned above).
Pharmacokinetics
The AUC and Cmax of rivaroxaban increased dose dependently with the 2.5–20-mg doses. However, these parameters were not significantly higher with the rivaroxaban 40 mg dose compared with the 20 mg dose (Table 2). AUC and Cmax values were normalized by dividing the values by the rivaroxaban dose (AUC/D and Cmax/D, respectively). Comparison of the LS-means ratios of these parameters confirmed that rivaroxaban exposure increased almost dose proportionally between 2.5 and 10 mg, and became less than dose proportional with the 20- and 40-mg doses. Rivaroxaban was absorbed rapidly, with a tmax of 1.25–2.25 h. The t1/2 of rivaroxaban was up to 7.9 h, and did not appear to be dose dependent (Table 2). Urinary excretion of the rivaroxaban 2.5–10 mg doses was approximately 26%, decreasing to 20% with the 20-mg dose, and 11% with the 40-mg dose.
Table 2.
Pharmacokinetic parameters of rivaroxaban after administration of single doses in healthy Chinese subjects (values presented as geometric means with geometric % coefficients of variation and 95% confidence intervals)
| Rivaroxaban dose | |||||
|---|---|---|---|---|---|
| 2.5 mg | 5 mg | 10 mg | 20 mg | 40 mg | |
| (n= 8) | (n= 8) | (n= 8) | (n= 8) | (n= 8) | |
| AUCτ (µg h−1 l−1) | 251.7 (0.57) | 410.6 (0.29) | 1022 (0.13) | 1 354 (0.10) | 1 402 (0.09) |
| [167.8, 335.5] | [352.5, 468.7] | [796.8, 1248] | [1 029, 1 678] | [1 161, 1 643] | |
| AUCτ(norm) (g h−1 l−1) | 6242 (0.02) | 5372 (0.02) | 5950 (0.02) | 3 987 (0.04) | 2 194 (0.06) |
| [4361, 8122] | [4244, 6500] | [4383, 7516] | [2 869, 5 106] | [1 800, 2 587] | |
| AUC/D (h l−1) | 0.1007 (1430) | 0.08212 (1462) | 0.1022 (1256) | 0.06768 (2 000) | 0.03505 (3 524) |
| [0.06709, 0.1342] | [0.07050, 0.09374] | [0.07968, 0.1248] | [0.05146, 0.08390] | [0.02902, 0.04108] | |
| Cmax (µg l−1) | 51.27 (3.40) | 67.21 (1.90) | 143.2 (0.91) | 204.4 (0.57) | 176.1 (0.70) |
| [25.41, 77.13] | [53.21, 81.21] | [110.6, 175.8] | [179.4, 229.3] | [142.9, 209.2] | |
| Cmax,norm (g l−1) | 1270 (0.14) | 879.5 (0.14) | 833.4 (0.16) | 601.9 (0.21) | 275.6 (0.46) |
| [671.6, 1868] | [719.7, 1039] | [619.2, 1047] | [500.4, 703.4] | [215.9, 335.3] | |
| Cmax/D (1 l−1) | 0.02051 (8504) | 0.01344 (9481) | 0.01432 (9131) | 0.01022 (11 419) | 0.00440 (28 156) |
| [0.01016, 0.03086] | [0.01064, 0.01624] | [0.01106, 0.01758] | [0.008972, 0.01146] | [0.003574, 0.005232] | |
| tmax* (h) | 2.00 (0.50–6.00) | 2.00 (1.00–4.00) | 2.25 (1.00–4.00) | 2.00 (0.50–3.00) | 1.25 (0.50–3.00) |
| [0.54, 3.46] | [1.00, 3.00] | [1.23, 3.27] | [1.19, 2.81] | [0.63, 1.87] | |
| t1/2 (h) | 3.38 (46.37) | 7.92 (18.11) | 7.57 (18.45) | 5.62 (24.62) | 7.03 (25.65) |
| [1.85, 4.92] | [5.33, 10.51] | [5.31, 9.82] | [4.06, 7.18] | [2.13, 11.92] | |
| CL/f (l h−1) | 9.93 (14.49) | 12.18 (9.86) | 9.78 (13.13) | 14.78 (9.16) | 28.53 (4.33) |
| [6.50, 13.37] | [10.14, 14.22] | [7.77, 11.79] | [10.44, 19.11] | [23.26, 33.80] | |
| Vz/F (l kg−1) | 0.78 (200.1) | 2.13 (75.54) | 1.84 (77.16) | 2.04 (70.85) | 4.62 (36.10) |
| [0.44, 1.13] | [1.25, 3.00] | [1.34, 2.33] | [1.36, 2.71] | [1.88, 7.35] | |
| Aeur (%) | 27.2 (5.94) | 23.6 (9.42) | 26.7 (5.25) | 19.6 (5.23) | 11.0 (3.26) |
| [22.2, 32.2] | [15.7, 31.5] | [22.3, 31.1] | [15.2, 24.0] | [8.27, 13.7] | |
Median (range). AUCτ, area under the concentration–time curve during the dosage interval τ; AUCτ(norm), AUCτ divided by dose (mg) per kg body weight; AUC/D, AUC divided by dose; Cmax, maximal drug concentration in plasma; Cmax,norm, Cmax divided by dose (mg) per kg body weight; Cmax/D, Cmax divided by dose; tmax, time to reach maximum drug concentration in plasma; t1/2, half-life; CL/f, total body clearance of drug from plasma calculated after oral administration; Vz/F, apparent volume of distribution during terminal phase; Aeur, amount of drug excreted via the urine.
Pharmacodynamics
Rivaroxaban inhibited FXa activity dose dependently up to 20 mg (FXa as a median percentage change from baseline, 45.92; 95% CI 44.64, 50.70); the effect with rivaroxaban 40 mg was not significantly greater than with rivaroxaban 20 mg (Figure 1a). Maximum inhibition occurred 1–3 h after dosing, reaching 48% with the rivaroxaban 40 mg dose. Inhibition of FXa activity reflected rivaroxaban plasma concentrations. Placebo had no effect on FXa activity.
Figure 1.
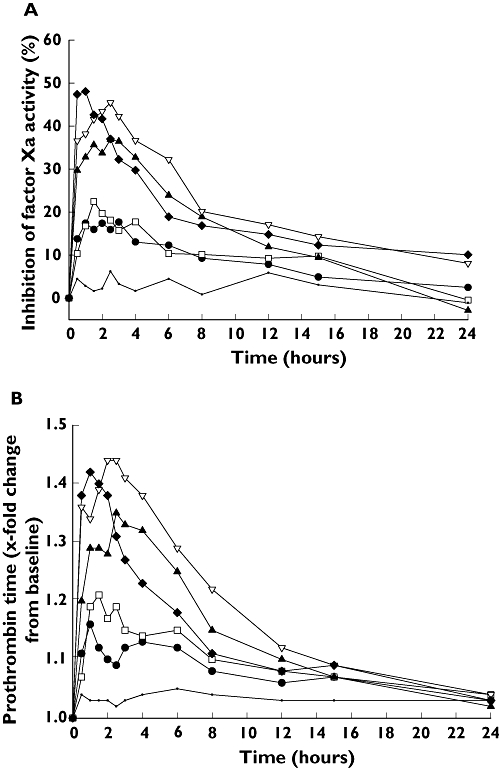
Inhibition of Factor Xa activity (A), shown as median percentage change from baseline, and prolongation of prothrombin time (B), shown as median relative change from baseline, in healthy, male Chinese subjects after single doses of rivaroxaban (n= 8 for each rivaroxaban dose and n= 10, placebo). Rivaroxaban 2.5 mg (—•—); Rivaroxaban 5 mg (—□—); Rivaroxaban 10 mg (—▴—); Rivaroxaban 20 mg (—▿—); Rivaroxaban 40 mg (—♦—); Placebo (—·—)
Maximum prolongation of PT followed a similar pattern to inhibition of FXa activity, with median prolongation of 1.51 (95% CI 1.47, 1.56) times baseline being observed with rivaroxaban 20 mg. PT had returned to baseline levels after 24 h with all rivaroxaban doses (Figure 1b). Similar results were observed for aPTT and HepTest: the median prolongations for aPTT and HepTest peaked at 1.37 and 1.73 times the baseline values, respectively. The highest aPTT prolongation values were observed in the 20-mg dosing group, at 2.5 h after study drug administration. The peak value of HepTest occurred in the highest dosing group (40 mg), 1.5 h after study drug administration. Rivaroxaban did not have any significant effects on thrombin or AT activity (data not shown).
Multiple-dose study
Subjects
Ten subjects were enrolled in each of the rivaroxaban 5, 10 and 30 mg b.i.d. dose groups and 11 in the rivaroxaban 20 mg b.i.d. dose group. A subject in the rivaroxaban 20 mg b.i.d. dose group withdrew consent after the first rivaroxaban dose; therefore, 41 subjects were included in the safety analysis and 40 in the pharmacokinetic and pharmacodynamic analyses. The mean age of the subjects was 34.7 years. There were no major differences between the groups in terms of demographic characteristics (Table 1).
Safety and tolerability
A total of 23 adverse events were reported by 16 subjects; of these, 10 were considered to be related to rivaroxaban (Table 3). Eight subjects had a transient increase in alanine aminotransferase levels (>×2 the upper limit of normal), three of whom had raised levels at screening. In addition, one subject had a transient increase in aspartate aminotransferase levels (>×2 the upper limit of normal). All adverse events were mild in severity, except for an incidence of glucose in the urine of one subject receiving rivaroxaban 5 mg b.i.d., which was considered to be of moderate severity, but was not considered to be drug related. All adverse events considered to be related to rivaroxaban resolved by the end of the study, without the need for treatment.
Table 3.
Treatment-emergent adverse events reported during multiple-dose administration of rivaroxaban in healthy Chinese subjects
| Rivaroxaban dose | |||||
|---|---|---|---|---|---|
| 5 mg b.i.d. | 10 mg b.i.d. | 20 mg b.i.d. | 30 mg b.i.d. | Placebo | |
| (n= 8) | (n= 8) | (n= 9) | (n= 8) | (n= 8) | |
| Patients with any event, n (%) | 5 (63) | 2 (25) | 3 (33) | 3 (38) | 3 (38) |
| Events, n (%) | |||||
| Gingival bleeding | – | – | – | 1 (13) | – |
| Catheter-site oedema | – | – | 1 (11) | – | – |
| Catheter-site pain | – | – | 1 (11) | – | – |
| Increased ALT | 3 (38) | 2 (25) | 2 (22) | 1 (13) | – |
| Increased AST | – | 1 (13) | – | – | – |
| Increased blood glucose | 1 (13) | – | – | – | – |
| Increased triglycerides | 2 (25) | 1 (13) | 1 (11) | – | 3 (38) |
| Glucose in urine | 1 (13) | – | – | – | – |
| Shoulder pain | 1 (13) | – | – | – | – |
| Haematuria | – | – | – | 1 (13) | – |
ALT, alanine aminotransferase; AST, aspartate aminotransferase.
No changes in vital signs or ECG parameters, including the QTc interval, were observed. Median bleeding times were not substantially prolonged with rivaroxaban after a single dose or at steady state (median relative change from baseline at steady state 1.06–1.29), compared with baseline.
Pharmacokinetics
Rivaroxaban AUCτ and Cmax increased dose proportionally after the first dose and at steady state for the 5–20 mg b.i.d. dose groups. For the 30 mg b.i.d. dose, the increases were slightly less than dose proportional (Figure 2; Table 4). Comparison of the LS-means ratios of AUCτ(norm) and Cmax,norm showed that all ratios were close to 1.0. The time to maximum plasma concentration was 2.0–2.5 h after rivaroxaban administration at steady state. The half-life of rivaroxaban at steady state was 4.6–5.8 h (Table 4).
Figure 2.
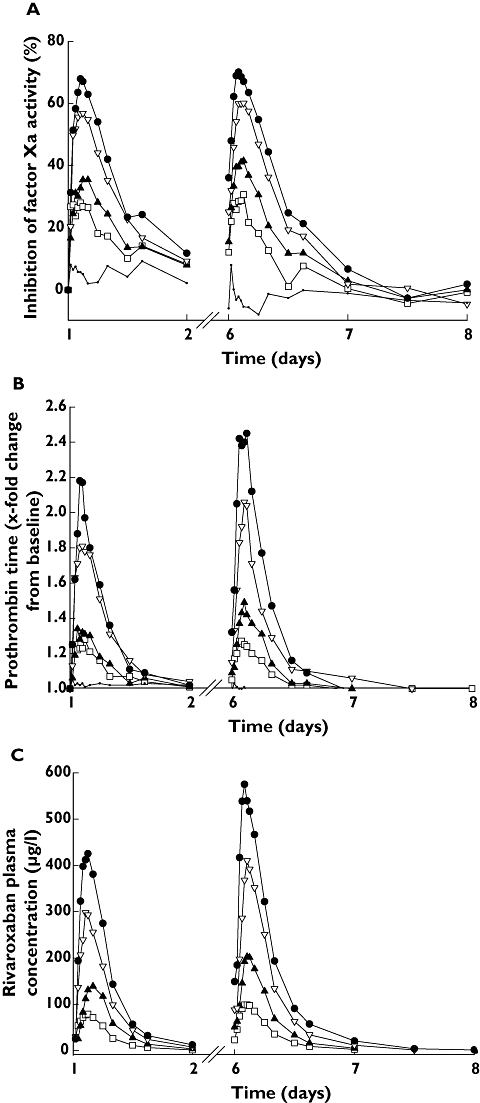
Inhibition of Factor Xa activity (A), shown as median percentage change from baseline, and prolongation of prothrombin time (B), shown as median relative change from baseline in healthy, male Chinese subjects after single (day 1; n= 8 for each rivaroxaban dose group and n= 10, placebo) and multiple doses of rivaroxaban (day 6; n= 8 for each rivaroxaban dose group and placebo). (C) Mean rivaroxaban plasma concentration–time profiles in healthy, male Chinese subjects (n= 8) after a single dose (day 1) and multiple dosing (day 6). Rivaroxaban 5 mg (—□—); Rivaroxaban 10 mg (—▴—); Rivaroxaban 20 mg (—▿—); Rivaroxaban 30 mg (—•—); Placebo (—·—)
Table 4.
Pharmacokinetic parameters of rivaroxaban at steady state in healthy Chinese subjects (values presented as geometric means with geometric % coefficients of variation and 95% confidence intervals)
| Rivaroxaban dose | ||||
|---|---|---|---|---|
| 5 mg b.i.d. | 10 mg b.i.d. | 20 mg b.i.d. | 30 mg b.i.d. | |
| (n= 8) | (n= 8) | (n= 8) | (n= 8) | |
| AUC0–12 (µg h−1 l−1) | 674.0 (14.59) | 1305 (18.11) | 2 527 (20.69) | 3601 (14.23) |
| [592.0, 756.0] | [1101, 1508] | [2 058, 2 997] | [3155, 4047] | |
| AUC0–12(norm) (ng h−1 l−1) | 8 757 (17.62) | 8304 (17.79) | 8504 (23.61) | 7840 (19.82) |
| [7 434, 10 080] | [7042, 9567] | [6 615, 10 393] | [6565, 9115] | |
| Cmax (µg l−1) | 115.4 (23.55) | 215.9 (19.51) | 415.1 (16.18) | 590.3 (8.68) |
| [91.10, 139.7] | [178.8, 252.9] | [355.9, 474.3] | [546.8, 633.8] | |
| Cmax,norm (ng l−1) | 1 499 (28.06) | 1374 (19.26) | 1 397 (19.06) | 1285 (13.41) |
| [1 120, 1 880] | [1143, 1606] | [1 150, 1 643] | [1136, 1435] | |
| tmax* (h) | 2.00 (1.00–3.00) | 2.50 (1.50–4.00) | 2.50 (1.50–3.00) | 2.25 (1.00–3.00) |
| [1.39, 2.61] | [1.87, 3.13] | [2.11, 2.89] | [1.70, 2.80] | |
| t1/2 (h) | 4.9 (18.86) | 5.1 (25.16) | 4.6 (11.68) | 5.8 (30.59) |
| [4.18, 5.66] | [3.90, 6.34] | [4.14, 5.03] | [4.34, 7.34] | |
| Aeur (%) | 52.0 (4.855) | 42.7 (12.83) | 38.3 (3.898) | 28.1 (3.836) |
| [47.94, 56.06] | [31.97, 53.43] | [35.04, 41.56] | [24.89, 31.31] | |
| RA4 (%) | 109.8 (13.30) | 113.4 (12.51) | 116.3 (15.79) | 113.7 (12.23) |
| [96.75, 122.9] | [101.7, 125.1] | [100.9, 131.7] | [101.8, 125.6] | |
Median (range). Aeur, amount of drug excreted via the urine; AUC0–12, area under the concentration–time curve during the 12-h dosage interval; AUC0–12(norm), AUC0–12 divided by dose (mg) per kg body weight; Cmax, maximal drug concentration in plasma; Cmax,norm, Cmax divided by dose (mg) per kg body weight; RA4, accumulation ratio (AUC0–12,day 6/AUC day1); tmax, time to maximum drug concentration in plasma; t1/2, half-life.
At steady state, urinary excretion of rivaroxaban decreased dose dependently from 52% in the 5 mg b.i.d. group to 28% in the 30 mg b.i.d. group (Table 4). There was no relevant accumulation of rivaroxaban after multiple dosing; values for the accumulation ratio RA4 were all close to 100% (Table 4).
Pharmacodynamics
Maximum inhibition of FXa activity increased dose dependently with rivaroxaban, and occurred 2.5–4 h after the first dose, and 2–3 h after administration at steady state (Figure 2a). At 20-mg dose, the median FXa inhibition as a median percentage change from baseline was 60.25 (95% CI 56.16, 63.05). Maximum inhibition of FXa activity was similar after the first dose and at steady state: 68 and 70%, respectively (Figure 2a).
Prolongation of PT with rivaroxaban followed a similar pattern to inhibition of FXa activity (Figure 2b). The greatest prolongations were observed with rivaroxaban 30 mg b.i.d., and were 2.18 times baseline after the first dose, and 2.45 times baseline after the last dose. Prolongation of aPTT and HepTest also followed a similar pattern to PT, and inhibition of FXa activity: greatest prolongations were 1.71 and 2.35 times baseline after the first dose, respectively.
Pharmacokinetic and pharmacodynamic correlation
There was a strong correlation between rivaroxaban plasma concentrations and both inhibition of FXa activity (Figure 3a) and prolongation of PT (Figure 3b; r= 0.942 and r= 0.931, respectively) across both studies.
Figure 3.
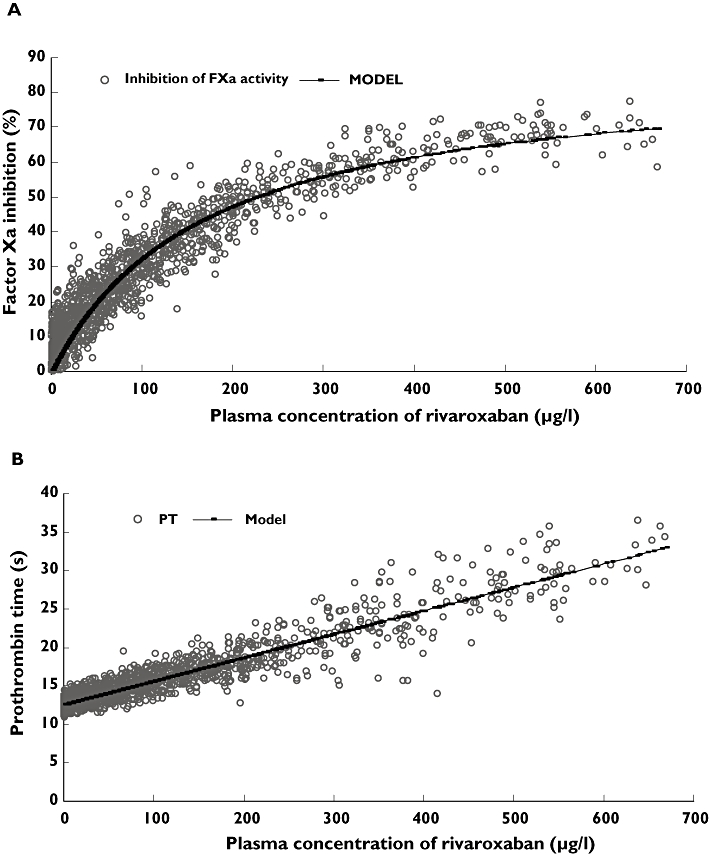
Correlation between plasma concentration of rivaroxaban and inhibition of Factor Xa (FXa) activity (A) and prothrombin time (PT) (B) after administration of single and multiple doses of rivaroxaban
Discussion
In these two dose-escalation studies in healthy, male Chinese subjects, rivaroxaban had predictable PK and PD. Rivaroxaban was well tolerated in both studies, with a low number of drug-related, treatment-emergent adverse events being reported. The incidences of adverse events were similar between rivaroxaban and placebo; there was no evidence of a dose-related increase in adverse events with rivaroxaban, and no signs or symptoms of bleeding were reported.
In the single-dose study, there were no elevations in liver enzyme parameters above the upper limit of normal related to rivaroxaban. The magnitude of the increases in alanine aminotransferase or aspartate aminotransferase levels in subjects in the multiple-dose study was low, and the elevations were transient. Furthermore, three subjects had elevated alanine aminotransferase levels at screening. Indeed, these elevations may have been the result of the subjects being confined within the trial unit for a prolonged period, an effect that has been observed in placebo recipients in numerous Phase I clinical studies [31, 32]. Similar transient elevations have been observed in other Phase I studies with rivaroxaban [19]. An important consideration for these results is how they compare with findings in similar studies conducted in White subjects. The adverse events reported in Chinese subjects were of a similar nature and frequency to those reported in the studies of rivaroxaban conducted in White subjects [18, 19], suggesting that rivaroxaban should have a similar safety profile in Chinese and White patients.
Rivaroxaban was absorbed rapidly in both studies, with Cmax occurring 1.25–2.5 h after administration. This rapid absorption is similar to previous observations in White subjects, in which maximum plasma concentrations were reached within 2.0–4.0 h [18, 19]. Indeed, the overall pharmacokinetic profile of rivaroxaban in Chinese and White subjects was similar, particularly in terms of key parameters such as AUC, Cmax, tmax and t1/2[18, 19]. Table 5 shows a comparison of key pharmacokinetic parameters in Chinese and White subjects based on a single dose of rivaroxaban (10 mg dose chosen for comparison based on expected clinical use).
Table 5.
Comparison of body mass index and key pharmacokinetic and pharmacodynamics parameters of rivaroxaban in Chinese and White subjects (values are geometric means with geometric % coefficients of variation) [18]
| Rivaroxaban 10 mg single dose | ||
|---|---|---|
| Chinese subjects (n= 8) | White subjects (n= 8) | |
| BMI (kg m−2) | 20.9 (0.08) | 24.9 (0.11) |
| AUCτ (µg h−1 l−1) | 1022 (0.13) | 1020 (14.9) |
| AUCτ(norm) (g h−1 l−1) | 5950 (0.02) | 8766 (18.0) |
| Cmax (µg l−1) | 143.2 (0.91) | 141 (15.5) |
| Cmax,norm (g l−1) | 833.4 (0.16) | 1211 (21.3) |
| tmax* (h) | 2.25 (1.00–4.00) | 2.00 (0.50–2.50) |
| t1/2 (h) | 7.57 (18.45) | 9.07 (61.8) |
| CL/f (l h−1) | 9.78 (13.13) | 9.8 (14.9) |
| Vz/F (l kg−1) | 1.84 (77.16) | 1.49 (66.3) |
| FXa Inhibition (%) maximum mean change from baseline | 40 (0.14) | 33 (0.21) |
| PT(x-fold change) mean change from baseline | 1.4 (0.06) | 1.3 (0.08) |
| aPTT(x-fold change) mean change from baseline | 1.3 (0.07) | 1.3 (0.04) |
| HepTest (x-fold change) mean change from baseline | 1.7 (0.13) | 1.3 (0.04) |
Median (range). AUCτ, area under the concentration–time curve during the dosage interval τ; AUCτ(norm), AUCτ divided by dose (mg) per kg body weight; BMI, body mass index; Cmax, maximal drug concentration in plasma; Cmax,norm, Cmax divided by dose (mg) per kg body weight; tmax, time to reach maximum drug concentration in plasma; t1/2, half-life; CL/f, total body clearance of drug from plasma calculated after oral administration; Vz/F, apparent volume of distribution during terminal phase.
Rivaroxaban exposure, in terms of AUC and Cmax, increased dose dependently with single doses up to 20 mg; however, there was almost no further increase in exposure with the 40-mg dose compared with the 20-mg dose. In the multiple-dose study, rivaroxaban exposure increased dose proportionally after the first dose and at steady state with the exception of the 30-mg b.i.d. dose. The slight differences in rivaroxaban exposure in the single- and multiple-dose studies may be because rivaroxaban was administered with food in the multiple-dose study and in the fasted state in the single-dose study. Similar differences between single dosing and multiple dosing have been observed in White subjects [18, 19]. Furthermore, food has been shown to reduce interindividual variability and increase the predictability of rivaroxaban PK [33]. The deviation from dose proportionality at higher rivaroxaban doses is thought to be due to the low solubility of rivaroxaban and thus incomplete absorption at these doses, and was also observed in the multiple-dose study conducted in Whites [34]. This theory is further supported by the amount of rivaroxaban excreted in urine, which decreased dose dependently in both studies (because unabsorbed rivaroxaban would be excreted via the faeces).
The t1/2 values for rivaroxaban observed in the single-dose study (3.4–7.9 h) were similar to those observed in the single-dose study in White subjects [18]. The t1/2 values for rivaroxaban observed in Chinese subjects after multiple doses (4.6–5.8 h at steady state) were slightly shorter than in White subjects receiving the same doses (5.7–9.2 h at steady state) [19]; however, whether this is clinically relevant will need to be investigated in further clinical studies. There was a tendency for the t1/2 to be higher with the higher rivaroxaban doses, which may be due to slower absorption of these doses. As in White subjects, rivaroxaban showed no signs of accumulation after multiple dosing [19].
The PK of rivaroxaban was affected by the generally smaller body size of the Chinese subjects, as would be expected compared with White subjects. As a result, values for AUCτ(norm) and Cmax,norm for the same single rivaroxaban doses were generally lower in Chinese compared with White subjects (5950 g h−1 l−1vs. 8766 g h−1 l−1, and 833 g l−1vs. 1211 g l−1, respectively) [18, 19]. The rate of absorption and elimination of rivaroxaban did not appear to be substantially different between the two populations. The differences in all of these parameters were small and are unlikely to be clinically relevant.
The effects of rivaroxaban on inhibition of FXa activity were predictable and closely mirrored the pharmacokinetic time course; close correlation between inhibition of FXa and rivaroxaban plasma concentrations was shown in both studies. Maximum effects on inhibition of FXa activity occurred 1–3 h after dosing in the single-dose study and 2–3 h after multiple doses. This matches the time course of inhibition of FXa activity observed in White subjects, in whom maximal inhibition occurred 1–4 h after single doses of rivaroxaban and after 3 h with multiple doses (Figure 4) [18, 19]. Similarly, the maximum levels of inhibition of FXa activity achieved in both studies in Chinese subjects were almost identical to those observed in White subjects: rivaroxaban 40 mg resulted in 48% inhibition of FXa activity in the Chinese single-dose study and 49% in the White single-dose study [18], whereas inhibition of FXa activity of 68% was observed after the first rivaroxaban 30-mg b.i.d. dose in both the Chinese and White multiple-dose studies [19]. The effect of rivaroxaban on global clotting tests (PT, aPTT, HepTest) followed a similar pattern to inhibition of FXa activity, with dose-dependent effects being observed. The effect of rivaroxaban on these tests was also predictable and correlated closely to inhibition of FXa in the single- and multiple-dose studies conducted in White subjects (PT shown in Figure 4) [18, 19].
Figure 4.

Comparison of inhibition of Factor Xa activity (A), shown as median percentage change from baseline, and prolongation of prothrombin time (B), shown as median relative change from baseline after administration of a single dose of rivaroxaban 10 or 20 mg, or placebo, in healthy, male Chinese and White subjects. Rivaroxaban 10 mg (Chinese; n = 8) (—▴—); Rivaroxaban 20 mg (Chinese; n = 8) (—▿—); Rivaroxaban 10 mg (Whites; n = 8) ( ); Rivaroxaban 20 mg (Whites; n = 7) (
); Rivaroxaban 20 mg (Whites; n = 7) ( ); Placebo (Chinese; n = 8) (—·—); Placebo (Whites; n = 25) (
); Placebo (Chinese; n = 8) (—·—); Placebo (Whites; n = 25) ( )
)
These studies have clearly demonstrated that rivaroxaban is well tolerated, with predictable, dose-proportional PK and dose-dependent anticoagulant activity in healthy Chinese subjects. Furthermore, the tolerability, PK and PD of rivaroxaban were very similar in Chinese and White subjects, suggesting that the metabolism and activity of rivaroxaban are not significantly affected by ethnicity. Rivaroxaban is currently in advanced development in numerous indications, and major clinical trials are ongoing. The effect of ethnicity on the efficacy and safety of rivaroxaban will be assessed as subgroup analyses of the ongoing studies; however, the findings presented here suggest that there are unlikely to be any clinically relevant differences between different ethnic groups. Ongoing and future studies in other ethnic study populations may add further evidence that rivaroxaban may be administered at the same fixed dose to patients, irrespective of their ethnic origin.
Acknowledgments
This study was sponsored by Bayer Schering Pharma AG. The authors acknowledge the contributions of Gabriele Rohde, Frank May and Stefan vom Lehn, who performed the rivaroxaban concentration assays; and Michael Zuehlsdorf, Angelica Roth, Anja Schafer and Ingo Loof, who performed the FXa, thrombin and antithrombin III activity assays. We acknowledge the contributions of Lin Peggy of APEX International (Taipei), who performed the statistical analysis. The authors would like to acknowledge Toby Allinson, who provided editorial assistance, funded by Bayer Schering Pharma AG, sponsors of the study.
Competing interests
D.K. is an employees of Bayer Schering Pharma AG. W.M. is an employee of Bayer Schering Pharma AG and holds shares in the company.
REFERENCES
- 1.Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the Seventh ACCP Conference on Antithrombotic and Thrombolytic therapy. Chest. 2004;126:401S–428S. doi: 10.1378/chest.126.3_suppl.401S. [DOI] [PubMed] [Google Scholar]
- 2.Geerts WH, Pineo GF, Heit JA, Bergqvist D, Lassen MR, Colwell CW, Ray JG. Prevention of venous thromboembolism: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:338S–400S. doi: 10.1378/chest.126.3_suppl.338S. [DOI] [PubMed] [Google Scholar]
- 3.Singer DE, Albers GW, Dalen JE, Go AS, Halperin JL, Manning WJ. Antithrombotic therapy in atrial fibrillation: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:429S–456S. doi: 10.1378/chest.126.3_suppl.429S. [DOI] [PubMed] [Google Scholar]
- 4.Harrington RA, Becker RC, Ezekowitz M, Meade TW, O'Connor CM, Vorchheimer DA, Guyatt GH. Antithrombotic therapy for coronary artery disease: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:513S–548S. doi: 10.1378/chest.126.3_suppl.513S. [DOI] [PubMed] [Google Scholar]
- 5.World Health Organization. The world is fast ageing – have we noticed? Available at http://www.who.int/ageing/en/ (last accessed 28 June 2006.
- 6.Miyasaka Y, Barnes ME, Gersh BJ, Cha SS, Bailey KR, Abhayaratna WP, Seward JB, Tsang TS. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation. 2006;114:119–25. doi: 10.1161/CIRCULATIONAHA.105.595140. [DOI] [PubMed] [Google Scholar]
- 7.Ansell J, Bergqvist D. Current options in the prevention of thromboembolic disease. Drugs. 2004;64(Suppl. 1):1–5. doi: 10.2165/00003495-200464001-00002. [DOI] [PubMed] [Google Scholar]
- 8.Agnelli G. Current issues in anticoagulation. Pathophysiol Haemost Thromb. 2005;34(Suppl. 1):2–9. doi: 10.1159/000083078. [DOI] [PubMed] [Google Scholar]
- 9.Ansell J, Hirsh J, Poller L, Bussey H, Jacobson A, Hylek E. The pharmacology and management of the vitamin K antagonists: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:204S–233S. doi: 10.1378/chest.126.3_suppl.204S. [DOI] [PubMed] [Google Scholar]
- 10.Hylek EM. Oral anticoagulants. Pharmacologic issues for use in the elderly. Clin Geriatr Med. 2001;17:1–13. doi: 10.1016/s0749-0690(05)70102-6. [DOI] [PubMed] [Google Scholar]
- 11.Ingelgard A, Hollowell J, Reddy P, Gold K, Tran K, Fitzmaurice D. What are the barriers to warfarin use in atrial fibrillation?: development of a questionnaire. J Thromb Thrombolysis. 2006;21:257–65. doi: 10.1007/s11239-006-5633-2. [DOI] [PubMed] [Google Scholar]
- 12.Spyropoulos AC. Managing oral anticoagulation requires expert experience and clinical evidence. J Thromb Thrombolysis. 2006;21:91–4. doi: 10.1007/s11239-006-5583-8. [DOI] [PubMed] [Google Scholar]
- 13.Eriksson BI, Quinlan DJ. Oral anticoagulants in development: focus on thromboprophylaxis in patients undergoing orthopaedic surgery. Drugs. 2006;66:1411–29. doi: 10.2165/00003495-200666110-00001. [DOI] [PubMed] [Google Scholar]
- 14.Kubitza D, Haas S. Novel factor Xa inhibitors for prevention and treatment of thromboembolic diseases. Expert Opin Investig Drugs. 2006;15:843–55. doi: 10.1517/13543784.15.8.843. [DOI] [PubMed] [Google Scholar]
- 15.Mann KG. Thrombin formation. Chest. 2003;124:4S–10S. doi: 10.1378/chest.124.3_suppl.4s. [DOI] [PubMed] [Google Scholar]
- 16.Mann KG, Brummel K, Butenas S. What is all that thrombin for? J Thromb Haemost. 2003;1:1504–14. doi: 10.1046/j.1538-7836.2003.00298.x. [DOI] [PubMed] [Google Scholar]
- 17.Perzborn E, Strassburger J, Wilmen A, Pohlmann J, Roehrig S, Schlemmer KH, Straub A. In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939 – an oral, direct Factor Xa inhibitor. J Thromb Haemost. 2005;3:514–21. doi: 10.1111/j.1538-7836.2005.01166.x. [DOI] [PubMed] [Google Scholar]
- 18.Kubitza D, Becka M, Voith B, Zuehlsdorf M, Wensing G. Safety, pharmacodynamics, and pharmacokinetics of single doses of BAY 59-7939, an oral, direct factor Xa inhibitor. Clin Pharmacol Ther. 2005;78:412–21. doi: 10.1016/j.clpt.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 19.Kubitza D, Becka M, Wensing G, Voith B, Zuehlsdorf M. Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939 – an oral, direct Factor Xa inhibitor – after multiple dosing in healthy male subjects. Eur J Clin Pharmacol. 2005;61:873–80. doi: 10.1007/s00228-005-0043-5. [DOI] [PubMed] [Google Scholar]
- 20.Kubitza D, Becka M, Mueck W, Zuehlsdorf M. The effect of extreme age, and gender, on the pharmacology and tolerability of rivaroxaban – an oral, direct Factor Xa inhibitor. Blood. 2006;108 Abstract 905. [Google Scholar]
- 21.Kubitza D, Becka M, Zuehlsdorf M, Mueck W. Body weight has limited influence on the safety, tolerability, pharmacokinetics, or pharmacodynamics of rivaroxaban (BAY 59-7939) in healthy subjects. J Clin Pharmacol. 2007;47:218–26. doi: 10.1177/0091270006296058. [DOI] [PubMed] [Google Scholar]
- 22.Eriksson BI, Borris LC, Dahl OE, Haas S, Huisman MV, Kakkar AK, Misselwitz F, Muehlhofer E, Kalebo P. Dose-escalation study of rivaroxaban (BAY 59-7939) – an oral, direct Factor Xa inhibitor – for the prevention of venous thromboembolism in patients undergoing total hip replacement. Thromb Res. 2007;120:685–93. doi: 10.1016/j.thromres.2006.12.025. [DOI] [PubMed] [Google Scholar]
- 23.Eriksson BI, Borris L, Dahl OE, Haas S, Huisman MV, Kakkar AK, Misselwitz F, Kalebo P. Oral, direct Factor Xa inhibition with BAY 59-7939 for the prevention of venous thromboembolism after total hip replacement. J Thromb Haemost. 2006;4:121–8. doi: 10.1111/j.1538-7836.2005.01657.x. [DOI] [PubMed] [Google Scholar]
- 24.Turpie AG, Fisher WD, Bauer KA, Kwong LM, Irwin MW, Kalebo P, Misselwitz F, Gent M. BAY 59-7939: an oral, direct Factor Xa inhibitor for the prevention of venous thromboembolism in patients after total knee replacement. A phase II dose-ranging study. J Thromb Haemost. 2005;3:2479–86. doi: 10.1111/j.1538-7836.2005.01602.x. [DOI] [PubMed] [Google Scholar]
- 25.Buller HR. Once-daily treatment with an oral, direct Factor Xa inhibitor – rivaroxaban (BAY 59-7939) – in patients with acute, symptomatic deep vein thrombosis. The EINSTEIN-DVT dose-finding study. Eur Heart J. 2006;27:761. Abstract Suppl. [Google Scholar]
- 26.Agnelli G, Gallus A, Goldhaber SZ, Haas S, Huisman MV, Hull RD, Kakkar AK, Misselwitz F, Schellong S. Treatment of proximal deep-vein thrombosis with the oral direct Factor Xa inhibitor rivaroxaban (BAY 59-7939): the ODIXa-DVT (oral direct Factor Xa inhibitor BAY 59-7939 in patients with acute symptomatic deep-vein thrombosis) study. Circulation. 2007;116:180–7. doi: 10.1161/CIRCULATIONAHA.106.668020. [DOI] [PubMed] [Google Scholar]
- 27.Deurenberg P, Deurenberg-Yap M, Guricci S. Asians are different from Caucasians and from each other in their body mass index/body fat per cent relationship. Obes Rev. 2002;3:141–6. doi: 10.1046/j.1467-789x.2002.00065.x. [DOI] [PubMed] [Google Scholar]
- 28.Radouco-Thomas S, Nosal G, Garcin F, Grumbach P, Radouco-Thomas C. A method for measurement of bleeding time: the hemostatic effect of some flavonoids. Life Sci. 1964;3:465–71. doi: 10.1016/0024-3205(64)90207-3. [DOI] [PubMed] [Google Scholar]
- 29.Bruschke G. Comparison of capillary resistance determination by Rumpel–Leede test with suction technic determination. Z Gesamte Inn Med. 1956;11:953–5. [PubMed] [Google Scholar]
- 30.Weinz C, Buetehorn U, Daehler HP, Kohlsdorfer C, Pleiss U, Sandmann S, Schlemmer KH, Schwarz T, Steinke W. Pharmacokinetics of BAY 59-7939 – an oral, direct Factor Xa inhibitor – in rats and dogs. Xenobiotica. 2005;35:891–910. doi: 10.1080/00498250500250493. [DOI] [PubMed] [Google Scholar]
- 31.Narjes H, Nehmiz G. Effect of hospitalisation on liver enzymes in healthy subjects. Eur J Clin Pharmacol. 2000;56:329–33. doi: 10.1007/s002280000142. [DOI] [PubMed] [Google Scholar]
- 32.Rosenzweig P, Miget N, Brohier S. Transaminase elevation on placebo during phase I trials: prevalence and significance. Br J Clin Pharmacol. 1999;48:19–23. doi: 10.1046/j.1365-2125.1999.00952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kubitza D, Becka M, Zuehlsdorf M, Mueck W. Effect of food, an antacid, and the H2 antagonist ranitidine on the absorption of BAY 59-7939 (rivaroxaban), an oral, direct Factor Xa inhibitor, in healthy subjects. J Clin Pharmacol. 2006;46:549–58. doi: 10.1177/0091270006286904. [DOI] [PubMed] [Google Scholar]
- 34.Mueck W, Becka M, Kubitza D, Voith B, Zuehlsdorf M. Population model of the pharmacokinetics and pharmacodynamics of rivaroxaban – an oral, direct Factor Xa inhibitor – in healthy subjects. Int J Clin Pharmacol Ther. 2007;45:335–44. doi: 10.5414/cpp45335. [DOI] [PubMed] [Google Scholar]