

Abstract
Cells of the innate immune system use Toll-like receptors (TLRs) to initiate the proinflammatory response to microbial infection. Recent studies have shown acute infections are associated with a transient increase in the risk of vascular thrombotic events. Although platelets play a central role in acute thrombosis and accumulating evidence demonstrates their role in inflammation and innate immunity, investigations into the expression and functionality of platelet TLRs have been limited. In the present study, we demonstrate that human platelets express TLR2, TLR1, and TLR6. Incubation of isolated platelets with Pam3CSK4, a synthetic TLR2/TLR1 agonist, directly induced platelet aggregation and adhesion to collagen. These functional responses were inhibited in TLR2-deficient mice and, in human platelets, by pretreatment with TLR2-blocking antibody. Stimulation of platelet TLR2 also increased P-selectin surface expression, activation of integrin αIIbβ3, generation of reactive oxygen species, and, in human whole blood, formation of platelet–neutrophil heterotypic aggregates. TLR2 stimulation also activated the phosphoinositide 3-kinase (PI3-K)/Akt signaling pathway in platelets, and inhibition of PI3-K significantly reduced Pam3CSK4-induced platelet responses. In vivo challenge with live Porphyromonas gingivalis, a Gram-negative pathogenic bacterium that uses TLR2 for innate immune signaling, also induced significant formation of platelet–neutrophil aggregates in wild-type but not TLR2-deficient mice. Together, these data provide the first demonstration that human platelets express functional TLR2 capable of recognizing bacterial components and activating the platelet thrombotic and/or inflammatory pathways. This work substantiates the role of platelets in the immune and inflammatory response and suggests a mechanism by which bacteria could directly activate platelets.
Keywords: platelets, Toll-like receptors, innate immunity
The innate immune system constitutes the first line of host defense against invasion by pathogenic microorganisms. Innate immune cells detect highly conserved bacterial, viral, and fungal components known as pathogen-associated molecular patterns (PAMPs). Recognition of PAMPs is primarily mediated by the family of Toll-like receptors (TLRs). To date, at least 11 members of the TLR family have been reported in humans (TLR1 through-11), with each member recognizing PAMPs of distinct microbial origin. TLR4, for instance, recognizes the Gram-negative cell wall component lipopolysaccharide (LPS), whereas TLR2 recognizes components of Gram-positive bacteria and, in cooperation with TLR1 and TLR6, bacterial lipoproteins.1 Binding of PAMPs to TLRs initiates a complex intracellular signaling cascade often resulting in activation of nuclear factor (NF)-κB, induction of proinflammatory genes, and downstream activation of the adaptive immune system.2 Thus, TLRs play a fundamental role in both the initiation and propagation of the inflammatory response to microbial infection.
Though primarily involved in hemostasis, platelets also participate in the immune and inflammatory response. Activated platelets release a variety of prothrombotic, proinflammatory, and antimicrobial mediators.3 In vitro and in vivo studies establish that platelets and bacteria physically interact, leading to direct platelet activation and aggregation.4–6 Recent clinical studies have demonstrated that bacterial infections are associated with a transient increase in the risk of acute thrombotic events such as myocardial infarction and stroke.7 Furthermore, platelets contribute significantly to the pathophysiology and high mortality rates of sepsis.8 Uncontrolled bacterial infection leads to widespread platelet activation, and, accordingly, platelet counts are decreased in septic patients, with the degree of thrombocytopenia correlating with the severity of the sepsis.9 Platelets, therefore, provide a fundamental link between the immune and thrombotic cascades, but the precise mechanism by which they interact with bacteria and their ligands is still unclear.
The expression, functionality, and signaling pathways of TLRs have been well studied in many hematologic and vascular cells, including neutrophils, monocytes/macrophages, and endothelial cells. Several groups have recently demonstrated that TLRs (specifically, TLR1, TLR2, TLR4, TLR6, TLR8, and TLR9) are expressed in human platelets.10,11 However, investigations of TLR functionality have focused mainly on TLR4 and the platelet response to LPS.12–18 Notably, LPS has been shown to induce thrombocytopenia and platelet accumulation in the lungs of wild-type but not TLR4-deficient mice.18 Another recent report demonstrates that LPS stimulation of platelet TLR4 induces platelet binding to and activation of adherent neutrophils.12 In the present study, we report for the first time that stimulation of platelet TLR2 by synthetic bacterial lipopeptide directly activates the platelet’s thrombotic and inflammatory response. Additionally, our data suggest that platelet TLR2 signals intracellularly through the phosphoinositide 3-kinase (PI3-K) signaling pathway. Finally, we report that a live bacterium, Porphyromonas gingivalis, induces a proinflammatory response in platelets in a TLR2-dependent manner. This work substantiates the functionality of platelet TLRs and suggests a possible mechanism by which select bacteria and bacterial components directly activate platelets.
Materials and Methods
Materials
All TLR-related antibodies, fluorescently labeled antibodies, and corresponding isotype controls were purchased from eBioscience (San Diego, Calif). FITC-conjugated anti-human PAC-1 mouse monoclonal antibody (mAb) and fluorescently labeled isotype control was purchased from BD Biosciences (Mountain View, Calf). Rabbit anti–phospho-AKT(Ser473), anti-AKT, anti–β-actin, and anti-p85 polyclonal antibodies were purchased from Cell Signaling Technology (Danvers, Mass). Mouse anti–platelet factor-4 polyclonal antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, Calif). Pam3CSK4 and Pam2CSK4 were purchased from InvivoGen (San Diego, Calif). Fibrinogen and thrombin were purchased from Enzyme Research Laboratories (South Bend, Ind); collagen was from Chrono-Log Corp (Havertown, Pa). LY294002 was purchased from EMB Chemicals (Gibbstown, NJ); 2′,7′-dichlorofluorescein diacetate (DCFH-DA) and calcein-AM were from Invitrogen (Carlsbad, Calif).
Platelet Isolation
Washed platelets were isolated from human whole blood by previously described methods.19 To avoid leukocyte contamination, only the top 75% of the platelet rich plasma was collected. Pelleted platelets were washed and resuspended in HEPES buffer, pH 7.4 (2×108 platelets/mL, unless otherwise noted). Where indicated, washed platelets were pretreated with the following antibodies or inhibitors (30 minutes; room temperature) before study: functional grade purified anti-TLR2 (Clone TL2.1) or anti-TLR1 (Clone GD2.F4) mAb (25 μg/mL); LY294002 (25 to 100 μmol/L); anti-P-selectin mAb (Clone AK-4) (10 μg/mL); and abciximab (10 μg/mL).
Platelet Lysate Preparation, Immunoprecipitation of TLR2, and Western Blot Analysis
Platelets were left quiescent or stimulated with Pam3CSK4 (0.1 to 10 μg/mL) before pelleting. After pelleting, supernatants were retained to analyze the platelet releasate. For Western blot analysis, platelets were lysed in 1% Triton-X cell lysis buffer with protease inhibitors; for immunoprecipitation, 1% NP-40 buffer with protease inhibitors. For immunoprecipitation, platelet lysates were precleared with isotype control, followed by incubation with Protein A/G agarose beads. Mouse anti-human TLR2 mAb or isotype control was added to precleared lysates at manufacturer-recommended concentrations. After overnight incubation (4°C), Protein A/G agarose beads were added and then pelleted and prepared for electrophoresis separation. For Western blot analysis, proteins of interest were analyzed similarly to published methods.10
Flow Cytometric Analysis of Platelet Surface Proteins
Studies were conducted using a FACScan flow cytometer with CellQuest Pro software (BD Biosciences, Mountain View, Calif). Briefly, PBS-suspended platelets were incubated with fluorescently labeled mAb or isotype control (30 minutes; room temperature). Where indicated, platelets were stimulated with Pam3CSK4 (5 to 10 μg/mL) before antibody staining. Unbound antibody was removed by washing with PBS, and samples were fixed with 1.5% formaldehyde in PBS. Samples were analyzed immediately on a flow cytometer, with at least 50 000 platelet events collected per sample. Antibody reactivity is reported as mean fluorescence intensity, which is calculated as mean fluorescence of specific antibody minus mean fluorescence of isotype control.
Double Immunofluorescence Staining
Resting or Pam3CSK4-activated (10 μg/mL) platelets were layered on poly-L-lysine–coated slides, fixed in ice-cold methanol:acetone (70:30), and permeabilized with 0.2% Triton-X. Slides were blocked in Protein Block Solution (Dako, Carpinteria, Calif) and incubated overnight (4°C) with appropriate primary antibodies, followed by incubation with secondary FITC- and Texas red–conjugated antibodies. As controls, some slides were incubated with either nonspecific IgG at the same concentrations as the primary antibodies or secondary antibody alone. Slides were examined by confocal microscopy (Leica TCS SP2 instrument; Leica Microsystems, Exton, Pa) at ×100 magnification, and images were captured using Leica Confocal Software.
Platelet Aggregation
Aggregation of washed platelets was optically monitored using a PAP-4 platelet aggregometer (Bio/Data Corporation, Horsham, Pa) as previously described.19 Percentage of aggregation was recorded 6 minutes after addition of Pam3CSK4 (1 to 10 μg/mL) or thrombin (0.5 U/mL). Platelet aggregation in whole blood was measured using a lumi-aggregometer (Chrono-Log Corporation, Havertown, Pa), as previously reported.20 Aggregation (ohms) was recorded 10 minutes after addition of Pam3CSK4 (10 μg/mL).
Measurement of Platelet–Leukocyte Aggregates by Two-Color Flow Cytometry
Immediately following whole blood aggregation, 30 μL of whole blood was added to a reaction mixture containing 30 μL PBS, 10 μL of FITC–anti-CD41 mAb (platelet-specific), and 10 μL of phycoerythrin (PE)-Cy7–anti-CD14 mAb (leukocyte-specific). Platelet–leukocyte aggregates were analyzed by flow cytometry as previously described.20 Briefly, monocytes and neutrophils were gated by light scattering and PE-Cy7–CD14 fluorescence and further gated into CD41-negative and CD41-positive populations. Isotype controls were used to compensate for nonspecific antibody binding. A minimum of 1000 monocytes or 5000 neutrophils were acquired, and the percentage of platelet-bound monocytes/neutrophils (CD14-positive/CD41-positive) was quantified.
Platelet Adhesion Assay
Adhesion assay was performed as previously described.21 Briefly, calcein-AM–labeled platelets were stimulated with Pam3CSK4 (10 μg/mL) and, using a cell adhesion flow chamber (Immunetics, Boston, Mass), were recirculated for 20 minutes over coverslips uniformly coated with collagen (1 μg/mL) or fibrinogen (1 mg/mL). Coverslips were then gently washed, mounted on a glass slide, and examined with a fluorescence microscope (Nikon) at ×20 magnification.
Platelet Production of Reactive Oxygen Species
Platelet reactive oxygen species (ROS) generation was measured by flow cytometry as previously described, with slight modifications.6 Isolated platelets were incubated with DCFH-DA (20 μmol/L), stimulated with Pam3CSK4 (10 μg/mL) for 10 minutes, and analyzed immediately by flow cytometry.
Murine Platelet Studies
TLR2−/−C57BL/6 and C57BL/6 control mice were originally purchased from The Jackson Laboratory (Bar Harbor, Me). The whole blood of 6 to 8 mice was pooled for each study, and mouse platelets were isolated using previously published methods.22 Platelets were resuspended in HEPES buffer (2×108 platelets/mL), and functional studies were performed identically to the human protocols.
Murine Infection Model
TLR2−/−C57BL/6 and C57BL/6 control mice were injected subcutaneously with saline or live P gingivalis strain 381 (1×109 CFU), cultured as previously described.23 Mice were euthanized 16 hours postinfection, whole blood was pooled, and the percentage of circulating platelet–leukocyte aggregates was measured as described above, except PE-Cy7–Ly-6G mAb (eBioscience) was used to label murine neutrophils.
Statistical Analysis
Numeric data are reported as means ±SD. Data were analyzed by Student’s t test using SigmaStat software, and statistical significance was assumed with a value of P<0.05.
Results
Human Platelets Express Functional TLR2
Previous reports have demonstrated that platelets express TLR2, TLR1, and TLR6, but the functionality of these receptors in platelets has not been investigated. In the present study, TLR2, TLR1, and TLR6 expression was further confirmed by immunoprecipitation and flow cytometry (Figure I in the online data supplement, available at http://circres.ahajournals.org). To determine whether these platelet-expressed TLRs are functional receptors, Pam3CSK4 (a synthetic triacylated bacterial lipopeptide known to interact with TLR2/TLR1 heterodimers) was added to stirred platelet suspensions.24,25 Pam3CSK4 dose-dependently induced platelet aggregation (Figure 1A). Pam2CSK4, a known TLR2/TLR6 ligand,24,26 also caused platelet aggregation, although not as consistently or effectively as the TLR2/TLR1 ligand (data not shown). To ensure that Pam3CSK4-induced platelet aggregation was dependent on TLRs, platelet suspensions were pretreated with functional grade anti-TLR2 mAb, anti-TLR1 mAb, or isotype control. Anti-TLR2 mAb has previously been shown to block TLR2-dependent cytokine production in other cell types.27,28 Neutralization of TLR2 significantly reduced Pam3CSK4-induced platelet aggregation (Figure 1B). TLR1 inhibition also reduced aggregation, although not as effectively as TLR2 blockage. Platelet aggregation was not affected by pretreatment with control antibody. Confocal microscopy also demonstrated that TLR2 and TLR1 colocalize under resting conditions, with moderate increases in colocalization in the presence of Pam3CSK4 (data not shown).
Figure 1.
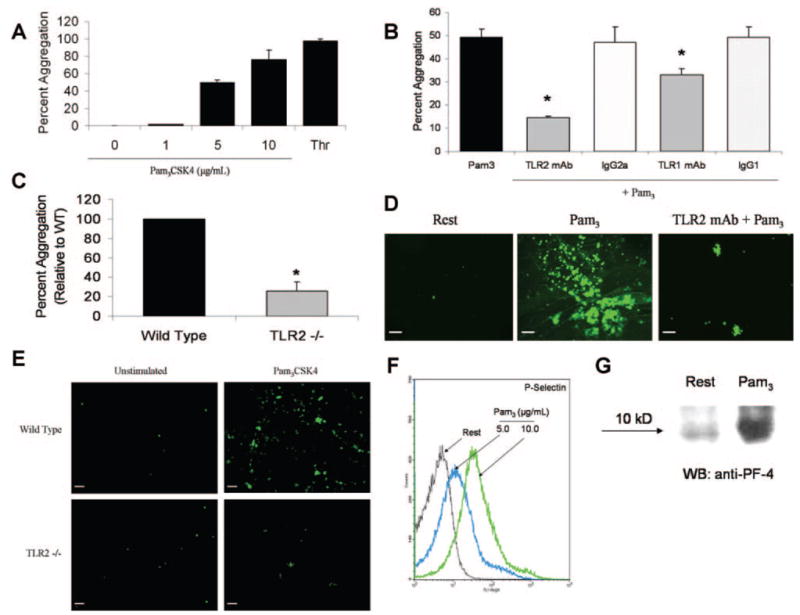
Pam3CSK4 induces platelet aggregation, adhesion, and secretion in a TLR2-dependent manner. A and B, Pam3CSK4 (1 to 10 μg/mL) or thrombin (0.5 U/mL) were added to stirred human platelets, and aggregation was monitored for 6 minutes. Several platelet suspensions (B) were preincubated with functional grade anti-TLR2 mAb, anti-TLR1 mAb, or equal concentrations of their respective isotype controls (25 μg/mL) before stimulation with Pam3CSK4 (5 μg/mL). Data are reported as maximum percentage of aggregation±SD (n=3) for all groups. *P<0.01 compared to Pam3CSK4 alone. C, Pam3CSK4 (30 μg/mL) was added to stirred murine platelets isolated from either control C57BL/6 or TLR2-deficient mice. Aggregation was monitored for 6 minutes, and maximum percentage of aggregation was recorded. Reported data are normalized to control/wild-type (n=5 for both groups). *P<0.001 compared to control/wild-type. D, Calcein-labeled human platelets (resting or Pam3CSK4-stimulated [10 μg/mL]) were recirculated over collagen-covered coverslips for 20 minutes in a cell adhesion flow chamber. For some studies, platelets were pretreated with TLR2-blocking antibody (25 μg/mL) before stimulation. Adherent cells were viewed on a fluorescent microscope at X20 magnification. Representative fluorescent micrographs are shown. Scale bar=5 μm. E, Calcein-labeled platelets isolated from control C57BL/6 or TLR2-deficient mice were left quiescent or stimulated with Pam3CSK4 (10 μg/mL) and recirculated over collagen-coated coverslips for 20 minutes. Representative fluorescent micrographs are shown. Scale bar=5 μm. F, Washed human platelets (resting or Pam3CSK4-stimulated) were labeled with FITC–anti–P-selectin mAb and analyzed by flow cytometry. A representative histogram of 3 independent experiments is shown. G, Isolated human platelets were left quiescent or stimulated with Pam3CSK4 (10 μg/mL). The supernatant/platelet releasate was harvested, and equal volumes of releasate protein were separated by gel electrophoresis and immunoblotted for platelet factor-4 (PF-4).
To confirm the role of TLR2 in Pam3CSK4-induced platelet aggregation, Pam3CSK4 was added to stirred platelet suspensions obtained from either wild-type or TLR2-deficient mice. Relative to human platelets, murine platelets appeared less sensitive to Pam3CSK4, possibly attributable to interspecies differences in platelet TLR expression or receptor affinity for the agonist. Nevertheless, TLR2-deficient platelets aggregated ≈75% less than wild-type platelets (10±1% versus 2.6±0.89%, P<0.001) (Figure 1C) in response to Pam3CSK4. By contrast, collagen-induced platelet aggregation (10 gμ/mL) was similar in wild-type and TLR2-deficient mice (data not shown).
We further investigated platelet TLR functionality by examining the effect of Pam3CSK4 on platelet adhesion and secretion. In a model of platelet adhesion under flow conditions, calcein-labeled human platelets were activated with Pam3CSK4 and recirculated over collagen-covered slides. Pam3CSK4 significantly increased platelet adhesion to the collagen matrix, whereas pretreatment with TLR2-blocking antibody considerably inhibited adhesion (Figure 1D). Similar results were obtained when Pam3CSK4-activated platelets were recirculated across fibrinogen, although the extent of adhesion was less pronounced (data not shown). Adhesion assays were also performed using washed platelets from both wild-type and TLR2-deficient mice. Wild-type platelets were more adherent to collagen following Pam3CSK4 (10 μg/mL) stimulation, whereas TLR2-deficient platelets treated with Pam3CSK4 showed no change in adhesion compared to resting (Figure 1E). The effect of TLR2 stimulation on platelet secretion was also investigated. Pam3CSK4 stimulation of washed human platelets dose-dependently increased surface expression of P-selectin (Figure 1F), and Western blot analysis of the platelet releasate indicated that Pam3CSK4 triggered significant release of platelet factor 4 (PF-4) (Figure 1G). Thus, Pam3CSK4 stimulation induces platelet secretion of proinflammatory mediators.
TLR2 Stimulation in Whole Blood Generates Platelet–Leukocyte Aggregates
In addition to secreting inflammatory mediators, platelets modulate the immune response by interacting with leukocytes.3 To investigate the ability of platelet TLR2 to generate platelet–leukocyte aggregates, we stimulated human whole blood with Pam3CSK4 (10 μg/mL) in a lumi-aggregometer. Interestingly, Pam3CSK4 did not induce platelet aggregation in whole blood (Figure 2). However, flow cytometric analysis of the whole blood samples indicated that Pam3CSK4 stimulated significant formation of platelet–neutrophil aggregates when compared to stirred, untreated samples (Figure 2). Pam3CSK4 was equally found to induce formation of platelet–monocyte coaggregates (data not shown). Generation of platelet–neutrophil aggregates was moderately but significantly inhibited by pretreatment with TLR2 antibody. In whole blood, in the presence of other TLR-expressing cells, higher concentrations of blocking antibody may be required to fully inhibit platelet TLR-induced responses. By contrast, anti–P-selectin mAb, which blocks the interaction between platelet P-selectin and neutrophilic P-selectin glycoprotein ligand-1, inhibited aggregate formation almost entirely (Figure 2). Control experiments using isolated platelets and neutrophils demonstrated that direct stimulation of platelet TLR2 is sufficient to induce platelet–neutrophil aggregates (supplemental Figure II). Thus, under nonaggregating conditions, stimulation of platelet TLR2 induces formation of platelet–neutrophil heterotypic aggregates in a P-selectin–dependent manner.
Figure 2.
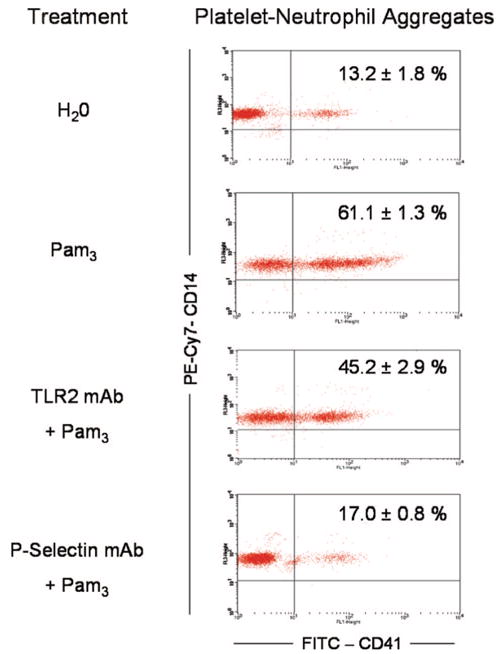
Pam3CSK4 does not induce platelet aggregation in human whole blood but stimulates formation of platelet–leukocyte aggregates. A, Human whole blood was diluted in 0.9% saline and stirred in a whole blood lumi-aggregometer. Pam3CSK4 (10 μg/mL) was added, and aggregation was monitored for 10 minutes. Some samples were treated with TLR2-blocking antibody (25 μg/mL) or P-selectin–blocking antibody (10 μg/mL) before stimulation. Immediately following aggregation, 30 μL of whole blood was extracted and added to a cocktail containing FITC–anti-CD41 and PE-Cy7–anti-CD14 mAbs. Samples were analyzed by flow cytometry. Dot plots representative of 3 independent experiments are shown. Events located in the upper right quadrant represent CD14-positive/CD41-positive neutrophils. The percentage of collected, platelet-positive neutrophils was quantified (n=3 for all groups).
TLR2 Stimulation Directly Activates the PI3-K/AKT Signaling Pathway
In other immune cells, a common end point of TLR stimulation is activation of nuclear factor κB and transcription of proinflammatory genes.1 However, in anucleate platelets, TLRs likely stimulate alternative signaling pathways. TLR stimulation has previously been shown to activate the PI3-K signaling pathway in a variety of immune cells,29 and in platelets, the PI3-K/Akt pathway regulates a broad range of functions, including platelet adhesion, secretion, and aggregation.30 We, therefore, investigated whether Pam3CSK4 stimulation of TLR2 activates the PI3-K/Akt pathway in human platelets. Pam3CSK4 dose-dependently increased Akt phosphorylation at serine 473 (Figure 3A), and preincubation with TLR2-blocking antibody reduced Pam3CSK4-induced Akt phosphorylation (Figure 3B). Preincubation with LY294002, a specific inhibitor of PI3-K, also dose-dependently inhibited Akt phosphorylation (Figure 3C).
Figure 3.

Pam3CSK4 stimulation of human platelets activates Akt in a TLR2/PI3-K-dependent manner. A through D, Platelets were left quiescent or stimulated with increasing concentrations of Pam3CSK4 (1 to 10 μg/mL) (A). Alternatively, platelets were treated with TLR2-blocking antibody (25 μg/mL) (B), LY294002 (25 to 100 μmol/L) (C), or abciximab (10 μg/mL) (D) before stimulation with Pam3CSK4 (10 μg/mL). For abciximab-treated samples, platelets were supplemented with fibrinogen (1 mg/mL) and stirred constantly. Equal amounts of platelet homogenate were resolved by SDS-PAGE and immunoblotted for phospho-Akt(Ser473), AKT, or β-actin. Representative blots are shown. E, Resting or Pam3CSK4-stimulated (10 μg/mL) platelets were stained with anti-TLR2 (green) and anti-p85 (red) primary antibodies. Slides were examined by confocal microscopy, and representative micrographs are shown. All images were adjusted to account for nonspecific binding of antibodies. Scale bar=2 μm. F, Resting or Pam3CSK4-treated (10 μg/mL) platelets were immunoprecipitated with anti-TLR2 mAb. Samples were resolved by SDS-PAGE and immunoblotted for p85 or TLR2. Representative blots are shown.
In platelets, PI3-K is although to play a role in shape change, secretion, and inside-out activation of integrin αIIB β3.30,31 PI3-K can also be activated downstream of integrin αIIBβ3, at which time it contributes to clot stability. To determine whether TLR2 stimulation activates the PI3-K/Akt pathway before or after integrin αIIBβ3 activation, we incubated platelets under aggregating conditions with abciximab, an mAb that blocks the interaction between fibrinogen and αIIBβ3, before addition of Pam3CSK4. Western blot analysis indicated that Pam3CSK4–induced Akt phosphorylation was not affected by the presence of abciximab (Figure 3D), thus suggesting TLR2 stimulation activates the PI3-K/Akt signaling pathway upstream of integrin αIIBβ3.
The mechanisms of PI3-K activation in TLR signaling are not completely understood, but it has been reported that the regulatory subunit of PI3-K, p85, can directly bind to the intracellular domain of TLRs.32 In platelets, Pam3CSK4 stimulation induced a dramatic increase in p85/TLR2 colocalization, as determined by confocal microscopy (Figure 3E). Immunoprecipitation studies further verified that p85/TLR2 interactions increased on Pam3CSK4 stimulation (Figure 3F). These data suggest that PI3-K is directly activated by TLR2 and may therefore play a fundamental role in transduction of the TLR signal in platelets.
PI3-K Regulates TLR2-Mediated Platelet Functional Responses
To assess the significance of the PI3-K signaling pathway in TLR2-mediated platelet responses, we treated platelets with LY294002 before various functional studies. Preincubation with LY294002 significantly and dose-dependently inhibited Pam3CSK4-induced platelet aggregation (Figure 4A). Moreover, pretreatment with LY294002 (25 μmol/L) prevented Pam3CSK4-induced platelet adhesion to collagen-coated slides (Figure 4B). PI3-K inhibition was found to reduce Pam3CSK4-induced increases in P-selectin surface expression (Figure 4C) and, subsequently, formation of platelet–neutrophil aggregates (Figure 4D). The effect of LY294002 on other Pam3CSK4-induced platelet responses was also investigated. TLR2 stimulation triggered inside-out activation of integrin αIIBβ3, as determined by increased surface binding of PAC-1, an mAb which recognizes only the active conformation of integrin αIIBβ3 (Figure 4E). This activation response was then inhibited by LY294002. Pam3CSK4 also stimulated platelet production of ROS, a response that is known to be downstream of PI3-K activation in platelets, and this production was similarly blocked by LY294002 (Figure 4F). Taken together, these data suggest that PI3-K plays an integral role in many TLR2-mediated functional responses, including platelet aggregation, adhesion, secretion, leukocyte interactions, integrin activation, and ROS generation.
Figure 4.

PI3-K regulates the thromboinflammatory response induced by stimulation of platelet TLR2. A through D, Human platelets were treated with vehicle (DMSO) or LY294002 (25 μmol/L) before stimulation with Pam3CSK4 (10 μg/mL). The effect of PI3-K inhibition on the TLR2-mediated functional responses was examined in assays of platelet aggregation (A), platelet adhesion (B), platelet secretion (C), and heterotypic aggregate formation (D). All assays were performed as described in preceding figures. E, Platelets (resting, Pam3CSK4-stimulated [10 μg/mL], or LY294002-pretreated [25 μmol/L]) were stained with FITC–anti–PAC-1 antibody. Samples were analyzed by flow cytometry, and mean fluorescence intensity was quantified as a measure of antibody binding. Data are reported as change in PAC-1 binding±SD relative to resting samples (n=5 for all groups). *P<0.05 compared to Pam3CSK4 alone. F, Platelets were pretreated with vehicle or LY294002 (25 μmol/L) before addition of DCFH-DA (20 μmol/L). Platelets were then stimulated with Pam3CSK4 (10 μg/mL) and analyzed by flow cytometry. Mean fluorescence intensity was quantified as a measure of DCF fluorescence/ROS production. Data are reported as change in mean fluorescence±SD relative to resting samples (n=5 for all groups). *P<0.05 compared to Pam3CSK4 alone.
TLR2 Participates in the In Vivo Platelet Response to P gingivalis
In macrophages and endothelial cells, TLR2 has previously been reported to mediate immune recognition of the Gram-negative bacterium, P gingivalis.23,33 Therefore, to determine whether in vivo challenge with live bacteria affects platelet function and, further, to investigate whether TLR2 participates in this platelet response, wild-type and TLR2-deficient mice were challenged with live P gingivalis. As shown, relative to uninfected controls in each group, P gingivalis infection increased circulating platelet–neutrophil aggregates in the whole blood of wild-type but not TLR2-deficient mice (Figure 5). Therefore, challenge with a specific bacterium, P gingivalis, induces a proinflammatory response in platelets in a manner modulated by TLR2.
Figure 5.

In vivo challenge with P gingivalis induces whole blood formation of platelet neutrophil aggregates in a TLR2-dependent manner. C57BL/6 or TLR2-deficient mice were challenged subcutaneously with live P gingivalis strain 381 (1 × 109 CFU) or saline (n=3 for all groups). Mice were euthanized 16 hours postinfection, blood from individual groups was pooled, and levels of platelet–leukocyte aggregates were measured as described above. Data are reported as the percentage of platelet-positive neutrophils, relative to control/uninfected (n=3 for each group). *P<0.05 compared to control/uninfected. Data are representative of 2 individual experiments.
Discussion
TLRs represent the most fundamental immune receptors, and their role in the recognition of foreign pathogens and the initiation of the inflammatory response is indispensable to host defense. TLRs have been studied extensively in many immune and vascular cells, including neutrophils, monocytes/macrophages, and endothelial cells, and, recently, their expression has also been described in platelets. Nonetheless, studies of TLR functionality in platelets have largely focused on LPS stimulation of platelet TLR4, and, to the best of our knowledge, there have been no studies positively demonstrating platelet TLR2 functionality. Here, we show that platelets express a subset of TLRs, namely TLR2, TLR1, and TLR6. Moreover, we report for the first time that platelet TLR2 is a functional receptor, because treatment with Pam3CSK4 (a synthetic triacylated bacterial lipopeptide widely used in other cell types as a prototypical TLR2 ligand) stimulates a variety of prothrombotic and proinflammatory platelet responses. These responses were significantly inhibited in human platelets by blocking TLR2 with mAb, and adhesion and aggregation were specifically inhibited in platelets from TLR2-deficient mice. Therefore, functional responses associated with Pam3CSK4 were determined to be TLR2-dependent and not attributable to other nonspecific effects of the agonist. Interestingly, Pam3CSK4 has previously been shown to induce platelet aggregation, albeit through an unknown mechanism, at similar concentrations to those used in this study.34 Another recent report did not detect platelet secretion or aggregation in response to Pam3CSK4, but this study was performed in platelet-rich plasma using lower ligand concentrations.35 Our group also did not observe changes in platelet response after stimulation with low-concentration Pam3CSK4 (1000 ng/mL). The physiological significance of the ligand concentrations presently used needs to be explored more fully, but platelet stimulation by high-concentration bacterial lipopeptide suggests an interesting scenario whereby platelets may act as sensors of acute infection, becoming activated in the presence of high bacterial concentrations but otherwise remaining quiescent to avoid an overwhelming immune response.
The presence of functional TLRs highlights the role of platelets as immunologic cells, critically participating in both inflammatory and thrombotic processes.36 Stimulation of platelet TLRs would logically trigger a combined prothrombotic and proinflammatory response. In the present study, TLR2 activation directly induced platelet aggregation, in addition to prompting increased platelet adhesion to collagen under flow conditions. Although adhesion and aggregation are primarily viewed as hemostatic responses, platelet adhesion at localized sites of vascular injury or bacterial infection represents a critical first step in the accumulation of platelets and the subsequent amplification of platelet-derived inflammatory signals. TLR2 stimulation did not cause platelet aggregation in human whole blood but did induce a significant increase in platelet–leukocyte interactions. These interactions were dependent on platelet secretion of P-selectin and possibly further regulated by PF-4, an α-granule protein which, in addition to possessing bactericidal properties, stimulates neutrophil chemotaxis and activation. Platelet–leukocyte interactions activate each cell synergistically, as demonstrated by a recent report showing that platelet–neutrophil interactions increase neutrophil superoxide generation and platelet release of soluble CD40 ligand.20 Formation of platelet–neutrophil heterotypic aggregates may be particularly important in the setting of sepsis, because neutrophil production of cytotoxic oxygen metabolites constitutes a major defense strategy against bacterial infection. Here, TLR2 stimulation also induced direct platelet ROS generation.
The signaling cascades which platelet TLRs use to generate a functional response have not been described. Here, we novelly demonstrate that stimulation of TLR2 dose-dependently activates the PI3-K/Akt signaling pathway in human platelets. PI3-K activation appears to occur directly, because the p85 regulatory subunit binds directly to TLR2 following Pam3CSK4-stimulation. TLR-mediated PI3-K activation has been demonstrated in many immune cells,29 but this mechanism of direct binding may be limited to TLR2, TLR1, and TLR6, because only these isoforms contain the putative binding site for p85 within their cytoplasmic tails.32 Activation of the PI3-K/Akt pathway appears to be critically important to TLR2-mediated platelet responses, because pretreatment with a specific PI3-K inhibitor decreased Pam3CSK4-induced platelet aggregation, adhesion, secretion, leukocyte interactions, integrin αIIBβ3 activation, and ROS production. In nucleated immune cells, PI3-K putatively act as a negative regulator of TLR signaling.29 In platelets, however, activation of PI3-K is well known to positively modulate a number of platelet functions, including adhesion, cytoskeletal remodeling, and aggregation.30 Thus, to stimulate a thrombotic and/or inflammatory response in platelets, it is logical that platelet TLRs would be coupled to PI3-K. Additional signaling pathways associated with TLRs, such p38 mitogen-activated protein kinase and extracellular signal-regulated kinase 1/2, might also become activated in platelets, but their activity and relative importance in TLR-mediated platelet responses remains unclear.
Many live bacteria, including P gingivalis, have previously been shown to stimulate platelet activity, but the exact mechanism of activation remains debated. In the present study, we demonstrate that in vivo challenge with live P gingivalis induces a platelet-dependent, proinflammatory response, namely the formation of platelet–neutrophil aggregates. This functional response was significantly reduced in TLR2-deficient mice, indicating that TLR2 participates in platelet recognition of P gingivalis. In other cell types, including macrophages and endothelial cells, TLR2 has been shown to mediate the immune response to P gingivalis through recognition of P gingivalis-derived LPS and fimbrial proteins.23,37–40 Whether these PAMPs also stimulate platelet TLR2 remains to be determined, but, regardless, the expression of functional TLRs represents a potential mechanism whereby live bacteria stimulate the platelet thromboinflammatory response.
In summary, our data indicate that human platelets express a subset of TLRs which are functional receptors capable of producing a measurable activation, aggregation, and signaling response. The physiological, and possibly pathophysiological, significance of these receptors in platelets remains to be elucidated, particularly in the setting of sepsis and atherosclerosis. Nevertheless, the expression of functional TLRs in platelets provides a possible mechanism by which platelets interact with bacteria and further highlights the importance of platelets in the immune and inflammatory responses.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported in part by NIH grants P50HL083801 (to J.E.F.), N01-HV-28178 (to J.E.F.), and R01-HL080387 (to C.A.G.).
Footnotes
Disclosures
None.
References
- 1.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 2.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 3.Weyrich AS, Zimmerman GA. Platelets: signaling cells in the immune continuum. Trends Immunol. 2004;25:489–495. doi: 10.1016/j.it.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 4.Joseph M. Immunopharmacology of Platelets. London, United Kingdom: Academic Press; 1995. [Google Scholar]
- 5.Sharma A, Novak EK, Sojar HT, Swank RT, Kuramitsu HK, Genco RJ. Porphyromonas gingivalis platelet aggregation activity: outer membrane vesicles are potent activators of murine platelets. Oral Microbiol Immunol. 2000;15:393–396. doi: 10.1034/j.1399-302x.2000.150610.x. [DOI] [PubMed] [Google Scholar]
- 6.Kalvegren H, Bylin H, Leanderson P, Richter A, Grenegard M, Bengtsson T. Chlamydia pneumoniae induces nitric oxide synthase and lipoxygenase-dependent production of reactive oxygen species in platelets. Effects on oxidation of low density lipoproteins. Thromb Haemost. 2005;94:327–335. doi: 10.1160/TH04-06-0360. [DOI] [PubMed] [Google Scholar]
- 7.Smeeth L, Thomas SL, Hall AJ, Hubbard R, Farrington P, Vallance P. Risk of myocardial infarction and stroke after acute infection or vaccination. N Engl J Med. 2004;351:2611–2618. doi: 10.1056/NEJMoa041747. [DOI] [PubMed] [Google Scholar]
- 8.Vincent JL, Yagushi A, Pradier O. Platelet function in sepsis. Crit Care Med. 2002;30:S313–S317. doi: 10.1097/00003246-200205001-00022. [DOI] [PubMed] [Google Scholar]
- 9.Mavrommatis AC, Theodoridis T, Orfanidou A, Roussos C, Christopoulou-Kokkinou V, Zakynthinos S. Coagulation system and platelets are fully activated in uncomplicated sepsis. Crit Care Med. 2000;28:451–457. doi: 10.1097/00003246-200002000-00027. [DOI] [PubMed] [Google Scholar]
- 10.Shiraki R, Inoue N, Kawasaki S, Takei A, Kadotani M, Ohnishi Y, Ejiri J, Kobayashi S, Hirata K, Kawashima S, Yokoyama M. Expression of Toll-like receptors on human platelets. Thromb Res. 2004;113:379–385. doi: 10.1016/j.thromres.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 11.Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of Toll-like receptor molecules on human platelets. Immunol Cell Biol. 2005;83:196–198. doi: 10.1111/j.1440-1711.2005.01314.x. [DOI] [PubMed] [Google Scholar]
- 12.Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, Keys EM, Allen-Vercoe E, Devinney R, Doig CJ, Green FH, Kubes P. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 13.Cognasse F, Lafarge S, Chavarin P, Acquart S, Garraud O. Lipopolysaccharide induces sCD40L release through human platelets TLR4, but not TLR2 and TLR9. Intensive Care Med. 2007;33:382–384. doi: 10.1007/s00134-006-0488-8. [DOI] [PubMed] [Google Scholar]
- 14.Jayachandran M, Brunn GJ, Karnicki K, Miller RS, Owen WG, Miller VM. In vivo effects of lipopolysaccharide and TLR4 on platelet production and activity: implications for thrombotic risk. J Appl Physiol. 2007;102:429–433. doi: 10.1152/japplphysiol.01576.2005. [DOI] [PubMed] [Google Scholar]
- 15.Stahl AL, Svensson M, Morgelin M, Svanborg C, Tarr PI, Mooney JC, Watkins SL, Johnson R, Karpman D. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood. 2006;108:167–176. doi: 10.1182/blood-2005-08-3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patrignani P, Di Febbo C, Tacconelli S, Moretta V, Baccante G, Sciulli MG, Ricciotti E, Capone ML, Antonucci I, Guglielmi MD, Stuppia L, Porreca E. Reduced thromboxane biosynthesis in carriers of toll-like receptor 4 polymorphisms in vivo. Blood. 2006;107:3572–3574. doi: 10.1182/blood-2005-12-4811. [DOI] [PubMed] [Google Scholar]
- 17.Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, Ni H, Lazarus AH, Freedman J, Semple JW. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood. 2006;107:637–641. doi: 10.1182/blood-2005-06-2202. [DOI] [PubMed] [Google Scholar]
- 18.Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional Toll-like receptor-4. Blood. 2005;106:2417–2423. doi: 10.1182/blood-2005-03-0916. [DOI] [PubMed] [Google Scholar]
- 19.Freedman JE, Farhat JH, Loscalzo J, Keaney JF., Jr alpha-tocopherol inhibits aggregation of human platelets by a protein kinase C-dependent mechanism. Circulation. 1996;94:2434–2440. doi: 10.1161/01.cir.94.10.2434. [DOI] [PubMed] [Google Scholar]
- 20.Vanichakarn P, Blair P, Wu C, Freedman JE, Chakrabarti S. Neutrophil CD40 enhances platelet-mediated inflammation. Thromb Res. 2008;122:346–358. doi: 10.1016/j.thromres.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 21.Chakrabarti S, Blair P, Wu C, Freedman JE. Redox state of dipyridamole is a critical determinant for its beneficial antioxidant and antiinflammatory effects. J Cardiovasc Pharmacol. 2007;50:449–457. doi: 10.1097/FJC.0b013e31813542db. [DOI] [PubMed] [Google Scholar]
- 22.Shivdasani RA, Fielder P, Keller GA, Orkin SH, de Sauvage FJ. Regulation of the serum concentration of thrombopoietin in thrombocytopenic NF-E2 knockout mice. Blood. 1997;90:1821–1827. [PubMed] [Google Scholar]
- 23.Gibson FC, III, Ukai T, Genco CA. Engagement of specific innate immune signaling pathways during Porphyromonas gingivalis induced chronic inflammation and atherosclerosis. Front Biosci. 2008;13:2041–2059. doi: 10.2741/2822. [DOI] [PubMed] [Google Scholar]
- 24.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci U S A. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, Akira S. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. 2002;169:10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 26.Hajjar AM, O’Mahony DS, Ozinsky A, Underhill DM, Aderem A, Klebanoff SJ, Wilson CB. Cutting edge: functional interactions between toll-like receptor (TLR) 2 and TLR1 or TLR6 in response to phenol-soluble modulin. J Immunol. 2001;166:15–19. doi: 10.4049/jimmunol.166.1.15. [DOI] [PubMed] [Google Scholar]
- 27.Flo TH, Halaas O, Lien E, Ryan L, Teti G, Golenbock DT, Sundan A, Espevik T. Human toll-like receptor 2 mediates monocyte activation by Listeria monocytogenes, but not by group B streptococci or lipopolysaccharide. J Immunol. 2000;164:2064–2069. doi: 10.4049/jimmunol.164.4.2064. [DOI] [PubMed] [Google Scholar]
- 28.Razonable RR, Henault M, Paya CV. Stimulation of toll-like receptor 2 with bleomycin results in cellular activation and secretion of pro-inflammatory cytokines and chemokines. Toxicol Appl Pharmacol. 2006;210:181–189. doi: 10.1016/j.taap.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 29.Hazeki K, Nigorikawa K, Hazeki O. Role of phosphoinositide 3-kinase in innate immunity. Biol Pharm Bull. 2007;30:1617–1623. doi: 10.1248/bpb.30.1617. [DOI] [PubMed] [Google Scholar]
- 30.Jackson SP, Yap CL, Anderson KE. Phosphoinositide 3-kinases and the regulation of platelet function. Biochem Soc Trans. 2004;32:387–392. doi: 10.1042/bst0320387. [DOI] [PubMed] [Google Scholar]
- 31.Freedman JE. Molecular regulation of platelet-dependent thrombosis. Circulation. 2005;112:2725–2734. doi: 10.1161/CIRCULATIONAHA.104.494468. [DOI] [PubMed] [Google Scholar]
- 32.Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, Godowski PJ, Ulevitch RJ, Knaus UG. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat Immunol. 2000;1:533–540. doi: 10.1038/82797. [DOI] [PubMed] [Google Scholar]
- 33.Burns E, Bachrach G, Shapira L, Nussbaum G. Cutting Edge: TLR2 is required for the innate response to Porphyromonas gingivalis: activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol. 2006;177:8296–8300. doi: 10.4049/jimmunol.177.12.8296. [DOI] [PubMed] [Google Scholar]
- 34.Berg M, Offermanns S, Seifert R, Schultz G. Synthetic lipopeptide Pam3CysSer(Lys)4 is an effective activator of human platelets. Am J Physiol. 1994;266:C1684–C1691. doi: 10.1152/ajpcell.1994.266.6.C1684. [DOI] [PubMed] [Google Scholar]
- 35.Ward JR, Bingle L, Judge HM, Brown SB, Storey RF, Whyte MK, Dower SK, Buttle DJ, Sabroe I. Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thromb Haemost. 2005;94:831–838. [PubMed] [Google Scholar]
- 36.von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. 2007;100:27–40. doi: 10.1161/01.RES.0000252802.25497.b7. [DOI] [PubMed] [Google Scholar]
- 37.Davey M, Liu X, Ukai T, Jain V, Gudino C, Gibson FC, III, Golenbock D, Visintin A, Genco CA. Bacterial fimbriae stimulate proinflammatory activation in the endothelium through distinct TLRs. J Immunol. 2008;180:2187–2195. doi: 10.4049/jimmunol.180.4.2187. [DOI] [PubMed] [Google Scholar]
- 38.Al-Qutub MN, Braham PH, Karimi-Naser LM, Liu X, Genco CA, Darveau RP. Hemin-dependent modulation of the lipid A structure of Porphyromonas gingivalis lipopolysaccharide. Infect Immun. 2006;74:4474–4485. doi: 10.1128/IAI.01924-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Darveau RP, Pham TT, Lemley K, Reife RA, Bainbridge BW, Coats SR, Howald WN, Way SS, Hajjar AM. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect Immun. 2004;72:5041–5051. doi: 10.1128/IAI.72.9.5041-5051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hajishengallis G, Tapping RI, Harokopakis E, Nishiyama S, Ratti P, Schifferle RE, Lyle EA, Triantafilou M, Triantafilou K, Yoshimura F. Differential interactions of fimbriae and lipopolysaccharide from Porphyromonas gingivalis with the Toll-like receptor 2-centred pattern recognition apparatus. Cell Microbiol. 2006;8:1557–1570. doi: 10.1111/j.1462-5822.2006.00730.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.