

Abstract
Background
The apolipoproteins (APOA1/C3/A4/A5) are key components in modulating lipoprotein metabolism. It is unknown whether variants at the APOA1/C3/A4/A5 gene cluster are associated with lipid response to pharmacologic intervention.
Methods and results
Plasma triglycerides (TGs) and high-density lipoprotein (HDL) levels were measured in 861 Genetics of Lipid-Lowering Drugs and Diet Network study participants who underwent a 3-week fenofibrate trial. We examined 18 common single nucleotide polymorphisms (SNPs) spanning the APOA1/C3/A4/A5 genes to investigate the effects of variants at the gene cluster on lipid response to fenofibrate treatment. We found that the minor alleles of the SNPs rs3135506 (APOA5_S19W), rs5104 (APOA4_N147S), rs4520 (APOC3_G34G), and rs5128 (APOC3_3U386) were associated with enhanced TG response to fenofibrate treatment (P = 0.0004–0.018). The minor allele of SNP rs2854117 (APOC3_M482) was associated with reduced rather than enhanced TG response (P = 0.026). The SNP rs3135506 (APOA5_S19W) was associated with HDL response, with minor allele related to reduced HDL response to fenofibrate (P = 0.002). Association analyses on haplotype provided corroborative evidence to single SNP association analyses. The common haplotypes H2, H3, and H5 were significantly associated with reduced TG response to fenofibrate.
Conclusion
The genetic variants at APOA1/C3/A4/A5 gene cluster may be useful markers to predict response of lipid-lowering therapy with fenofibrate. Further studies to replicate/confirm our findings are warranted.
Keywords: apolipoproteins gene cluster, fenofibrate, hypertriglyceridemia, pharmacogenetics
Introduction
Coronary heart disease is an important threat to public health in the world [1]. Elevated plasma triglyceride (TG) and reduced high-density lipoprotein cholesterol (HDL-C) concentrations are major risk factors for coronary heart disease [2,3]. Plasma TG response to pharmacological and behavioral interventions manipulation is highly variable [4-6]. Intensive and sustained lifestyle modifications such as physical activity and diets especially when associated with weight loss, reduced TG levels, but for many individuals pharmacological interventions are needed to achieve substantial reduction in TGs. Fenofibrate is an efficacious therapeutic agent for hypertriglyceridemia by reducing plasma TG levels by 35–50% and raising HDL-C levels by 10–20% [7].
Emerging studies suggest that genetic variation may serve as a useful marker to assess therapeutic efficacy of lipid-lowering treatments [8]. The apolipoproteins (APOA1/C3/A4/A5) are key components in modulating lipoprotein metabolism [9]. The APOA1/C3/A4 genes are located on human chromosome 11q23 and extend approximately to 17 kb. Approximately 30 kb distal to APOA1/C3/A4 is the fourth member, APOA5 [10]. Human mutants and overexpression as well as gene knockout mouse models showed that each member of the gene cluster plays an important role in lipid homeostasis [9]. A few studies tested associations between apolipoprotein genes and lipid levels in the fasting and/or postprandial states, but largely focused on a specific gene or polymorphism [9,11]. Owing to the functional and positional relevance, it is necessary to examine the entire APOA1/C3/A4/A5 gene cluster as a whole [12-14]. In particular, no study was carried out to understand the pharmacogenetic implications of this gene cluster on lipid response.
Our objective was to test for the association between the APOA1/C3/A4/A5 gene cluster and lipid response to fenofibrate treatment in patients participating in the National Heart, Lung, and Blood Institute Genetics of Lipid-Lowering Drugs and Diet Network (GOLDN) study. We genotyped 18 single nucleotide polymorphisms (SNPs) spanning the gene cluster to determine the linkage disequilibrium (LD) structure across the region. The single SNPs and haplotypes association analyses were performed for association with lipid response to fenofibrate treatment.
Methods
Patients and intervention
GOLDN is part of the PROGENI (PROgram for GENetic Interaction) Network, a group of family intervention studies focusing on gene–environment interactions. The participants in the GOLDN study were mainly re-recruited from two National Heart, Lung, and Blood Institute Family Heart Study field centers: Minneapolis, Minnesota, and Salt Lake City, Utah. Nearly, all participants were of European ancestry. Our earlier studies demonstrated that Caucasians in Utah and Minnesota were homogeneous and thus pooling data across centers will not threaten the validity of this study [15].
Eligibility criteria were: (i) ≥ 18 years of age; (ii) fasting TGs less than 1500mg/dl; (iii) willing to participate in the study and attend the scheduled clinic exams; (iv) member of a family with at least two members in a sibship; (v) aspartate aminotransferase and alanine aminotransferase results within normal range; and (vi) creatinine ≤ 2.0mg/dl. Exclusion criteria were: (i) history of liver, kidney, pancreas, gall bladder disease, or malabsorption; (ii) current pregnancy; (iii) insulin use; (iv) use of lipid-lowering drugs (including prescription, over-the-counter and nutraceuticals; volunteers taking these agents were withdrawn from them at least 4 weeks before the study with physician’s approval; (v) use of warfarin; (vi) women of childbearing potential not using an acceptable form of contraception; (vii) known hypersensitivity to fenofibrate; and (viii) history of pancreatitis within 12 months before enrolment.
After signing consent forms and Health Insurance Portability and Accountability Act authorizations, all family members underwent a baseline-screening visit. The screening visit included a fasting blood draw and pregnancy test, if applicable. If at least two members in a sibship qualified after the screening visit, all willing family members were asked to participate. The day before the first clinical exam, participants came to the clinic for a fasting blood draw. The clinical exam was then performed on the next day and included anthropometric and blood pressure measurements. The fenofibrate intervention consisted of a 3-week treatment period, in which participants took fenofibrate (160mg) daily. Lipids were measured twice after a minimum fast of 10 h on the last 2 days of the treatment period.
Biochemical analyses
In this study, only the information of TG and HDL-C responses were included. This is because fenofibrate is not usually prescribed for normalization of low-density lipoprotein cholesterol (LDL-C) or total cholesterol levels and thus our hypothesis here with fenofibrate was specifically targeted to TG and HDL. Venous blood was drawn after an overnight fast, and plasma was centrifuged within 20min of collection at 2000 g for 15 min at 4°C. Plasma samples of each participant were stored at 4°C until the completion of the treatment period. All samples from a single participant were analyzed within the same batch. TGs were measured using a glycerol-blanked enzymatic method (Trig/GB; Roche Diagnostics Corporation, Indianapolis, Indiana, USA) on the Roche/Hitachi 911 Automatic Analyzer (Roche Diagnostics Corporation). Cholesterol was measured on the Hitachi 911 using a cholesterol esterase, cholesterol oxidase reaction (Chol R1; Roche Diagnostics Corporation). The same reaction was also used to measure HDL-C after precipitation of non-HDL-C with magnesium/dextran.
Single nucleotide polymorphism selection and genotyping
SNPs were identified through searching public databases such as dbSNP (http://www.ncbi.nlm.nih.gov/SNP/) and others. We selected 18 SNPs at the APOA1/C3/A4/A5 gene cluster based on the following criteria in the order of importance: (i) validation status, that is, experimentally validated in Caucasians; (ii) functional relevance and importance, namely the potential ‘functional’ SNPs residing within the transcription factor binding sites in the 5′ promoter region, in the mRNA stability regulatory protein binding sites in 3′ untranslated region, in exons that change amino acid sequences, or in exon–intron boundaries that alter mRNA splicing; (iii) degree of heterozygosity, that is, minor allele frequencies greater than 0.05; (iv) tagging SNPs; and (v) earlier evidence of association with lipid measurements. The SNPs selected should meet at least one of the above criteria.
SNPs were genotyped using the 5′ nuclease allelic discrimination Taqman assay with allelic specific probes on the ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, California, USA). PCR reactions were carried out in a 5 μl final volume, consisting of 2.5 μl of 2 × TaqMan Universal PCR Master mix, 0.125 μl of SNP genotyping assay mix (Applied Biosystems), and 10 ng of DNA with cycling conditions of 95°C for 10 min, followed by 50 cycles of 92°C for 15 s and 60°C for 1 min. Internal controls and repetitive experiments were performed to ensure genotyping quality. The overall genotyping error and missing rate was approximately 1%. We used the GRR software [16] to detect pedigree errors by graphically inspecting the distribution for marker allele sharing among pairs of family members or all pairs of individuals. The SNP allele frequencies were estimated by a maximum-likelihood method [17]. For each SNP, a χ2 test was used to examine deviation of SNP genotypes from the Hardy–Weinberg equilibrium (HWE) [18]. In the assessment of significance of the HWE test, only the unrelated individuals from the total sample were used.
Linkage disequilibrium and haplotype analyses
LD and haplotype analyses were performed using Haploview [19]. LD structure was assessed by D′ for pairs of SNPs [20]. Haplotype blocks were defined using the four-gamete rule [19]. We selected haplotype-tagging SNPs (htSNPs) according to the pairwise correlation method using software tagger (http://www.broad.mit.edu/mpg/tagger/). We set the pairwise correlation criterion as a minimum r2 of 0.80. In addition, we used other methods to aid the selection of tag SNPs, such as , a method of selecting htSNPs to predict common haplotypes [21], and , a method of selecting htSNPs to predict unmeasured SNPs [21].
Association analysis
Genotype–phenotype association analyses were performed using a linear mixed model, which was implemented in SAS (version 9.1, SAS Institute, Cary, North Carolina, USA). In the mixed model, genotypes were treated as fixed effects and the dependencies among members within each family were treated as random effects. Age, age2, age3, sex, and field center were included as covariates.
The lipid changes were estimated using a growth curve model [22,23]. The model is defined as follows:
where yit is the phenotype of individual i measured at time point t. α and β are the intercept and slope for the population level effects (fixed effects) respectively, whereas γiα and γiβ are for the individual level effects (random effects), respectively. εit is the residual. The model was implemented in the PROC MIXED procedure in SAS (SAS Institute Inc., Cary, North Carolina, USA). As the growth curve slope (β+γiβ) eliminates the noise εit, it represents an accurate and stable estimate of lipid change.
Haplotypes were reconstructed based on the 10 tag SNPs. Haplotype inference was performed using MERLIN [24], which is preferable for family data [25]. Merlin can accommodate LD in haplotype inference [26]. Association analysis for haplotypes was conducted using a mixed model similar to that for single SNPs. In addition, a global test was performed to examine whether phenotype differences existed among all haplotypes simultaneously. In haplotype analyses, a general mode of inheritance was used (i.e. genotypes as categorical variable with three classes).
The proportion of variance explained by the significant SNPs was estimated using regression analyses, in which the variance is the difference of R2 between the models including SNP and those without. In association analysis, all P values less than 0.05 in our study were considered nominally significant, which were further subject to Bonferroni’s correction to account for multiple comparisons. The conservative significance threshold for a single test was assessed at a type I error rate of 0.05/N. N was the number of tested markers.
Results
Study sample characteristics
Table 1 presents the baseline characteristics of the study sample. A total of 861 participants (427 men and 434 women) from 148 families having both phenotype and genotype data were included in the study. The mean age of men was 50.6 years, and that of women was 51.1 years. The values of TGs and HDL are shown classified by sex (Table 1). For both men and women, the measurements of TG were significantly lower after fenofibrate treatment than those before treatment (all P < 0.001). In contrast, HDL-C increased significantly after fenofibrate treatment (P < 0.001).
Table 1.
Characteristics of study participants
| Men (N=427) |
Women (N=434) |
|||
|---|---|---|---|---|
| Prefenofibrate | Postfenofibrate | Prefenofibrate | Postfenofibrate | |
| Age (years) | 50.6 ± 15.9 | 51.1 ± 15.8 | ||
| BMI (kg/m2) | 28.72 ± 4.86 | 28.48 ± 6.33 | ||
| Triglyceride (mg/dl) | 153.41 ± 142.01 | 99.18 ± 59.83 | 125.19 ± 82.22 | 80.67 ± 47.30 |
| HDL cholesterol (mg/dl) | 41.57 ± 9.84 | 43.67 ± 9.99 | 52.29 ± 13.68 | 55.03 ± 14.23 |
Values are listed as mean ± SD or as number (raw data).
HDL, high-density lipoprotein.
Linkage disequilibrium pattern and haplotypes
On the basis of HapMap SNP data (http://www.hapmap.org) in Caucasian population, the selected 18 SNPs can tag more than 80% SNPs in the APOA1/C3/A4/A5 gene cluster locus, if D′ of 0.8 is used for block partitioning. Therefore, these 18 SNPs can cover and tag the majority of APOA1/C3/A4/A5 gene cluster locus. Information of the 18 SNPs is summarized in Table 2. All the SNPs except APOA4_Q360H were in HWE (P=0.004). We excluded APOA4_Q360H from further association analysis. Figure 1 depicts the genomic organization of the gene cluster, LD structure, and haplotype blocks. Five haplotype blocks of strong LD were identified in the region, with APOA1, APOC3, APOA4, and APOA5 falling into separate blocks. In each block, a small number (three to six) of common haplotypes represented greater than 95% of all haplotypes. Ten tag SNPs, namely, APOA5_S19W, APOA5_-M1123, APOA4_N147S, APOA4_T29T, APOA4_M35, APOC3_M640, APOC3_M482, APOC3_G34G, APOC3-_3U386, and APOA1_M2803, were selected to capture the common genetic variation at the gene cluster.
Table 2.
Properties of the 18 SNPs in the APOA1/C3/A4/A5 gene cluster
| Gene | DbSNP No. | SNP name | Variant type | Chromosome position (bp) |
Intermarker distances (bp) |
Allele | MAF | % Geno | HWE P value |
|---|---|---|---|---|---|---|---|---|---|
| APOA5 | rs3135506 | APOA5_S19W | Coding nonsynonymous (Trp/Ser) | 116167617 | – | C/G | 0.068 | 100.0 | 0.493 |
| rs662799 | APOA5_M1123 | 5′ Flanking | 116168917 | 1300 | G/A | 0.062 | 99.8 | 0.398 | |
| APOA4 | rs1263177 | APOA4_A5INTER | 3′ Flanking | 116180028 | 11 111 | C/T | 0.353 | 99.7 | 0.107 |
| rs5110 | APOA4_Q360H | Coding nonsynonymous (His/Gln) | 116196844 | 16 816 | T/G | 0.088 | 99.8 | 0.004 | |
| rs675 | APOA4_T347S | Coding nonsynonymous (Ser/Thr) | 116196885 | 41 | A/T | 0.183 | 99.9 | 0.582 | |
| rs5104 | APOA4_N147S | Coding nonsynonymous (Asn/Ser) | 116197544 | 659 | G/A | 0.130 | 99.8 | 0.966 | |
| rs5092 | APOA4_T29T | Coding, synonymous | 116198674 | 1130 | G/A | 0.173 | 99.8 | 0.142 | |
| rs5090 | APOA4_M35 | 5′ Flanking | 116199265 | 591 | C/G | 0.051 | 100.0 | 0.832 | |
| APOC3 | rs2542051 | APOC3_M2886 | 5′ Flanking | 116202948 | 3683 | C/A | 0.348 | 99.8 | 0.463 |
| rs2542052 | APOC3_M640 | 5′ Flanking | 116205194 | 2246 | A/C | 0.360 | 99.9 | 0.228 | |
| rs2854117 | APOC3_M482 | 5′ Flanking | 116205352 | 158 | A/G | 0.250 | 99.8 | 0.469 | |
| rs2854116 | APOC3_M455 | 5′ Flanking | 116205379 | 27 | G/A | 0.356 | 99.7 | 0.160 | |
| rs4520 | APOC3_G34G | Coding, synonymous | 116206745 | 1366 | T/C | 0.262 | 100.0 | 0.393 | |
| rs5128 | APOC3_3U386 | Exon, untranslated | 116208850 | 2105 | C/G | 0.094 | 99.9 | 0.384 | |
| APOA1 | rs670 | APOA1_M75 | 5′ Flanking | 116213623 | 4773 | A/G | 0.160 | 99.9 | 0.136 |
| rs613808 | APOA1_M2630 | 5′ Flanking | 116216178 | 2555 | A/G | 0.292 | 100.0 | 0.482 | |
| rs2727784 | APOA1_M2803 | 5′ Flanking | 116216351 | 173 | G/A | 0.353 | 99.8 | 0.521 | |
| rs11216158 | APOA1_M3012 | 5′ Flanking | 116216560 | 209 | A/G | 0.128 | 99.8 | 0.652 |
APOA4_Q360H (rs5110) was excluded from the association analysis because of deviation from the Hardy–Weinberg equilibrium.
Boldface indicates tag SNPs. Bold italic indicates minor alleles.
% Geno, completeness of genotyping call; Asn, asparagine; Gln, glutamine; HET, heterozygosity; His, histidine; HWE, Hardy–Weinberg equilibrium; MAF, minor allele frequency; Ser, serine; SNP, single nucleotide polymorphism; Thr, threonine; Trp, tryptophan.
Fig. 1.
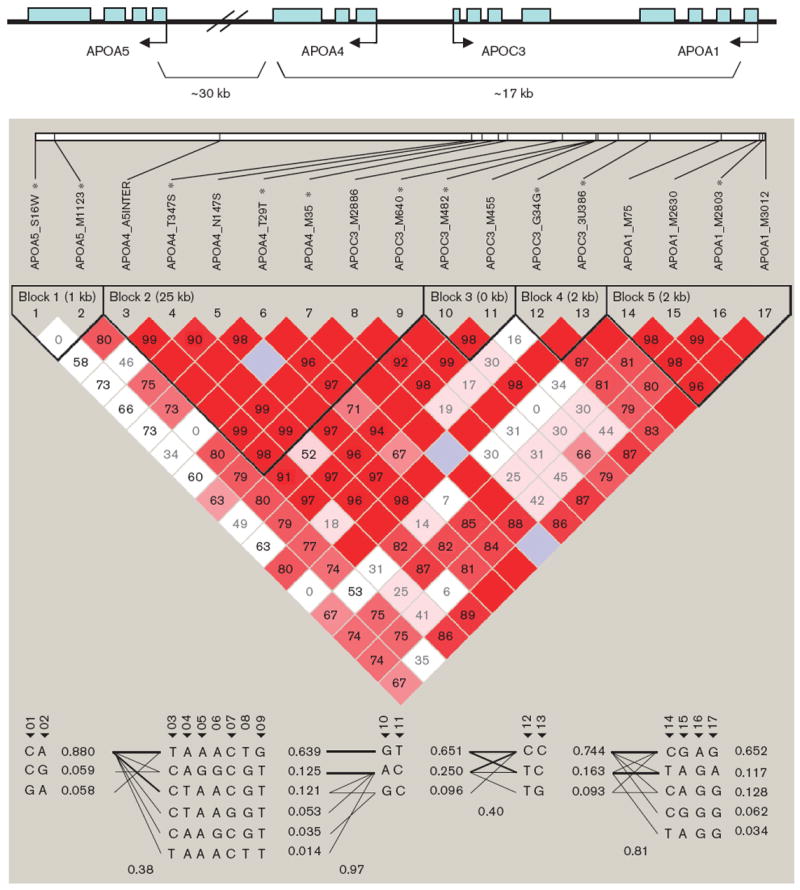
Linkage disequilibrium structure and haplotype blocks of the APOA1/C3/A4/A5 gene cluster.
Association analysis
Tables 3 and 4 summarize the results of association analyses for TG and HDL-C response to fenofibrate, respectively. Note that the lipid responses here were estimated from growth curve models. For the ease of interpretation of our results, we also provided the absolute changes in TG and HDL levels, respectively, by genotype in Tables 3 and 4 for the SNPs showing significant association. The minor alleles of APOA5_S19W, APOA4_N147S, APOC3_G34G, and APOC3_3U386 were nominally significantly associated with enhanced TG response (P= 0.018–0.0004). The most pronounced association was observed for APOA5_S19W, which remained significant even after conservative Bonferroni’s correction (P=0.0004). APOA5_S19W explains 3.1% of variance of TG in drug response. APOA5_M1123 showed marginal evidence of significance (P=0.054). The minor allele of SNP APOC3_M482 showed reduced rather than enhanced TG response (P=0.026). In association analysis for HDL response, we only found significance for APOA5_S19W (P=0.002), whose minor allele is related to reduced HDL response. APOA5_S19W explains 2.6% of variance of HDL-C in drug response. Figure. 2a and b show the growth curve change and absolute change, respectively, for APOA5_S19W genotypes. In association analyses, age, sex, and field center were all significant in the model and were adjusted as covariates accordingly.
Table 3.
Association analysis of APOA1/C3/A4/A5 SNPs with TG response to fenofibratea
| Phenotypic mean ± SE |
||||||
|---|---|---|---|---|---|---|
| SNP | Alleles | MAF | 1/1 (n) | 1/2 (n) | 2/2 (n) | P |
| APOA5_S19W | C/G | 0.068 | − 2.052 ± 0.109 (− 44.9 ± 57.4) (763) | − 3.236 ± 0.559 (− 77.5 ± 107.2) (95) | − 8.562 ± 0.583 (− 212.3 ± 78.5) (3) | 0.0004b |
| APOA5_M1123 | G/A | 0.062 | − 2.134 ± 0.123 (762) | − 2.883 ± 0.376 (97) | − 3.021 ± 0.357 (2) | 0.054 |
| APOA4_A5INTER | C/T | 0.353 | − 2.372 ± 0.285 (370) | − 2.313 ± 0.176 (395) | − 2.040 ± 0.165 (96) | 0.394 |
| APOA4_T347S | A/T | 0.183 | − 2.305 ± 0.159 (588) | − 1.947 ± 0.140 (245) | − 2.172 ± 0.373 (28) | 0.186 |
| APOA4_N147S | G/A | 0.130 | − 1.945 ± 0.109 (− 43.3 ± 54.5) (656) | − 2.813 ± 0.735 (− 50.1 ± 61.2) (189) | − 3.070 ± 0.335 (− 68.7 ± 93.5) (16) | 0.004 |
| APOA4_T29T | G/A | 0.173 | − 2.009 ± 0.123 (591) | − 2.608 ± 0.277 (249) | − 3.060 ± 0.768 (21) | 0.067 |
| APOA4_M35 | C/G | 0.051 | − 2.216 ± 0.132 (781) | − 2.106 ± 0.210 (80) | 0.654 | |
| APOC3_M2886 | C/A | 0.348 | − 2.313 ± 0.290 (382) | − 2.342 ± 0.187 (382) | − 2.032 ± 0.159 (97) | 0.338 |
| APOC3_M640 | A/C | 0.360 | − 2.051 ± 0.158 (368) | − 2.337 ± 0.174 (392) | − 2.218 ± 0.291 (98) | 0.387 |
| APOC3_M482 | A/G | 0.250 | − 2.712 ± 0.368 (− 57.7 ± 50.4) (505) | − 2.549 ± 0.221 (− 57.1 ± 79.4) (306) | − 1.950 ± 0.131 (− 43.3 ± 57.0) (46) | 0.026 |
| APOC3_M455 | G/A | 0.356 | − 2.275 ± 0.298 (376) | − 2.276 ± 0.174 (391) | − 2.106 ± 0.169 (94) | 0.736 |
| APOC3_G34G | T/C | 0.262 | − 1.903 ± 0.108 (− 40.8 ± 46.3) (480) | − 2.462 ± 0.207 (− 56.7 ± 76.2) (318) | − 3.282 ± 0.703 (− 71.7 ± 112.1) (63) | 0.006 |
| APOC3_3U386 | C/G | 0.094 | − 2.013 ± 0.110 (− 44.6 ± 54.7) (718) | − 3.218 ± 0.437 (− 64.8 ± 82.7) (138) | − 3.994 ± 1.627 (− 71.0 ± 103.6) (5) | 0.018 |
| APOA1_M75 | A/G | 0.160 | − 2.161 ± 0.120 (603) | − 2.198 ± 0.257 (226) | − 3.136 ± 0.660 (32) | 0.354 |
| APOA1_M2630 | A/G | 0.292 | − 3.088 ± 0.710 (433) | − 2.254 ± 0.191 (359) | − 2.029 ± 0.120 (69) | 0.194 |
| APOA1_M2803 | G/A | 0.353 | − 1.999 ± 0.136 (374) | − 2.249 ± 0.172 (389) | − 2.827 ± 0.531 (98) | 0.173 |
| APOA1_M3012 | A/G | 0.128 | − 3.642 ± 0.898 (657) | − 2.078 ± 0.270 (181) | − 2.197 ± 0.123 (21) | 0.259 |
Bold italic indicates minor alleles. P values less than 0.05 are shown in boldface.
MAF, minor allele frequeny; SNP, single nucleotide polymorphism; TG, triglyceride.
Response to fenofibrate (phenotype) was represented by slope estimated from growth curve models. Numbers in brackets are absolute changes of TG (mg/dl) in response to fenofibrate.
Significant associations after Bonferroni’s correction. The significance threshold is P < 0.003 (0.05/17).
Table 4.
Association analysis of of APOA1/C3/A4/A5 SNPs with HDL response to fenofibratea
| Phenotypic mean ± SE |
||||||
|---|---|---|---|---|---|---|
| SNP | Alleles | MAF | 1/1 (n) | 1/2 (n) | 2/2 (n) | P |
| APOA5_S19W | C/G | 0.068 | 0.120 ± 0.001 (2.72 ± 5.70) (763) | 0.117 ± 0.002 (4.49 ± 5.48) (95) | 0.096 ± 0.006 (6.66 ± 4.72) (3) | 0.002b |
| APOA5_M1123 | G/A | 0.062 | 0.120 ± 0.001 (762) | 0.116 ± 0.003 (97) | 0.117 ± 0.002 (2) | 0.270 |
| APOA4_A5INTER | C/T | 0.353 | 0.120 ± 0.003 (370) | 0.121 ± 0.001 (395) | 0.119 ± 0.001 (96) | 0.510 |
| APOA4_T347S | A/T | 0.183 | 0.120 ± 0.001 (588) | 0.120 ± 0.001 (245) | 0.118 ± 0.006 (28) | 0.947 |
| APOA4_N147S | G/A | 0.130 | 0.119 ± 0.001 (656) | 0.122 ± 0.002 (189) | 0.122 ± 0.007 (16) | 0.487 |
| APOA4_T29T | G/A | 0.173 | 0.119 ± 0.001 (591) | 0.121 ± 0.001 (249) | 0.122 ± 0.006 (21) | 0.553 |
| APOA4_M35 | C/G | 0.051 | 0.120 ± 0.001 (781) | 0.117 ± 0.003 (80) | 0.325 | |
| APOC3_M2886 | C/A | 0.348 | 0.121 ± 0.002 (382) | 0.121 ± 0.001 (382) | 0.119 ± 0.001 (97) | 0.502 |
| APOC3_M640 | A/C | 0.360 | 0.118 ± 0.001 (368) | 0.121 ± 0.001 (392) | 0.121 ± 0.002 (98) | 0.273 |
| APOC3_M482 | A/G | 0.250 | 0.124 ± 0.004 (505) | 0.120 ± 0.001 (306) | 0.119 ± 0.001 (46) | 0.441 |
| APOC3_M455 | G/A | 0.356 | 0.120 ± 0.002 (376) | 0.121 ± 0.001 (391) | 0.119 ± 0.001 (94) | 0.468 |
| APOC3_G34G | T/C | 0.262 | 0.119 ± 0.001 (480) | 0.121 ± 0.001 (318) | 0.120 ± 0.004 (63) | 0.772 |
| APOC3_3U386 | C/G | 0.094 | 0.120 ± 0.001 (718) | 0.118 ± 0.002 (138) | 0.135 ± 0.012 (5) | 0.354 |
| APOA1_M75 | A/G | 0.160 | 0.118 ± 0.001 (603) | 0.122 ± 0.001 (226) | 0.125 ± 0.006 (32) | 0.216 |
| APOA1_M2630 | A/G | 0.292 | 0.122 ± 0.003 (433) | 0.122 ± 0.001 (359) | 0.118 ± 0.001 (69) | 0.112 |
| APOA1_M2803 | G/A | 0.353 | 0.118 ± 0.001 (374) | 0.122 ± 0.001 (389) | 0.120 ± 0.002 (98) | 0.132 |
| APOA1_M3012 | A/G | 0.128 | 0.120 ± 0.008 (657) | 0.122 ± 0.002 (181) | 0.119 ± 0.001 (21) | 0.355 |
Numbers in brackets are absolute changes of HDL (mg/dl) in response to fenofibrate. Bold italic indicates minor alleles. P values less than 0.05 are shown in boldface.
MAF, minor allele frequency; SNP, single nucleotide polymorphism; HDL, high-density lipoprotein.
Response to fenofibrate (phenotype) was represented by slope estimated from growth curve models.
Indicates significant associations after Bonferroni’s correction. The significance threshold is P < 0.003 (0.05/17).
Fig. 2.
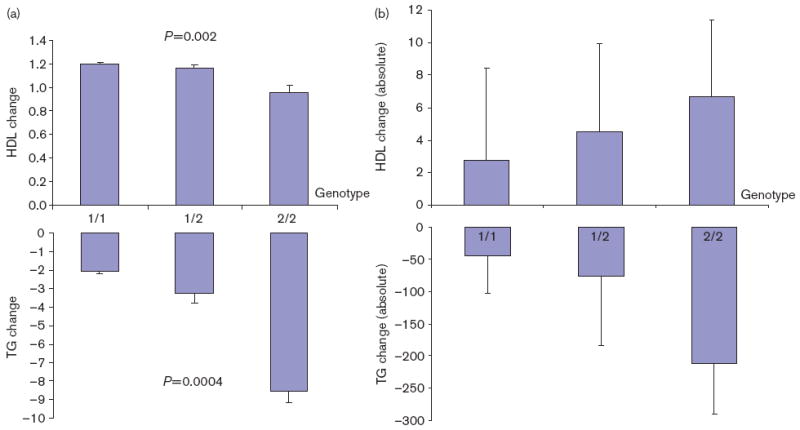
(a) Phenotypic changes with APOA5_S19W genotypes. Changes (mean ± SE) of high-density lipoprotein (HDL) cholesterol and triglyceride (TG) (y-axis) were growth curve slopes estimated from the mixed models. (b) Absolute changes of TG and HDL with APOA5_S19W genotypes.
Table 5 summarizes the information on common haplotypes, frequencies, P values for association, and the mean trait values for the different haplotype combinations. Association analysis was conducted for haplotypes with frequency of greater than 0.04, with an intention to maximize the statistical power and to minimize potential bias because of small number of patients bearing a specific haplotype. A total of six such common haplotypes were identified, with the most common haplotype 1 (CAAACGGCCA) accounting for 0.462 of total frequencies. Another two common haplotypes 2 (CAAACTGCCA) and 3 (CAAACGGTCG) have frequencies of 0.071 and 0.067, respectively. The haplotypes 2, 3, and 5 were significantly associated with reduced TG response to fenofibrate. For HDL, the haplotypes 2, 4, and 5 were associated with reduced responsiveness.
Table 5.
Association of APOA1/C3/A4/A5 common haplotypes with lipid response to fenofibratea
| Haplotype–genotype mean (SE) for TG response |
Haplotype–genotype mean (SE) for HDL cholesterol response |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Haplotypeb | Haplotype frequency |
+ / + c | ± d | − / − e | Haplotype P |
Global P |
+ / + c | ± d | − / − e | Haplotype P |
Global P |
| Hap1 CAAACGGCCA | 0.462 | − 1.995 (0.211) | − 2.293 (0.149) | − 2.113 (0.207) | 0.429 | 0.003* | 0.112 (0.008) | 0.128 (0.007) | 0.132 (0.007) | 0.202 | 0.014 |
| Hap2 CAAACTGCCA | 0.071 | − 0.743 (0.167) | − 1.851 (0.186) | − 2.230 (0.134) | < 0.0001* | 0.113 (0.005) | 0.111 (0.010) | 0.128 (0.004) | 0.031 | ||
| Hap3 CAAACGGTCG | 0.067 | − 0.256 (0.999) | − 1.773 (0.333) | − 2.238 (0.137) | 0.035 | 0.123 (0.046) | 0.126 (0.015) | 0.126 (0.004) | 0.997 | ||
| Hap4 CAAAGTACCG | 0.048 | − 1.172 (0.476) | − 2.476 (0.315) | − 2.150 (0.131) | 0.077 | 0.052 (0.022) | 0.121 (0.017) | 0.126 (0.004) | 0.011 | ||
| Hap5 CGGGCTATGG | 0.044 | − 1.052 (0.162) | − 2.136 (0.121) | − 2.732 (0.508) | < 0.0001* | 0.077 (0.006) | 0.143 (0.015) | 0.125 (0.004) | < .0001* | ||
| Hap6 GAAACGGTCG | 0.044 | − 4.940 (1.818) | − 3.951 (1.731) | − 2.088 (0.108) | 0.231 | 0.159 (0.032) | 0.195 (0.026) | 0.123 (0.004) | 0.032 | ||
HDL, high-density lipoprotein; TG, triglyceride.
Response to fenofibrate (phenotype) was represented by slope estimate from growth curve models.
Haplotypes were determined using the Haploview software for tag SNPs following the order of APOA5_S19W, APOA5_M1123, APOA4_N147S, APOA4_T29T, APOA4_M35, APOC3_M640, APOC3_M482, APOC3_G34G, APOC3_3U386, and APOA1_M2803. To maximize the statistical power, we only tested associations for the haplotypes with estimated frequencies greater than 0.04.
Two copies of the haplotype of interest.
One copy of the haplotype of interest.
Any other haplotype combination.
P < 0.05 are shown in boldface; indicates significant associations after Bonferroni’s correction. The significance threshold is P < 0.008 (0.05/6).
To investigate whether the association findings apply to a patient population with clinically significant hypertriglyceridemia, we performed further analyses in the subsample of 192 subjects whose TGs level greater than 200 mg/dl (i.e. hypertriglyceridemia). We could not, however, find any significant SNP–phenotype associations.
Discussion
This is the first study to investigate the APOA1/C3/A4/A5 gene cluster in relation to lipid response to fenofibrate treatment. We found significant associations between the SNPs of the gene cluster and lipid response.
The most pronounced association was for APOA5_S19W, which was associated with both TG and HDL-C response to fenofibrate. APOA5_S19W alters codon 19 of the predicted amino-terminal signal sequence of APOA5, which substitutes a serine residue with tryptophan. As serine 19 can be placed at the −5 position for the preapolipoprotein [27], a tryptophan residue so close to the cleavage site of the APOA5 signal sequence could considerably reduce the processing of this preprotein. Although no functional studies and induced mutations in mice have been carried out to prove its actual role in lipid metabolism, studies using protein structure prediction algorithm [28] suggested that APOA5_S19W may cause a change in the secondary structure of APOA5, with a concomitant change in tertiary structure [29]. We hypothesize that this APOA5 structure alteration may lead to altered gene function and thus lipid response to fenofibrate treatment.
APOC3_M482 is located at the insulin response element in the promoter region of APOC3 and confers decreased responsiveness to insulin in in-vitro assays [30]. APOC3_M482 is in strong LD with APOC3_3U386, which is significantly associated with TG response in this study. APOC3_3U386 was associated with combined hyperlipidemia and hypertriglyceridemia [31]. APO-A4_N147S is a coding variant that results in a nonsynonymous change at position 147 of APOA4 from asparagine to serine. Studies showed that APOA4 may control intravascular transport of dietary lipids and was associated with postprandial clearance of TG-rich lipoproteins [32]. We postulate that APOA4_N147S may cause altered conformation and function of APOA4 and thus differential response to fenofibrate treatment.
Pharmacogenetic studies of lipid-lowering therapy, in particular with fibrate, are relatively rare. In the Lopid Coronary Angiography Trial study, no association was observed between APOA5 variants and lipid response and disease progress to gemfibrozil (another fibrate) treatment [33]. The Lopid Coronary Angiography Trial study was conducted in men who had undergone coronary artery bypass grafting with HDL-C ≤ 42.57 mg/dl, whereas our study samples were randomly selected and may represent the general population. Moreover, our sample size is larger and thus has higher statistical power.
A few pharmacogenetic studies of lipid-lowering therapy with statin were reported [34]. An APOA1 promoter polymorphism was found to influence basal HDL-C and its response to pravastatin therapy [35]. As statins are 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors and have different metabolic mechanisms from that of fibrates, it is difficult to compare the results between statin-based studies and ours here with fibrates. Currently, statins are regarded as first-line therapy for lipid disorder and seem to have eclipsed the clinical use of fibrates. Emerging evidence, however, emphasizes the necessity of refocusing on fibrates in the poststatin era [36]. First, statins are primarily designed for correcting hypercholesterolemia as the sole abnormality, but up to half of coronary patients are normocholesterolemic. Second, clinical endpoint trials suggest that the relative risk reduction with statins over a period of 5 years is at best 35% [36], suggesting that a significant proportion of patients sustain cardiovascular events independent of LDL-C-lowering or a direct effect of statins on atherogenesis. Finally, up to a third of coronary patients have elevated plasma TG levels with low HDL-C and normal or near-normal LDL-C [36]. Given these, our findings here may have important clinical implications.
We were not able to find significant association in analyses of subsample with hypertriglyceridemia (a TG level > 200 mg/dl). A possible reason is that the SNP effects on lipid response to fenofibrate may be different between those who have higher TG levels and those who have lower TG levels. Another possible reason is that the largely decrease sample size (192 patients) may not be sufficiently powerful to detect the modest effects that truly exist.
The strengths of this study lie in: a rigorous fenofibrate trial, a large family-based sample, and comprehensive survey of the entire gene cluster. Our study has, however, some potential limitations. First, although multiple SNPs were genotyped, they were not sufficient to exhaustively cover the entire gene cluster. The LD pattern and haplotype block structure of the gene cluster in this study sample are, however, comparable with some earlier studies in which denser SNPs were genotyped [12,14]. Second, validation/replication in an independent sample are generally required to confirm association findings. Currently, large-scale fibrate intervention studies are, however, extremely rare in the field (largely because of the high costs involved and low visit adherence). Therefore, we lack available data set for validation/replication at this stage. Third, a number of tests were performed because of multiple SNPs involved. After correction for multiple testing, some significant associations only reached nominal significance or even disappeared. As the tested SNPs are in LD, Bonferroni’s correction may, however, be too conservative. Finally, the GOLDN project is a genetic epidemiology study, which alone is not sufficient to define a functional variant unambiguously.
In summary, we found that the APOA1/C3/A4/A5 gene cluster is associated with lipid response to fenofibrate treatment, suggesting that variation in this gene cluster could be useful to tailor lipid-lowering therapies. Further studies in other populations and/or molecular functional studies are necessary to validate/replicate our findings.
Acknowledgments
The authors thank the families for their participation in this research. This study was supported by NIH Heart, Lung, and Blood Institute Grant U 01 HL72524, Genetic and Environmental Determinants of Triglycerides. They acknowledge Abbott Laboratories (Abbott Park, Illinois, USA) for their supply of study medication for this project. Dr David B. Allison is supported by NIH Grant 3P30DK056336. Dr Kui Zhang is supported by NIH Grant R01GM74913. Dr Guimin Gao is supported by NIH Grant R01GM073766.
References
- 1.Watkins LO. Epidemiology and burden of cardiovascular disease. Clin Cardiol. 2004;27(Suppl 3):III2–III6. doi: 10.1002/clc.4960271503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krauss RM. Triglycerides and atherogenic lipoproteins: rationale for lipid management. Am J Med. 1998;105:58S–62S. doi: 10.1016/s0002-9343(98)00213-7. [DOI] [PubMed] [Google Scholar]
- 3.Malloy MJ, Kane JP. A risk factor for atherosclerosis: triglyceride-rich lipoproteins. Adv Intern Med. 2001;47:111–136. [PubMed] [Google Scholar]
- 4.Suter PM, Gerritsen-Zehnder M, Hasler E, Gurtler M, Vetter W, Hanseler E. Effect of alcohol on postprandial lipemia with and without preprandial exercise. J Am Coll Nutr. 2001;20:58–64. doi: 10.1080/07315724.2001.10719015. [DOI] [PubMed] [Google Scholar]
- 5.Durstine JL, Grandjean PW, Davis PG, Ferguson MA, Alderson NL, DuBose KD. Blood lipid and lipoprotein adaptations to exercise: a quantitative analysis. Sports Med. 2001;31:1033–1062. doi: 10.2165/00007256-200131150-00002. [DOI] [PubMed] [Google Scholar]
- 6.Kuller LH, Simkin-Silverman LR, Wing RR, Meilahn EN, Ives DG. Women’s Healthy Lifestyle Project: a randomized clinical trial: results at 54 months. Circulation. 2001;103:32–37. doi: 10.1161/01.cir.103.1.32. [DOI] [PubMed] [Google Scholar]
- 7.Knopp RH, Brown WV, Dujovne CA, Farquhar JW, Feldman EB, Goldberg AC, et al. Effects of fenofibrate on plasma lipoproteins in hypercholesterolemia and combined hyperlipidemia. Am J Med. 1987;83:50–59. doi: 10.1016/0002-9343(87)90871-0. [DOI] [PubMed] [Google Scholar]
- 8.Ordovas JM. Pharmacogenetics of lipid diseases. Hum Genomics. 2004;1:111–125. doi: 10.1186/1479-7364-1-2-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lai CQ, Parnell LD, Ordovas JM. The APOA1/C3/A4/A5 gene cluster, lipid metabolism and cardiovascular disease risk. Curr Opin Lipidol. 2005;16:153–166. doi: 10.1097/01.mol.0000162320.54795.68. [DOI] [PubMed] [Google Scholar]
- 10.Pennacchio LA, Olivier M, Hubacek JA, Cohen JC, Cox DR, Fruchart JC, et al. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294:169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- 11.Lai CQ, Arnett DK, Corella D, Straka RJ, Tsai MY, Peacock JM, et al. Fenofibrate effect on triglyceride and postprandial response of apolipoprotein A5 variants: the GOLDN study. Arterioscler Thromb Vasc Biol. 2007;27:1417–1425. doi: 10.1161/ATVBAHA.107.140103. [DOI] [PubMed] [Google Scholar]
- 12.Fullerton SM, Buchanan AV, Sonpar VA, Taylor SL, Smith JD, Carlson CS, et al. The effects of scale: variation in the APOA1/C3/A4/A5 gene cluster. Hum Genet. 2004;115:36–56. doi: 10.1007/s00439-004-1106-x. [DOI] [PubMed] [Google Scholar]
- 13.Groenendijk M, Cantor RM, de Bruin TW, linga-Thie GM. The apoAI-CIII-AIV gene cluster. Atherosclerosis. 2001;157:1–11. doi: 10.1016/s0021-9150(01)00539-1. [DOI] [PubMed] [Google Scholar]
- 14.Olivier M, Wang X, Cole R, Gau B, Kim J, Rubin EM, et al. Haplotype analysis of the apolipoprotein gene cluster on human chromosome 11. Genomics. 2004;83:912–923. doi: 10.1016/j.ygeno.2003.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pankow JS, Province MA, Hunt SC, Arnett DK. Regarding ‘Testing for population subdivision and association in four case-control studies’. Am J Hum Genet. 2002;71:1478–1480. doi: 10.1086/344582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 17.Boehnke M. Allele frequency estimation from data on relatives. Am J Hum Genet. 1991;48:22–25. [PMC free article] [PubMed] [Google Scholar]
- 18.Fisher RA. Statistical methods for research workers. Edinburgh: Oliver and Boyd; 1934. [Google Scholar]
- 19.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 20.Lewontin RC. The interaction of selection and linkage. II. Optimum models. Genetics. 1964;50:757–782. doi: 10.1093/genetics/50.4.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stram DO, Haiman CA, Hirschhorn JN, Altshuler D, Kolonel LN, Henderson BE, et al. Choosing haplotype-tagging SNPS based on unphased genotype data using a preliminary sample of unrelated subjects with an example from the Multiethnic Cohort Study. Hum Hered. 2003;55:27–36. doi: 10.1159/000071807. [DOI] [PubMed] [Google Scholar]
- 22.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38:963–974. [PubMed] [Google Scholar]
- 23.Corbett J, Kraja A, Borecki IB, Province MA. Use of a random coefficient regression (RCR) model to estimate growth parameters. BMC Genet. 2003;4(Suppl 1):S5. doi: 10.1186/1471-2156-4-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 25.Zhang K, Zhao H. A comparison of several methods for haplotype frequency estimation and haplotype reconstruction for tightly linked markers from general pedigrees. Genet Epidemiol. 2006;30:423–437. doi: 10.1002/gepi.20154. [DOI] [PubMed] [Google Scholar]
- 26.Abecasis GR, Wigginton JE. Handling marker-marker linkage disequilibrium: pedigree analysis with clustered markers. Am J Hum Genet. 2005;77:754–767. doi: 10.1086/497345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nielsen H, Engelbrecht J, Brunak S, von HG. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 28.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 29.Lai CQ, Demissie S, Cupples LA, Zhu Y, Adiconis X, Parnell LD, et al. Influence of the APOA5 locus on plasma triglyceride, lipoprotein subclasses, and CVD risk in the Framingham Heart Study. J Lipid Res. 2004;45:2096–2105. doi: 10.1194/jlr.M400192-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Li WW, Dammerman MM, Smith JD, Metzger S, Breslow JL, Leff T. Common genetic variation in the promoter of the human apo CIII gene abolishes regulation by insulin and may contribute to hypertriglyceridemia. J Clin Invest. 1995;96:2601–2605. doi: 10.1172/JCI118324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eichenbaum-Voline S, Olivier M, Jones EL, Naoumova RP, Jones B, Gau B, et al. Linkage and association between distinct variants of the APOA1/C3/A4/A5 gene cluster and familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol. 2004;24:167–174. doi: 10.1161/01.ATV.0000099881.83261.D4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hockey KJ, Anderson RA, Cook VR, Hantgan RR, Weinberg RB. Effect of the apolipoprotein A-IV Q360H polymorphism on postprandial plasma triglyceride clearance. J Lipid Res. 2001;42:211–217. [PubMed] [Google Scholar]
- 33.Talmud PJ, Martin S, Taskinen MR, Frick MH, Nieminen MS, Kesaniemi YA, et al. APOA5 gene variants, lipoprotein particle distribution, and progression of coronary heart disease: results from the LOCAT study. J Lipid Res. 2004;45:750–756. doi: 10.1194/jlr.M300458-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Schmitz G, Langmann T. Pharmacogenomics of cholesterol-lowering therapy. Vascul Pharmacol. 2006;44:75–89. doi: 10.1016/j.vph.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 35.Lahoz C, Pena R, Mostaza JM, Jimenez J, Subirats E, Pinto X, et al. Apo A-I promoter polymorphism influences basal HDL-cholesterol and its response to pravastatin therapy. Atherosclerosis. 2003;168:289–295. doi: 10.1016/s0021-9150(03)00094-7. [DOI] [PubMed] [Google Scholar]
- 36.Watts GF, Dimmitt SB. Fibrates, dyslipoproteinaemia and cardiovascular disease. Curr Opin Lipidol. 1999;10:561–574. doi: 10.1097/00041433-199912000-00011. [DOI] [PubMed] [Google Scholar]