

Abstract
Emanuel syndrome is characterized by multiple congenital anomalies and developmental disability. It is caused by the presence of a supernumerary derivative chromosome that contains material from chromosomes 11 and 22. The origin of this imbalance is 3:1 malsegregation of a parental balanced translocation between chromosomes 11 and 22, which is the most common recurrent reciprocal translocation in humans. Little has been published on the clinical features of this syndrome since the 1980s and information on natural history is limited. We designed a questionnaire to collect information from families recruited through an international online support group, Chromosome 22 Central. Data gathered include information on congenital anomalies, medical and surgical history, developmental and behavioural issues, and current abilities. We received information on 63 individuals with Emanuel syndrome, ranging in age from newborn to adulthood. As previously recognized, congenital anomalies were common, the most frequent being ear pits (76%), micrognathia (60%), heart malformations (57%), and cleft palate (54%). Our data suggest that vision and hearing impairment, seizures, failure to thrive and recurrent infections, particularly otitis media, are common in this syndrome. Psychomotor development is uniformly delayed, however the majority of individuals (over 70%) eventually learn to walk with support. Language development and ability for self-care are also very impaired. This study provides new information on the clinical spectrum and natural history of Emanuel syndrome for families and physicians caring for these individuals.
Keywords: Emanuel syndrome, Translocation, congenital anomalies, der22
Introduction
Emanuel syndrome (OMIM 609029), also known as supernumerary der(22)t(11;22) syndrome, is characterized by multiple congenital anomalies, craniofacial dysmorphism and significant cognitive handicap [Fraccaro et al., 1980; Zackai and Emanuel, 1980; Iselius et al., 1983; Emanuel et al., 1976; Lin et al., 1986]. Affected individuals have an unbalanced chromosome complement as a result of 3:1 meiotic segregation of a parental balanced translocation between chromosomes 11 and 22, which is the most common recurrent reciprocal translocation in humans. Carriers are typically ascertained following investigation for multiple miscarriages, infertility, or after the birth of a child with Emanuel syndrome [Fraccaro et al., 1980]. Carriers of the balanced t(11;22)(q23.3;q11.2) translocation have up to a 10% chance of conceiving a child with this syndrome who survives to term [Fraccaro et al., 1980; Zackai and Emanuel, 1980; Iselius et al., 1983; Emanuel et al., 1976].
Most of the clinical information about this syndrome was published prior to the mid-1980s [Fraccaro et al., 1980; Zackai and Emanuel, 1980; Iselius et al., 1983; Emanuel et al., 1976; Lin et al., 1986]. Congenital anomalies are well documented and include heart defects, cleft palate, genitourinary tract malformations, and intestinal atresias. Craniofacial dysmorphism has also been well described. Development is significantly delayed in infancy; however, the existing literature contains limited information on outcomes beyond the first few years of life. While the true infant mortality rate in Emanuel syndrome is unknown, long-term survival is possible.
Chromosome 22 Central (www.c22c.org) provides support for individuals and families affected by chromosome 22 disorders from more than 40 countries, with 82 current members having at least one child with Emanuel syndrome. Using this online support group to recruit participants, we surveyed parents of individuals with Emanuel syndrome regarding pregnancy and delivery, congenital anomalies, medical and surgical history, developmental milestones, and current abilities. Although limited by biases inherent in questionnaire studies, this is the largest and only current clinical study on Emanuel syndrome that addresses the natural history of the condition. Given the recurrent nature of the 11;22 translocation in humans, the results of this study are useful for reproductive counselling for known translocation carriers, and particularly valuable for parents and health care providers of individuals with Emanuel syndrome.
Subjects and Methods
We reviewed all available case reports and case series in the English language literature on individuals with supernumerary derivative 22 syndrome or partial 11/22 trisomy. Only cases with a confirmed diagnosis of Emanuel syndrome, based on chromosome studies (ie, 47,XX or XY,+der(22)t(11;22)) were considered. Using the information in the case reports, we developed a questionnaire for parents of individuals with a diagnosis of Emanuel syndrome to survey the clinical features, with particular emphasis on areas for which we felt there was insufficient data available. These latter areas included health care issues, developmental milestones and growth beyond infancy, ability for self-care, and behaviour. Ethics approval for this study was obtained from the Children's Hospital of Eastern Ontario Research Ethics Board.
The questionnaire was divided into four sections: 1) Demographics, 2) Prenatal and Neonatal Period, 3) Medical and Surgical Issues, and 4) Growth, Development and Behavior. In each section, the respondent (parent or primary caregiver) was asked a series of questions, to be answered by selection from a list of options, and/or an “other” category, in which answers could be supplied in free-form. We asked for photographs of the affected individual(s) in the family. We did not ask respondents to provide medical records. Recruitment of study participants was done through the Chromosome 22 Central online parent support group. Each active member of the support group was sent either a paper or email version of the questionnaire, with a consent form to be signed and mailed or faxed back to the study group. Eighty-two members were sent one questionnaire for each affected child in the family. Sixty-five completed surveys were received. Two surveys were excluded from the study because there was not enough available information; one because the diagnosis was made prenatally and the pregnancy was terminated, and the second because the child died on the first day of life. A database with all available data from each questionnaire was generated.
Results
Study Population
A total of 63 surveys were included in the study. The majority of surveys (48%) were from the U.S., with the remaining surveys from Canada (11%), the United Kingdom (10%), Australia, France, Italy, Norway, Spain and Chile. The respondents were mostly biological mothers of an individual with Emanuel syndrome (95%); two respondents were biological fathers and one was an adoptive mother. The sex distribution of the affected individuals was 28 males (44%) and 35 females (56%). The age distribution is shown in Table I. Five respondents had a child who was deceased. Of the deceased, the age at death was less than one month (n=2), 5 months (n=2) and 33 years (n=1). The remaining 58 respondents ranged in age from 9 months to 33 years. Seventy-one percent of respondents had children who are (or were at time of death) 6 years of age or older.
Table I.
Age distribution of participants with Emanuel syndrome
| Age Group | Number reported | Percentage |
|---|---|---|
| Under 1 year | 5 | 8 |
| 1-5 years | 13 | 21 |
| 6-12 years | 13 | 21 |
| 13-19 years | 16 | 25 |
| 20-30 years | 11 | 17 |
| 30-40 years | 5 | 8 |
Carrier status of parents
The parent carrying the 11;22 translocation was known in 95% of cases. The overwhelming majority (90%) of carriers were mothers. Only 5% of respondents indicated that the father was the translocation carrier (n=3). In 5% of cases the carrier parent was unknown, either because the parents had not yet been tested, or the child was adopted. There were no cases in which both parents were tested and neither found to carry the translocation.
Diagnosis and Counseling
The diagnosis of Emanuel syndrome was made within the first month of the child's life in 48% of cases. A further 30% received the correct diagnosis between one month and one year of their child's birth. Only one respondent received the diagnosis prenatally. For those who did not receive the correct diagnosis in the first month of life, respondents were asked whether they were given an explanation for their child's difficulties in the newborn period. Other explanations initially given for neonatal difficulties were Pierre Robin sequence (n=4), cat eye syndrome (n=2), prematurity (n=3), cri du chat syndrome (n=1) and DiGeorge syndrome (n=1). Eighteen percent reported that they were not given a specific explanation for their baby's difficulties in the newborn period.
Parents were asked to comment on the information they were given about prognosis and natural history at the time of diagnosis. In general, those with children born before 1996 indicated that they were given very little information, or the information they received was uniformly pessimistic. Some parents were told their child would survive one to two years at most. Others were given articles from medical journals, which were difficult to understand and provided limited natural history data aside from death or profound developmental disability. Those with children born in the late 1990s found it easier to find information on their own, primarily through internet searches. Respondents repeatedly cited the Chromosome 22 Central website and online support group as their best and/or only source of information on the syndrome.
Pregnancy and Neonatal Period
Overall, 81% of respondents reported no pregnancy complications. The most commonly reported pregnancy complication was intrauterine growth restriction (24%). Other complications reported included decreased fetal movements (18%), oligohydramnios (16%), breech position (14.5%), vaginal bleeding (11%), and prematurity (9.5%). Ultrasound anomalies were reported in 16%, and included heart, brain and kidney malformations.
Birth weight ranged from 1192 g (delivered at 32 weeks) to 3.5 kg, with an average birth weight of 2668 g (5 lbs, 14 oz). The majority (69%) had a birth weight within two standard deviations of the mean birth weight of a 40 week gestation newborn. The length of hospital stay in the neonatal period was greater than one week in 70%, and greater than one month in 25%. The most common reasons given for prolonged hospital stay were hypotonia (51%), feeding issues (44%), need for surgery (30%) or oxygen therapy (36%), and jaundice (34%). Less common reasons reported included seizures (10%) and infection (11%). Twelve respondents (20%) reported that their child needed to be artificially ventilated during the neonatal period. The reasons given for artificial ventilation included diaphragmatic hernia (n=4), apnea (n=4), prematurity (n=4), pneumothorax (n=3), infection (n=4), pulmonary hypertension secondary to meconium aspiration (n=1), and bilateral vocal cord paralysis (n=1).
Congenital Anomalies
At least one major congenital anomaly was reported in 87%. The most frequently reported major anomalies are listed in Table II. The types of cleft anomalies reported were Pierre Robin sequence (34%), soft palate cleft only (28%), complete cleft palate (24%), submucous cleft or bifid uvula only (14%). Thirty-two percent had surgery to repair a cleft palate. There were no reports of associated cleft lip. Cardiac surgery was required in 30% of those with heart malformations. Renal malformations were relatively common; the types of renal anomalies were variable. There were no reports of nephrolithiasis or renal insufficiency.
Table II.
Congenital anomalies most commonly reported in individuals with Emanuel syndrome
| Category | Specific Anomaly | Percentage |
|---|---|---|
| Craniofacial | Ear pits | 76 |
| Preauricular tags | 33 | |
| Other ear differences1 | 30 | |
| Cleft palate | 54 | |
| Complete | 24 | |
| Pierre Robin sequence | 34 | |
| Soft palate only | 28 | |
| Submucous/bifid uvula only | 14 | |
| Micrognathia | 60 | |
| Excess nuchal skin | 24 | |
| Cardiovascular | Overall | 57 |
| ASD | 45 | |
| VSD | 13 | |
| PDA | 11 | |
| Other2 | 10 | |
| Gastrointestinal | Imperforate anus | 14 |
| Inguinal hernia | 14 | |
| Intestinal malrotation | 8 | |
| Renal | Overall | 36 |
| Small kidney(s) | 17 | |
| Single kidney | 12 | |
| Other3 | 7 | |
| Genital | Small penis | 64 |
| Cryptorchidism | 46 | |
| Other | Sacral dimple | 24 |
| Diaphragmatic hernia | 8 |
Includes abnormalities in ear shape, size, and position
Includes pulmonic stenosis, total anomalous pulmonary venous return, and coarctation of the aorta
Includes vesicoureteral reflux, cystic kidney, urethral valve abnormality and duplicated collecting system
All respondents (100%) reported at least one minor congenital anomaly in their child; see Table II for those most commonly reported.
Dysmorphism
Dysmorphic features were assessed by review of photographs of affected individuals submitted by their parents. We obtained good quality photographs of 36 individuals ranging in age from newborn to young adult, and were able to compare photographs of the same individual at different stages (infancy, childhood, and/or adulthood) in nine cases. The facial phenotype is variable. The most common features (Fig. 1) are hooded eyelids, deep-set eyes, upslanting palpebral fissures, low-hanging columnella, and micrognathia. Facial asymmetry is common, often observed with unilateral ptosis. There is a tendency for the face shape to be square due to a broad mandible. With age, the face lengthens as expected and micrognathia becomes less obvious (Fig. 2). There was not, as previously reported [Medne et al., 2007], a tendency for coarsening of the facial features with age in most individuals.
Fig. 1.
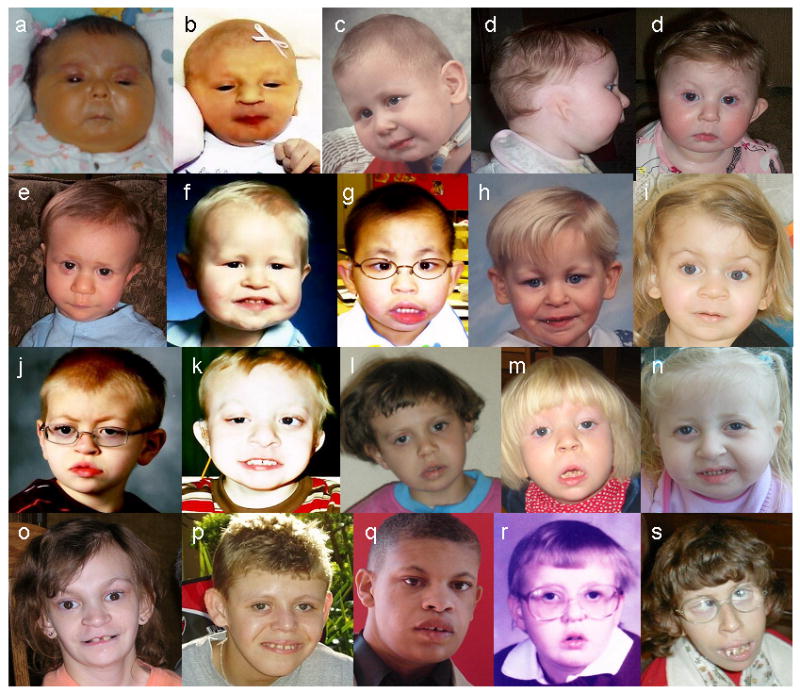
Individuals with Emanuel syndrome at various ages: newborn (a and b), preschool age (c to f), school age (g to n) and adolescent (o to s). The common dysmorphic features are demonstrated, including micrognathia (most obvious in infancy and preschool age), hooded eyelids, deep-set eyes, upslanting palpebral fissures, low-hanging columnella, and long philtrum. Some individuals have facial asymmetry and/or unilateral ptosis (for example, Patients g and k).
Fig. 2.
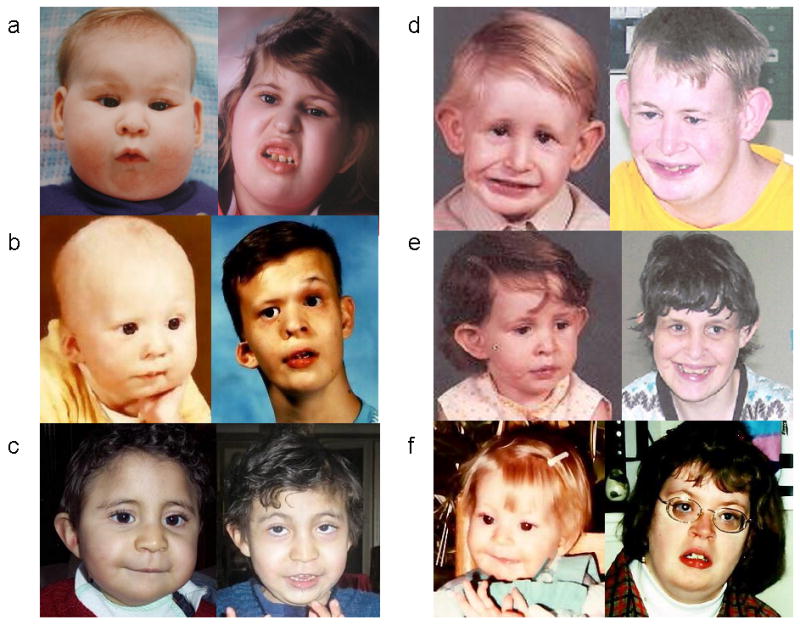
Six individuals with Emanuel syndrome, demonstrating evolution of facial features with age. Approximate ages are: a) 12 months and 13 years; b) 8 months and 15 years; c) 24 months and 8 years; d and e) 4 years and 28 years; f) 30 months and 20 years. Patients d and e are siblings. Note that, as expected, there is facial elongation and growth of the mandible with age. Features remain fine, without significant coarsening.
Medical Issues
Table III lists the most commonly reported medical issues. Feeding issues were common. Only 37% of respondents indicated that their child was eating age-appropriate foods. Nineteen percent were exclusively G-tube fed. Forty-four percent of those who fed orally require specially prepared food textures (e.g. pureed). The most commonly reported gastrointestinal complaints are found in Table III. Thirty-one percent of respondents indicated that their child is currently taking a daily laxative or stool softener; 24% are on a daily antacid medication. Eight percent have undergone a surgical procedure for treatment of gastroesophageal reflux (fundoplication).
Table III. Medical issues most commonly reported in individuals with Emanuel syndrome.
| Category | Issue | Percentage |
|---|---|---|
| Vision/Ocular | Myopia | 38 |
| Strabismus | 33 | |
| Astigmatism | 13 | |
| Ptosis | 8 | |
| Hyperopia | 7 | |
| Other1 | 22 | |
| Hearing Loss | Any degree | 72 |
| Mild to moderate | 56 | |
| Moderate to severe | 28 | |
| Severe to profound | 15 | |
| Dentition | Delayed eruption of primary teeth | 80 |
| Misaligned teeth | 56 | |
| Crowded teeth | 37 | |
| Missing teeth | 22 | |
| Gastrointestinal | Drooling | 74 |
| Constipation | 63 | |
| Failure to thrive | 62 | |
| Choking and/or swallowing problems | 54 | |
| Gastroesophageal reflux | 54 | |
| Respiratory | Recurrent chest infections | 48 |
| Aspiration | 37 | |
| Snoring | 32 | |
| Cough | 27 | |
| Supplemental oxygen2 | 18 | |
| Musculoskeletal | Ankle instability | 48 |
| Hip subluxation or dislocation | 47 | |
| Scoliosis | 32 | |
| Requiring surgery | 5 | |
| Requiring bracing | 42 | |
| Kyphosis | 30 | |
| Joint contractures | 15 | |
| Torticollis | 15 | |
| Immunity | Recurrent ear infections | 96 |
| Recurrent pneumonia | 47 | |
| Recurrent sinusitis | 33 | |
| Recurrent urinary tract infection | 18 | |
| Recurrent oral candidiasis | 11 | |
| Low immunoglobulins | 19 | |
| Treated with IVIG | 9 |
Includes Duane anomaly, optic atrophy, 6th nerve palsy, tear duct hypoplasia, cataracts, nystagmus and glaucoma
Outside of newborn period
Endocrinopathies were not commonly reported. Hypothyroidism was reported in 10%. One individual had “parathyroid disease” diagnosed in adulthood.
While respiratory issues were common, only two individuals required tracheostomy; one was short term secondary to tracheal trauma during intubation for surgery. Twenty-six percent reported greater than five hospitalizations for respiratory issues in the individual's lifetime.
Two individuals were reported to have hypertension (one was 32 months old, the other was 15 years old), but the cause and type of hypertension was unspecified. There was one voluntary report of pulmonary hypertension, and one report of cardiomyopathy (type unspecified). One respondent reported that their child has a “leaky valve.”
Growth and Puberty
Eighty percent of respondents reported their child's current height and weight; 50% had weight below the 3rd centile for age, and 73% had height below 3rd centile for age. Sixty-two percent reported that their child had been diagnosed with “failure to thrive.”
Of 17 females over the age of 9 years, 15 (88%) had begun menstruating. The age at menarche ranged from 9 years to18 years, with the majority (71%) between 11 and 13 years. Two girls who had not started menstruation were 16 and 22 years old.
Neurologic issues, Development and Behavior
The most commonly reported neurological abnormalities are found in Table IV. Sixty-five percent of the respondents indicated that their child had had an MRI or CT scan of the brain. However, 37% indicated that they were unsure of the results of the scan. Of 26 respondents who knew the results of their child's head imaging, 27% reported ventriculomegaly, 23% reported atrophy, 19% reported absent or hypoplastic corpus callosum, and 19% reported white matter abnormalities. Dandy-Walker malformation was reported in two patients (8%) and Chiari malformation in a further two patients (8%). Three respondents (6%) indicated that their child had a surgical shunt placement for hydrocephalus.
Table IV.
Neurological abnormalities most commonly reported by parents in individuals with Emanuel syndrome
| Functional neurologic anomaly | Percentage |
|---|---|
| Developmental delay | 100 |
| Hypotonia | 65 |
| Seizures | 48 |
| Requiring anticonvulsants | 34 |
| Structural neurologic anomaly | |
| Microcephaly | 23 |
| Atrophy | 23 |
| Ventriculomegaly | 27 |
| Hydrocephalus | 11 |
| Requiring shunt placement | 6 |
| Absent or hypoplastic corpus callosum | 19 |
| White matter abnormalities | 19 |
| Dandy-Walker malformation | 8 |
| Chiari malformation (type unspecified) | 8 |
Global developmental delay was present in 100%. Respondents were asked the age at which their child attained various developmental milestones (Table V). Gross motor delays were significant. Sixty-five percent of individuals over one year of age could not support their own weight in standing. While 71% could walk with support, only 27% of respondents indicated that their child could walk unsupported. The average age of achieving supported walking was 61 months (just over 5 years), with a range of 18 months to 10 years.
Table V.
Age at attainment of developmental milestones in individuals with Emanuel syndrome
| Developmental milestone | Mean Age (months) |
Range (months) |
Mode Age Range (months) |
|---|---|---|---|
| Hold head up | 8 | 2-28 | 5-8 |
| Smile | 7 | 2-36 | 2-4 |
| Sitting independently | 20 | 8-36 | 13-24 |
| Crawling1 | 34 | 20-51 | 20-24 |
| Standing unsupported | 71 | 24-150 | 24-48 |
| Walking with support | 61 | 18-120 | 37-48 |
| Walking unsupported | 84 | 41-150 | 97-150 |
83% of respondents indicated that their child did not learn to crawl
Language skills were significantly impaired. Seventy-seven percent of parents reported that their child has no speech. Of those with some speech, the average age of first word was 44 months (3.7 years), with a range of 15 months to 8 years. Twenty percent indicated that their child uses at least one word to communicate. Forty-eight percent knew at least one word in sign language. Two individuals could speak in short phrases. Although the majority of these children are non-verbal, many parents commented that they feel strongly that their child's understanding is much better than their ability to communicate.
Fine motor and self-help skills were also significantly impaired. Eighty-four percent need to be dressed and undressed by a care provider. Only 5% of individuals of a developmentally appropriate age could do up a zipper. Sixty-three percent needed to be fed by a care provider. Only 32% of individuals of a developmentally appropriate age could use a spoon or fork appropriately. Forty-three percent of individuals over five years of age were incontinent; 21% were fully or partially toilet-trained, while 36% were toilet-regulated. Seventy-four percent of school-aged children attended a class or school for the disabled; the remainder were home-schooled (7%) or in a regular class with individualized assistance (9%).
Behavioral issues were not common overall. The most common “problem” behaviours were anxiety (16%), screaming (16%), self-harm (14%), and tactile defensiveness (this was not asked about specifically in the questionnaire, but many parents commented on it). Although there were no specific questions asked about personality characteristics, there was a free-form section in which parents could tell us anything about their child they felt was important. Although this data is limited, many parents commented that their child is generally happy and sociable, enjoys music and being around people, and has a good sense of humour.
Discussion
The earliest descriptions of Emanuel syndrome appear in the English-language literature in the 1960s and 70s. Although G-banding was not yet available, there are several case reports of siblings with multiple malformations and developmental disability with “an extra small acrocentric chromosome resembling that in the G group, exclusive of Down syndrome or XYY,” which was thought to be trisomy 22 [Uchida et al., 1968; Penchaszadeh and Coco, 1975; Alfi et al., 1975; Emanuel et al., 1976; Shokeir, 1978]. Once chromosomal banding techniques became available, the atypical nature of the extra chromosome was noted, and eventually it was recognized to be a derivative chromosome of 22 and 11 material derived from a balanced translocation carrier parent [Kessel and Pfeiffer, 1977; Feldman and Sparkes, 1978; Nakai et al., 1979]. The characterization of the breakpoints on chromosomes 11 and 22 demonstrated that t(11;22) is a recurrent translocation, and it is the most common recurring reciprocal translocation in humans [Zackai and Emanuel, 1980; Schinzel et al., 1981; Griffin et al., 1986; Budarf et al., 1989; Shaikh et al., 1999; Edelmann et al., 1999; Tapia-Perez et al., 2000; Kurahashi et al., 2000c]. The breakpoints of the t(11;22) translocation are within palindromic AT-rich repeats (PATRRs) on chromosomes 11 and 22, suggesting that hairpin/cruciform structures mediate double-strand breaks in meiosis leading to recombination between 11q23 and 22q11, resulting in this recurrent translocation [Kurahashi et al., 2000a, b; Kurahashi et al., 2001; Kurahashi et al., 2004].
In 2004, founding members of the online parent support group, Chromosome 22 Central, successfully lobbied to have Emanuel syndrome added as an entry in the OMIM database. Prior to this, there was concern among parents about the disparate names given to their children's condition (for example, supernumerary derivative 22 and partial 11q trisomy), which impeded parents' ability to find online support. The eponym “Emanuel syndrome” was therefore suggested by the parent group in recognition of Dr. Emanuel's cytogenetic work and molecular characterization of the breakpoints, as well as her consistent involvement with the support group.
Over 100 individuals with Emanuel syndrome have been reported [Fraccaro et al., 1980; Zackai and Emanuel 1980; Biederman et al., 1980; Pihko et al., 1981; Schinzel et al., 1981; Iselius et al., 1983; Lin et al., 1986]. However, the most recent large case series was published 25 years ago [Iselius et al., 1983]. While the characteristic dysmorphic features and congenital anomalies most frequently associated with ES have been well described, information about natural history is more limited, making the task of providing prognostic information and anticipatory guidance rather difficult. This study reports data from the largest cohort of individuals with Emanuel syndrome ever described in the literature.
Pregnancy and perinatal issues have not been described previously for this syndrome. Overall, the frequency of pregnancy complications was low (19%) and prematurity was not common. Intrauterine growth restriction was the most commonly reported complication (24%) and birth weights averaged in the lower range of normal. These data imply that, not surprisingly, growth deficiency begins prenatally in these children. Ultrasound abnormalities during pregnancy were only reported in 16%, which is surprising given the high rate of congenital anomalies in this syndrome. Given that many of our study subjects are adolescents and adults, this likely reflects the more limited availability of ultrasound technology in the past. Nonetheless, this suggests that a normal prenatal ultrasound does not rule out Emanuel syndrome, and therefore carrier parents who wish to have prenatal diagnosis need to have more invasive testing such as chorionic villus sampling or amniocentesis.
Congenital anomalies associated with Emanuel syndrome have been well described. The most commonly reported congenital anomalies reported by parents in our study are listed in Table II. Heart defects were reported in 57% of our study subjects, and the most common lesions were atrial septal defect (ASD), ventricular septal defect (VSD) and patent ductus arteriosus (PDA). This is similar to the results of a literature review by Lin et al. [1986], in which 62% of reported cases had a congenital heart malformation, with ASD, VSD and PDA being the three most common. The apparent high frequency of ASDs and VSDs included both isolated defects, and those that were associated with more significant congenital heart defects. Other heart malformations that were reported in our study included coarctation of the aorta, pulmonic stenosis, and total anomalous pulmonary venous return (TAPVR). We had no reports of tetralogy of Fallot, truncus arteriosus, transposition of great arteries, or tricuspid atresia, which have been reported previously [Giraud et al., 1975; Fraccaro et al., 1980; Pangalos et al., 1980; Lin et al., 1986]. The vast majority of individuals with congenital heart defects, both in our study and in the literature, have acyanotic lesions. Our results also confirm the reported incidence of cleft palate (54% in our study vs 53% [Fraccaro et al., 1980]) and imperforate anus (14% vs 13% [Fraccaro et al., 1980]). Renal malformations were reported to occur in 19% in the largest study [Fraccaro et al., 1980] but were seen in 36% in our cohort; this increase is likely secondary to improved imaging techniques. Congenital anomalies reported by parents in our study that have not previously been reported include pyloric stenosis and choanal atresia (Table II). Intestinal malrotation was reported in 8%; this malformation has been reported only once previously [Prieto et al., 2007].
The incidence of structural brain anomalies in individuals with Emanuel syndrome is unknown, as it has not been studied systematically. Pallotta et al [1996] reviewed the reported central nervous system (CNS) anomalies in Emanuel syndrome, and determined that 30% of reported individuals have some CNS abnormality. They found the most frequent anomalies to be consistent with a developmental field defect affecting the midline, such as arrhinencephaly, Dandy-Walker malformation, hypoplasia of the corpus callosum, pons and cerebellar vermis, dilatation of the third and fourth ventricles, and trigonocephaly. Our study is inconclusive with regard to CNS anomalies in Emanuel syndrome, as we relied on reports from parents and not medical records. While 65% of respondents indicated that their child had had some form of brain imaging, they were generally not aware of the specific findings. The most common brain anomalies reported in our study were ventriculomegaly, atrophy, white matter abnormalities and hypoplastic corpus callosum (Table IV). Microcephaly was reported in only 23% of our subjects, while other literature states that 100% of individuals with Emanuel syndrome have microcephaly [Medne et al., 2007]. This most likely represents underreporting of microcephaly by parents in our study.
The malformations seen in individuals with Emanuel syndrome overlap with those of cat eye syndrome (CES). These conditions have in common extra chromosome 22 material spanning the proximal p and q arms. CES is usually associated with a supernumerary bisatellited marker chromosome containing material of chromosome 22 (idic(22)(pter->q11.2::q11.2->pter)), which results in partial tetrasomy 22 [McDermid et al., 1986]. Both conditions have in common a high frequency of preauricular pits and skin tags, anorectal anomalies, and congenital heart defects [Rosias et al., 2001]. Iris colobomata, however, which are a cardinal feature of CES, are not reported in Emanuel syndrome. Unlike ES, the majority of individuals with CES have mild or no intellectual impairment [Rosias et al., 2001]. The most obvious explanation for this discrepancy is the presence in individuals with ES of partial 11q trisomy. Indeed, the published cases of isolated 11q trisomy have almost universally reported severe intellectual and physical disability [Pihko et al., 1981; Zhao et al., 2003]. In addition, it is notable that some congenital anomalies seen in ES, such as congenital diaphragmatic hernia, hip dysplasia, cleft palate, heart and kidney malformations, and structural brain malformations have also been reported in individuals with 11q trisomy [Pihko et al., 1981; Zhao et al., 2003; Klassens et al., 2006]. Further study of the genomic imbalances present in individuals with ES, CES and 11q trisomy will be useful in delineating the causative genes for the shared anomalies.
Facial dysmorphism in individuals with Emanuel syndrome is well described in infants and very young children, however information about how the facial phenotype evolves with age is limited. Medne et al. [2007] suggest that the facial features of individuals with ES coarsen over time. Our review of the available photographs from individuals participating in this study did not demonstrate coarsening of facial features. The faces of individuals with ES lengthen over time with micrognathia becoming less pronounced, as would be expected (Fig. 2). There was significant variability in the facial appearance of individuals with ES. The most common facial features observed include hooded eyelids, deep-set eyes, upslanting palpebral fissures, low-hanging columnella, facial asymmetry and ear anomalies. We cannot comment more specifically on the ear anomalies in our study participants, as we did not have the appropriate views in the photographs we obtained from parents. In childhood the combination of deep-set eyes, long philtrum and micrognathia is seen in many, but not all, children.
Fifty percent of our subjects were over the age of 13 years, which allowed us to gather useful natural history information on Emanuel syndrome. The medical issues most commonly reported in our subjects with Emanuel syndrome are summarized in Table III. Most of these have been previously unrecognized or under-recognized, probably due to the small numbers of older children and adults reported in the literature. One of the most clinically relevant findings was the high incidence (72%) of hearing loss. The degree of hearing loss was mild to moderate in the majority. Our questionnaire did not differentiate between sensorineural and conductive hearing loss. However, there was a very high rate of recurrent ear infections and tympanostomy tube placement in these children, so it is likely that conductive hearing loss would be a significant contributing factor. Similarly, visual problems have been underreported in the literature. In our study, vision is impaired in at least one-third of subjects, with myopia and strabismus being the most commonly reported problems. The case report literature has not emphasized seizures as a frequent feature of ES. Our data suggest that seizures are common (48%) but unfortunately we were not able to determine the type of seizure(s) experienced in all cases, so this figure likely includes febrile seizures and all other types of seizures. Gastrointestinal problems, such as gastroesophageal reflux, impaired swallowing and chronic constipation, are common in children with severe developmental disability. Therefore it is not surprising that over half of respondents identified these as ongoing issues for their children. A significant percentage (at least 20%) of individuals in our study required a gastric feeding tube (G-tube) at some point in their lives. Failure to thrive is a common problem in the neonatal period and into childhood. In neonates, poor feeding due to hypotonia and the presence of cardiac and gastrointestinal malformations are the most likely cause. Aside from feeding issues, which are ongoing in childhood, recurrent infections may be partly responsible for failure to thrive as these children get older. Chronic and recurrent ear infections were especially prevalent (96%) in our study subjects. Low immunoglobulins have been reported in children with ES [Tovo et al., 1986]. Nineteen percent of respondents in our study reported low immunoglobulins and 9% had been treated with intravenous immunoglobulin (IVIG) with reported benefit. However, no conclusions can be drawn about the use of IVIG in children with Emanuel syndrome, as the numbers are small.
As expected from the literature, 100% of children with Emanuel syndrome have global developmental delay and intellectual disability. We were able to more precisely define the extent of these disabilities and determine the average developmental trajectory, by asking parents about age of developmental milestone attainment (Table V) and current abilities. While most children do not independently ambulate, over 70% of our subjects eventually learned to walk with support. This ability is largely overlooked in the literature. Expressive language is significantly impaired, with rudimentary speech acquisition in only 20%. However, our study indicates that receptive language is less impaired (at least from the point of view of the parents), which has been noted previously [Medne et al., 2007], although this is anecdotal.
There are two important limitations to this study. Questionnaire studies in general are subject to recall or reporting bias. The accuracy of the medical history information provided by parents about their children will vary depending on a number of factors including the age of the child, whether the child is still living, whether the parent kept records, the complexity of the medical needs of the child, and the education and socioeconomic status of the parents. We cannot verify that the information given by parents is accurate, as we did not collect medical records on our study subjects. Instead, we endeavored to design the questionnaire in simple language with primarily “yes or no” questions and kept the questions that would require additional medical knowledge to a minimum. The results of this study may also be influenced by ascertainment bias, as recruitment was limited to those whose parent or guardian belongs to an online support group. It is possible that parents of the more severely affected children are more likely to seek this type of support. This would bias our results in favor of a poorer prognosis. On the other hand, the majority of those participating in this study have a child with Emanuel syndrome still living, which excludes parents whose children died earlier in life. Thus, our findings apply primarily to long-term survivors, who in general may have had fewer life-threatening congenital anomalies.
Our study has identified several healthcare issues that can now be anticipated for individuals with Emanuel syndrome. Vision and hearing impairment and potential seizure activity can be identified and addressed early. Failure to thrive may be ameliorated with special attention to gastrointestinal issues and prompt treatment of infection. Children with recurrent infections may benefit from an assessment by an immunologist. Speech and physical therapy are of utmost importance for optimizing development and quality of life; this study has clearly demonstrated that, at least in some individuals, limited communication and ambulation is possible and should be strived for.
This study summarizes the clinical features and long-term outcome of a cohort of 63 individuals with Emanuel syndrome ranging in age from infancy to 33 years. The information gained from this survey will be very useful to carriers of the 11;22 translocation in making reproductive decisions, and to health care providers taking care of children and adults with such a rare condition. Most importantly, new parents of children with Emanuel syndrome can be provided with more accurate and up-to-date prognostic information, which may help ease the anxiety of parenting a child with a rare diagnosis.
Acknowledgments
The authors wish to express gratitude to all families who participated. We also thank: Dr. Judith Allanson for her help with assessment of photographs for dysmorphic features; Laura Munoz for translating surveys into Spanish, and Dr. Nick Barrowman for helping set up the clinical database. Grant support was received from the Children's Hospital of Eastern Ontario Research Institute (MTC and KMB) and CA39926 from the National Institutes of Health (BSE).
References
- Alfi OS, Sanger RG, Donnell GM. Trisomy 22: A clinically identifiable syndrome. Birth Defects: Original Article Series. 1975;11:241–245. [PubMed] [Google Scholar]
- Biederman BM, Lin CC, Lowry RB, Somerville R. Tertiary trisomy (22q11q), 47,+der(22),t(11;22) Hum Genet. 1980;53:173–177. doi: 10.1007/BF00273491. [DOI] [PubMed] [Google Scholar]
- Budarf M, Sellinger B, Griffin C, Emanuel BS. Comparative mapping of the constitutional and tumor-associated 11;22 translocations. Am J Hum Genet. 1989;45:128–139. [PMC free article] [PubMed] [Google Scholar]
- Edelmann L, Spiteri E, McCain N, Goldberg R, Pandita RK, Duong S, Fox J, Blumenthal D, Lalani SR, Shaffer LG, Morrow BE. A common breakpoint on 11q23 in carriers of the constitutional t(11;22) translocation. Am J Hum Genet. 1999;65:1608–1616. doi: 10.1086/302689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuel BS, Zackai EH, Aronson MM, Mellman WJ, Moorhead PS. Abnormal chromosome 22 and recurrence of trisomy-22 syndrome. J Med Genet. 1976;13(6):501–6. doi: 10.1136/jmg.13.6.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman GM, Sparkes RS. The problem of partial trisomy 22 reconsidered. Hum Genet. 1978;45:97–101. doi: 10.1007/BF00277580. [DOI] [PubMed] [Google Scholar]
- Fraccaro M, Lindsten J, Ford CE, Iselius L. The 11q;22q translocation: A European collaborative analysis of 43 cases. Hum Genet. 1980;56:21–51. doi: 10.1007/BF00281567. [DOI] [PubMed] [Google Scholar]
- Giraud F, Mattei JF, Mattei MG, Bernard R. Trisomie partielle 11q et translocation familiale 11-22. Humangenetik. 1975;28:343–347. [PubMed] [Google Scholar]
- Griffin CA, McKeon C, Israel MA, Gegonne A, Ghysdael J, Stehelin D, Douglass EC, Green AA, Emanuel BS. Comparison of constitutional and tumor-associated 11;22 translocations: Nonidentical breakpoints on chromosomes 11 and 22. Proc Natl Acad Sci. 1986;83:6122–6126. doi: 10.1073/pnas.83.16.6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iselius L, Lindsten J, Aurias A, Fraccaro M, Bastard C, Bottelli AM, Bui TH, Caufin D, Dalpra L, Delendi N, Dutrillaux B, Fukushima Y, Geraedts JPM, De Grouchy J, Gyftodimou J, Hanley AL, Hansmann I, Ishii T, Jalbert P, Jingelski S, Kajii T, von Koskull H, Niikawa N, Noel B, Pasquali F, Probeck HD, Robinson A, Roncarati E, Sachs E, Scappaticci S, Schwinger E, Simoni G, Veenema H, Vigi V, Volpato S, Wegner RD, Welch JP, Winsor EJT, Zhang S, Zuffardi O. The 11q;22q translocation: A collaborative study of 20 new cases and analysis of 110 families. Hum Genet. 1983;64:343–55. doi: 10.1007/BF00292366. [DOI] [PubMed] [Google Scholar]
- Kessel E, Pfeiffer RA. 47,XY,+der(11;22)(q23;q12) following balanced translocation t(11;22)(q23;q12)mat. Remarks on the problem of trisomy 22. Hum Genet. 1977;37(1):111–116. doi: 10.1007/BF00293781. [DOI] [PubMed] [Google Scholar]
- Klassens M, Scott DA, van Dooren M, Hochstenbach R, Eussen HJ, Cai WW, Galjaard RJ, Wouters C, Poot M, Laudy J, Lee B, Tibboel D, de Klein A. Congenital diaphragmatic hernia associated with duplication of 11q23-qter. Am J Med Genet. 2006;140A:1580–1586. doi: 10.1002/ajmg.a.31321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi H, Shaikh TH, Emanuel BS. Alu-mediated PCR artefacts and the constitutional t(11;22) breakpoint. Hum Mol Genet. 2000a;9:2727–2732. doi: 10.1093/hmg/9.18.2727. [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Shaikh TH, Hu P, Roe B, Emanuel BS, Budarf ML. Regions of genomic instability on 22q11 and 11q23 as the etiology for the recurrent constitutional t(11;22) Hum Mol Genet. 2000b;9:1665–1670. doi: 10.1093/hmg/9.11.1665. [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Shaikh TH, Zackai EH, Celle L, Driscoll DA, Budarf ML, Emanuel BS. Tightly clustered 11q23 and 22q11 breakpoints permit PCR-based detection of the recurrent constitutional t(11;22) Am J Hum Genet. 2000c;67:763–768. doi: 10.1086/303054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi H, Emanuel BS. Long AT-rich palindromes and the constitutional t(11;22) breakpoint. Hum Mol Genet. 2001;10:2605–2617. doi: 10.1093/hmg/10.23.2605. [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Inagaki H, Yamada K, Ohye T, Taniguchi M, Emanuel BS, Toda T. Cruciform DNA structure underlies the etiology for palindrome-mediated human chromosomal translocations. J Biol Chem. 2004;279:35377–35383. doi: 10.1074/jbc.M400354200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AL, Bernar J, Chin AJ, Sparkes RS, Emanuel BS, Zackai EH. Congenital heart disease in supernumerary der(22), t(11;22) syndrome. Clin Genet. 1986;29:269–275. doi: 10.1111/j.1399-0004.1986.tb01254.x. [DOI] [PubMed] [Google Scholar]
- McDermid HE, Duncan AM, Brasch KR, Holden JJ, Magenis E, Sheehy R, Burn J, Kardon N, Noel B, Schinzel A. Characterization of the supernumerary chromosome in cat eye syndrome. Science. 1986;232:646–648. doi: 10.1126/science.3961499. [DOI] [PubMed] [Google Scholar]
- Medne L, Zackai EH, Emanuel BS. Emanuel Syndrome. [December 22, 2008];GeneReviews at GeneTests: Medical Genetics Information Resource (database online) 2007 Apr 20; Copyright, University of Washington, Seattle. 1997-2008. Available at http://www.genetests.org.
- Nakai H, Yamamoto Y, Kuroki Y. Partial trisomy of 11 and 22 due to familial translocation t(11:22)(q23;q11), inherited in three generations. Hum Genet. 1979;51:349–355. doi: 10.1007/BF00283408. [DOI] [PubMed] [Google Scholar]
- Pallotta E, Fusilli P, Ehresmann T, Cinti R, Verrotti A, Morgese G. Cerebral defects confirm midline developmental field disturbances in supernumerary der(22),t(11;22) syndrome. Clin Genet. 1996;50:411–416. doi: 10.1111/j.1399-0004.1996.tb02398.x. [DOI] [PubMed] [Google Scholar]
- Pangalos C, Courturier J, Bartsocas C, Theodorou S. Trisomie partielle 11q par malsegregation d'une translocation maternelle t(11;22)(q23;q11.1) Nouv Presse Med. 1980;9:3065–3067. [PubMed] [Google Scholar]
- Penchaszadeh VB, Coco R. Trisomy 22: Two new cases and delineation of the phenotype. J Med Genet. 1975;12:193–199. doi: 10.1136/jmg.12.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihko H, Therman E, Uchida IA. Partial 11q trisomy syndrome. Hum Genet. 1981;58:129–134. doi: 10.1007/BF00278696. [DOI] [PubMed] [Google Scholar]
- Prieto JC, Garcia NM, Elder FF, Zinn AR, Baker LA. Phenotypic expansion of the supernumerary derivative (22) chromosome syndrome: VACTERL and Hirschsprung's disease. J of Ped Surg. 2007;42:1928–1032. doi: 10.1016/j.jpedsurg.2007.07.030. [DOI] [PubMed] [Google Scholar]
- Rosias PR, Sijstermans JM, Theunissen PM, Pulles-Heintzberger CF, De Die-Smulders CE, Engelen JJ, Van Der Meer SB. Phenotypic variability of the cat eye syndrome. Case report and review of the literature. Genet Couns. 2001;12:273–282. [PubMed] [Google Scholar]
- Schinzel A, Schmid W, Auf der Maur P, Moser H, Degenhardt KH, Geisler M, Grubisic A. Incomplete trisomy 22. I. Familial 11/22 translocation with 3:1 meiotic disjunction. Delineation of a common clinical picture and report of nine new cases from six families. Hum Genet. 1981;56:249–262. doi: 10.1007/BF00274675. [DOI] [PubMed] [Google Scholar]
- Shaikh TM, Budarf ML, Celle L, Zackai EH, Emanuel BS. Clustered 11q23 and 22q11 breakpoints and 3:1 meiotic malsegregation in multiple unrelated t(11;22) families. Am J Hum Genet. 1999;65:1595–1607. doi: 10.1086/302666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokeir MKH. Complete Trisomy 22. Clin Genet. 1978;14:139–146. doi: 10.1111/j.1399-0004.1978.tb02119.x. [DOI] [PubMed] [Google Scholar]
- Tapia-Paez I, O'Brien KP, Kost-Alimova M, Sahlen S, Kedra D, Bruder CEG, Andersson B, Roe BA, Hu P, Imreh S, Blennow E, Dumanski JP. Fine mapping of the constitutional translocation t(11;22)(q23;q11) Hum Genet. 2000;106:506–516. doi: 10.1007/s004390000287. [DOI] [PubMed] [Google Scholar]
- Tovo PA, Davi G, Fraceschini P, Delpiano A. Thymic hormone dependent immunodeficiency in an infant with partial trisomy of chromosome 22. Thymus. 1986;8:313–318. [PubMed] [Google Scholar]
- Uchida IA, Ray M, McRae KN, Besant DF. Familial occurrence of trisomy 22. Am J Hum Genet. 1968;20:107–118. [PMC free article] [PubMed] [Google Scholar]
- Zackai EH, Emanuel BS. Site-specific reciprocal translocation, t(11;22)(q23;q11), in several unrelated families with 3:1 meiotic disjunction. Am J Med Genet. 1980;7:507–521. doi: 10.1002/ajmg.1320070412. [DOI] [PubMed] [Google Scholar]
- Zhao H, Rope AF, Saal HM, Blough-Pfau AI, Hopkin RJ. Upper airway malformation associated with partial trisomy 11q. Am J Med Genet. 2003;120A:331–337. doi: 10.1002/ajmg.a.20134. [DOI] [PubMed] [Google Scholar]