

Abstract
Oncogenes contribute to tumorigenesis by promoting growth and inhibiting apoptosis. Here we examine the function of Sch9, the Saccharomyces cerevisiae homologue of the mammalian Akt and S6 kinase, in DNA damage and genomic instability during aging in nondividing cells. Attenuation of age-dependent increases in base substitutions, small DNA insertions/deletions, and gross chromosomal rearrangements (GCRs) in sch9Δ mutants is associated with increased mitochondrial superoxide dismutase (MnSOD) expression, decreased DNA oxidation, reduced REV1 expression and translesion synthesis, and elevated resistance to oxidative stress-induced mutagenesis. Deletion of REV1, the lack of components of the error-prone Polζ, or the overexpression of SOD1 or SOD2 is sufficient to reduce age-dependent point mutations in SCH9 overexpressors, but REV1 deficiency causes a major increase in GCRs. These results suggest that the proto-oncogene homologue Sch9 promotes the accumulation of superoxide-dependent DNA damage in nondividing cells, which induces error-prone DNA repair that generates point mutations to avoid GCRs and cell death during the first round of replication.
Introduction
Mutations that activate the Akt and Ras proto-oncogenes or the upstream insulin-like growth factor 1 (IGF-I) receptor are among the most frequently detected in human cancers (Rodriguez-Viciana et al., 1994, 2005; Toker and Yoeli-Lerner, 2006). Because of their roles in stimulating cellular proliferation and inhibiting apoptosis, these oncogenes are widely believed to contribute to cancer by allowing damaged cells to survive, grow, and metastasize (DePinho, 2000; Pollak et al., 2004; Anisimov et al., 2005; Toker and Yoeli-Lerner, 2006). Mutations that cause the down-regulation of homologues of Ras, Akt, or IGF-I receptors extend life spans in organisms ranging from yeast to mice (Longo and Finch, 2003), and deficiency in IGF-I or IGF-I–like signaling (IIS) is associated with decreased neoplastic death in mice (Vergara et al., 2004; Anisimov et al., 2005) or resistance to germline tumors in Caenorhabditis elegans (Pinkston et al., 2006). In agreement with the widely accepted role for IGF-I, Akt, and Ras in cancer, Pinkston et al. (2006) proposed that the resistance of the C. elegans daf-2/insulin-receptor mutants to gld-1–dependent germline tumors is attributed to decreased cell division and increased apoptosis within the tumors. Although defects in cell division and apoptosis undoubtedly play a major role in mutagenesis and cancer, our previous studies in Saccharomyces cerevisiae provided evidence for a link between Sch9, homologue of mammalian S6K and Akt (Toda et al., 1988; Urban et al., 2007), and age-dependent spontaneous mutations in nondividing yeast cells (Fabrizio et al., 2004, 2005), which raises the possibility that proto-oncogene homologues may contribute to DNA damage during aging independently of cell division (Lombard et al., 2005).
The present study investigates the mechanisms underlying the role of Sch9 in increasing DNA damage and genomic instability. We present data indicating that, in chronologically aging yeast, DNA damage occurs primarily in quiescent cells. The deletion of the proto-oncogene homologue SCH9 postpones/attenuates age-dependent mutagenesis by up-regulating the transcription factor Gis1, increasing the expression of mitochondrial SOD2, preventing superoxide-dependent DNA oxidation, and reducing the expression of REV1 and translesion synthesis (TLS) activity. We propose that both superoxide and the error-prone Rev1/Polζ-dependent TLS are required for the major portion of age-dependent mutations.
Results
Sch9 promotes age-dependent genomic instability
The yeast chronological life span (Fabrizio and Longo, 2003, 2007) has provided a paradigm for the identification of key pathways responsible for the regulation of life span and age-dependent damage (Fabrizio et al., 2001). The combination of this simple model organism with several mutator assays (Madia et al., 2007) has proven to be a valuable system to address the mechanisms underlying age-dependent genomic instability.
Using the canavanine resistance assay (Canr), which detects mutations that abolish CAN1 function (Chen and Kolodner, 1999), we have previously reported that chronologically aging yeast displayed a progressive increase of spontaneous mutation frequency (Fabrizio et al., 2004). More recently, we demonstrated that this age-dependent increase of genomic instability encompassed various categories of mutations: gross chromosomal rearrangements (GCRs; Canr5FOAr), point mutations (trp1-289 reversions, trp− → Trp+) and small insertion/deletion mutations (lys2-BglII → Lys+; Madia et al., 2008). The deletion of SCH9 not only extends life span (Fig. 1 A) but also attenuates/delays the age-dependent increase in spontaneous Canr mutation frequency (Fig. 1 B; Fabrizio et al., 2004, 2005) and prevents the premature genomic instability caused by the Sgs1 helicase deficiency (Madia et al., 2008).
Figure 1.

Attenuated age-dependent genomic instability in cells lacking the oncogene homologue SCH9. (A) Chronological survival. Data represent the mean ± SEM (error bars), n = 12–14. (B) Mutation frequency over time in the CAN1 gene (measured as Canr mutants/106 cells, n = 20). (C) Cumulative mutation frequency, measured as Canr mutants/106 cells, calculated from day 1 to day 7 or to day 15 in the wild type (DBY746) and mutants lacking SCH9. Day 7 and day 15 represent the ∼50% survival point of the wild type and sch9Δ population, respectively. n = 20. **, P < 0.01; ***, P < 0.001; ANOVA test, Tukey's test versus wild type (WT) on day 7. (D) GCR frequency (Canr5FOAr mutants/108 cells, n = 10–11). (E) Age-dependent base substitutions (trp1-289 reversion) frequency (n = 8–9). (F) Small DNA insertion/deletion mutations in the lysΔBglII background (EH150 strains, n = 8–9). (G) Budding index of chronologically aging cells (n = 6). (H) Cell cycle profile (FACS) during chronological aging. Strains shown are wild type (DBY746) and sch9Δ. Data represent the mean ± SEM (error bars). *, P < 0.05; **, P < 0.01; ***, P < 0.001; two-tailed t test, sch9Δ versus WT at the indicated time points.
Given the homology to the mammalian AKT and S6K and their implication in tumorigenesis, we investigated the role of Sch9 in the regulation of genomic instability and the mechanism linking its activity to DNA mutations. We determined the cumulative number of Canr mutations from day 1 to day 7 in the wild type and to day 15 in sch9Δ cells, which represent the ∼50% survival point of each population, respectively (Fig. 1 C). The data indicated that in SCH9-deficient cells, the protection against the generation of age-dependent mutations was not simply the result of postponed mortality (P = 0.0013, cumulative Canr mutations in the wild type on day 7 vs. sch9Δ on day 15; Fig. 1 C). Consistent with the observed low amount of Canr mutations (Fig. 1 B), the deletion of SCH9 caused a significant reduction (10-fold compared with wild-type cells) in GCRs over the 13-d period of a chronological life span study (Fig. 1 D). Notably, only ∼1% of the wild-type cells survived to day 13 (Fig. 1 A), which indicates that GCRs are very high in the small subpopulation of old cells that survived to advanced ages.
We then extended the analysis to other types of DNA lesions by examining the occurrence of a specific single base-substitution mutation (T to C: trp1-289 reversions, trp− → Trp+; Capizzi and Jameson, 1973) and small insertion/deletion mutations (lys2-BglII → Lys+); the latter may serve as an indicator for nonhomologous end-joining activity (Heidenreich and Wintersberger, 1998; Heidenreich et al., 2003). Interestingly, SCH9-deficient cells displayed a twofold reduction of single point mutations (Fig. 1 E) as well as a fivefold reduction of the frequency of small DNA insertion/deletion (Lys+) mutations compared with wild-type cells (Fig. 1 F). The results presented in Fig. 1 (A–F) suggest that the lack of SCH9 protects chronologically aging yeast cells against a wide variety of mutations.
SCH9 deletion enhances G1 arrest and reduces age-dependent regrowth
The inability to properly exit the cell cycle can lead to senescence and cancer (Campisi, 2001), and can contribute to mutagenesis. Our previous studies and those from other laboratories suggest that life span–extending mutations cause a switch from a pro-growth to a pro-maintenance mode (Longo, 2003). To determine whether the sch9Δ mutation increases protection against age-dependent genomic instability by promoting G1 arrest, we assessed cell cycle profiles and budding indices of wild-type and sch9Δ mutants. The deletion of SCH9 indeed reduced the budding index from ∼2–3% to 1% on days 3, 5, and 7 (Fig. 1 G). FACS analysis confirmed that sch9Δ cells (98% of the population, starting from day 1) were arrested in the G1 phase (Figs. 1 H and S1 A). Notably, the low budding index as well as the high percentage of G1 arrested cells in sch9Δ cultures are in agreement with our earlier work indicating that deletion of SCH9 caused a major reduction in age-dependent “regrowth,” which appears to depend on adaptive mutations and resembles the differentiation process that leads to tumorigenesis in mammalian cells (Fabrizio et al., 2003, 2004, 2005). The age-dependent early regrowth frequency (days 7–17; Madia et al., 2008) in a large number of cultures of three different genetic backgrounds confirmed that mutants lacking SCH9 were unable to regrow, whereas wild-type populations display the regrowth phenomenon (Table S1).
Quiescent cells contribute to age-dependent mutagenesis
The inability of aging cells to properly arrest in G1 could lead to a higher mutation frequency (Bartkova et al., 2005; Houtgraaf et al., 2006; Burhans and Weinberger, 2007; Löbrich and Jeggo, 2007). Sch9 may increase mutations during chronological aging by promoting replication and cell division before the cell is able to repair DNA damage, causing “replication stress” (Weinberger et al., 2007). To test this possibility, we isolated quiescent and nonquiescent cells from cultures aging chronologically using the Percoll density gradient fractionation (Fig. 2 A; Allen et al., 2006; Aragon et al., 2008; Madia et al., 2008). This method allowed the separation of two distinct subpopulations: the upper fraction, mainly composed of a heterogeneous population of nonquiescent cells, including dividing mother cells, nondividing, apoptotic, and necrotic cells; and a lower fraction mainly composed of quiescent cells (Fig. 2, A and B; and Fig. S1 B; Allen et al., 2006; Aragon et al., 2008). The budding indices were also measured in both the quiescent and nonquiescent cell fraction (Fig. 2 D).
Figure 2.

Genomic instability arises from quiescent cells during chronological aging. (A) Density-gradient separation of nonquiescent (upper) and quiescent (lower) cells from chronologically aging cultures. (B) Calcofluor staining of upper and lower fractions from wild-type and sch9Δ mutant cultures. The arrows indicate budding cells. Bar, 10 µm. (C) Percentage of upper and lower fractions in the culture. (D) Budding indices of upper and lower fractions (n = 6–7). (E) Mutation frequency in CAN1 gene (Canr mutants/106 cells) in the upper and lower fractions (n = 6–7). (F) Mutation frequency in the CAN1 gene before and after allowing the cells to undergo one population doubling during a standard chronological life span study (n = 4). Error bars indicate ±SEM. (C–E) Data represent the mean ± SEM (error bars); *, P < 0.05; **, P < 0.01; two-tailed t test, sch9Δ versus wild type (WT). ^, P < 0.05; ^^, P < 0.01; two-tailed t test, day 5 or day 7 versus day 3 of the same strain.
On day 3, 60% of wild-type cells were in the upper nonquiescent fraction (Fig. 2 C). This fraction increased gradually from day 3 to day 7. Interestingly, the Canr mutation frequency in the nonquiescent wild-type subpopulation was significantly lower than that in the quiescent subpopulation (Fig. 2 E). Furthermore, the frequency of mutations in the nonquiescent subpopulation remained unchanged over time, whereas an age-dependent increase of mutations was observed in the quiescent fraction (P < 0.05: day 7 vs. day 3; Fig. 2 E). This result indicates that quiescent cells may accumulate DNA damage that leads to mutations. In contrast to the wild type, SCH9-deficient cells were mostly quiescent, as indicated by FACS analysis (Figs. 1 H and S1 B). From day 3 to day 7, >90% of the SCH9-deficient cells separated in the lower fraction compared with 10–40% of wild-type cells (Fig. 2 C). The deletion of SCH9 caused a decrease of mutation frequency in the quiescent fraction compared with that of wild type, and abolished the age-dependent increase in mutations (Fig. 2 E). The latter data suggest that the effect of SCH9 deletion on the cell cycle state does not play a major role in age-dependent mutations.
To test whether mutations may occur during the multiple rounds of cell divisions required to generate a colony and not during either the aging of the nondividing population or the very first round of replication, we allowed old wild-type and sch9Δ cells to undergo one population doubling before treatment with the toxic l-canavanine sulfate. If the DNA damage did not occur during aging but during multiple rounds of cell division after exit from G0, we would expect the additional population doubling of the 2 × 107 cells to cause a major increase in mutation frequency. The frequency of mutations normalized for population size, instead, remained the same for cells exposed to canavanine before or after one population doubling (Fig. 2 F), which suggests that at least the initial DNA damage in the CAN1 gene occurs in the nondividing cells.
Gis1, a stress-response transcription factor downstream of Sch9, regulates the expression of REV1 and DNA mutations
The anti-aging and stress-resistance effects of the sch9Δ mutations depend on the protein kinase Rim15 and its downstream stress response transcription factors Gis1, Msn2, and Msn4 (Fabrizio et al., 2001, 2003; Wei et al., 2008). Deficiency in Gis1 led to an elevation of mutation frequency in the wild type (Fig. 3 B) and reversed the protective effect of sch9Δ on age-dependent mutations starting from day 9 (Fig. 3, A and B). To investigate further the mediators of the Sch9 effect on DNA mutations, we obtained the global gene expression profiles of 2.5-d-old sch9Δ and wild-type cells. Among the DNA repair genes, REV1, encoding the deoxycytidyl transferase that is involved in TLS and the repair of abasic sites, and the recombination repair genes RAD51 and RAD54 were among the most down-regulated (Fig. 3 C and Table S2) in the long-lived sch9Δ mutants.
Figure 3.

Stress response transcription factor Gis1 is required for life span extension and attenuated genomic instability associated with SCH9 deficiency. (A) Chronological survival (n = 11) and (B) mutation frequency in the CAN1 gene (n = 11) of the wild type (WT; DBY746) and sch9Δ mutants. ^, P < 0.05; ^^, P < 0.01; two-tailed t test, sch9Δ gis1Δ versus sch9Δ at the indicated time points. (C) mRNA levels of REV1, REV3, REV7, and recombination genes involved in DNA repair. WT (DBY746) and sch9Δ were grown in SDC media. Cells were collected on day 2.5 for mRNA extraction and microarray analysis. Data represent mean ± SEM (error bars) of percentage change, sch9Δ versus WT (n = 3). For complete microarray data, see Table S2.
REV1 mRNA level was 40% lower in sch9Δ mutant cells compared with wild-type cells on day 3 (Fig. 4 A). Deletion of GIS1 in both wild-type and sch9Δ mutant cells was associated with significant increase in the REV1 mRNA level (Fig. 4 A), suggesting that Sch9 modulates Rev1 expression/activity partially through the down-regulation of the transcription factor Gis1. However, the deletion of SCH9 caused a major reduction of REV1 expression and of mutation frequency during the first 7 d of survival even in GIS1-deficient cells, which suggests that part of the protective effect is Gis1 independent. Other transcription factors such as Msn2 and Msn4 activated in sch9Δ mutants might be partially responsible for the effect on life span and mutations (Fig. 3, A and B).
Figure 4.

The age-dependent increase in genomic instability is mediated by Rev1. (A) mRNA levels of REV1 in wild-type (WT; DBY746), sch9Δ, gis1Δ, and gis1Δ sch9Δ cells. mRNA extracted from 3-d-old cells were subjected to quantitative real-time PCR analysis. **, P < 0.01, mutant versus WT; ^^, P < 0.01, mutant versus sch9Δ, n = 12, ANOVA test, Tukey's test. (B) Chronological survival and (C) mutation frequency over time in the CAN1 gene of wild type, sch9Δ, rev1Δ, and rev1D sch9Δ in both the DBY746 and BY4741 genetic backgrounds. ***, P < 0.001; rev1Δ versus WT. ^, P < 0.05; rev1Δ sch9Δ versus sch9Δ, two-tailed t test at indicated time points (n = 9). (D) Chronological survival and (E) mutation frequency over time in the CAN1 gene of wild-type, sch9Δ, rev1Δ, and cells overexpressing SCH9 or REV1. *, P < 0.05; **, P < 0.01; ***, P < 0.001; mutant versus WT. ^, P < 0.05; ^^, P < 0.01; sch9Δ REV1ox versus sch9Δ. a, P < 0.05; rev1ΔSCH9ox versus SCH9ox, two-tailed t test at indicated time points (n = 6–9). (F) Chronological survival and (G) GCRs in the wild type and rev1Δ mutants. **, P < 0.01; ***, P < 0.001; rev1Δ versus WT, two-tailed t test at indicated time points (n = 4). Error bars indicate ±SEM.
The Y superfamily DNA polymerase Rev1 and Polζ mediate Sch9-dependent mutations during aging
To gain further insight into the types of age-dependent mutations, we analyzed the mutation spectra in Canr mutant colonies arising from chronologically aging wild-type and sch9Δ cultures on day 7, when the difference in mutation frequency was elevated and 50% of the wild-type population was still alive (Fig. 1 C and Table I). Sequencing data of Canr mutants from wild-type cultures indicated a high occurrence of multiple mutations within a short stretch of a 500–1,000-bps DNA sequence (6 out of 10 mutated CAN1 genes): base substitutions accompanied with frameshift mutations caused by small deletion or insertion of homopolymeric runs (sequences of 3–4 or more identical nucleotides, often As and/or Ts; Table I). In agreement with the age-dependent increase in oxidative stress (Moraes et al., 1990; Kreutzer and Essigmann, 1998; Wang et al., 1998), we observed a high occurrence of G → T (transversion) or C → T (transition) mutations in wild-type cells, which was not seen in the sch9Δ cells (Table I). It is not clear why 5 out of 10 sequenced CAN1 genes from sch9Δ mutant colonies contained a C → A base substitution at position 11 away from ATG (Table I). One possibility is that this base may constitute a mutation hot spot. Another possibility is that the mutation generated during the growth phase expanded with the population. Multiple mutational events were not observed in cells lacking SCH9. Only 2 out of 10 Canr mutants from sch9Δ cultures contained multiple substitution/deletion/insertion mutations on day 7 (Table I).
Table I.
Mutations spectra observed in Canr mutant colonies from 7-d-old wild-type, sch9Δ, and rev1Δ cultures
| Strains | Clone | Type | Base change | Mutations | Position from ATG | Sequence |
| WTa | 1 | Base substitution | C → T | Proline-leucine | 656 | 5′-TGTTCCCTGTC-3′ |
| WTa | 2 | Base substitution | C → T | Proline-serine | 937 | 5′-GCTGCAAACCCC-3′ |
| WTa | 3 | Base substitution | T → G | Asparagine-lysine | 1,173 | 5′-AAATTCAAATA-3′ |
| WTa | 3 | Insertion | G | Frameshift | 1,710 | 5′-GAGGCGAATT-3′ |
| WTa | 4 | Base substitution | G → C | Alanine-proline | 709 | 5′-TTTTAGCCATTA-3′ |
| WTa | 4 | Deletion | TG | Frameshift | 1,098–1,099 | 5′-TTGCTATTGAGA-3′ |
| WTa | 5 | Base substitution | G → T | Glycine-valine | 353 | 5′-ACGCCGGCCCA-3′ |
| WTa | 5 | Insertion | A | Frameshift | 1,341 | 5′-CAAAAGTTTTC-3′ |
| WTa | 6 | Deletion | AT | Frameshift | 1,129–1,130 | 5′-GTCACATATCTT-3′ |
| WTa | 6 | Insertion | T | Frameshift (T3-T4) | 1,086 | 5′-CCTTTTATTATT-3′ |
| WTa | 7 | Base substitution | G → T | Tryptophan-cysteine | 531 | 5′-GTTTTCTTGGCA-3′ |
| WTa | 7 | Deletion | A | Frame-shift (A3-A2) | 663 | 5′-AAATATTACGGT-3′ |
| WTa | 7 | Insertion | T | Frameshift (T3-T4) | 1,086 | 5′-CCTTTTATTATT-3′ |
| WTa | 8 | Base substitution | G → A | Glutamic A–lysine | 679 | 5′-AATTCGAGTTCT-3′ |
| WTa | 8 | Base substitution | G → T | Valine-phenylalanine | 907 | 5′-AACTATTTGGTA-3′ |
| WTa | 8 | Deletion | A | Frameshift | 1,217 | 5′-ATCAAAGAACAC-3′ |
| WTa | 9 | Duplication | NA | 248 bp | 184–431 | NA |
| WTa | 10 | No PCR | NA | NA | NA | NA |
| sch9Δ | 1 | Base substitution | C → A | Serine-STOP | 11 | 5′-AAATTCAAAAG-3′ |
| sch9Δ | 2 | Base substitution | C → A | Serine-STOP | 11 | 5′-AAATTCAAAAG-3′ |
| sch9Δ | 3 | Base substitution | C → A | Serine-STOP | 11 | 5′-AAATTCAAAAG-3′ |
| sch9Δ | 4 | Base substitution | C → A | Serine-STOP | 11 | 5′-AAATTCAAAAG-3′ |
| sch9Δ | 5 | Base substitution | C → A | Serine-STOP | 1,244 | 5′-CTGTCAAGGAC-3′ |
| sch9Δ | 6 | Base substitution | T → C | Phenylalanine-leucine | 682 | 5′-ATTCGAGTTCT-3′ |
| sch9Δ | 7 | Insertion | T | Frameshift (T6-T7) | 1,386 | 5′-CTTTTTTTGCAT-3′ |
| sch9Δ | 8 | Base substitution | T → G | Isoleucine-serine | 305 | 5′-GGTACTATTGGT-3′ |
| sch9Δ | 8 | Insertion | T | Frameshift | 1,013 | 5′-TATTATTTCATT-3′ |
| sch9Δ | 9 | Base substitution | C → A | Serine-STOP | 11 | 5′-AAATTCAAAAG-3′ |
| sch9Δ | 9 | Base substitution | C → A | No change | 1,347 | 5′-TTTTCGAATGG-3′ |
| sch9Δ | 10 | Insertion | T | Frameshift (T4-T5) | 1,022 | 5′-GGACTTTTTAGT-3′ |
| sch9Δ | 10 | Insertion | T | Frameshift (T6-T7) | 1,386 | 5′-CTTTTTTTGCAT-3′ |
| rev1Δ | 1 | Base substitution | C → T | Arginine-serine | 1,195 | 5′-GGTTCCCGTAT-3′ |
| rev1Δ | 2 | Insertion | A | Frameshift (A3-A4) | 702 | 5′-CAAAAGTTTA-3′ |
| rev1Δ | 3 | Deletion | G | Frameshift | 422 | 5′-GGGTGAAATGG-3′ |
| rev1Δ | 3 | Deletion | AAA | A3-A0 | 425-427 | 5′-GGGTGAAATGG-3′ |
| rev1Δ | 4 | Deletion | G | Frameshift | 422 | 5′-GGGTGAAATGG-3′ |
| rev1Δ | 4 | Deletion | AAA | A3-A0 | 425-427 | 5′-GGGTGAAATGG-3′ |
| rev1Δ | 5 | No PCR | NA | NA | NA | NA |
| rev1Δ | 6 | Base substitution | G → A | Tryptophan-STOP | 522 | 5′-GTATTGGTTTTC-3′ |
| rev1Δ | 7 | Base substitution | C → T | Serine-leucine | 1,214 | 5′-TATCAAAGAAC-3′ |
| rev1Δ | 8 | Base substitution | C → T | Histidine-tyrosine | 274 | 5′-CAAAGACATATT-3′ |
Letters in bold indicate base substitutions; bold and italics indicate insertions; underline indicates deletions. NA, not applicable.
from Madia et al. (2008).
The mutation pattern observed in wild-type cells was consistent with the recruitment to the DNA lesion site of error-prone polymerases, which is analogous to SOS-induced mutagenesis in Escherichia coli mediated by PolIV and PolV of the Y superfamily of polymerases (Tang et al., 2000; Kobayashi et al., 2002). This family includes Rev1 and Rad30 (Polη) in S. cerevisiae (Ohmori et al., 2001). Most spontaneous and damage-induced mutagenesis during growth phase was found to depend on Rev1 and the DNA polymerase zeta (Polζ, which belongs to the B superfamily of DNA polymerases; Quah et al., 1980). Sloppy DNA synthesis by error-prone polymerases could then have been responsible for the multiple mutations observed in the aging wild-type cells (Table I). The absence of multiple mutation events in the CAN1 gene in 7-d-old rev1Δ mutants (Table I) suggests a role for Rev1 in the sloppy DNA synthesis leading to the secondary mutations.
Considering that REV1 was among the most down-regulated DNA repair-related genes in sch9Δ (Fig. 3 C and Table S2) and that its mRNA expression was significantly increased in the absence of the downstream Sch9 effector Gis1 (Fig. 4 A), we tested whether the Rev1 protein mediated the effect of Sch9 on age-dependent genomic instability. In the DBY746 background, spontaneous age-dependent mutation frequency started at a much lower level and did not increase in cells lacking REV1 (Fig. 4 C). Similar results were obtained in the BY4741 background (Fig. 4, B and C). rev1Δ cells died earlier than wild-type cells in the DBY746 (Fig. 4 B) but not in the BY4741 genetic background (Fig. 4 B), indicating that its role in the repair of double strand breaks and other lesions could be important for survival. In agreement with the role of Rev1 in mediating the Sch9-dependent mutagenesis, the deletion of both SCH9 and REV1 did not further decrease the age-dependent mutation frequency (Fig. 4 C). In contrast, overexpression of SCH9, which models mammalian oncogenic mutations, caused a doubling of the frequency of age-dependent mutations (Fig. 4 E). The deletion of REV1 reversed the hypermutagenic phenotype of mutants overexpressing SCH9, although it also led to early death (Fig. 4, D and E).
REV1 overexpression increased Canr mutation frequency (Fig. 4 E), and overexpression of REV1 in sch9Δ mutants reversed the protective effect of this longevity mutation only on day 9 and day 11, indicating that sch9Δ exerts additional protection independently of Rev1 (Fig. 4 E). Interestingly, larger lesions such as GCRs occurred more frequently in cells lacking REV1 (10–25-fold higher than in wild-type cells; Fig. 4 G), which suggests that, during aging, the Rev1 protein contributes to the generation of point mutations as part of an emergency response aimed at preventing DNA rearrangements and deletions, which could be lethal.
Polζ consists of a catalytic subunit Rev3 and an accessory Rev7, which associates with Rev1 and is involved mainly in DNA lesion bypass (Gan et al., 2008). To test whether the Polζ was also responsible for an increase in age-dependent mutations, we studied mutants lacking either REV3 or REV7. Cells deficient of Rev3 or Rev7 lived shorter than wild-type cells (Fig. 5, A and C). In both DBY746 and BY4741 genetic backgrounds, Rev3-deficient cells showed a reduction of age-dependent Canr mutations similar to that observed in the rev1Δ mutants (Fig. 5, B and D; and Fig. 4 C). In the BY4741 background, Rev7 deficiency also caused a major reduction in mutation during aging (Fig. 5 B). However, deficiency in Rad30 (Polη), which is mainly involved in the error-free DNA damage bypass (Johnson et al., 1999; Washington et al., 1999; Prakash et al., 2000), caused an elevated age-dependent increase of mutation frequency (Fig. 5 D). These data strongly suggest that the error-prone Rev1–Polζ complex plays a central role in the generation of point mutations and small insertions/deletions, but prevents GCRs during chronological aging.
Figure 5.

TLS mediates the age-dependent increase in genomic instability. (A) Chronological survival and (B) mutation frequency (Canr) of the wild type (BY4741) and mutants deficient of Polζ, rev3Δ, and rev7Δ. *, P < 0.05; **, P < 0.01; mutants versus wild type (WT) two-tailed t test at the indicated time points (n = 4). (C) Chronological survival and (D) mutation frequency (Canr) of the wild type (DBY746), rev3Δ, and rad30Δ mutants. *, P < 0.05; **, P < 0.01; ***, P < 0.001; mutants versus WT, two-tailed t test at the indicated time points (n = 6–10). Error bars indicate ±SEM. (E) Abolished TLS in Sch9-deficient cells. Nuclear extracts from 3-d-old stationary phase wild-type (DBY746) and sch9Δ mutant cells were incubated with undamaged or abasic site-containing DNA templates for 30 min at 30°C. Primer extension products were analyzed on 19% polyacrylamide gels. There was no lesion bypass observed in sch9Δ mutants. Free primers are indicated by the arrow. TLS products are indicated by solid (undamaged) or broken (damaged) lines. On the right, the percentage of incorporation opposite the abasic site is given.
To test further the hypothesis that error-prone polymerases mediate age-dependent mutations, we investigated the TLS in 3-d-old wild-type and sch9Δ mutant cultures. Nuclear extracts from both wild-type and sch9Δ mutant cells were capable of extending the 32P-labeled primer on undamaged DNA templates (Fig. 5 E). In contrast to wild-type cells, nuclear extracts from sch9Δ mutants were deficient in lesion bypass activity on the DNA templates containing an abasic site (Fig. 5 E). About 5% TLS efficiency was observed in the wild-type extract when used at 5, 10, or 20 µg, which is in agreement with the lesion bypass efficiency observed in vivo (Pagès et al., 2008). Furthermore, in the nuclear extract from the wild type, ∼20–25% of all extended products represented the incorporation opposite the abasic site, whereas <5% incorporation opposite the lesion was observed in sch9Δ (Fig. 5 E).
SCH9 deletion protects against DNA oxidation
The Rev1/Polζ-dependent TLS is activated in response to DNA damage. Thus, we asked whether SCH9 deficiency might also protect against oxidative damage to DNA. Cells were treated with 1 mM H2O2 on days 1 and 3, and Can1 mutation frequency was monitored for 13 d. In the wild-type strain (DBY746), the initial oxidative treatment led to a fourfold increase in Canr mutations, which continued to increase until day 9, when the great majority of cells were dead (Fig. 6, A and B). In H2O2-treated sch9Δ cells, mutation frequency and viability, instead, were unaffected or slightly lower, respectively, suggesting that the SCH9 deletion protects cells against oxidation-induced DNA damage/mutagenesis and cell death (Fig. 6, A and B). Indeed, a sixfold increase of the 8-hydroxy-2′ deoxyguanidine (8-OHdG) level was observed when comparing day 1 versus day 7 in wild-type cells, whereas Sch9-deficient cells showed significantly lower age-dependent accumulation of 8-OHdG (Fig. 6 C). Furthermore, sch9Δ mutant cells were protected from oxidative damage (Fig. 6 D), which is in agreement with the lack of Canr mutation frequency increase during chronological aging (Fig. 6 B). In vivo, 8-OHdG is a prevalent oxidative lesion that, when unrepaired, can lead to G-to-T transversion (Grollman and Moriya, 1993; Moriya, 1993; Le Page et al., 1995). Polζ incorporates A (fives times more likely than C) opposite of 8-OHdG, thus leading to a G → T transversion (Prakash et al., 2005), which is one of the most common mutations found in chronologically aging cells (Table I).
Figure 6.

The lack of SCH9 protects against oxidative stress-induced genomic instability. (A) Chronological survival and (B) mutation frequency (Canr) of the wild type (DBY476) and sch9Δ mutants. Cells were treated with 1 mM H2O2 at day 1 and day 3. *, P < 0.05; **, P < 0.01; sch9Δ versus wild type (WT), two-tailed t test at the indicated time points (n = 4). (C) 8-OHdG contents in the wild type (DBY746) and sch9Δ mutants during chronological aging. (D) Day 3 SDC wild-type and sch9Δ cultures were treated with 1 mM H2O2 for 48 h. The mean of two independent experiments in duplicate is presented. (E–G) Wild-type (DBY746) and sch9Δ mutant cells were exposed to 100 mM H2O2 for 30 min. Survival (E), mutation frequency (F; Canr), and GCR (G; Canr5FOAr) were monitored at 30 min, 2 h, and 5 h after the end of H2O2 treatment. (H–J) Wild-type (DBY746) and sch9Δ mutant cells were exposed to 0.02% MMS for 50 min. Error bars indicate ±SEM. Survival (H), mutation frequency (I; Canr), and GCR (J; Canr5FOAr) were monitored at 30 min and 5 h after the end of MMS treatment. For E–J, data represent mean ± SEM (error bars), n = 3. *, P < 0.05; **, P < 0.01; two-tailed t test at the indicated time points, sch9Δ versus WT.
Increased protection against oxidation- but not alkylation-dependent DNA damage in sch9Δ mutants
To test whether the lack of SCH9 decreased age-dependent genomic instability by improving DNA repair, we monitored the mutagenesis induced by either oxidation (hydrogen peroxide [H2O2]) or alkylation (methyl methane sulfonate [MMS]) in nondividing cells. Survival, Canr, and GCRs frequencies in 3-d-old wild-type and sch9Δ cells were determined after exposure to 100 mM H2O2 for 30 min or to 0.02% of MMS for 50 min. After treatment, cells were washed four times and allowed to repair DNA without dividing in toxin-free expired synthetic dextrose complete (SDC) medium obtained from parallel aging cultures. Canr mutations and GCRs were measured 30 min, 2 h, and 5 h after the end of each treatment. The exposure to hydrogen peroxide had little or no effect on sch9Δ viability (10% cell death), but killed 90% of the wild-type cells (Fig. 6 E). 2 h after treatment with H2O2, Canr mutation frequency in sch9Δ showed only a minor increase and remained sixfold lower than that of wild-type cells (Fig. 6 F). Similarly, the sch9Δ mutant did not show any increase in GCRs frequency in contrast to the wild type, which displayed a >100-fold increase in GCRs by 5 h (Fig. 6 G).
In contrast to its role in protecting against oxidative stress-induced mutations, the sch9Δ mutation did not protect against alkylation-induced DNA damage (Fig. 6, H, I and J). Wild-type cells were slightly more sensitive to 0.02% MMS, and sch9Δ only gave a small amount of protection against mutations and GCRs in response to MMS (Fig. 6, I and J). These results indicate that sch9Δ mutant cells are not well protected from alkylating DNA damage but are partially protected from alkylation-induced point mutations and small insertions/deletions (Fig. 6 I).
The role of superoxide in age-dependent mutations
Based on the age-dependent increase of 8-OHdG levels (Fig. 6 C), the spectra of Canr mutations (Table I), the involvement of the Rev1–Polζ complex in age-related mutagenesis (Figs. 4 and 5), and the protection against oxidative damage to DNA exerted by deficiency in SCH9 (Fig. 6), we studied the role of superoxide and oxidative DNA damage in Sch9- and age-dependent genomic instability.
The life span extension associated with deficiencies in Sch9 and Ras2/cAMP/PKA requires the expression of the mitochondrial superoxide dismutase (MnSOD; Fabrizio et al., 2003) and of the stress response transcription factors Msn2/4 and Gis1 (Pedruzzi et al., 2000; Wei et al., 2008). Both the stress response element (STRE) and post-diauxic shift motif (PDS) are present in the promoter region of SOD2, which is up-regulated in cells lacking Sch9 (Fig. S2 A). The genome-wide expression profiling also indicated that deletion of SCH9 causes a metabolic switch from the TCA cycle and respiration to glycolysis and glycerol biosynthesis, as well as a general reduction of mRNA levels of proteins targeted to mitochondria (Wei et al., 2009), which is in agreement with the reduced generation and/or level of superoxide in sch9Δ mutants.
Overexpression of the cytosolic Cu, Zn superoxide dismutase SOD1 (introduction of plasmid YEp352-SOD1ox[URA3] induced an eightfold increase of SOD1 mRNA level compared with the wild type; Figs. S2 B and S3), was able to completely abolish the detrimental effect of the overexpression of SCH9 on either age-dependent spontaneous or paraquat-induced genomic instability (Fig. 7, A and B). Moreover, the overexpression of either SOD1 or SOD2 (introduction of plasmid YEp351-SOD2ox[LEU2] induced a fourfold increase of the SOD2 mRNA level compared with the wild type; Figs. S2 B and S3) was effective in attenuating age-dependent mutation frequency increase in Gis1-deficient cells and down-regulating the REV1 mRNA level (Fig. 8, A–C). To test whether the combination of both a high superoxide and REV1 level contribute to age-dependent mutations, we treated rev1Δ mutants and REV1 overexpressors with 1 mM paraquat (Fig. 9, A–C). REV1 overexpression did not increase further paraquat-dependent mutagenesis, possibly because the superoxide generated by paraquat might have maximized REV1 expression (Fig. 9 C). SOD1 and SOD2 overexpression did cause a significant decrease in the REV1 mRNA level (Fig. 9 D). These results indicate that the Sch9-dependent accumulation of oxidative DNA damage is the major source of age-dependent genomic instability in nondividing cells. We propose that during the first round of replication, the Rev1–Polζ complex processes the oxidation-induced abasic sites and generates base substitutions to avoid GCRs and possibly cell death (Fig. 9 E).
Figure 7.

SOD1 overexpression attenuates age-dependent oxidative stress and SCH9 overexpression–induced genomic instability. (A) Mutation frequency (Canr) of cells during normal chronological aging. (B) Mutation frequency (Canr) of cells treated with 1 mM paraquat on day 1 and on day 3. Strains shown are wild type (WT; DBY476), sch9Δ, and cell overexpression of SOD1 and/or SCH9. *, P < 0.05; mutant versus WT. ^, P < 0.01; mutant versus sch9Δ, two-tailed t test at the indicated time points (n = 5). Error bars indicate ±SEM.
Figure 8.
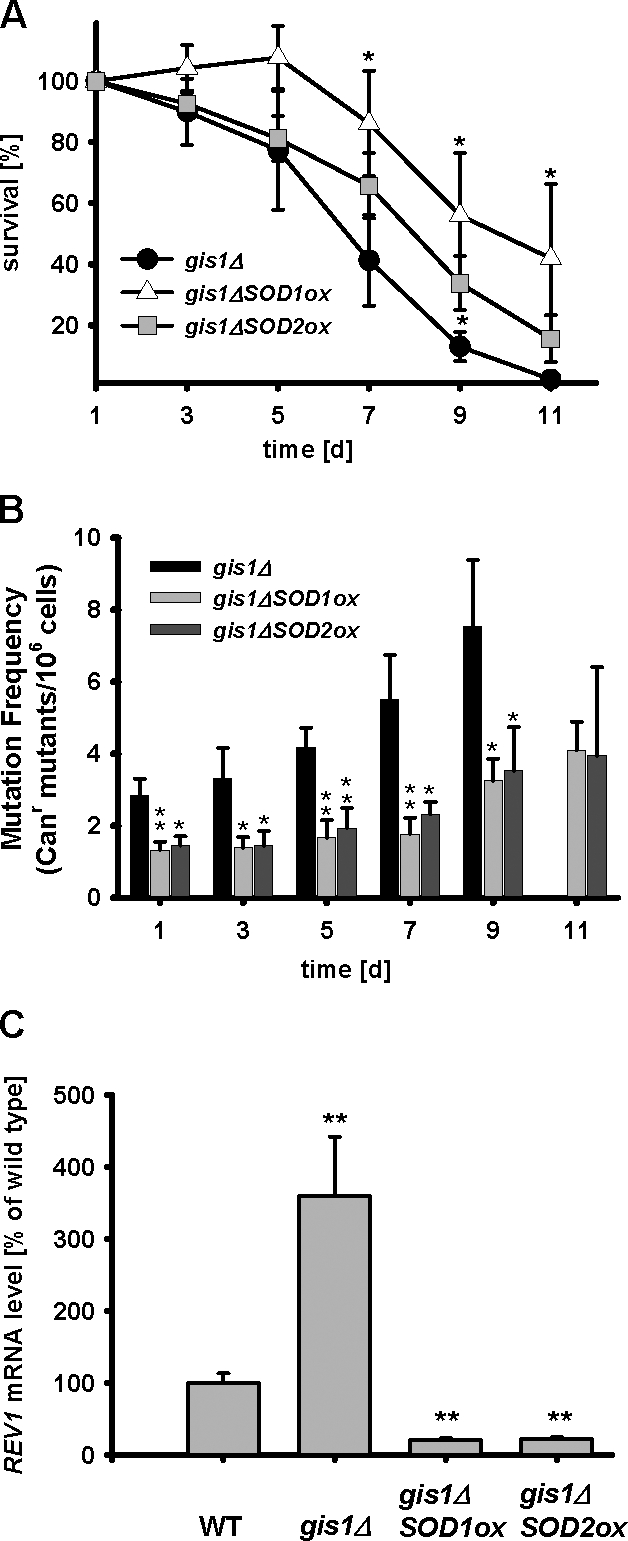
Oxidative stress and genomic instability. (A and B) Overexpression of SOD1 or SOD2 extends life span and reduces genomic stability associated with gis1Δ mutants. *, P < 0.05; **, P < 0.01; compared with gis1Δ, two-tailed t test at the indicated time points (n = 4). (C) mRNA levels of REV1 in wild-type (WT; DBY746), gis1Δ, and gis1Δ cells overexpressing either SOD1 or SOD2. mRNA extracted from 3-d-old cells was subjected to quantitative real-time PCR analysis. **, P < 0.01; mutant versus WT (n = 12, ANOVA test, Tukey's test). Error bars indicate ±SEM.
Figure 9.

Rev1 mediates oxidative stress induced genomic instability. (A–C) Mutation frequency (Canr) of the wild type (DBY476), rev1Δ, and cells overexpressing REV1 (REV1ox) during chronological aging and under oxidative stress (1 mM paraquat at day 1 and at day 3). *, P < 0.05; **, P < 0.01; rev1Δ versus wild-type (WT) untreated, two-tailed t test at the indicated time points (n = 4). Error bars indicate ±SEM. (D) REV1 mRNA levels at day 3. REV1 mRNA level were normalized to ACT1 and represent mean ± SEM (error bars) of the percentage of wild type. **, P < 0.01; compared with WT. ^^, P < 0.01; compared with sch9Δ (two-tailed t test, n = 8–12). (E) Modulation of oxidative stress and genomic instability by Sch9. During chronological aging, the oncogene homologue SCH9 inhibits the expression of genes involved in glycolysis and promotes respiration. Sch9 also regulates the error-prone Polζ complex component Rev1 through the stress response transcription factor Gis1. Accumulation of oxidative DNA damage during aging activates Rev1-mediated TLS, which leads to increased base substitutions and reduced GCRs in the first round of DNA replication when cells exit the arrested state.
Discussion
Age-dependent DNA damage and cancer are widely believed to depend on mutations associated with the number of divisions completed by a cell. Here, in the study of a unicellular eukaryote, the frequencies of Canr mutations and GCRs (Fig.1 B and D) reached 3- and 20-fold, respectively, higher levels in cells aging under nondividing conditions than in the population resulting from the many divisions necessary for the expansion from the initial 100 thousand to 1 billion cells contained in each aging study (we refer to the Canr and GCRs frequencies reached in the cell population at the end of the growth phase on day 1).
Mutated Ras, Akt, and PTEN are widely believed to contribute to cancer by promoting growth and preventing apoptosis (Pollak et al., 2004). Based on mammalian studies, inhibition of Ras and Akt is predicted to slow down growth and promote the apoptosis of cancer cells. Studies by our laboratory and others have shown that yeast homologues of mammalian Ras and Akt/S6K promote aging, oxidative stress, and age-dependent mutations (Fabrizio et al., 2001, 2003, 2004; Hlavatá and Nyström, 2003). Although Sch9 and Ras promote growth in S. cerevisiae, we show that Sch9 can promote DNA damage during aging in nondividing cells. Interestingly, the separation of aging wild-type cultures by a density gradient revealed that quiescent cells, otherwise considered stable and protected, exhibit an elevated DNA mutation frequency compared with nonquiescent cells (Fig. 2 E). Our data suggest that DNA damage and mutations including GCRs occur mostly in old cells and are associated with a shorter life span, but do not provide conclusive evidence for the role of DNA damage as a major factor in the aging process (Vijg, 2008). Sch9 appears to be responsible for the major portion of age-dependent increase in DNA mutations because cells lacking SCH9 displayed reduced point mutations, small insertions/deletions, and GCRs, whereas cells overexpressing SCH9, which models mammalian Akt-activating oncogene mutations (PTEN etc), displayed a significant increase in mutations. The effect of Sch9 on age-dependent mutations did not appear to be simply due to its effect on survival because, at a comparable percent of survival, the cumulative frequency of mutations in sch9Δ was less than half of that in wild-type cells (Fig. 1 C).
We observed a higher occurrence of multiple mutations within a short stretch of DNA in Canr mutants from 7-d-old wild-type cultures than in sch9Δ mutants (Table I). This mutation profile was reminiscent of that caused by the error-prone PolIV and PolV polymerases, which are involved in increased mutability in nondividing E. coli (McKenzie et al., 2001; Kobayashi et al., 2002), and of that caused by eukaryotic TLS polymerases (Rev1, Polζ, and Rad30; Waters et al., 2009). In the sch9Δ mutants, base substitutions were mostly C → A, followed in one case only by a T insertion (Table I), suggesting that the lack of SCH9 not only reduced the frequency of spontaneous age-dependent mutations but also prevented multiple mutations within a short stretch of DNA sequence.
REV1 was among the most down-regulated DNA repair-related genes in the sch9Δ mutant, and its expression (mRNA level) was reduced 40% compared with the wild type on day 3 (Figs. 3 C and 4A). The lack of REV1 prevented the mutation frequency increase associated with aging or induced by SCH9 overexpression, but did not affect the mutation frequency of sch9Δ (Fig. 4). Rev1 expression is threefold higher during G2/M than in G1 phase (Waters and Walker, 2006; Sabbioneda et al., 2007). Based on these results and on our FACS data (Fig. S1 A), we would have expected a smaller reduction of the REV1 mRNA level than the 60% measured in Sch9-deficient cells if its regulation was only dependent on the cell cycle. These results indicate additional regulation/activation of REV1 expression in aging cells.
rev1Δ mutants showed an 80% decrease in mutation frequency compared with wild-type cells (Fig. 4 C). Rev1 is known to insert C opposite the abasic site (Nelson et al., 1996), but it has also been shown to insert all four nucleotides to repair a gap (Haracska et al., 2002). This mutagenic role of Rev1 has been confirmed in vivo (Auerbach et al., 2005). The base substitutions observed in the CAN1 gene (Table I) were consistent with the deoxycytidine monophosphate transferase activity of Rev1 or its modulation of thymidine monophosphate insertions opposite an abasic site, although part of them are likely to be generated independently of TLS. Furthermore, Rev1 can play a structural role in aiding polymerases bypass lesion sites, which does not depend on its nucleotidyl transferase activity (Nelson et al., 2000; Lawrence, 2004). In this case, the insertion of the nucleotide opposite an abasic site is catalyzed by polymerase δ (Haracska et al., 2001).
The deletion of REV1 in wild-type cells blocked the generation of multiple mutations/insertions/deletions within a short stretch of DNA sequence (Table I). Previous studies from the laboratories of S. Jinks-Robertson and T.A. Kunkel (Kozmin et al., 2003; Minesinger et al., 2006) described spontaneous “complex” mutational events, in which a frameshift is accompanied by one or more base substitutions. These events depended on the mutagenic TLS polymerase Polζ activity, which functions in association with Rev1 (Minesinger et al., 2006). Our results show that age-dependent mutagenesis was reduced in cells lacking either REV3 or REV7, the two subunits of Polζ polymerase, but was increased in mutants lacking RAD30, which is regarded as an error-free DNA damage bypass enzyme (Johnson et al., 1999; Washington et al., 1999; Prakash et al., 2000).
These findings support the hypothesis that the majority of age-dependent mutations are generated through the activity of the Rev1–Polζ error-prone complex, whose activity may be elevated in aging cells (Fig. 5 E). The down-regulation of Rev1 expression alone, however, may not explain the complete absence of TLS activity in sch9Δ mutants. Rev1 is not absolutely required for TLS in yeast (Pagès et al., 2008); we cannot exclude the possibility that the deficiency and/or modification in other important TLS components, such as proliferating cell nuclear antigen ubiquitination and/or Polζ itself, contributed to the complete absence of TLS in sch9Δ mutant cells (Fig. 5 E).
Mutants lacking SCH9 were highly protected against exogenous oxidative stress (Fabrizio et al., 2001). This study shows that the deficiency in SCH9 protected against endogenous age-dependent oxidative damage to DNA, as indicated by the level of 8-OHdG (Fig. 6 C). The elevated expression of SOD2 and protection of the DNA against oxidative damage in sch9Δ mutants, together with the effect of the overexpression of both SOD1 and SOD2 in reducing point mutations, indicate that Sch9 promotes mutations by a superoxide-dependent mechanism. We propose that superoxide and the oxidized DNA cause an increased expression of REV1 and activation of the error-prone Polζ, which result in point mutations during the first round of replication in order to prevent double strand breaks and GCRs (Fig. 9 E).
Although the deletion of SCH9 causes Gis1-dependent repression of REV1 expression (Fig. 4 A), Sch9 and Gis1 may not necessarily function in the same pathway as Rev1/Polζ. The superoxide production and DNA oxidation regulated by Sch9 may instead directly lead to the activation of the error-prone system (Brunet et al., 2004; Shen et al., 2007). In fact, the overexpression of either SOD1 or SOD2 was sufficient to decrease REV1 expression (Fig. 9 D).
Recent work in neurons and mice suggests that somatic mutations associated with Huntington's disease occur during the removal of oxidation lesions by an error-prone mechanism dependent on the base excision repair enzyme Ogg1 (Kovtun et al., 2007). Our data raise the possibility that down-regulation of the mammalian Akt/S6K signaling pathway implicated in life span regulation may also regulate oxidative damage as well as error-prone polymerases that can cause DNA damage and diseases during aging. Notably, down-regulation of Rev1 in human cells reduced UV-dependent mutations (Gibbs et al., 2000), which suggests that the role of this protein is at least partially conserved from yeast to humans. The connection between oncogene homologs, Rev1, and age-dependent genomic instability observed in the simple S. cerevisiae model has not been demonstrated in mammals. However, the recent papers showing reduced age-dependent cancer incidence in mice deficient in IGF-I (Vergara et al., 2004) or Akt (Skeen et al., 2006), and the reduction in tumors in C. elegans deficient in the IGF-I–like receptor DAF-2 (Pinkston et al., 2006) are consistent with our previous and present results indicating a link between these oncogenes, mutagenesis, and cancer (Longo et al., 2008). It will be important to determine whether these conserved antiaging pathways may regulate, in addition to cell growth and apoptosis, oxidation damage of DNA and error-prone DNA repair proteins in both dividing and nondividing cells.
Materials and methods
Yeast strains
The experiments were performed in DBY746 (MATα,leu2-3,112,his3Δ1,trp1-289,ura3-52,GAL+), provided by D. Botstein, Massachusetts Institute of Technology, Cambridge, MA. The strain BY4741 (MATa,his3Δ1leu2Δ0,met15Δ0,ura3Δ0; Thermo Fisher Scientific) was used to confirm the results obtained with the DBY746 strain.
Strain EH150 (MATa,lys2ΔBglII,trp1-Δ,his3-Δ200,ura3-52,ade2-1o) was used for the small insertion/deletion mutation assay (provided by E. Heidenreich, Institute of Cancer Research, Medical University of Vienna, Vienna, Austria). The sch9Δ mutant has been described previously (Fabrizio et al., 2001). All the mutant strains were originated in the different backgrounds by one-step gene replacement according to Brachmann et al. (1998). K.A. Morano (University of Texas Medical School, Houston, TX) provided low copy plasmid pRS416-HA3-SCH9. Multicopy 2 μ plasmid carrying REV1 was obtained from Thermo Fisher Scientific. Overexpressor plasmids for SOD2 and SOD1 were constructed in multicopy vectors (YEp352 and YEp351, respectively) and have been described previously (Fabrizio et al., 2003).
Growth conditions
Yeast chronological life span was monitored in expired SDC medium by measuring colony-forming units (CFUs) every 48 h. The number of CFUs at day 1 was considered to be the initial survival (100%) and was used to determine the age-dependent mortality (Fabrizio and Longo, 2003).
Can1 mutation frequency measurements
Spontaneous mutation frequency was evaluated by measuring the frequency of mutations of the CAN1 (YEL063) gene. In brief, overnight inoculations were diluted in liquid SDC medium and incubated at 30°C. The cells' viability was measured every 2 d starting at day 1 by plating appropriate dilutions onto yeast extract peptone dextrose (YPD) medium plates and counting the CFUs. To identify the canavanine-resistant mutants (Canr) in the liquid culture, an appropriate number of cells (starting amount of 2 × 107 cells) was harvested by centrifugation, washed once with sterile water, and plated on selective medium (SDC-Arg supplemented with 60 µg/ml l-canavanine sulfate). Mutant colonies were counted after 3–4 d. The mutation frequency was expressed as the ratio of Canr to total viable cells.
Large-scale measurement of GCRs
To detect GCRs, a DBY476 background strain was generated by the replacement of HXT13 (YEL069), encoding for a highly redundant hexose transporter, with a URA3 cassette (Chen and Kolodner, 1999). HXT13 is located 7.5 kb telomeric to CAN1 on chromosome V. The experiment was conducted similarly to that described for the Can1 mutations, but the detection for the loss of both CAN1 and URA3 was performed on SDC-Arg plates containing 1 mg/ml 5-fluoroorotic acid (5FOA) and 60 µg/ml l-canavanine. Mutant colonies were counted after 3–4 d.
Measurement of age-dependent small insertion/deletion mutations
Based on the experimental design proposed by Heidenreich et al., (2003) and Heidenreich and Wintersberger (1998), we generated sch9Δ mutants in a Lys− strain (EH150) in which a lys2ΔBglII mutation was constructed by filling in a BglII restriction site of the LYS2 gene. The resulting +4 shift in the open reading frame causes an auxotrophy for lysine that can be reversed by small age-dependent insertion/deletion mutations. Cells were plated onto selective SDC-Lys medium. The experiments were performed similarly to the one described for the Can1 mutations.
Measurement of age-dependent single-base substitution mutations
To monitor the frequency of reversion of a base substitution, we used the strain DBY746 that carries a trp 1–289 amber mutation (C → T at residue 403 of the coding sequence) and measured the frequency of trp1-289 to Trp+ reversions (Capizzi and Jameson, 1973). Cells were plated onto selective SDC-Trp medium. The experiments were performed similarly to the one described for the Can1 mutations.
Isolation of quiescent and nonquiescent cells
Percoll density gradient (GE Healthcare) was prepared using the protocol described by Allen et al., (2006). Straight after the separation, cells were plated on YPD and on SDC-Arg medium supplemented with 60 µg/ml l-canavanine sulfate solid media to measure viability (CFUs) and determine mutation frequency, respectively (Madia et al., 2008). Morphological characterization was performed with a microscope (DM IRB; Leica) equipped for phase contrast and fluorescence light microscopy. Cells were stained either with 0.1 mg/ml phloxin B (Sigma-Aldrich) or 25 µM Calcofluor White N2R fluorescent brightener 28 (Invitrogen). Lower- and upper-fraction cells were also examined microscopically, without previous sonication, for the presence of new buds.
FACS analysis and budding index
Cells from exponentially proliferating cultures of wild-type and sch9Δ strains were inoculated into SDC medium at an initial density of 5 × 105/ml, and continuously cultured for 7 d at 30°C with rotary shaking. Aliquots of cells removed from each culture at the indicated times were collected to measure budding index and to measure DNA content by flow cytometry, as described previously (Madia et al., 2008). Data were calculated by MODFIT (Verity Software House).
8-OHdG measurement
8-OHdG content was estimated by an ELISA assay, using a 8-OHdG Check kit (Cosmo Bio Co., Ltd). Nuclear DNA was isolated from wild-type and sch9Δ mutant cells by mechanical release of DNA from cells disrupted with glass beads. About 50 × 108 cells were washed off the medium and suspended in 4 ml of lysis buffer (Tris-EDTA buffer, 1% SDS, 2% Triton X-100, and 100 mM NaCl). DNA was extracted using the standard phenol/chloroform method and finally suspended in Tris-EDTA buffer. 200 µg of DNA were digested with nuclease P1 and alkaline phosphatase prior to the ELISA assay, according to the manufacture's instructions.
CAN1 sequencing
Canavanine-resistant clones from wild-type (DBY746) and sch9Δ strains were collected during a chronological life span study. Genomic DNA was isolated using standard glass beads/chloroform-phenol procedure. Two primer sets were used for PCR amplification to cover the CAN1/YEL063C open reading frame. PCR products were gel purified and sequenced using the amplification primers (both directions). Additional primers were used to confirm the sequencing results when necessary (Madia et al., 2008). All primers were synthesized by Integrated DNA Technologies, Inc.; sequencing was performed by Laragen, Inc. Identification of mutation was performed using Mutation Surveyor version 3.00 (Softgenetics, LLC).
Microarray gene expression analysis
Day 2.5 wild-type and sch9Δ cultures were used to extract total RNA according to the acid phenol method. Total RNA from three independent cultures of each strain was used as a template to synthesize complementary RNA (cRNA). cRNA was hybridized to GeneChip Yeast Genome 2.0 array (Affymetrix). For each wild-type sample, three replicates of a mutant were normalized with respect to the wild-type reference array. Then, the four arrays were summarized by the median polishing method in robust multi-array average (RMA). This leads to three estimates of expression fold changes of the mutant versus the wild type. In total, we had nine estimates of expression fold changes from three wild-type references, and the median was taken as the final estimate (Cheng et al., 2007a,b). The change in expression level between a baseline and an experimental array is included in Table S2.
Quantification of mRNA by real-time PCR
Total RNA was extracted with a standard phenol/chloroform method. First-strand cDNA was synthesized using SuperScript II reverse transcription (Invitrogen) and random primers. Real-time PCR was performed using the BIO-RAD Opticon 2 detection system in the presence of SYBR-green I dye (Bio-Rad laboratory). The forward and reverse primers for REV1 were 5′-GCGAAAAGGATAGTCGCTTG-3′ and 5′-CTTCCATGCGGAGAGATGAT-3′, respectively. The forward and reverse primers for SOD1 were 5′-TTTGATCAAGCTTATCGGTCCT-3′ and 5′-TTGGTTAGACCAATGACACCAC-3′, respectively. The forward and reverse primers for SOD2 were 5′-CTACAACCAGGATACCGTCACA-3′ and 5′-CTTCTGGATGCTTCTTTCCAGT-3′, respectively. The forward and reverse primers for the housekeeping gene ACT1 were 5′-AGCTCCAATGAACCCTAAATCA-3′ and 5′-ACGACGTGAGTAACACCATCAC-3′, respectively. The concentration (ng/µl) of both genes was calculated by reference to the respective standard curve. Relative gene expression was expressed as a ratio of REV1 gene concentration to ACT1 concentration, and the values given represent the percentage of wild type of the mean gene expression ± SEM.
TLS in yeast cell-free nuclear extracts
Yeast cell-free nuclear extracts were prepared according to the established protocol (Wang, 2006). In brief, yeast cells from indicated strains (wild type or sch9Δ) were grown at 30°C in SDC medium until day 3. Cells were harvested by centrifugation for 10 min, washed in water, and suspended at 0.1 g/ml in 0.1M EDTA, pH 8.0/10 mM dithiothreitol buffer. After a 30-min incubation, cells were pelleted and suspended at 1 g/ml in 1 M sorbitol solution, and yeast lytic enzyme (Zymo Research Corporation) was added at 1.4–2.8 mg/g of cells. Spheroplasts were resuspended in Ficoll buffer and lysed with a Teflon glass homogenizer. Nuclei were recovered from the supernatant by at least four consecutive centrifugations and resuspended in lysis buffer. After precipitation with ammonium sulfate, proteins were dialyzed in Hepes buffer. Precipitates formed during dialysis were removed by centrifugation, and the resulting nuclear extracts were stored at −70°C. Extract concentration was quantified using a Pierce BCA protein assay (Thermo Fisher Scientific).
48-mer undamaged or damaged oligonucleotide substrate (48 mer) 5′-TCGATACTGGTACTAATGATTAACGACTXAAGCACGTCCGTACCATCG-3′ (X is either T for the undamaged or tetrahydrofurane-type of abasic site for the damaged template) and a 12-mer primer 5′-CGATGGTACGGA-3′ were synthesized on a 3400 DNA Synthesizer (Applied Biosystems) and purified by denaturing PAGE. TLS reactions (50 µl volume) were performed in a TLS buffer (20 mM Hepes, pH 7.8, 7 mM MgCl2, 1 mM DTT, and 25 mM NaCl) containing 200 µM deoxynucleotide triphosphate, 100 nM of 32P-labeled primer annealed to the template (undamaged or damaged), and the indicated amount of wild-type or sch9Δ nuclear extract (0, 5, 10, or 20 µg). After incubation at 30°C for 30 min, reactions were terminated by addition of 10 mM EDTA and 0.4 mg/ml proteinase K, and incubated for an additional 30 min at 37°C. TLS reaction products were purified by extracting twice with phenol:chloroform:isoamyl alcohol, separated on a 19% denaturing PAGE. Gel band intensities were measured by phosphorimaging with ImageQuant software (GE Healthcare).
Statistical analysis
Longevity curves and mutation frequency curves were analyzed by an unpaired two-tailed t test (P-values were P < 0.001, P < 0.01, and P < 0.05) on the data for each pair of strains at each day. RT-PCR data were analyzed by a one-way analysis of variance (ANOVA) test.
Online supplemental material
Fig. S1 shows the percentage distribution in each cell cycle compartment, as resulted from the FACS analysis from wild-type and SCH9-deficient cultures at early time points, and the cell cycle profiles of quiescent and nonquiescent fractions from wild-type and SCH9-deficient cultures after density gradient separation. Fig. S2 shows the mRNA SODs and REV1 levels in 3-d-old wild-type, sch9Δ, gis1Δ, and gis1Δsch9Δ mutants and in cells overexpressing SCH9 and mRNA SODs and REV1 levels in 3-d-old wild-type and sch9Δ mutants cells treated with 1 mM paraquat for 48 h. Fig. S3 shows the survival and age-dependent mutation frequency of plasmid vectors. Table S1 shows the percentage of early regrowth in wild-type and Sch9-deficient cells. Table S2 shows the microarray gene expression analysis of DNA repair genes in sch9Δ cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200906011/DC1.
Acknowledgments
We thank Dr. E. Heidenreich for providing strain EH150 and Dr. Kevin A. Morano for providing the SCH9 overexpressing plasmid. We thank Dr. Michael Lieber for very helpful comments and Drs. William C. Burhans and Martin Weinberger for their assistance in FACS analysis.
This work was supported in part by an American Federation for Aging Research grant and by National Institutes of Health grants AG20642, AG025135, and GM075308.
Footnotes
Abbreviations used in this paper:
- 5FOA
- 5-fluoroorotic acid
- 8-OHdG
- 8-hydroxy-2′deoxyguanidine
- ANOVA
- analysis of variance
- CFU
- colony-forming unit
- GCR
- gross chromosomal rearrangement
- IGF-I
- insulin-like growth factor 1
- MMS
- methyl methane sulfonate
- SDC
- synthetic dextrose complete
- SOD
- superoxide dismutase
- TLS
- translesion synthesis
References
- Allen C., Büttner S., Aragon A.D., Thomas J.A., Meirelles O., Jaetao J.E., Benn D., Ruby S.W., Veenhuis M., Madeo F., Werner-Washburne M. 2006. Isolation of quiescent and nonquiescent cells from yeast stationary-phase cultures.J. Cell Biol. 174:89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimov V.N., Berstein L.M., Popovich I.G., Zabezhinski M.A., Egormin P.A., Tyndyk M.L., Anikin I.V., Semenchenko A.V., Yashin A.I. 2005. Central and peripheral effects of insulin/IGF-1 signaling in aging and cancer: antidiabetic drugs as geroprotectors and anticarcinogens.Ann. N. Y. Acad. Sci. 1057:220–234 [DOI] [PubMed] [Google Scholar]
- Aragon A.D., Rodriguez A.L., Meirelles O., Roy S., Davidson G.S., Tapia P.H., Allen C., Joe R., Benn D., Werner-Washburne M. 2008. Characterization of differentiated quiescent and nonquiescent cells in yeast stationary-phase cultures.Mol. Biol. Cell. 19:1271–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach P., Bennett R.A., Bailey E.A., Krokan H.E., Demple B. 2005. Mutagenic specificity of endogenously generated abasic sites in Saccharomyces cerevisiae chromosomal DNA.Proc. Natl. Acad. Sci. USA. 102:17711–17716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J., Horejsí Z., Koed K., Krämer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J.M., Lukas C., et al. 2005. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis.Nature. 434:864–870 [DOI] [PubMed] [Google Scholar]
- Brachmann C.B., Davies A., Cost G.J., Caputo E., Li J., Hieter P., Boeke J.D. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications.Yeast. 14:115–132 [DOI] [PubMed] [Google Scholar]
- Brunet A., Sweeney L.B., Sturgill J.F., Chua K.F., Greer P.L., Lin Y., Tran H., Ross S.E., Mostoslavsky R., Cohen H.Y., et al. 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase.Science. 303:2011–2015 [DOI] [PubMed] [Google Scholar]
- Burhans W.C., Weinberger M. 2007. DNA replication stress, genome instability and aging.Nucleic Acids Res. 35:7545–7556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. 2001. From cells to organisms: can we learn about aging from cells in culture? Exp. Gerontol. 36:607–618 [DOI] [PubMed] [Google Scholar]
- Capizzi R.L., Jameson J.W. 1973. A table for the estimation of the spontaneous mutation rate of cells in culture.Mutat. Res. 17:147–148 [DOI] [PubMed] [Google Scholar]
- Chen C., Kolodner R.D. 1999. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants.Nat. Genet. 23:81–85 [DOI] [PubMed] [Google Scholar]
- Cheng C., Fabrizio P., Ge H., Longo V.D., Li L.M. 2007a. Inference of transcription modification in long-live yeast strains from their expression profiles.BMC Genomics. 8:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C., Fabrizio P., Ge H., Wei M., Longo V.D., Li L.M. 2007b. Significant and systematic expression differentiation in long-lived yeast strains.PLoS One. 2:e1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePinho R.A. 2000. The age of cancer.Nature. 408:248–254 [DOI] [PubMed] [Google Scholar]
- Fabrizio P., Longo V.D. 2003. The chronological life span of Saccharomyces cerevisiae.Aging Cell. 2:73–81 [DOI] [PubMed] [Google Scholar]
- Fabrizio P., Longo V.D. 2007. The chronological life span of Saccharomyces cerevisiae.Methods Mol. Biol. 371:89–95 [DOI] [PubMed] [Google Scholar]
- Fabrizio P., Pozza F., Pletcher S.D., Gendron C.M., Longo V.D. 2001. Regulation of longevity and stress resistance by Sch9 in yeast.Science. 292:288–290 [DOI] [PubMed] [Google Scholar]
- Fabrizio P., Liou L.L., Moy V.N., Diaspro A., Valentine J.S., Gralla E.B., Longo V.D. 2003. SOD2 functions downstream of Sch9 to extend longevity in yeast.Genetics. 163:35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P., Battistella L., Vardavas R., Gattazzo C., Liou L.L., Diaspro A., Dossen J.W., Gralla E.B., Longo V.D. 2004. Superoxide is a mediator of an altruistic aging program in Saccharomyces cerevisiae.J. Cell Biol. 166:1055–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P., Gattazzo C., Battistella L., Wei M., Cheng C., McGrew K., Longo V.D. 2005. Sir2 blocks extreme life-span extension.Cell. 123:655–667 [DOI] [PubMed] [Google Scholar]
- Gan G.N., Wittschieben J.P., Wittschieben B.O., Wood R.D. 2008. DNA polymerase zeta (pol zeta) in higher eukaryotes.Cell Res. 18:174–183 [DOI] [PubMed] [Google Scholar]
- Gibbs P.E., Wang X.D., Li Z., McManus T.P., McGregor W.G., Lawrence C.W., Maher V.M. 2000. The function of the human homolog of Saccharomyces cerevisiae REV1 is required for mutagenesis induced by UV light.Proc. Natl. Acad. Sci. USA. 97:4186–4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grollman A.P., Moriya M. 1993. Mutagenesis by 8-oxoguanine: an enemy within.Trends Genet. 9:246–249 [DOI] [PubMed] [Google Scholar]
- Haracska L., Unk I., Johnson R.E., Johansson E., Burgers P.M., Prakash S., Prakash L. 2001. Roles of yeast DNA polymerases delta and zeta and of Rev1 in the bypass of abasic sites.Genes Dev. 15:945–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haracska L., Prakash S., Prakash L. 2002. Yeast Rev1 protein is a G template-specific DNA polymerase.J. Biol. Chem. 277:15546–15551 [DOI] [PubMed] [Google Scholar]
- Heidenreich E., Wintersberger U. 1998. Replication-dependent and selection-induced mutations in respiration-competent and respiration-deficient strains of Saccharomyces cerevisiae.Mol. Gen. Genet. 260:395–400 [DOI] [PubMed] [Google Scholar]
- Heidenreich E., Novotny R., Kneidinger B., Holzmann V., Wintersberger U. 2003. Non-homologous end joining as an important mutagenic process in cell cycle-arrested cells.EMBO J. 22:2274–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlavatá L., Nyström T. 2003. Ras proteins control mitochondrial biogenesis and function in Saccharomyces cerevisiae.Folia Microbiol. (Praha). 48:725–730 [DOI] [PubMed] [Google Scholar]
- Houtgraaf J.H., Versmissen J., van der Giessen W.J. 2006. A concise review of DNA damage checkpoints and repair in mammalian cells.Cardiovasc. Revasc. Med. 7:165–172 [DOI] [PubMed] [Google Scholar]
- Johnson R.E., Washington M.T., Prakash S., Prakash L. 1999. Bridging the gap: a family of novel DNA polymerases that replicate faulty DNA.Proc. Natl. Acad. Sci. USA. 96:12224–12226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S., Valentine M.R., Pham P., O'Donnell M., Goodman M.F. 2002. Fidelity of Escherichia coli DNA polymerase IV. Preferential generation of small deletion mutations by dNTP-stabilized misalignment.J. Biol. Chem. 277:34198–34207 [DOI] [PubMed] [Google Scholar]
- Kovtun I.V., Liu Y., Bjoras M., Klungland A., Wilson S.H., McMurray C.T. 2007. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells.Nature. 447:447–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozmin S.G., Pavlov Y.I., Kunkel T.A., Sage E. 2003. Roles of Saccharomyces cerevisiae DNA polymerases Poleta and Polzeta in response to irradiation by simulated sunlight.Nucleic Acids Res. 31:4541–4552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzer D.A., Essigmann J.M. 1998. Oxidized, deaminated cytosines are a source of C —> T transitions in vivo.Proc. Natl. Acad. Sci. USA. 95:3578–3582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence C.W. 2004. Cellular functions of DNA polymerase zeta and Rev1 protein.Adv. Protein Chem. 69:167–203 [DOI] [PubMed] [Google Scholar]
- Le Page F., Margot A., Grollman A.P., Sarasin A., Gentil A. 1995. Mutagenicity of a unique 8-oxoguanine in a human Ha-ras sequence in mammalian cells.Carcinogenesis. 16:2779–2784 [DOI] [PubMed] [Google Scholar]
- Löbrich M., Jeggo P.A. 2007. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction.Nat. Rev. Cancer. 7:861–869 [DOI] [PubMed] [Google Scholar]
- Lombard D.B., Chua K.F., Mostoslavsky R., Franco S., Gostissa M., Alt F.W. 2005. DNA repair, genome stability, and aging.Cell. 120:497–512 [DOI] [PubMed] [Google Scholar]
- Longo V.D. 2003. The Ras and Sch9 pathways regulate stress resistance and longevity.Exp. Gerontol. 38:807–811 [DOI] [PubMed] [Google Scholar]
- Longo V.D., Finch C.E. 2003. Evolutionary medicine: from dwarf model systems to healthy centenarians? Science. 299:1342–1346 [DOI] [PubMed] [Google Scholar]
- Longo V.D., Lieber M.R., Vijg J. 2008. Turning anti-ageing genes against cancer.Nat. Rev. Mol. Cell Biol. 9:903–910 [DOI] [PubMed] [Google Scholar]
- Madia F., Gattazzo C., Fabrizio P., Longo V.D. 2007. A simple model system for age-dependent DNA damage and cancer.Mech. Ageing Dev. 128:45–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madia F., Gattazzo C., Wei M., Fabrizio P., Burhans W.C., Weinberger M., Galbani A., Smith J.R., Nguyen C., Huey S., et al. 2008. Longevity mutation in SCH9 prevents recombination errors and premature genomic instability in a Werner/Bloom model system.J. Cell Biol. 180:67–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie G.J., Lee P.L., Lombardo M.J., Hastings P.J., Rosenberg S.M. 2001. SOS mutator DNA polymerase IV functions in adaptive mutation and not adaptive amplification.Mol. Cell. 7:571–579 [DOI] [PubMed] [Google Scholar]
- Minesinger B.K., Abdulovic A.L., Ou T.M., Jinks-Robertson S. 2006. The effect of oxidative metabolism on spontaneous Pol zeta-dependent translesion synthesis in Saccharomyces cerevisiae.DNA Repair (Amst.). 5:226–234 [DOI] [PubMed] [Google Scholar]
- Moraes E.C., Keyse S.M., Tyrrell R.M. 1990. Mutagenesis by hydrogen peroxide treatment of mammalian cells: a molecular analysis.Carcinogenesis. 11:283–293 [DOI] [PubMed] [Google Scholar]
- Moriya M. 1993. Single-stranded shuttle phagemid for mutagenesis studies in mammalian cells: 8-oxoguanine in DNA induces targeted G.C—>T.A transversions in simian kidney cells.Proc. Natl. Acad. Sci. USA. 90:1122–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson J.R., Lawrence C.W., Hinkle D.C. 1996. Deoxycytidyl transferase activity of yeast REV1 protein.Nature. 382:729–731 [DOI] [PubMed] [Google Scholar]
- Nelson J.R., Gibbs P.E., Nowicka A.M., Hinkle D.C., Lawrence C.W. 2000. Evidence for a second function for Saccharomyces cerevisiae Rev1p.Mol. Microbiol. 37:549–554 [DOI] [PubMed] [Google Scholar]
- Ohmori H., Friedberg E.C., Fuchs R.P., Goodman M.F., Hanaoka F., Hinkle D., Kunkel T.A., Lawrence C.W., Livneh Z., Nohmi T., et al. 2001. The Y-family of DNA polymerases.Mol. Cell. 8:7–8 [DOI] [PubMed] [Google Scholar]
- Pagès V., Johnson R.E., Prakash L., Prakash S. 2008. Mutational specificity and genetic control of replicative bypass of an abasic site in yeast.Proc. Natl. Acad. Sci. USA. 105:1170–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedruzzi I., Bürckert N., Egger P., De Virgilio C. 2000. Saccharomyces cerevisiae Ras/cAMP pathway controls post-diauxic shift element-dependent transcription through the zinc finger protein Gis1.EMBO J. 19:2569–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkston J.M., Garigan D., Hansen M., Kenyon C. 2006. Mutations that increase the life span of C. elegans inhibit tumor growth.Science. 313:971–975 [DOI] [PubMed] [Google Scholar]
- Pollak M.N., Schernhammer E.S., Hankinson S.E. 2004. Insulin-like growth factors and neoplasia.Nat. Rev. Cancer. 4:505–518 [DOI] [PubMed] [Google Scholar]
- Prakash S., Johnson R.E., Washington M.T., Haracska L., Kondratick C.M., Prakash L. 2000. Role of yeast and human DNA polymerase eta in error-free replication of damaged DNA.Cold Spring Harb. Symp. Quant. Biol. 65:51–59 [DOI] [PubMed] [Google Scholar]
- Prakash S., Johnson R.E., Prakash L. 2005. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function.Annu. Rev. Biochem. 74:317–353 [DOI] [PubMed] [Google Scholar]
- Quah S.K., von Borstel R.C., Hastings P.J. 1980. The origin of spontaneous mutation in Saccharomyces cerevisiae.Genetics. 96:819–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P., Warne P.H., Dhand R., Vanhaesebroeck B., Gout I., Fry M.J., Waterfield M.D., Downward J. 1994. Phosphatidylinositol-3-OH kinase as a direct target of Ras.Nature. 370:527–532 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P., Tetsu O., Oda K., Okada J., Rauen K., McCormick F. 2005. Cancer targets in the Ras pathway.Cold Spring Harb. Symp. Quant. Biol. 70:461–467 [DOI] [PubMed] [Google Scholar]
- Sabbioneda S., Bortolomai I., Giannattasio M., Plevani P., Muzi-Falconi M. 2007. Yeast Rev1 is cell cycle regulated, phosphorylated in response to DNA damage and its binding to chromosomes is dependent upon MEC1.DNA Repair (Amst.). 6:121–127 [DOI] [PubMed] [Google Scholar]
- Shen W.H., Balajee A.S., Wang J., Wu H., Eng C., Pandolfi P.P., Yin Y. 2007. Essential role for nuclear PTEN in maintaining chromosomal integrity.Cell. 128:157–170 [DOI] [PubMed] [Google Scholar]
- Skeen J.E., Bhaskar P.T., Chen C.C., Chen W.S., Peng X.D., Nogueira V., Hahn-Windgassen A., Kiyokawa H., Hay N. 2006. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner.Cancer Cell. 10:269–280 [DOI] [PubMed] [Google Scholar]
- Tang M., Pham P., Shen X., Taylor J.S., O'Donnell M., Woodgate R., Goodman M.F. 2000. Roles of E. coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis.Nature. 404:1014–1018 [DOI] [PubMed] [Google Scholar]
- Toda T., Cameron S., Sass P., Wigler M. 1988. SCH9, a gene of Saccharomyces cerevisiae that encodes a protein distinct from, but functionally and structurally related to, cAMP-dependent protein kinase catalytic subunits.Genes Dev. 2:517–527 [DOI] [PubMed] [Google Scholar]
- Toker A., Yoeli-Lerner M. 2006. Akt signaling and cancer: surviving but not moving on.Cancer Res. 66:3963–3966 [DOI] [PubMed] [Google Scholar]
- Urban J., Soulard A., Huber A., Lippman S., Mukhopadhyay D., Deloche O., Wanke V., Anrather D., Ammerer G., Riezman H., et al. 2007. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae.Mol. Cell. 26:663–674 [DOI] [PubMed] [Google Scholar]
- Vergara M., Smith-Wheelock M., Harper J.M., Sigler R., Miller R.A. 2004. Hormone-treated snell dwarf mice regain fertility but remain long lived and disease resistant.J. Gerontol. A Biol. Sci. Med. Sci. 59:1244–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijg J. 2008. The role of DNA damage and repair in aging: new approaches to an old problem.Mech. Ageing Dev. 129:498–502 [DOI] [PubMed] [Google Scholar]
- Wang Z. 2006. Controlled expression of recombinant genes and preparation of cell-free extracts in yeast.Methods Mol. Biol. 313:317–331 [DOI] [PubMed] [Google Scholar]
- Wang D., Kreutzer D.A., Essigmann J.M. 1998. Mutagenicity and repair of oxidative DNA damage: insights from studies using defined lesions.Mutat. Res. 400:99–115 [DOI] [PubMed] [Google Scholar]
- Washington M.T., Johnson R.E., Prakash S., Prakash L. 1999. Fidelity and processivity of Saccharomyces cerevisiae DNA polymerase eta.J. Biol. Chem. 274:36835–36838 [DOI] [PubMed] [Google Scholar]
- Waters L.S., Walker G.C. 2006. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase.Proc. Natl. Acad. Sci. USA. 103:8971–8976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters L.S., Minesinger B.K., Wiltrout M.E., D'Souza S., Woodruff R.V., Walker G.C. 2009. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance.Microbiol. Mol. Biol. Rev. 73:134–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M., Fabrizio P., Hu J., Ge H., Cheng C., Li L., Longo V.D. 2008. Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9.PLoS Genet. 4:e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M., Fabrizio P., Madia F., Hu J., Ge H., Li L.M., Longo V.D. 2009. Tor1/Sch9-regulated carbon source substitution is as effective as calorie restriction in life span extension.PLoS Genet. 5:e1000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger M., Feng L., Paul A., Smith D.L., Jr., Hontz R.D., Smith J.S., Vujcic M., Singh K.K., Huberman J.A., Burhans W.C. 2007. DNA replication stress is a determinant of chronological lifespan in budding yeast.PLoS One. 2:e748. [DOI] [PMC free article] [PubMed] [Google Scholar]