

Abstract

Botulinum neurotoxins (BoNTs), etiological agents of the deadly food poisoning disease botulism, are the most toxic proteins currently known. By using in situ lead identification chemistry we have uncovered the first class of inhibitors that display nanomolar potency. From a 15 µM lead compound, structure activity relationship studies were performed granting the most potent BoNT/A inhibitor reported to date that displays an inhibition constant of 300 nM.
Botulinum neurotoxin (BoNT), an agent responsible for the deadly food poisoning disease botulism and a dreaded biological weapon, is one of the most toxic proteins currently known (~100 billion times more toxic than cyanide).1 Clostridium botulinum is classified into seven strains (A–G) each of which can cause flaccid muscle paralysis and subsequent death by blocking the release of a neurotransmitter, acetylcholine, at neuromuscular junctions.2 Structurally, BoNT consists of three functional domains; catalytic, translocation, and binding; BoNT toxicity results from the catalytic activity of its light chain, a Zn(II) endopeptidase.
The catalytic domain of BoNT is a compact globule consisting of a mixture of α-helices, β-sheets and strands with a gorge-like zinc containing metalloprotease active site (15–20Å deep depending on serotype).3 The metalloprotease activity is responsible for BoNT’s neurotoxicity through the hydrolytic cleavage of one of three SNARE (soluble NSF-attachment protein receptor) proteins that are involved in neuronal synaptic vesicle function. Moreover, the hydrolytic cleavage sites of these SNARE proteins (SNAP-25, VAMP, Sb-1) differ across the BoNT serotypes; however, any degradation of these SNARE proteins disables the exocytosis of acetylcholine, resulting in paralysis and potentially death.2 Current therapy for BoNT intoxication involves “passive immunization” with equine antitoxin.4 Unfortunately, treatment must start shortly after intoxication, and several safety concerns exist2 over the use of antitoxins in the general population.5 Therefore, inhibition of the catalytic light chain protease with a small molecule inhibitor may provide an attractive approach to counter the effects of botulism poisoning.
BoNT serotype A (BoNT/A) is the most toxic form of BoNT’s and is considered the most threatening for biological attacks due to a prolonged half-life in vivo and ease of its production.4 While there are reports of success treating BoNT/A toxicity with multiple monoclonal antibodies6 as antitoxins this is of limited therapeutic utility since the antibodies must be administered prior to, or shortly after, toxin exposure (<12 hrs).
Presently, there are only modest small molecule, non-peptidic, protease inhibitors for BoNT/A with IC50 values in the range of > 20 µM.7 We established a high-throughput screen for the identification of inhibitors of BoNT/A LC protease.8 Using this screen we have analyzed a library of hydroxamate-based compounds generated using in situ chemistry to reveal the lead structure 1, (Figure 1). Herein, we report upon the synthesis and structure activity relationship studies of these BoNT/A inhibitors.
Figure 1.

Structures of lead compounds identified from in situ screen.
Recently, a convenient method for the preparation of hydroxamates from readily available esters was reported.9 In this general procedure a diverse array of acids can easily be converted to hydroxamates with hydoxylamine in the presence of a catalytic amount of potassium cyanide. In addition, several reports have been disclosed for the preparation of hydroxamates;10 however, these procedures have strict substrate requirements.
With these thoughts in mind, we set out to generate a library of diverse hydroxamates from a readily available carboxylic acid library. Namely, 150 carboxylic acids purchased from Aldrich Chemical Company were randomly chosen and converted to their ester by treatment with diazomethane (Scheme 1). After removal of the solvent, reactions were subjected to a mixture of THF:MeOH:HONH2 1:1:2 with a catalytic amount of KCN overnight. Again, the solvent was removed and the crude products were reconstituted with DMSO to prepare stock solutions of a final concentration of 10 mM for screening. To check the quality of the library 30 compounds were randomly selected and analyzed by ES-MS, in all cases the expected masses corresponding to the products were found (data not shown).
Scheme 1.

Synthesis and screening of in situ hydroxamate library.
Using a high-throughput screen,8 namely, a 17 amino acid substrate fluorescence resonance energy transfer (FRET) assay developed in our laboratory, the library of hydroxamates were analyzed at a concentration of 50 µM. Any inhibitors found to display 50% inhibition were considered “hits” and evaluated further. From the initial screen five compounds were found to give 50% or more inhibition in the FRET-based assay. These compounds were resynthesized, purified and validated using the FRET-based assay. Of the five lead compounds, only compound 1 showed potent activity; para-chloro-cinnamic hydroxamte (1) displayed an IC50 of 15 µM, and thus this simple high-throughput screen uncovered an interesting lead for further development.
To refine this positive lead we synthesized a series of 12 compounds so as to further explore the structure activity relationship of compound 1. However, the method outlined in Scheme 1 was found to be inefficient for the isolation of the desired compounds due to purification difficulties. Therefore, we sought out a more convenient method for the expeditious generation and purification of the hydroxamate compounds. Solid-phase organic synthesis (SPOS) lends itself to these requirements nicely due to reactions proceeding in high yield, and purification being significantly simplified. Several groups have demonstrated successful hydroxamate syntheses using solid phase routes11 and hydroxyamine resins.12 However, we found these procedures to be insufficient for our purposes. 2-Chlorotrityl resin was purchased from Chemical and Biopharmaceutical Laboratories of Patras S. A., (CBL), and used in the synthesis of hydroxamate inhibitors as outlined in Scheme 2. It is important to note that several resins were explored using this methodology, however, the CBL was the only resin found to give satisfactory results. This resin was treated with N-hydroxyphthalimide for 2 d in DMF at room temperature for the quantitative conversion to 6. The phthalimide was successfully removed with exposure to anhydrous hydrazine to give the hydroxylamine resin 7, ready for the coupling of a variety of acids. Next, the desired carboxylic acids were coupled to the resin 7 using standard diisopropylcarbodiimide, and 6-chlorohydroxybenzotriazole protocols. The desired hydroxamates were recovered by treatment of the resin with 5% TFA and removal of the solvent.
Scheme 2.

Solid-phase synthesis of hydroxamic acids
These compounds were further individually purified via silica gel, then reconstituted in DMSO for evaluation of BoNT/A LC inhibition.
In order to investigate the structure activity relationship of compound 1 we first synthesized a group of para substituted cinnamic hydroxamates. Unexpectedly, the chloro substitution in the para position appears to be highly conserved as seen in Table 1 compounds 10–16. For example, when a bulky functionality such as a t-butyl, compund 14, is introduced to the para position, inhibition is completely lost. Moreover, inhibition is compromised when introducing electron withdrawing or donating groups (compounds 10–16). Thus, we next examined other possible chloro substitution patterns of the lead compound 1. Gratifyingly, when evaluating different chloro substitution patterns, (compounds 17 and 18), the orthopara cinnamic hydroxamate, 18, displayed an IC50 of 0.41 µM. Furthermore, it appears the trans olefin motif is also conserved as seen in compounds 19, 20, and 21. It is important to note, the cis-olefin form of , 1, was not examined due to instability in solution.13 However, strikingly, when the trans olefin is substituted for an alkyne, 20, inhibition was attenuated by greater than 175-fold. The reduced form of 18 to the alkane, compound 19, also reduced potency significantly.
Table 1.
IC50 values.a
| compound | structure | IC50 µM |
|---|---|---|
| 10 | 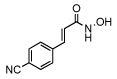 |
61 ± 4 |
| 11 | 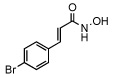 |
40 ± 5 |
| 12 |  |
54 ± 8 |
| 13 |  |
79 ± 8 |
| 14 | 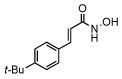 |
>100 |
| 15 | 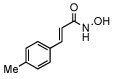 |
87 ± 6 |
| 16 |  |
72 ± 5 |
| 17 |  |
75 ± 4 |
| 18 | 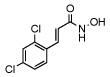 |
0.41 ± 0.03 |
| 19 | 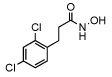 |
3.1 ± 0.9 |
| 20 |  |
73 ± 5 |
| 21 |  |
>100 |
Assays were conducted at various inhibitor concentrations at 22.5 °C, pH 7.4 in 40 mM HEPES 0.01% (W/V) Tween® 20, 5 µM SNAPtide substrate and 200 nM enzyme.
To further kinetically characterize 18, we evaluated it in an assay system with the native SNAP-25 (141–206) substrate using HPLC for analysis. It is important to note, that the SNAP-25 (141–206) has no structural modification, thus the structural integrity of the molecule has not been compromised. Using this substrate, a Ki of 300 nM ± 12 nM was found, and also displayed the expected competitive mode of inhibition (data not shown). Moreover, these findings demonstrate that a 10 contiguous carbon chain previously thought to be a strict requirement for BoNT/A LC inhibition14 is in fact not a prerequisite and small organic molecules can be used for possible drug leads.
In total, a simple in situ synthesis and screen was developed for the identification of non-peptidic protease inhibitors of BoNT/A LC. The power of using this strategy was that no prior bias was placed on the selection of input acids, yet, a desirable lead was uncovered. We note that compound 18, Table 1, is the most potent protease inhibitor described to date for BoNT/A and is currently being investigated in cell and mouse assays for anti-botulism effects and will be reported in due course.
Supplementary Material
Experimental synthetic procedures and NMR, HRMS data for the library members and intermediates and screening procedures. This material is available free of charge via the internet at http://pubs.acs.org.
Acknowledgments
This work was supported by the National Institute of Health BT010-04 and The Skaggs Institute for Chemical Biology.
References
- 1.Singh BR. Nature. 2000;7:617–619. doi: 10.1038/77900. [DOI] [PubMed] [Google Scholar]
- 2.Simpson LL. Annu. Rev. Pharmacol. Toxicol. 2004;44:167–193. doi: 10.1146/annurev.pharmtox.44.101802.121554. [DOI] [PubMed] [Google Scholar]
- 3.Lacy BD, Tepp W, Cohen AC, DasGupta BR, Stevens RC. Nat. Struct. Biol. 1998;5:898–902. doi: 10.1038/2338. [DOI] [PubMed] [Google Scholar]
- 4.Hicks RP, Hartell MG, Nichols DA, Bhattacharjee AK, Von Hamont JE, Skillman DR. Curr. Med. Chem. 2005;12:667–690. doi: 10.2174/0929867053202223. [DOI] [PubMed] [Google Scholar]
- 5.Arnon SS, Schechter R, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Hauer J, Layton M, Lillibridge S, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Swerdlow DL, Tonat K. JAMA. 2001;285:1059–1070. doi: 10.1001/jama.285.8.1059. [DOI] [PubMed] [Google Scholar]
- 6.(a) Amersdorfer P, Wong C, Smith T, Chen S, Deshpande S, Sheridan R, Marks JD. Vaccine. 2002;20:1640–1648. doi: 10.1016/s0264-410x(01)00482-0. [DOI] [PubMed] [Google Scholar]; (b) Nowakowski A, Wang C, Powers DB, Amersdorfer P, Smith TJ, Montgomery VA, Sheridan R, Blake R, Smith LA, Marks JD. Proc. Natl. Acad. Sci. USA. 2002;99:11346–11350. doi: 10.1073/pnas.172229899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Burnett JC, Schmidt JJ, Connor F, McGrath CF, Nguyen TL, Hermone AR, Panchal RG, Vennerstrom JL, Kodukula K, Zaharevitz DW, Gussio R, Bavari S. Bioorg. Med. Chem. 2005;13:333–341. doi: 10.1016/j.bmc.2004.10.026. [DOI] [PubMed] [Google Scholar]; (b) Burnett JC, Schmidt JJ, Stafford RG, Panchal RG, Nguyen TL, Hermone AR, Vennerstrom JL, McGrath CF, Lane DJ, Sausville EA, Zaharevitz DW, Gussio R, Bavari S. Biochem. Biophys. Res. Commun. 2003;310:84–93. doi: 10.1016/j.bbrc.2003.08.112. [DOI] [PubMed] [Google Scholar]
- 8.Boldt GE, Kennedy JP, Hixon MS, McAllister LA, Barbieri JT, Tzipori S, Janda KD. J. Comb. Chem. 2006 doi: 10.1021/cc060010h. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho CY, Strobel E, Ralbovsky J, Galemmo RA. J. Org. Chem. 2005;70:4873–4875. doi: 10.1021/jo050036f. [DOI] [PubMed] [Google Scholar]
- 10.(a) Burns CJ, Groneberg RD, Salvino JM, McGeehan G, Condon SM, Morris R, Morrissette M, Mathew R, Darnbrough S, Neuenschwander K, Scotese A, Djuric S, Ullrich J, Labaudiniere R. Angew. Chem., Int. Ed. 1998;37:2848–2850. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2848::AID-ANIE2848>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]; (b) Mori K, Koseki K. Tetrahedron. 1988;44:6013–6020. [Google Scholar]; (c) Spengler J, Burger K. Synthesis. 1998;1:67–70. [Google Scholar]
- 11.(a) Dankwardt SM. Synlett. 1998;7:761. [Google Scholar]; (b) Dankwardt SM, Billedeau RJ, Lawley LK, Abbot SC, Martin RL, Chan CS, Van Wart HE, Walker KE. Bioorg. Med. Chem. Lett. 2000;10:2513–2516. doi: 10.1016/s0960-894x(00)00525-4. [DOI] [PubMed] [Google Scholar]
- 12.(a) Barlaam B, Koza P, Berriot J. Tetrahedron. 1999;55:7221–7232. [Google Scholar]; (b) Mellor SL, McGuire C, Chan WC. Tetrahedron Lett. 1997;38:3311–3314. [Google Scholar]; (c) Floyd CD, Lewis CN, Patel SR, Whittaker M. Tetrahedron Lett. 1996;37:8045–8048. [Google Scholar]; (d) Ede NJ, James IW, Krywuth BM, Griffiths RM, Eagle SN, Gubbins B, Leitch JA, Sampson WR, Bray AM. Lett. Pep. Sci. 1999;6:157–163. [Google Scholar]; (e) Bauer U, Ho W-B, Koskinen AMP. Tetrahedron Lett. 1997:7233–7236. [Google Scholar]
- 13.Cis-2,4, dichloro-cinnamic acid was found to be unstable during coupling.
- 14.Park JG, Sill PC, Makiyi EF, Garcia-Sosa AT, Millard CB, Schmidt JJ, Pang YP. Bioorg. Med. Chem. 2006;14:395–408. doi: 10.1016/j.bmc.2005.08.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental synthetic procedures and NMR, HRMS data for the library members and intermediates and screening procedures. This material is available free of charge via the internet at http://pubs.acs.org.