

Abstract
A number of mouse models expressing mutant huntingtin (htt) with an expanded polyglutamine (polyQ) domain are useful for studying the pathogenesis of Huntington's disease (HD) and identifying appropriate therapies. However, these models exhibit neurological phenotypes that differ in their severity and nature. Understanding how transgenic htt leads to variable neuropathology in animal models would shed light on the pathogenesis of HD and help us to choose HD models for investigation. By comparing the expression of mutant htt at the transcriptional and protein levels in transgenic mice expressing N-terminal or full-length mutant htt, we found that the accumulation and aggregation of mutant htt in the brain is determined by htt context. HD mouse models demonstrating more severe phenotypes show earlier accumulation of N-terminal mutant htt fragments, which leads to the formation of htt aggregates that are primarily present in neuronal nuclei and processes, as well as glial cells. Similarly, transgenic monkeys expressing exon-1 htt with a 147-glutamine repeat (147Q) died early and showed abundant neuropil aggregates in swelling neuronal processes. Fractionation of HD150Q knock-in mice brains revealed an age-dependent accumulation of N-terminal mutant htt fragments in the nucleus and synaptosomes, and this accumulation was most pronounced in the striatum due to decreased proteasomal activity. Our findings suggest that the neuropathological phenotypes of HD stem largely from the accumulation of N-terminal mutant htt fragments and that this accumulation is determined by htt context and cell-type-dependent clearance of mutant htt.
INTRODUCTION
Huntington's disease (HD) is caused by expansion of a polyglutamine (polyQ) tract in the N-terminal region of huntingtin (htt) (1,2). Htt is a large, 350 kDa protein that is ubiquitously expressed and interacts with many other proteins (3). However, a pathologic feature of HD is selective neurodegeneration that preferentially occurs in the striatum and extends to various brain regions as the disease progresses (4,5). Exactly how mutant htt induces this selective neurodegeneration remains unclear.
Various HD mouse models have been established. These models include transgenic mice (R6/2 and N171-82Q) expressing N-terminal mutant htt (6,7), full-length mutant htt transgenic mice [YAC128 (yeast artificial chromosome transgenic mice)] (8), and HD repeat knock-in (KI) mice (9–11). HD mouse models provide clear evidence that small N-terminal htt fragments with expanded polyQ tracts become misfolded and form aggregates. Consistently, polyQ-containing N-terminal htt fragments form aggregates in HD cellular models (12–14) and in HD patient brains (15,16).
R6/2 and N171-82Q mice display severe neurological symptoms and earlier death, and stereological analysis revealed that the estimated striatal neuronal count is reduced by 25% in the R6/2 mice relative to wild-type (WT) control mice (17). Shortstop (SS) mice, which express the first 117 amino acids of htt with a 120-glutamine repeat (120Q), do not show phenotypes despite the presence of abundant htt aggregates in the brain (18). YAC128 mice express full-length mutant human htt with a 120Q, however, display selective neuronal loss in the striatum and neurological symptoms (8). Importantly, eliminating caspase-6 cleavage in mutant htt can ameliorate HD pathology in transgenic YAC mice (19). We have recently established transgenic monkeys expressing exon-1 mutant htt, which exhibit HD-like symptoms and die early (20). All these facts support the notion that cleavage of full-length mutant htt is a critical step toward HD neuropathology (21,22). Although obvious neurological symptoms or neuropathology have not been found in some transgenic mouse models expressing small N-terminal htt fragments (18,23,24), a careful analysis of HD150Q KI mice at 22 months demonstrated that these HD mice develop the well-characterized molecular phenotypes seen in R6/2 mice at the age of 12 weeks (25). Hence an important question is how mutant htt causes the variable neuropathology in different HD mouse models.
In this study, we compared the expression and distribution of transgenic htt in different HD animal models. Our findings show that protein context of htt in HD models greatly influences the accumulation of N-terminal htt and neurological phenotypes. Also, the age-dependent and cell-type-specific accumulation of N-terminal htt fragments correlates with selective neuropathology. These findings offer new insight into the pathogenesis of HD and help us investigate different HD mouse models.
RESULTS
Transcriptional levels of mutant htt in various HD mouse models
We examined several HD mouse models expressing different forms of mutant htt that have been well characterized in terms of their neuropathology and phenotypes (Table 1). Among these, R6/2 mice express exon-1 (1–67 amino acids) of human htt containing 115–150Q under the control of the human HD promoter (6). N171-82Q mice express the first 171 amino acids of human htt with 82Q under the control of mouse prion promoter (7). Three YAC transgenic mouse models were also analyzed; these express the first 117 amino acids of human N-terminal htt (SS) (18), full-length human htt (YAC128) (8), and caspase-6-resistant full-length mutant htt (C6R) (19). All of the YAC transgenic mice carry a 120–133Q repeat in human htt that is expressed under the control of the human HD promoter. In addition, we examined HD150Q KI mice that express full-length mouse htt carrying a 150Q repeat under the control of the endogenous mouse HD promoter (10). PCR (polymerase chain reaction) genotyping verified that these HD mice have CAG repeat expansions of the expected sizes as reported previously. Due to the instability of the CAG repeat, R6/2 mice maintained by us showed an ∼110–120 CAG repeat (Supplementary Material, Fig. S1).
Table 1.
Varying phenotypes of HD mouse models
| Model | Htt | PolyQ repeat | Promoter (mouse strain) | Phenotypes detected at weeks |
|||
|---|---|---|---|---|---|---|---|
| Early death | Fail to gain weight | Motor deficita | Degeneration | ||||
| R6/2 | 1–67 | 115–156 | Human htt (CBAxC57BL/6) | 12–14 | 7 | 5–6 | 10 (17,56,57) |
| SS | 1–117 | 120 | YAC human htt (FVB/N) | No | No | ND | ND (18) |
| N171 | 1–171 | 82 | Mouse prion (C3HxC57BL/6) | 24–30 | 8–10 | 12 | 16–20 (58,59) |
| YAC | 1–3144 | 120 | YAC human htt (FVB/N) | No | No | 8–12 | 36–48 (8,31) |
| C6R | 1–3144 | 133 | YAC human htt (FVB/N) | No | No | ND | ND (19) |
| KI | 1–3144 | 150 | Mouse htt (C57BL/6J) | No | 48 | 20–32 (25) | 100 (60) |
ND, not detected.
aMotor deficit include decreased mobility, abnormal grip strength, or poor rotarod performance.
We first compared the transcription levels of mutant htt in these mice. Because the sizes of the transcripts for mutant htt in these HD mouse models differ significantly (from exon-1 to full-length) and this size difference can greatly influence RNA (ribonucleic acid) transfer onto the blot membrane and probe labeling intensity, it is difficult to use northern blotting to compare the levels of transcripts of different sizes. Also, detecting exon-1 htt transcript may require a probe that contains the CAG repeat, which would give a different labeling intensity for different repeat lengths. In addition, northern blotting is less quantifiable than quantitative reverse transcription(qRT)–PCR. Given these difficulties with northern blotting, we elected to perform RT–PCR and quantitative PCR to analyze the expression levels of transcripts of mutant htt. We isolated RNAs from the prefrontal cortical region in different HD mice brains that had been frozen and stored at −80°C. RT was carried out using oligo dT, random primers, and primers specific for htt. The housekeeping gene (glyceraldehyde-3-phosphate dehydrogenase, GAPDH) was also amplified in the same reaction to serve as an internal control for assessing the quantity of RNA and to rule out variations due to RNA preparation and PCR amplification.
All the primers used for PCR yielded PCR products lacking the CAG repeat, thereby avoiding any influence of repeat length on PCR efficiency. One set of primers was specific to human htt (459S and 565A Fig. 1A). The other set of primers was derived from the common sequences of human and mouse htt (670S and 790A Fig. 1A). Thus, the common primers also amplify the endogenous mouse htt transcripts, which serve as another control for evaluating the relative levels of additional transgenic htt transcripts. Because both exon-1 htt and transgenic htt in SS mice lack such common sequences, their transcriptional levels cannot be detected by the common primers. Controls without reverse transcriptase showed no detectable levels in PCR results with either the primers specific to human htt or the primers common to both mouse and human htt, indicating that the PCR products are derived from transcripts.
Figure 1.
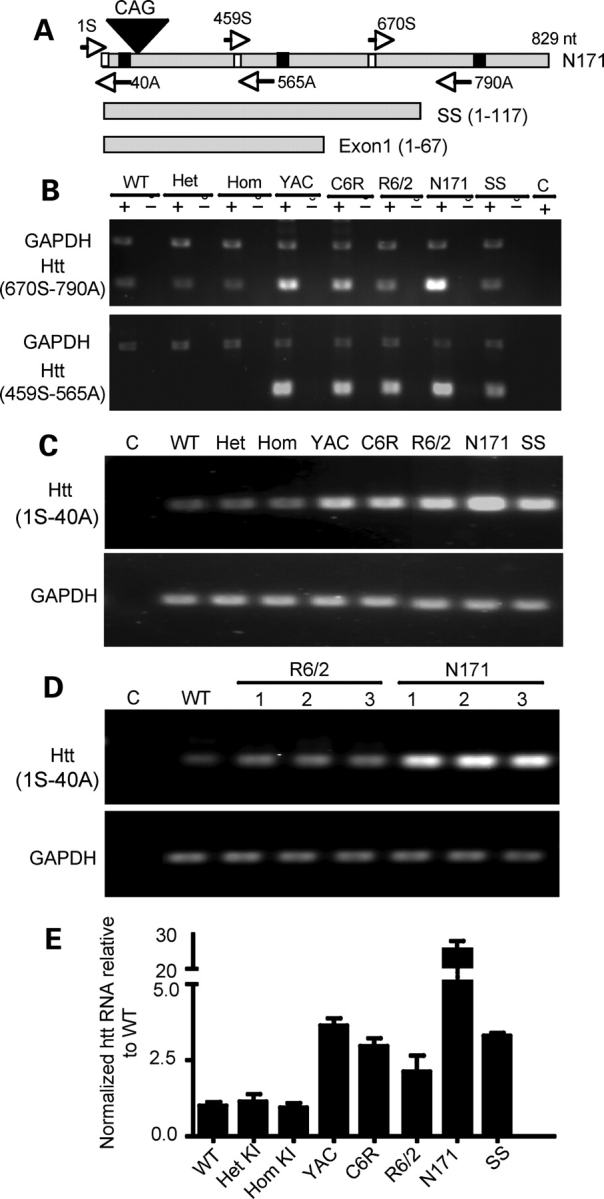
Transcript expression of mutant htt in various HD mouse models. (A) Primers used for RT–PCR. The forward primer 670S and the reverse primer 790A are capable of amplifying both mouse and human htt cDNAs, whereas the forward primer 459S and the reverse primer 565A are specific for human htt. Note that 790A cannot be used to amplify transgenic htt in SS and exon-1 htt. (B) RT–PCR results obtained with 670S and 790A primers for mouse and human htt (upper panel) or the primers (459S and 565A) specific to human htt (lower panel). RT–PCR was performed with (+) or without (−) reverse transcriptase. The same PCR reactions also included primers to amplify GAPDH. Control (C) is PCR without a reverse transcriptase. (C) Gel result of qRT–PCR using primers (1S and 40A). PCR of GAPDH was also performed to verify the quantity of cDNA used in each reaction (lower panel). (D) Repeating PCR of the brain tissues from different mice confirmed that N171-82Q transcripts are higher than transgenic htt transcripts in R6/2 mice (lower panel). (E) qRT–PCR data showing the relative levels (mean ± SE., n = 3) of mutant htt transcripts in the cerebral cortex tissues of various HD mice. KI, HD150Q knock-in; YAC, YAC128 transgenic mice; C6R, caspase-6-resistant YAC transgenic mice; SS, shortstop. Agarose gel [1.5% for (B) and 2.2% for (C–D)] electrophoresis was performed to reveal PCR products.
As expected, use of the primers common to mouse and human htt detected equal levels of endogenous mouse htt in WT, heterozygous or homozygous KI, R6/2 and SS mice brains (upper panel in Fig. 1B). N171-82Q, C6R and YAC128 transgenic mouse samples showed increased levels of PCR products due to the expression of additional transgenic human htt. The N171-82Q mouse brain sample displayed the highest transcription level, as GAPDH levels are similar among all HD mouse brain samples (upper panel in Fig. 1B). We then used primers specific for human htt in PCR to analyze the levels of transgenic human htt. Transcripts in N171-82Q mice also appeared to be higher than in other mice, such as SS and R6/2 (lower panel in Fig. 1B).
To quantify the levels of htt transcripts, we performed real-time PCR using different primers (S1 and A40) that generate small PCR products for quantification and are also capable of amplifying both mouse and human htt. We first verified that quantitative PCR could reveal equal amounts of endogenous GAPDH (lower panel in Fig. 1C) while detecting different levels of transgenic htt in various HD mouse models (upper panel in Fig. 1C). Analysis of multiple samples of R6/2 and N171-82Q mice brains confirmed the higher levels of N171-82Q transcripts (Fig. 1D). With this validation, we performed quantitative PCR and found that WT, heterozygous and homozygous KI mice brains have similar levels of htt. Transcripts of transgenic htt in other HD mouse models are all above the endogenous htt levels seen in WT or KI mice brains. Again, much higher levels of htt transcripts were detected in N171-82Q mice brains (Fig. 1E). The results suggest that the descending order for levels of transcripts is N171-82Q, YAC128, SS, C6/R and R6/2.
Expression of mutant htt at the protein level in HD mice brains
Next we compared the expression of mutant htt at the protein level in HD mice brains. Because polyQ expansion alters protein conformation and immunoreactivity to antibodies, we employed three commonly used antibodies in western blotting. One is the mouse antibody 1C2, which reacts with expanded polyQ tracts (26). The second is the mouse antibody 2166, which recognizes an epitope between residues 414 and 503 in htt (27). The third is rabbit EM48, which was generated previously by us using the first 256 amino acids of truncated htt without the polyQ tract (16). As expected, 1C2 only recognized mutant htt with an expanded polyQ, as the same immunoreactive products were not present in the WT mouse sample (Fig. 2A). Noticeably, there were multiple smaller 1C2 fragments in all HD mice that express full-length mutant htt, supporting the previous findings that cleavage and processing of mutant htt yield multiple N-terminal htt fragments (19,21,22,28). The sizes of N-terminal htt fragments in C6R brains appear to be larger than those in YAC128 brains. This is because the CAG repeat in the transgenic htt gene in C6R mice is larger than that in YAC128 mice (Supplementary Material, Fig. S1), such that the resulting larger polyQ tract yielded slower migration of htt fragments in the SDS gel. Small bands (65 and 55 kDa) in YAC128 and C6R mice were not seen in HD150Q KI mice brains, though other htt bands in HD150Q KI mice brains show intense immunoreactive signals with 1C2. Thus, the small htt fragments in YAC transgenic mice might be generated from cleavage of specific human htt amino acids. For transgenic mice expressing N-terminal htt fragments, 1C2 seemed to react more intensely with SS htt than exon-1 htt and N171-82Q htt.
Figure 2.

Western blot analysis of htt expression in the cerebral cortex of various HD mouse models. Whole cell lysates of cerebral cortex tissues from WT and different HD mouse models were analyzed via western blotting with 1C2 (A), 2166 (B), and EM48 (C) antibodies. KI, HD150Q knock-in; YAC, YAC128 transgenic mice; C6R, caspase-6-resistant YAC transgenic mice; SS, shortstop. R6/2 (1-month-old) and other HD mice (3-month-old) were examined. For 1C2 and 2166 immunoblots, short exposed blots (lower panels) are shown to distinguish between endogenous and transgenic htt (arrows). mEM48 was used for immunostaining of N-terminal htt fragments, and the blot was also probed with anti-tubulin.
The mouse antibody 2166 has also been widely used to detect mutant htt. However, there is no size difference in a number of small immunoreactive bands between WT and HD mice brains (Fig. 2B). This suggests that 2166 is unable to specifically recognize N-terminal htt fragments containing a polyQ repeat. Despite this, this antibody appears to react equally with full-length normal and mutant htt (lower panel in Fig. 2B), as similar 2166 immunoreactive signals for normal and mutant htt were seen in a heterozygous KI mouse brain that carries a single copy of normal and mutant htt gene. Based on this and the weaker 2166 staining of mutant htt in full-length htt transgenic mice, the level of full-length mutant htt in YAC128 and C6R mice does not appear to exceed the level of endogenous full-length mouse htt.
EM48 also labeled transgenic full-length htt in YAC128 mice brains on the western blot, but not endogenous mouse htt and mutant htt in HD150Q KI mice (Fig. 2C), suggesting that it preferentially reacts with human mutant htt; its western blot also shows that C6R mice carry a larger polyQ tract in transgenic htt at a lower level than do YAC128 mice. EM48 was unable to detect smaller htt products that can be seen with 1C2 staining. Using mouse EM48 (mEM48), we also obtained a similar result (data not shown). mEM48 appeared to label truncated htt better on western blots and strongly labeled misfolded or aggregated htt that remained in the stacking gel (Fig. 2C right panel). In contrast to 1C2, mEM48 labeled more intensively htt in R6/2 mouse brain than SS htt. It also detected the soluble htt fragments in N171-82Q mouse brain. Although the soluble form of SS htt is clearly detected by 1C2 and EM48 and carries more polyQ repeats than N171-82Q htt, mEM48 labeled more aggregated htt in N171-82Q mice than in SS mice. Thus, protein context clearly determines the aggregation of mutant htt in the brain.
Cellular localization of mutant htt in HD mouse brain
We next performed immunostaining of HD mice brains using rabbit EM48, which is more specific to polyQ-expanded htt in immunohistochemisty. We focused on the cerebral cingulate cortex, dorsomedial striatum, and white matter in the corpus callosum to compare the expression of mutant htt in these regions. At 12 months of age, mutant htt in full-length htt mouse models accumulates abundantly in the nucleus, allowing for a definitive comparison of the accumulation of N-terminal htt fragments. Rabbit EM48 is able to react with htt aggregates in immunohistochemistry, despite its negative staining of mouse mutant htt on western blots. Low-magnification (×10 objective lens) micrographs show significant EM48 labeling of the cortex and striatum of HD150Q KI mice compared with WT mice (Supplementary Material, Fig. S2). As htt is also expressed in glial cells (29), the white matter of HD150Q KI mice shows clear EM48 labeling. Because EM48 labels human htt much better than mouse htt, its staining is more pronounced in YAC128 mice than in KI mice. As reported previously (19), C6R mice brains display delayed nuclear accumulation of mutant htt as compared with YAC128 mice brains (Supplementary Material, Fig. S2A). For transgenic mice expressing N-terminal mutant htt, EM48 immunohistochemistry labeled the brains of R6/2 mice most intensively. It also intensively labeled N171-82Q mice at the age of 4 months. SS htt transgenic mice, however, show only weak EM48 labeling at 3 months. Even at the age of 12 months, EM48 staining in SS mice is still weaker than in N171-82Q and R6/2 mice (Supplementary Material, Fig. S2B). This difference also confirms the western blotting result that showed less aggregated htt detected in SS mice (Fig. 2).
Higher magnification (×40 objective lens) micrographs show that mutant htt forms neuronal nuclear inclusions and neuropil aggregates and also accumulates in glial cells in the white matter of YAC128 mice (Fig. 3A). The presence of htt aggregates in glial cells has been revealed by immunofluorescent double staining and electron microscopy in our previous study (29). Similar staining patterns were found in C6R mice and YAC128 mice. Under the same staining conditions, EM48 labeled htt aggregates in KI mice, but more diffuse nuclear EM48 labeling was seen in YAC128 mice, again suggesting that EM48 is more sensitive to mutant human htt.
Figure 3.
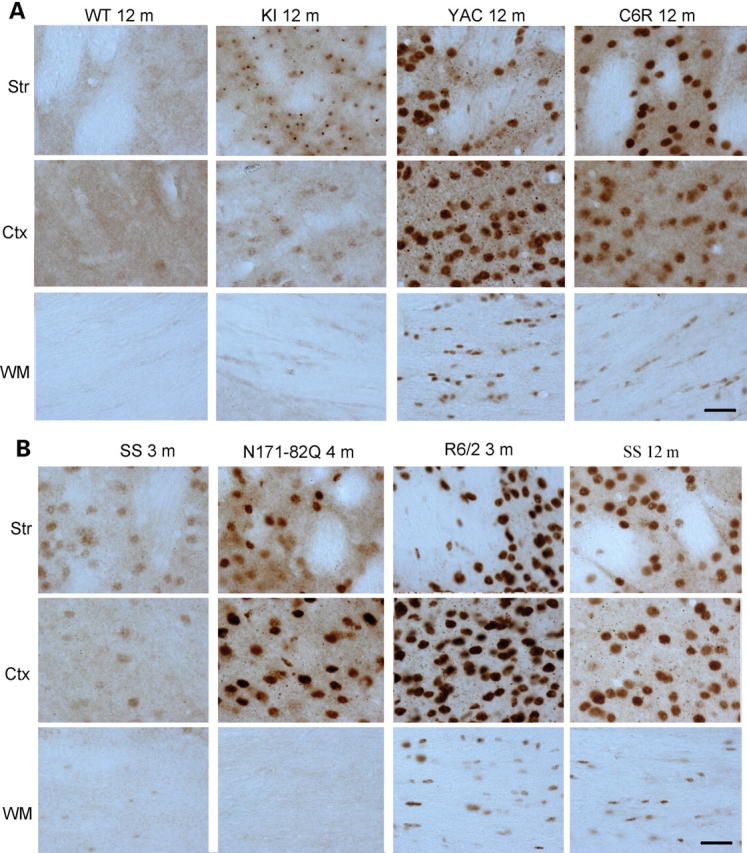
Micrographs of EM48 immunohistochemistry. (A) EM48 staining of 12-month-old WT or HD mice expressing full-length mutant htt. KI, HD150Q knock-in; YAC, YAC128 transgenic mice; C6R, caspase-6-resistant YAC transgenic mice. (B) EM48 staining of transgenic mice expressing N-terminal mutant htt at the age of 3, 4, or 12 months as indicated. SS, shortstop transgenic mice; Ctx, cerebral cingulate cortex; Str, dorsomedial striatum; WM, white matter in the middle portion of the corpus callosum. Scale bars, 20 µm.
In transgenic mice expressing N-terminal mutant htt, the strongest EM48 staining is seen in the cortex, white matter, and striatum in R6/2 mice brains (Fig. 3B). In N171-82Q mice, EM48 labeling was primarily localized to neuronal cells, with no specific labeling of white matter. This difference is perhaps due to the use of the neuronal prion promoter in N171-82Q mice, which drives transgenic htt expression primarily in neurons. Although SS mice do show weaker EM48 staining, EM48-positive nuclei were present in neuronal and glial cells.
We also performed immunostaining with 1C2, which reacts with the polyQ domain, an epitope that is different from the one of EM48. Although 1C2 immunohistochemistry requires formic acid treatment of brain sections and its labeling signal is weaker than EM48 staining, the 1C2 labeling pattern is similar to EM48 staining (Supplementary Material, Fig. S3). Basically, HD mice expressing N-terminal mutant htt, such as R6/2 and SS mice, display nuclear htt staining in neuronal cells and glial cells. N171-82Q mice only show 1C2 labeling in neuronal cells, though their 1C2 signal is stronger than that in SS mice (Fig. 4). For HD mice expressing full-length mutant htt, YAC128 mice show the most intensive nuclear 1C2 staining compared with the other mice. 1C2 labeling is present in neuronal nuclei, neuropils, and glial cells in YAC128, C6R and HD150Q KI mice brains (Fig. 4). High magnification micrographs of EM48 staining at ×63 clearly show nuclear htt and neuropil aggregates in the striatum of R6/2 and N171-82Q mice (Fig. 5A). As only N-terminal mutant htt fragments form aggregates, the density of htt aggregates in the nuclei and neuronal processes reflects the accumulation of N-terminal mutant htt fragments. Again, the striatum of SS mice show fewer htt aggregates than that of R6/2 and N171-82Q mice. For mouse models expressing full-length mutant htt, YAC128 mice display more aggregates in the nuclei and neuropil than HD150Q KI mice under the same immunostaining conditions. Compared with neuronal htt aggregates, mutant htt forms much smaller aggregates in glial cells (Fig. 5B). Since the transcriptional levels and polyQ repeats of full-length mutant htt in YAC128 and HD150Q KI mice are not markedly different, the difference between EM48 or 1C2 immunohistochemistry in these mouse models suggests that human and mouse htt sequences confer different immunoreactivity to EM48 and 1C2.
Figure 4.
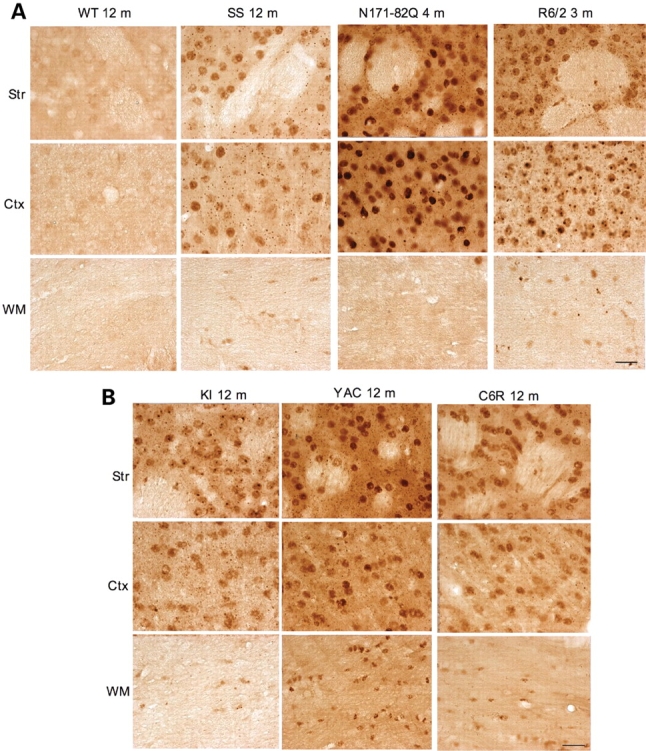
Micrographs of 1C2 immunohistochemistry. (A) 1C2 staining of WT or transgenic mice expressing N-terminal mutant htt (B) at the age of 3, 4, or 12 months as indicated. (B) 1C2 staining of 12-month-old HD mice expressing full-length mutant htt. KI, HD150Q knock-in; YAC, YAC128 transgenic mice; C6R, caspase-6-resistant YAC transgenic mice; SS, shortstop transgenic mice; Ctx, cerebral cingulate cortex; Str, dorsomedial striatum; WM, white matter in the middle portion of the corpus callosum. Scale bars, 20 µm.
Figure 5.
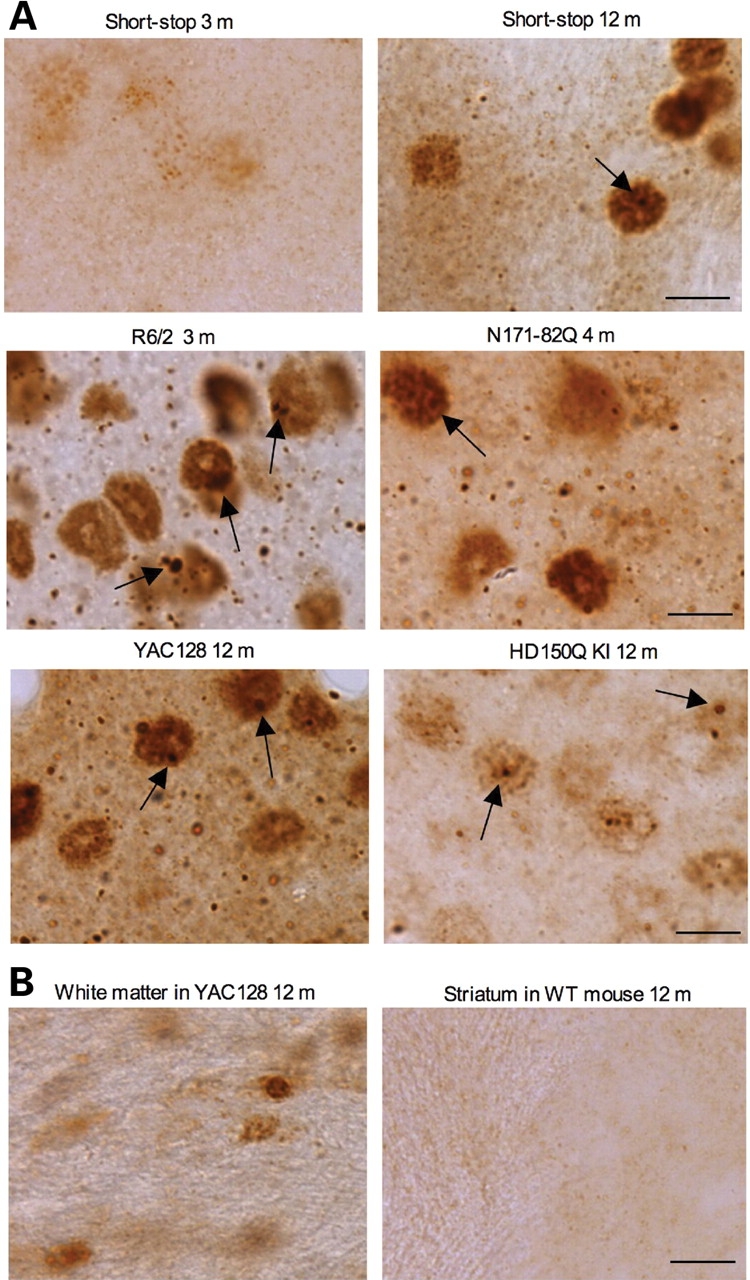
High-magnification (×63) micrographs of EM48 immunohistochemistry. (A) Rabbit EM48 staining of the dorsomedial striatum of shortstop mouse (3‐ and 12‐month‐old), R6/2 mouse (3‐month‐old), N171-82Q mouse (4‐month‐old), HD150Q KI mouse (12‐month‐old), YAC128 mouse (12‐month‐old). (B) Rabbit EM48 staining of glial cells in the white matter of YAC128 mouse (12‐month‐old) and the striatum of WT mouse (12‐month‐old). Note that mutant htt forms nuclear inclusions (arrows) and neuropil aggregates that are out of neuronal cell bodies. Mutant htt also accumulates in the nuclei of glial cells. Scale bar, 10 µm.
Expression of N-terminal htt fragments in transgenic monkeys
We recently established transgenic rhesus monkeys that express exon-1 htt containing a 65–88Q tract under the control of the ubiquitin promoter (20). Using the same transgenic strategy to introduce mutant htt genes into monkey oocytes, we also expressed exon-1 htt with a 147Q tract in transgenic monkeys. While transgenic monkeys expressing the shorter polyQ tracts (65–88Q) could develop to full term or survived after birth (20), transgenic monkeys expressing a large repeat (147Q) died after 4 months gestation, well before full-term fetal development at 5 months, indicating toxicity from the large polyQ tract. Examining the brains of two 150Q transgenic monkeys (rHD147Q-2 and -3) revealed the widespread expression of mutant htt in various brain regions, including the striatum and cortex (Fig. 6A). The distribution and accumulation of mutant htt in 147Q transgenic monkey brains are virtually the same as in the 65–88Q transgenic monkeys (20). However, there were some interesting phenomena observed in the HD monkey brains. First, we did not find large EM48-positive nuclear inclusions, as seen in HD patients and mouse models. Instead, mutant htt is quite diffuse and abundant in the nucleus of transgenic monkey brains (Fig. 6B–D). As fetal brain development in the monkey takes much longer time than in rodents (5 months versus 21 days), mutant htt only form large nuclear aggregates in adult neurons in primates. Second, we observed many aggregates outside the cell body, which likely localize to neuronal processes. Many neuronal processes show discontinued EM48 staining with aggregates, suggesting that they are fragmented axons or dendrites (Fig. 6C–E). Noticeably, mutant htt forms aggregates in the distal region of axonal-like processes that show swelling and fragmentation (Fig. 6C–E). This evidence suggests that the accumulation of N-terminal htt fragments in axons or dendrites can lead to the disruption of the integrity of neuronal processes.
Figure 6.
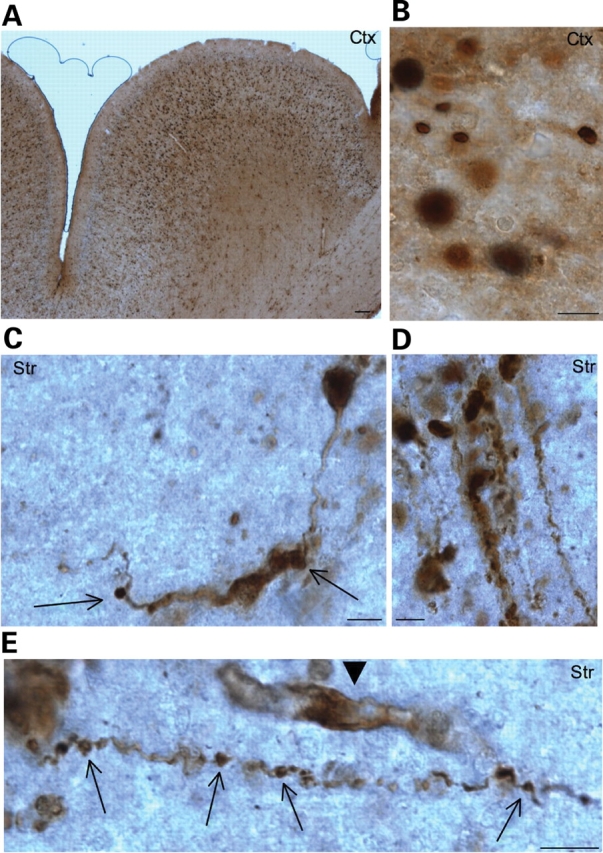
Expression of N-terminal mutant htt in transgenic monkey brains. EM48 immunostaining of the cerebral cortex (A and B) and striatum (C–E) of transgenic monkeys [rHD147Q-2 (A, B, C, E) and rHD147Q-3 (D)] that expressed exon-1 htt with 147Q. A low-magnification (×10 objective lens) graph is shown in (A), and high-magnification (×63 objective lens) graphs are shown in (B–E). Note that mutant htt is abundant in the nucleus and also forms aggregates in neuronal processes. Arrows indicate aggregates in the neuronal processes with a disrupted or swelling appearance. Arrowhead indicates a glial-like cell that was also labeled by EM48. Scale bars, 5 µm.
Accumulation of N-terminal htt fragments in striatal neurons
To more quantitatively assess the accumulation of N-terminal mutant htt in HD brains, we used fractionation and western blotting to analyze the levels of N-terminal htt fragments in HD150Q KI mice. These mice express full-length mutant htt under the control of the endogenous mouse htt promoter, and thereby avoid the potential influence due to the chromosomal integration of transgenic DNAs. We first examined the cerebral cortex from HD150Q KI mice at 4, 14 and 24 months of age. Western blotting was performed using 1C2, as this antibody specifically detects mutant htt (Fig. 7A). Full-length mutant htt was found in the total cell lysates (T), cytosolic fraction (C) and synaptosomal (S) fractions. The minimum level of full-length mutant htt was seen in the nuclear fraction, and the highest level in the cytosolic fraction, supporting the idea that full-length htt is predominantly distributed in the cytoplasm. As age increases, more N-terminal htt fragments were found in older HD150Q KI mice. In addition, more N-terminal htt fragments accumulate in the fractions isolated from HD150Q KI striatal tissues, and this accumulation is age-dependent, or correlates with disease progression (Fig. 7B). These findings are consistent with the preferential accumulation of mutant htt in the nuclei of striatal neurons in HD mice expressing full-length htt (9,10,30,31).
Figure 7.
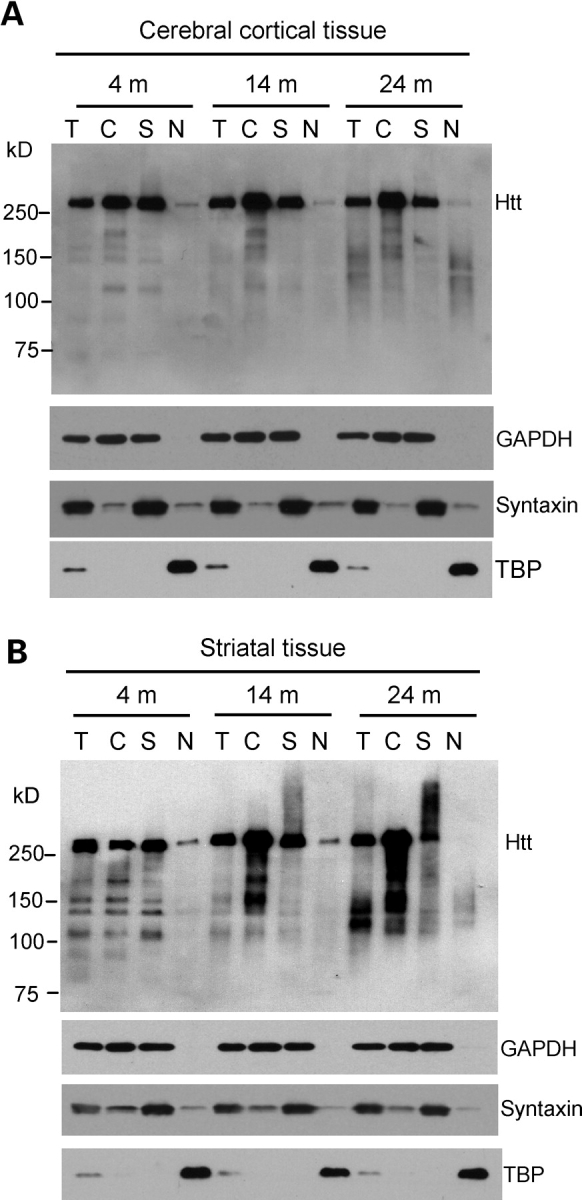
Accumulation of N-terminal htt fragments in the brains of HD150Q knock-in mice. (A and B) Western blotting of total cell lysates (T) and cytosolic (C), synaptosomal (S), and nuclear (N) fractions from the cerebral cortex (A) and striatum (B) of HD150Q KI mice at 4, 14 and 24 months of age. The blots were probed with 1C2 antibody for htt and antibodies for the cytoplasmic protein GAPDH, the synaptic protein syntaxin, or the nuclear protein TBP.
The accumulation of more htt fragments in the striatum suggests that straital neurons may have a decreased capacity to remove toxic htt fragments. As the ubiquitin-proteasomal system (UPS) plays a critical role in clearing polyQ proteins (1,32,33), we compared the proteasomal (chymotrypsin- and trypsin-like) activity in the cortical and striatal regions and found there is an age-dependent decrease of proteasomal activity in the brain (Fig. 8A–B). We did not observe any significant difference in proteasomal activity between WT and HD KI mice brains. Importantly, both chymotrypsin- and trypsin-like activity is significantly lower in striatal tissue than in cortical tissue from WT or HD KI mice brains (Fig. 8A–B). However, biochemical assay of brain homogenates derived from mixed populations of cells cannot define UPS activity in neurons. We therefore cultured primary neurons from the rat striatum and cortex and expressed a UPS function reporter (GFPu), a green fluorescent protein that carries the sequences for UPS-mediated degradation (32), in cultured neurons via adenoviral infection. For the control, we co-expressed red fluorescent reporter (RFP) that does not carry the UPS degradation sequences. Thus, the increased ratio of GFPu to RFP reflects decreased UPS activity in cultured neurons and is unlikely influenced by transgene expression variation in different cells. Using this assay, we also found that striatal neurons display decreased UPS activity as compared with cortical neurons (Fig. 8C). Quantitative analysis of the ratio of GFPu to RFP verified that striatal neurons show increased GFPu/RFP (1.18 ± 0.09 mean ± SE, n = 13) compared with cortical neurons (0.59 ± 0.02 mean ± SE, n = 13, P < 0.001). As the increased GFPu/RFP ratio reflects decreased proteasomal activity, these findings suggest that the accumulation of N-terminal htt fragments is determined by cell-type-dependent clearance of mutant htt.
Figure 8.
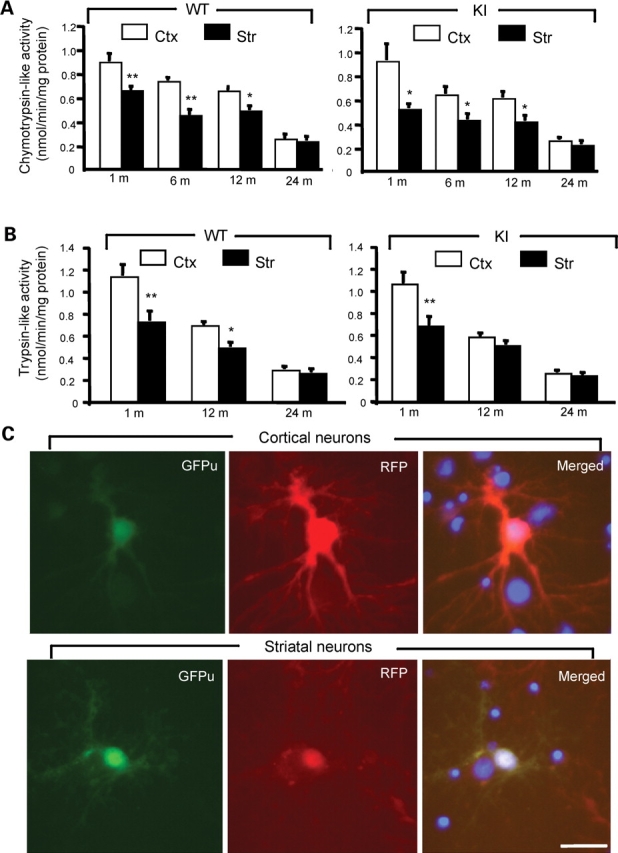
Decreased proteasomal activity in striatal neurons. (A and B) Biochemical assays of chymotrypsin-like (A) and trypsin-like (B) activity in homogenates from the brain cortex (Ctx) and striatum (Str) of WT and HD150Q KI mice at different ages. The data (mean ± SE) were obtained from 4–8 mice each group. (C) Expression of GFPu/RFP in cultured cortical and striatal neurons (DIV 12) from rat brains. The merged images (right panels) also show the nuclei of cells that did not express transgenic GFPu/RFP. The ratio of GFPu to RFP was described in the text. Scale bar, 10 µm. *P<0.05; **P<0.01 as compared to the cortex sample.
DISCUSSION
In the present study we compared the expression of mutant htt in different HD mouse models under the same experimental conditions. Such comparison revealed several interesting findings. First, the transcription levels of mutant htt do not necessarily correlate with the protein levels of mutant htt or the abundance of htt aggregates. Secondly, although the protein levels of mutant htt are in general correlated with the reported severity of neurological symptoms in HD mice, it is protein context that appears to influence htt distribution and neurological symptoms far more. Thirdly, the preferential accumulation of N-terminal htt fragments in the striatum is the likely cause of selective vulnerability seen in the striatum in HD.
Our RT–PCR analysis revealed that N171-82Q mice, among all the HD mice examined, express the highest level of transcripts, suggesting that the prion promoter may drive transgene expression more efficiently than other promoters. However, western blots probed with 1C2 or EM48 did not reveal a high level of the N171-82Q protein. Schilling et al. (7) used a different antibody, AP194, which reacts with the first 17 amino acids that are conserved in human and mouse htt, also found that the N171-82Q protein level is ∼5-fold less than endogenous mouse htt in the same N171-82Q mice. Interestingly, the N171-82Q protein forms far more 1C2- and EM48-immunoreactive aggregates in the brain than SS transgenic htt even though it carries a shorter polyQ tract (82Q) than SS htt (120Q). It is possible that the soluble form of N171-82Q (1–171 amino acids) is less stable and can more readily form htt aggregates. On the other hand, SS htt (1–117 amino acids) may be more stable in its soluble form, but form less aggregated htt because of its unique conformation. These findings suggest that the expression levels of transgenic truncated htt are not necessarily correlated with htt aggregation or its accumulation. Rather, the protein context of N-terminal htt fragments is important for their processing, subcellular localization, and aggregation in the brain. There is no dramatic difference in the expression levels of full-length mutant htt between YAC128 and HD150Q KI mice. The amino acids of human and mouse htt share ∼91% identity (34). Since YAC128 mice express human htt, whereas HD150Q KI mice express an expanded polyQ tract in endogenous mouse htt, the protein context of mutant htt in these mice could account for the differences in neuropathology. In addition, chromosomal position of transgenic htt and two copies of the endogenous mouse htt gene in YAC transgenic mice could also influence the expression of transgenic htt and its related neurological phenotypes.
Protein context has been shown to be an important influence on the neuropathology of HD mice (35). It is evident that smaller htt fragments are prone to misfolding and aggregation, and also more toxic than full-length mutant htt (36). Consistently, R6/2 and N171-82 mice develop more severe neurological symptoms and die earlier. Further, C6R mice expressing full-length mutant htt, in which the caspase-6 cleavage site is mutated, show delayed nuclear accumulation of htt without obvious neuropathology (19). Removal of the caspase-6 site in htt could alter htt proteolysis by other proteases, conformation, and/or interactions with other proteins, leading to a protective effect (37). These findings also support the idea that protein context determines the accumulation and toxicity of mutant htt. In support of this idea a previous study reported that although 1C2 western blotting demonstrates a much higher level of mutant htt in HdhCAG(150) or HD150Q KI mouse brain than that in HprtCAG(146) KI mouse brain that expresses a small expanded polyQ fragment, HprtCAG(146) mice show a more widespread accumulation of nuclear polyQ aggregates and severe neurological phenotypes (38).
Our results demonstrated that small N-terminal htt fragments are capable of accumulating in the nucleus and neuronal process to form aggregates that are recognized by EM48 and 1C2. Consistently, the brains of HD mice that have more severe neurological phenotypes show more accumulation of N-terminal htt fragments at an earlier age. For example, R6/2 and N171-82Q display more htt aggregates in their brains at much earlier ages (4–12 weeks). In contrast, transgenic mice, which express N-terminal htt but show obvious aggregates at 6 months of age or later, do not suffer early death and severe symptoms (18,23). For HD mice expressing full-length mutant htt, YAC128 mice demonstrate more severe phenotypes and earlier nuclear htt accumulation than C6R mice and HD KI mice. It should be pointed out that htt aggregates are not necessarily causative of neuropathology and symptoms; soluble mutant htt can induce neuropathology prior to the formation of aggregates, and htt aggregates are found in mice that do not show obvious clinical symptoms (18,23,24). Neurological symptoms can also be detected prior to the formation of htt aggregates in the nuclei (8,11). In addition, cytoplasmic mutant htt forms neuropil aggregates and can elicit neurological symptoms (39). Consistently, HD mice showing more severe neurological phenotypes also display more abundant neuropil aggregates. The abundance of htt aggregates reflects the accumulation of N-terminal htt fragments, and some N-terminal htt fragments have been shown to be toxic in R6/2 and N171-82Q mice.
The accumulation of N-terminal mutant htt fragments is not limited to neuronal nuclei and processes, but also occurs in glial cells (29). It should also be pointed out that overexpression of N-terminal htt fragments in neurons is sufficient to induce neuropathology in N171-82Q mice (7). However, given the important role of glial cells and cell–cell interactions in HD (23), HD pathology is also determined by cell-type-specific effects of mutant htt, in addition to the accumulation of toxic htt fragments.
Examining the brains of transgenic HD monkeys that express exon-1 mutant htt also revealed an abundant accumulation of transgenic htt. These monkeys died following a premature birth at 4 months gestation. Although the cause of the deaths remains to be determined, it is clear that a large polyQ tract leads to earlier death in transgenic monkeys. The finding also indicates that N-terminal htt fragments are toxic and pathogenic. Although we did not observe the large nuclear inclusions in HD monkey brains, our recent studies using western blotting have revealed the presence of misfolded or oligomerized htt in transgenic monkey brains (20). Importantly, transgenic monkey brain samples provided clear evidence for the axonal degeneration-like phenomenon that is closely associated with the accumulation of mutant htt in neuronal processes. This finding also supports the early observation of axonal degeneration in HD mouse models (30,35) and in HD patient brains (16,40). Such pathological changes are likely due to the effects of mutant htt on axonal trafficking (41–45) and nerve terminal function (3,46).
In the present study we also verified that multiple N-terminal htt fragments are generated in the brains of mice expressing full-length mutant htt. Some of these fragments are apparently larger than exon-1 htt and N171-82Q htt. Because these htt fragments contain different amino acids, their protein context is likely to influence their processing, localization, clearance, and toxicity. Given that both exon-1 and N171-82Q htt, which lack the conserved nuclear localization sequences, can abundantly accumulate in the nucleus to form aggregates, it is likely that small htt fragments passively enter the nucleus, and an expanded polyQ prevents their export (47). The presence of full-length mutant htt in synaptosomes also supports the idea that htt can be transported in neuronal processes (41,48) or plays a role in axonal trafficking (49).
In HD KI and YAC128 mice, the striatum displays a much earlier nuclear accumulation of mutant htt and more aggregates than other brain regions (9,10,30,31). Such early accumulation of mutant htt in the striatum is well correlated with the preferential neuronal loss seen in the striatum of HD patients. Fractionation of HD150Q KI mice offers biochemical evidence for the increased accumulation of htt fragments in medium spiny neurons, the major population of neurons in the striatum. As multiple htt fragments are accumulated, a possible mechanism is that medium spiny neurons are less able to clear misfolded htt and htt fragments. While the UPS activity in the HD mouse brain is not decreased as compared with normal mice (50,51), striatal proteasomal activity is intrinsically lower than cortical UPS, a phenomenon that is also observed in normal rat brains (52). Using the GFPu reporter, we confirmed that isolated striatal neurons show less UPS activity than cultured cortical neurons. Given that the medium spiny neurons are more vulnerable to excitotoxicity and oxidative stress (53) and that oxidative stress can impair UPS function (54), the UPS activity in striatal neurons may be more readily decreased during aging. As HD mice expressing full-length mutant htt show the preferential accumulation of mutant htt in striatal medium spiny neurons, the age-dependent and intrinsic lower UPS activity in medium spiny neurons is more likely to be important for the accumulation of degraded htt products that are generated from full-length mutant htt via proteolysis. Although it remains to be investigated whether there is cell-type-specific processing of mutant htt in striatal neurons, the finding that N-terminal mutant htt fragments preferentially accumulate in striatal neurons implies a pathogenic mechanism for HD. Findings from the present studies also promise to aid in evaluating possible therapeutic effects based on the expression and distribution of mutant htt in different HD mouse models.
MATERIALS AND METHODS
Animals
Hdh(CAG)150 KI (HD150Q KI) mice were generated previously (10) and maintained on the SV129/B6 background. Transgenic mice expressing a 171-amino-acid N-terminal fragment htt containing an 82-polyQ (82Q) stretch (N171-82Q-81 line) under the mouse prion promoter (7) were maintained on a hybrid (B6C3F1/J) background. Transgenic mice (R6/2) were generated using a human htt promoter to express exon-1 mutant htt and were maintained on the CBA×C57BL/6 background (6). R6/2 and N171-82Q mice were obtained from The Jackson Laboratory, and HD150Q KI mice were bred and maintained in the animal facility at Emory University in accordance with institutional guidelines. YAC128, SS and C6R transgenic mice, which express human mutant htt, were generated on the FVB/N background (8,18) and maintained at the University of British Columbia, Vancouver, BC, Canada. Transgenic rhesus monkeys were generated as described recently (20). Basically, lentiviruses carrying exon-1 of the htt gene containing 147 CAG repeats and the GFP gene under the control of the ubiquitin promoter were microinjected into the perivitelline space of metaphase II-arrested oocytes from rhesus monkey, followed by in vitro fertilization and embryo transfer into surrogate females. Using EM48 immunohistochemistry we examined the brains of two transgenic monkeys, which were born prematurely at 4 months gestation (prior to full-term of 5 months) and died after birth.
Antibodies and reagents
Rabbit (EM48) and mouse (mEM48) antibodies against N-terminal human htt were generated previously (16,28). Antibodies against an internal epitope of htt (2166) and expanded polyQ (1C2) and GAPDH were obtained from Chemicon (Temecula, CA, USA). Other antibodies used were against the following proteins: α-tubulin (Sigma, St Louis, MO, USA), syntaxin (Sigma), and TBP (Santa Cruz, CA, USA). Secondary antibodies were peroxidase-conjugated donkey anti -mouse, -rabbit, -rat, -guinea pig, -goat, or -sheep IgG (H + L) from Jackson ImmunoResearch (West Grove, PA, USA; all used at 1:5000).
PCR and qRT–PCR
Genotyping of transgenic mice expressing mutant human htt was performed using the sense primer HD-32S: 5′-CTACGAGTCCCTCAAGTCCTTCCAGC-3′ and the antisense primer HD177A: 5′-GACGCAGCAGCGGCTGTGCCTG-3′, which flanked the CAG repeat and specifically amplified human htt. For genotyping of HD 150Q KI mice, the primers (OPD-8, 5′-CCCATTCATTGCCTTGCTG-3′and HD 01–3, 5′-GCGGCTGAGGGGGTTGA-3′) were used. These primers amplify normal and transgenic mouse htt sequences. For RT–PCR, total RNA was isolated from mouse cortex with the RNeasy Lipid Tissue Mini Kit (Qiagen Inc., CA, USA). RT reactions were performed with 2 µg of total RNA and the Superscript First-Strand Synthesis System Kit (Invitrogen, Carlsbad, CA, USA) using Oligo dT, random primers, and primers for htt. Primers specific for human htt were 459S 5′-GCCGCCTCCTCAGCTTCCTCAG-3′ and 565A 5′-GTCGGTGCAGCGGCTCCTC-3′. Primers universal for both mouse and human htt were 670S 5′-AACTCTCCAGAATTTCAGAAAC-3′ and 790A 5′-GAAGATTAGAATCCATCAAAGC-3′. Primers for GAPDH were GAPDH-141S 5′-ACGACCCCTTCATTGACCTC-3′ and GAPDH-498A 5′-GGGGGCTAAGCAGTTGGTGG-3′. PCR was carried out in a 25 µl reaction volume. The following PCR conditions were used for mouse and human htt: 94°C for 3 min, followed by 30 cycles of 45 s at 94°C, 45 s at the annealing temperature 55°C, and 60 s at 72°C. For specific amplification of human htt, the annealing temperature was 64°C. The last cycle was followed by a final elongation step at 72°C for 10 min.
Htt mRNA levels in different mouse models were also determined in triplicate by quantitative fluorogenic RT (qRT)–PCR using the reverse transcripts described earlier. The quantitative PCR requires amplification of smaller PCR products (∼60 nucleotides) for quantification, such that the following primers were used: primers specific for mouse and human htt were HD1S (forward) 5′-ATGGCGACCCTGGAAAAGCT-3′ and HD40A (reverse) 5′-TGCTGCTGGAAGGACTTGAG-3′; GAPDH mRNA expression served as an internal control and was detected with the forward primer (GAPDH-530S 5′-CTCGCCAAGGTCATCCATGA-3′) and the reverse primer (GAPDH-577A 5′-TGATGGCATGGACTGTGGTC-3′). qRT–PCRs consisted of 4 µl of diluted cDNA reaction mixture (equivalent to 3 ng of RNA template), 12.5 µl of Power SYBR Green PCR Master Mix (Applied Biosystems), and 200 nM of each forward and reverse primer in a final volume of 25 µl. The qRT–PCR was performed with Applied Biosystems 7500 Fast Real-Time PCR Systems. Htt mRNA levels were calculated and normalized to the internal GAPDH mRNA standards.
Immunohistochemistry
Mice were anesthetized and perfused intracardially with phosphate-buffered saline (PBS, pH 7.2) for 30 s followed by 4% paraformaldehyde in 0.1 M phosphate buffer (PB) at pH 7.2. Brains were removed, cryoprotected in 30% sucrose at 4°C, and sectioned at 40 µm using a freezing microtome. Free-floating sections were preblocked in 4% normal goat serum in PBS, 0.1% Triton X, and then incubated with EM48 antibody (16,30) at 4°C for 48 h. The rabbit EM48 (1:2000) immunoreactive product was visualized with the Avidin–Biotin Complex kit (Vector ABC Elite, Burlingame, CA, USA). For 1C2 immunocytochemistry, fixed mouse brain tissues sections were treated with 88% formic acid for 10 min at room temperature, and then subjected to immunohistochemistry with 1C2 (1:20,000 dilution). Immunostaining of transgenic monkey brains was performed as described (20) using mouse monoclonal EM48 at 1:200 dilution. Light micrographs were taken using a Zeiss (Oberkochen, Germany) microscope (Axiovert 200 MOT) equipped with a digital camera (Orca-100; Hamamatsu, Bridgewater, NJ, USA) and the image acquisition software Openlab (Improvision, Lexington, MA, USA). A ×20 (LD Achroplan 20×/0.4 NA) or ×63 (63×/0.75 NA) objective lens was used for light microscopy.
Subcellular fractionation and western blotting
Mice were sacrificed by cervical dislocation and their cortices were used to isolate cytoplasmic, nuclear, and synaptosomal fractions via the previously described method (55). Cortical or striatal tissues from HD KI mice and control littermates were homogenized in homogenizing buffer (0.3 g/ml in 225 mm mannitol, 75 mm sucrose, 10 mm MOPS, 1 mm EGTA, 1:1000 protease inhibitor cocktail, pH 7.2.) at 4°C using a Teflon-glass homogenizer. Fifty µl of the homogenate was served as total cell lysate. The remaining homogenate was centrifuged at 1300 g for 5 min to yield the crude nuclear pellet (P1). The supernatant (S1) was transferred to a new tube and centrifuged at 13000 g for 15 min, resulting in the cytosolic fraction (S2). The resulting pellet (P2) was resuspended in 15% Percoll in homogenizing buffer and carefully layered over a 26 and 40% discontinuous Percoll gradient in ultracentrifuge tubes. The gradient was centrifuged at 44500 g at 4°C for 25 min. The synaptosomal layer (at the interface of 15 and 26% Percoll layers) was removed, washed with homogenizing buffer, and resuspended in RIPA buffer (50 mm Tris pH 8.0, 150 mm NaCl, 1 mm EDTA pH 8.0, 1 mm EGTA pH 8.0, 0.1% SDS, 0.5% DOC, 1% Triton X-100). Crude nuclei (P1) were washed once each with buffer A (250 mm sucrose, 25 mm KCl, 5 mm MgCl2, 10 mm HEPES, 0.6% NP40) and buffer B (250 mm sucrose, 10 mm KCl, 10 mm MgCl2, 10 mm HEPES), then resuspended in RIPA buffer. All fractions were assayed for protein amount using a detergent-compatible protein assay kit (Bio-Rad). Twenty µg of proteins from each of whole cell lysates, cytosolic, synaptosomal, and nuclear fractions were subjected to western blotting. The fractions were dissolved in SDS sample buffer and resolved by electrophoresis with a 4–12% tris-glycine acrylamide gel. Western blotting was performed with antibodies 2166 (1:1000), anti-syntaxin (1:5000), rabbit EM48 (1:800), 1C2 (1:5000), anti-GAPDH (1:3000), and anti-TBP (1:2000).
Assays of the ubiquitin-proteasomal activity
For determining proteasome activity, clear whole-cell extracts were adjusted to 0.5 mg/ml total protein by dilution with homogenization buffer. All assays were done in triplicate. Chymotrypsin-like activity of 20S β5 was determined using the substrate Suc-LLVY-aminomethylcoumarin (AMC) (Bilmol; 40 µM) and trypsin-like activity of 20S β2 was determined using the substrate Boc-LRR-AMC (Bilmol; 100 µM). Equal amounts (10 µg) of the extracts were incubated with corresponding substrates in 200 µl proteasome activity assay buffer [0.05 M Tris-HCl, pH 8.0, 0.5 mm EDTA, 1 mm ATP, 1 mm DTT] for 30–60 min at 37°C. The reactions were stopped by adding 0.8 ml of cold water and placing the reaction mixtures on ice for at least 10 min. The free AMC fluorescence was quantified using the CytoFluor multi-well plate reader (FLUOstar, BMG) with excitation and emission wavelengths at 380 and 460 nm, respectively. All readings were standardized using the fluorescence intensity of an equal volume of free 7-amino-4-methyl-coumarin (AMC) solution (40 mm), normalized by the protein concentrations and expressed as nmol/min/mg protein (28).
To determine proteasomal activity in cultured neurons, a CL-1 degron sequence (32) was added to the C-terminus of GFP in the PRK vector to generate PRK-GFPu construct. cDNA sequences for DsRed (RFP) were used to replace GFPu to generate PRK-RFP construct. The DNA fragments encoding these reporters were inserted into the shuttle vector of the pAdEasy vector system for expressing adenoviral reporters (Qbiogene, Carlsbad, CA). Adenovirus amplification, purification, and infection of cultured neurons were performed according to the method used in our previous study (29,46). All viral stocks were adjusted to 109 VP/ml. Fluorescence micrographs were taken using a Zeiss microscope (Axiovert 200 MOT) equipped with a digital camera (Orca-100; Hamamatsu, Bridgewater, NJ, USA). Quantification of fluorescence signals of cultured cells was performed using the Openlab software (Improvision, Lexington, MA, USA).
Statistical analysis
Data are expressed as mean ± SE and were analyzed using student's t-test for statistical significance (P < 0.05) with Prism software (version 4, GraphPad Software, San Diego, CA, USA). The specific tests used are noted in the text and figure legends.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
FUNDING
This work was supported by NIH grants AG019206, NS041669 (X.-J.L.), NS045016 (S.H.L.), R24RR018827 (A.W.S.C.), and the Huntington Disease Society of America (M.R.H), Cure HD Initiative (M.R.H), Canadian Institutes of Health Research (M.R.H) and Michael Smith Foundation for Health Research (R.K.G.).
ACKNOWLEDGEMENTS
We thank Z.H. Fang for technical assistance and Cheryl Strauss for critical reading of the manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Landles C., Bates G.P. Huntingtin and the molecular pathogenesis of Huntington's disease. Fourth in molecular medicine review series. EMBO Rep. 2004;5:958–963. doi: 10.1038/sj.embor.7400250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gusella J.F., Macdonald M.E. Huntington's disease: seeing the pathogenic process through a genetic lens. Trends Biochem. Sci. 2006;31:533–540. doi: 10.1016/j.tibs.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 3.Li S.H., Li X.J. Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet. 2004;20:146–154. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 4.Vonsattel J.P., Myers R.H., Stevens T.J., Ferrante R.J., Bird E.D., Richardson E.P., Jr Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Martin J.B., Gusella J.F. Huntington's disease. Pathogenesis and management. N. Engl. J. Med. 1986;315:1267–1276. doi: 10.1056/NEJM198611133152006. [DOI] [PubMed] [Google Scholar]
- 6.Davies S.W., Turmaine M., Cozens B.A., DiFiglia M., Sharp A.H., Ross C.A., Scherzinger E., Wanker E.E., Mangiarini L., Bates G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 7.Schilling G., Becher M.W., Sharp A.H., Jinnah H.A., Duan K., Kotzuk J.A., Slunt H.H., Ratovitski T., Cooper J.K., Jenkins N.A., et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 8.Slow E.J., van Raamsdonk J., Rogers D., Coleman S.H., Graham R.K., Deng Y., Oh R., Bissada N., Hossain S.M., Yang Y.Z., et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington's disease. Hum. Mol. Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- 9.Wheeler V.C., White J.K., Gutekunst C.A., Vrbanac V., Weaver M., Li X.J., Li S.H., Yi H., Vonsattel J.P., Gusella J.F., et al. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum. Mol. Genet. 2000;9:503–513. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- 10.Lin C.H., Tallaksen-Greene S., Chien W.M., Cearley J.A., Jackson W.S., Crouse A.B., Ren S., Li X.J., Albin R.L., Detloff P.J. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum. Mol. Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- 11.Menalled L.B., Sison J.D., Wu Y., Olivieri M., Li X.J., Li H., Zeitlin S., Chesselet M.F. Early motor dysfunction and striosomal distribution of huntingtin microaggregates in Huntington's disease knock-in mice. J. Neurosci. 2002;22:8266–8276. doi: 10.1523/JNEUROSCI.22-18-08266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim Y.J., Yi Y., Sapp E., Wang Y., Cuiffo B., Kegel K.B., Qin Z.H., Aronin N., DiFiglia M. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington's disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc. Natl Acad. Sci. USA. 2001;98:12784–12789. doi: 10.1073/pnas.221451398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gafni J., Ellerby L.M. Calpain activation in Huntington's disease. J. Neurosci. 2002;22:4842–4849. doi: 10.1523/JNEUROSCI.22-12-04842.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lunkes A., Lindenberg K.S., Ben-Haiem L., Weber C., Devys D., Landwehrmeyer G.B., Mandel J.L., Trottier Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell. 2002;10:259–269. doi: 10.1016/s1097-2765(02)00602-0. [DOI] [PubMed] [Google Scholar]
- 15.DiFiglia M., Sapp E., Chase K.O., Davies S.W., Bates G.P., Vonsattel J.P., Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 16.Gutekunst C.A., Li S.H., Yi H., Mulroy J.S., Kuemmerle S., Jones R., Rye D., Ferrante R.J., Hersch S.M., Li X.J. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J. Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stack E.C., Kubilus J.K., Smith K., Cormier K., Del Signore S.J., Guelin E., Ryu H., Hersch S.M., Ferrante R.J. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington's disease transgenic mice. J. Comp. Neurol. 2005;490:354–370. doi: 10.1002/cne.20680. [DOI] [PubMed] [Google Scholar]
- 18.Slow E.J., Graham R.K., Osmand A.P., Devon R.S., Lu G., Deng Y., Pearson J., Vaid K., Bissada N., Wetzel R., et al. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc. Natl Acad. Sci. USA. 2005;102:11402–11407. doi: 10.1073/pnas.0503634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graham R.K., Deng Y., Slow E.J., Haigh B., Bissada N., Lu G., Pearson J., Shehadeh J., Bertram L., Murphy Z., et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 20.Yang S.H., Cheng P.H., Banta H., Piotrowska-Nitsche K., Yang J.J., Cheng E.C., Snyder B., Larkin K., Liu J., Orkin J., et al. Towards a transgenic model of Huntington's disease in a non-human primate. Nature. 2008;453:921–924. doi: 10.1038/nature06975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wellington C.L., Ellerby L.M., Gutekunst C.A., Rogers D., Warby S., Graham R.K., Loubser O., van Raamsdonk J., Singaraja R., Yang Y.Z., et al. Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington's disease. J. Neurosci. 2002;22:7862–7872. doi: 10.1523/JNEUROSCI.22-18-07862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellerby L.M., Orr H.T. Neurodegenerative disease: cut to the chase. Nature. 2006;442:641–642. doi: 10.1038/442641a. [DOI] [PubMed] [Google Scholar]
- 23.Gu X., Li C., Wei W., Lo V., Gong S., Li S.H., Iwasato T., Itohara S., Li X.J., Mody I., et al. Pathological cell–cell interactions elicited by a neuropathogenic form of mutant huntingtin contribute to cortical pathogenesis in HD mice. Neuron. 2005;46:433–444. doi: 10.1016/j.neuron.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 24.Gu X., Andre V.M., Cepeda C., Li S.H., Li X.J., Levine M.S., Yang X.W. Pathological cell–cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington's disease. Mol. Neurodegener. 2007;2:8. doi: 10.1186/1750-1326-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woodman B., Butler R., Landles C., Lupton M.K., Tse J., Hockly E., Moffitt H., Sathasivam K., Bates G.P. The Hdh(Q150/Q150) knock-in mouse model of HD and the R6/2 exon 1 model develop comparable and widespread molecular phenotypes. Brain Res. Bull. 2007;72:83–97. doi: 10.1016/j.brainresbull.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 26.Trottier Y., Lutz Y., Stevanin G., Imbert G., Devys D., Cancel G., Saudou F., Weber C., David G., Tora L., et al. Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature. 1995;378:403–406. doi: 10.1038/378403a0. [DOI] [PubMed] [Google Scholar]
- 27.Dyer R.B., McMurray C.T. Mutant protein in Huntington's disease is resistant to proteolysis in affected brain. Nat. Genet. 2001;29:270–278. doi: 10.1038/ng745. [DOI] [PubMed] [Google Scholar]
- 28.Zhou H., Cao F., Wang Z., Yu Z.X., Nguyen H.P., Evans J., Li S.H., Li X.J. Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J. Cell Biol. 2003;163:109–118. doi: 10.1083/jcb.200306038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shin J.Y., Fang Z.H., Yu Z.X., Wang C.E., Li S.H., Li X.J. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 2005;171:1001–1012. doi: 10.1083/jcb.200508072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H., Li S.H., Yu Z.X., Shelbourne P., Li X.J. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington's disease mice. J. Neurosci. 2001;21:8473–8481. doi: 10.1523/JNEUROSCI.21-21-08473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Raamsdonk J.M., Murphy Z., Slow E.J., Leavitt B.R., Hayden M.R. Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington's disease. Hum. Mol. Genet. 2005;14:3823–3835. doi: 10.1093/hmg/ddi407. [DOI] [PubMed] [Google Scholar]
- 32.Bence N.F., Sampat R.M., Kopito R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 33.Goldberg A.L. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 34.Matsuyama N., Hadano S., Onoe K., Osuga H., Showguchi-Miyata J., Gondo Y., Ikeda J.E. Identification and characterization of the miniature pig Huntington's disease gene homolog: evidence for conservation and polymorphism in the CAG triplet repeat. Genomics. 2000;69:72–85. doi: 10.1006/geno.2000.6317. [DOI] [PubMed] [Google Scholar]
- 35.Yu Z.X., Li S.H., Evans J., Pillarisetti A., Li H., Li X.J. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J. Neurosci. 2003;23:2193–2202. doi: 10.1523/JNEUROSCI.23-06-02193.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hackam A.S., Singaraja R., Wellington C.L., Metzler M., McCutcheon K., Zhang T., Kalchman M., Hayden M.R. The influence of huntingtin protein size on nuclear localization and cellular toxicity. J. Cell Biol. 1998;141:1097–1105. doi: 10.1083/jcb.141.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fryer J.D., Zoghbi H.Y. Huntingtin’s critical cleavage. Nat. Neurosci. 2006;9:1088–1089. doi: 10.1038/nn0906-1088. [DOI] [PubMed] [Google Scholar]
- 38.Tallaksen-Greene S.J., Crouse A.B., Hunter J.M., Detloff P.J., Albin R.L. Neuronal intranuclear inclusions and neuropil aggregates in HdhCAG(150) knockin mice. Neuroscience. 2005;131:843–852. doi: 10.1016/j.neuroscience.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 39.Wang C.E., Zhou H., McGuire J.R., Cerullo V., Lee B., Li S.H., Li X.J. Suppression of neuropil aggregates and neurological symptoms by an intracellular antibody implicates the cytoplasmic toxicity of mutant huntingtin. J. Cell. Biol. 2008;181:803–816. doi: 10.1083/jcb.200710158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sapp E., Penney J., Young A., Aronin N., Vonsattel J.P., DiFiglia M. Axonal transport of N-terminal huntingtin suggests early pathology of corticostriatal projections in Huntington's disease. J. Neuropathol. Exp. Neurol. 1999;58:165–173. doi: 10.1097/00005072-199902000-00006. [DOI] [PubMed] [Google Scholar]
- 41.Gunawardena S., Her L.S., Brusch R.G., Laymon R.A., Niesman I.R., Gordesky-Gold B., Sintasath L., Bonini N.M., Goldstein L.S. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- 42.Szebenyi G., Morfini G.A., Babcock A., Gould M., Selkoe K., Stenoien D.L., Young M., Faber P.W., MacDonald M.E., McPhaul M.J., et al. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. 2003;40:41–52. doi: 10.1016/s0896-6273(03)00569-5. [DOI] [PubMed] [Google Scholar]
- 43.Lee W.C., Yoshihara M., Littleton J.T. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington's disease. Proc. Natl Acad. Sci. USA. 2004;101:3224–3229. doi: 10.1073/pnas.0400243101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang D.T., Rintoul G.L., Pandipati S., Reynolds I.J. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol. Dis. 2006;22:388–400. doi: 10.1016/j.nbd.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 45.Orr A.L., Li S., Wang C.E., Li H., Wang J., Rong J., Xu X., Mastroberardino P.G., Greenamyre J.T., Li X.J. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 2008;28:2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J., Wang C.E., Orr A., Tydlacka S., Li S.H., Li X.J. Impaired ubiquitin-proteasome system activity in the synapses of Huntington's disease mice. J. Cell Biol. 2008;180:1177–1189. doi: 10.1083/jcb.200709080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cornett J., Cao F., Wang C.E., Ross C.A., Bates G.P., Li S.H., Li X.J. Polyglutamine expansion of huntingtin impairs its nuclear export. Nat. Genet. 2005;37:198–204. doi: 10.1038/ng1503. [DOI] [PubMed] [Google Scholar]
- 48.Trushina E., Dyer R.B., Badger J.D., II, Ure D., Eide L., Tran D.D., Vrieze B.T., Legendre-Guillemin V., McPherson P.S., Mandavilli B.S., et al. Mutant huntingtin impairs axonal trafficking in Mammalian neurons in vivo and in vitro. Mol. Cell Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gauthier L.R., Charrin B.C., Borrell-Pages M., Dompierre J.P., Rangone H., Cordelieres F.P., De Mey J., MacDonald M.E., Lessmann V., Humbert S., et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 50.Diaz-Hernandez M., Hernandez F., Martin-Aparicio E., Gomez-Ramos P., Moran M.A., Castano J.G., Ferrer I., Avila J., Lucas J.J. Neuronal induction of the immunoproteasome in Huntington's disease. J. Neurosci. 2003;23:11653–11661. doi: 10.1523/JNEUROSCI.23-37-11653.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bett J.S., Goellner G.M., Woodman B., Pratt G., Rechsteiner M., Bates G.P. Proteasome impairment does not contribute to pathogenesis in R6/2 Huntington's disease mice: exclusion of proteasome activator REGgamma as a therapeutic target. Hum. Mol. Genet. 2006;15:33–44. doi: 10.1093/hmg/ddi423. [DOI] [PubMed] [Google Scholar]
- 52.Zeng B.Y., Medhurst A.D., Jackson M., Rose S., Jenner P. Proteasomal activity in brain differs between species and brain regions and changes with age. Mech. Ageing Dev. 2005;126:760–766. doi: 10.1016/j.mad.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 53.Browne S.E., Beal M.F. Oxidative damage in Huntington's disease pathogenesis. Antioxid. Redox Signal. 2006;8:2061–2073. doi: 10.1089/ars.2006.8.2061. [DOI] [PubMed] [Google Scholar]
- 54.Breusing N., Grune T. Regulation of proteasome-mediated protein degradation during oxidative stress and aging. Biol. Chem. 2008;389:203–209. doi: 10.1515/BC.2008.029. [DOI] [PubMed] [Google Scholar]
- 55.Phillips G.R., Huang J.K., Wang Y., Tanaka H., Shapiro L., Zhang W., Shan W.S., Arndt K., Frank M., Gordon R.E., et al. The presynaptic particle web: ultrastructure, composition, dissolution, and reconstitution. Neuron. 2001;32:63–77. doi: 10.1016/s0896-6273(01)00450-0. [DOI] [PubMed] [Google Scholar]
- 56.Turmaine M., Raza A., Mahal A., Mangiarini L., Bates G.P., Davies S.W. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc. Natl Acad. Sci. USA. 2000;97:8093–8097. doi: 10.1073/pnas.110078997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wade A., Jacobs P., Morton A.J. Atrophy and degeneration in sciatic nerve of presymptomatic mice carrying the Huntington's disease mutation. Brain Res. 2008;1188:61–68. doi: 10.1016/j.brainres.2007.06.059. [DOI] [PubMed] [Google Scholar]
- 58.Li S.H., Yu Z.X., Li C.L., Nguyen H.P., Zhou Y.X., Deng C., Li X.J. Lack of huntingtin-associated protein-1 causes neuronal death resembling hypothalamic degeneration in Huntington's disease. J. Neurosci. 2003;23:6956–6964. doi: 10.1523/JNEUROSCI.23-17-06956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saydoff J.A., Garcia R.A., Browne S.E., Liu L., Sheng J., Brenneman D., Hu Z., Cardin S., Gonzalez A., von Borstel R.W., et al. Oral uridine pro-drug PN401 is neuroprotective in the R6/2 and N171–82Q mouse models of Huntington's disease. Neurobiol. Dis. 2006;24:455–465. doi: 10.1016/j.nbd.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 60.Heng M.Y., Tallaksen-Greene S.J., Detloff P.J., Albin R.L. Longitudinal evaluation of the Hdh(CAG)150 knock-in murine model of Huntington's disease. J. Neurosci. 2007;27:8989–8998. doi: 10.1523/JNEUROSCI.1830-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.