

Abstract
We investigated the role of PPAR γ coactivator 1α (PGC-1α) in muscle dysfunction in Huntington's disease (HD). We observed reduced PGC-1α and target genes expression in muscle of HD transgenic mice. We produced chronic energy deprivation in HD mice by administering the catabolic stressor β-guanidinopropionic acid (GPA), a creatine analogue that reduces ATP levels, activates AMP-activated protein kinase (AMPK), which in turn activates PGC-1α. Treatment with GPA resulted in increased expression of AMPK, PGC-1α target genes, genes for oxidative phosphorylation, electron transport chain and mitochondrial biogenesis, increased oxidative muscle fibers, numbers of mitochondria and motor performance in wild-type, but not in HD mice. In muscle biopsies from HD patients, there was decreased PGC-1α, PGC-1β and oxidative fibers. Oxygen consumption, PGC-1α, NRF1 and response to GPA were significantly reduced in myoblasts from HD patients. Knockdown of mutant huntingtin resulted in increased PGC-1α expression in HD myoblast. Lastly, adenoviral-mediated delivery of PGC-1α resulted increased expression of PGC-1α and markers for oxidative muscle fibers and reversal of blunted response for GPA in HD mice. These findings show that impaired function of PGC-1α plays a critical role in muscle dysfunction in HD, and that treatment with agents to enhance PGC-1α function could exert therapeutic benefits. Furthermore, muscle may provide a readily accessible tissue in which to monitor therapeutic interventions.
INTRODUCTION
A characteristic feature of Huntington's disease (HD) is weight loss despite sustained caloric intake, which occurs early in the course of the illness (1–3). Using whole body indirect calorimetry, early HD patients are in negative energy balance. In transgenic mouse models of HD, similar observations were made and the weight loss was shown to be due to a loss of muscle bulk (4). This appears to be due to a myopathy which may be due to metabolic dysfunction (5). Well-recognized metabolic deficits occur in the brain, muscle and cellular models of HD. There is glucose hypometabolism on positron emission tomography imaging, even in presymptomatic gene carriers (6–9). NMR spectroscopy reveals increased lactate in cortex and basal ganglia (10) and impaired phosphocreatine (PCr) and ATP production in muscle (11–13). Biochemical studies show reduced activities of complex II–III and aconitase in human HD brain (14–16).
In human lymphoblastoid cell lines, ATP/ADP ratios are reduced, and decrease with increasing CAG repeat length (17). In striatal cells from mutant huntingtin (Htt) knock-in mouse embryos, mitochondrial respiration and ATP production are significantly impaired (18). The mitochondrial toxins 3-nitropropionic acid and malonate that selectively inhibit succinate dehydrogenase (SDH) and complex II, induce in human, primates and rodents, a clinical and pathological phenotype that very closely resembles HD (19–22).
There are several mechanisms by which the mutation could result in mitochondrial dysfunction and impair mitochondrial trafficking (23). First, Htt may interact directly with mitochondria. Lymphoblast mitochondria from HD patients and brain mitochondria from HD transgenic mice depolarize at lower calcium loads than control mitochondria, and localize mutant Htt by electron microscopy (23–26).
Another mechanism through which mutant Htt may affect mitochondrial function is by altering transcription (27,28). Htt interacts with a number of transcription factors, including p53, cAMP response element binding protein (CREB), TAFII130 and SP1 (29–32). A link to the transcriptional coactivator PGC-1α was first suggested by observations in PGC-1α-deficient mice (33,34). Since the discovery of PGC-1α, there has been an explosion of new information regarding its biological role in energy homeostasis, adaptive thermogenesis, α-oxidation of fatty acids and glucose metabolism (35,36). Originally identified as a PPAR-γ-interacting protein in brown adipose tissue, PGC-1α and a close homologue, PGC-1β are highly expressed in brown adipose tissue, heart and slow-twitch skeletal muscle, tissues known for their high mitochondrial content and energy demand (36).
PGC-1α's ability to activate a diverse set of metabolic programs in different tissues depends on its ability to form heteromeric complexes with a variety of transcription factors, including nuclear respiratory factors, NRF-1 and NRF-2, and the nuclear hormone receptors, PPARα, PPARδ, estrogen related receptor α (ERRα) and thyroid receptor (37). Of particular interest are NRF-1, NRF-2 and ERRα since they regulate the expression of many nuclear-encoded mitochondrial genes, including those encoding cytochrome c (Cyt C), complexes I–V and mitochondrial transcription factor A (Tfam) (38).
PGC-1α knockout mice exhibit impaired mitochondrial function, a hyperkinetic movement disorder and striatal degeneration, all features also observed in HD (33,34). Furthermore, impaired PGC-1α function and levels occur in striatal cell lines, transgenic mouse models of HD and in postmortem brain tissue from HD patients (39,40).
PGC-1α plays a critical role in muscle in inducing mitochondrial biogenesis and in influencing whether muscle contains slow-twitch oxidative or fast-twitch glycolytic fibers (41). A myopathy occurs in both HD patients and transgenic mice (4,25,42–46). Furthermore, phosphorous NMR spectroscopy showed impaired PCr and ATP production in muscle of HD patients, including presymptomatic gene carriers (11–13). Specific cAMP reduction implicates energy deficits conditions in HD mice (47). We, therefore, investigated whether impaired PGC-1α activity contributes to the myopathy and muscle wasting that occurs in HD. In the present studies, we provide evidence of impaired PGC-1α activity in muscle of HD transgenic mice, myoblasts from HD patients and muscle biopsies from HD patients, both at baseline and following a metabolic challenge with β-guanidinopropionic acid (GPA), which depletes PCr and ATP, leading to activation of AMPK and PGC-1α.
RESULTS
Decreased expression of PGC-1α and downstream target genes in muscles of HD mouse
We examined the relative expression of PGC-1α and its downstream target genes such as PGC-1β, NRF-1 and Tfam in different muscle types in 26-week-old (wild-type) WT and NLS-N171-82Q HD mice. There was significantly decreased expression of PGC-1α (56%) and Tfam (35%) in type-I ‘slow-twitch’ fiber enriched soleus muscle from HD mice, as when compared with WT mice, whereas PGC-1β and NRF-1 were non-significantly decreased 12 and 8% (Fig. 1A). In gastrocnemius (GM), a mixed fiber type muscle, Tfam mRNA expression was significantly decreased by 28% (Fig. 1B). There was significantly decreased mRNA expression of PGC-1α (34%), PGC-1β (32%) and Tfam (44%) in the glycolytic ‘fast-twitch’ fiber enriched muscle extensor digitorum longus (EDL) in HD mice when compared with WT mice (Fig. 1C). Decreased PGC-1α mRNA expression was confirmed by western blot, where reduced PGC-1α protein levels were observed in soleus and EDL (Fig. 1D). These results indicate impaired PGC-1α transcription in muscle of HD transgenic mice.
Figure 1.

Huntington's disease transgenic mice display transcriptional impairment of PGC-1α and its downstream target genes, AMP kinase pathway and impaired energy metabolism in muscles. (A–C) Total RNA was isolated from the type I fibers enriched soleus, type II fibers enriched gastrocnemius (GM) and extensor digitorum longus (EDL) muscles from wild-type (WT; black bar) and NLS-N171-82Q Huntington's disease mice (HD; gray bar). Quantitative real-time PCR analysis was performed for relative mRNA expression of PGC-1α, PGC-1β, NRF-1 and Tfam and normalized to β-actin. In this and all other figures data are expressed as mean ± SEM. *P < 0.05, **P < 0.01 (n = 5 in each group). (D) Analysis of PGC-1α protein levels by western blot in LDE, GM and soleus muscle of WT and HD mice. β-actin was used as loading control. (E–G) ATP and PCr levels are decreased in NLS-N171-82Q HD mice under chronic energy deprivation conditions. To create an artificial model of chronic energy deprivation conditions, WT and HD mice were treated for 10 weeks with the creatine analogue β-guanidinopropionic acid (GPA), which reduces the levels of high energy phosphate metabolites. After GPA treatment, HPLC measurement of high energy phosphate metabolites such as PCr, AMP, ADP and ATP was carried out in soleus muscles from WT and HD mice. *P < 0.05, **P < 0.01, ***P < 0.001 (n = 5 in each group). (H) Relative mRNA expression of AMP-activated protein kinase (AMPK) in soleus muscles from WT and NLS-N171-82Q HD mice treated with NS or GPA. AMPK mRNA levels were normalized to β-actin. *P < 0.05, **P < 0.01 (n = 5 in each group). (I and J) Analysis of AMPK, phosphorylated AMPK (pAMPK) and LKB-1 protein levels by western blots in extracts of soleus muscles from WT and HD mice treated with NS or GPA. Relative density expressed after normalization with β-actin. n = 5 mice were used from each group for each protein, with the average value of the WT+NS group set to 1. Representative blots showing 2–3 samples from each group. *P < 0.05.
Impaired AMP kinase pathway and energy metabolism in HD mice under energy deprivation conditions
In muscle of symptomatic and presymptomatic HD patients, there is a reduced ATP/PCr ratio, decreased PCr to inorganic phosphate ratio and impaired complex-I activity, which suggests abnormal skeletal muscle mitochondrial energy metabolism (12,13). In the present study, we created artificial chronic energy deprivation in NLS-N171-82Q HD mice by injecting the creatine analogue GPA for 10 weeks. GPA is a catabolic stressor which competes for creatine transport in skeletal muscle, depletes intracellular PCr and creatine levels and reduces creatine kinase activity (48,49). Chronic GPA treatment creates a chronic energy stress condition in skeletal muscle that resembles several muscle adaptations which occur during endurance exercise training, such as increased muscle oxidative capacity (50), increased GLUT-4 content (51), and hexokinase and citrate synthase activities (50). We found no significant difference between food intake in WT+NS (NS, normal saline) versus WT+GPA groups and HD+NS versus HD+GPA groups (data not shown). Interestingly, despite similar food intake, decreased body weight was observed in the GPA-treated groups (Supplementary Material, Fig. S1).
We measured the levels of muscle high-energy phosphate metabolites such as ATP and PCr by HPLC in WT and HD mice treated with either GPA or NS. We found significantly decreased levels of ATP in skeletal muscle of HD mice under basal conditions. GPA treatment caused a significant decrease in ATP levels in WT and HD mice (Fig. 1G). However, the reduction of ATP was more pronounced in the HD+GPA group when compared with WT+GPA. No significant changes were observed in levels of creatine, PCr, AMP and ADP in HD mice under basal conditions when compared with WT mice (Fig. 1E and F). Following GPA treatment, creatine and PCr levels were significantly reduced, whereas AMP and ADP were unchanged in HD mice when compared with WT mice (Fig. 1E and F).
During energy deprivation conditions (reduced ATP) such as starvation, ischemia and chronic metabolic stress, AMP-activated protein kinase (AMPK) serves as an energy sensor for whole body energy regulation. AMPK is activated by decreases in both the ATP/AMP ratio and PCr content. After activation it increases glucose transport, fatty acid oxidation and mitochondrial biogenesis (52). In skeletal muscle, AMPK increases phosphorylation of PGC-1α (53). We found a significant 20 and 32% decrease of AMPK mRNA expression and protein levels, respectively, in skeletal muscle isolated from untreated HD mice when compared with WT mice (Fig. 1H–J). Following GPA treatment, AMPK mRNA (20%) and protein levels (45%) were significantly increased in WT mice, but unchanged in HD mice (Fig. 1H–J). AMPK is mainly regulated allosterically by AMP and by reversible phosphorylation. Phosphorylation of AMPK at Thr172 on the activation loop of the catalytic α-subunit by one or more upstream AMPK kinases (AMPKK) leads to activation of AMPK (54). We examined pAMPK protein expression using western blots. Thr172 phosphorylation was unchanged in HD mice under basal conditions, compared with WT mice (Fig. 1I and J). Treatment with GPA increased phosphorylation of AMPK in WT mice, which was unchanged in GPA-treated HD mice. The content of LKB1, a major upstream AMPKK in skeletal muscle, was unchanged in HD mice under basal conditions. GPA treatment in WT mice non-significantly increased LKB1 content by (21%) and in HD mice by (10%) (Fig. 1I and J). These results show an impaired ability to activate the AMPK pathway in skeletal muscle of HD mice under energy deprivation conditions.
Reduction in oxidative capacity of muscle in energy deprivation conditions in HD mice
Mammalian skeletal muscle responds to chronically increased energy demand such as during repeated mechanical overload and endurance training, by converting glycolytic muscle fibers (fast-twitch or fatigable fibers) into oxidative fibers (slow-twitch or fatigue resistant fibers). Oxidative fibers (type I; red fibers) contain large amounts of myoglobin, are rich in mitochondria, generate ATP by the aerobic system, split ATP at a slow rate and have slow contraction velocity, whereas glycolytic fibers (type IIB; white fibers) contain low myoglobin content, few mitochondria, generate ATP through glycolysis and split ATP very quickly. We found that GM and soleus muscles from WT mice showed the distinct red color characteristics of oxidative muscles and are redder than HD mice muscles, which have a paler appearance (Fig. 2A). The GPA treatment caused a dark red appearance of GM and soleus muscle of WT mice, which was unchanged in HD mice muscles (Fig. 2A). These results show more oxidative muscles fibers in WT mice under basal and GPA treatment conditions, compared with HD mice.
Figure 2.
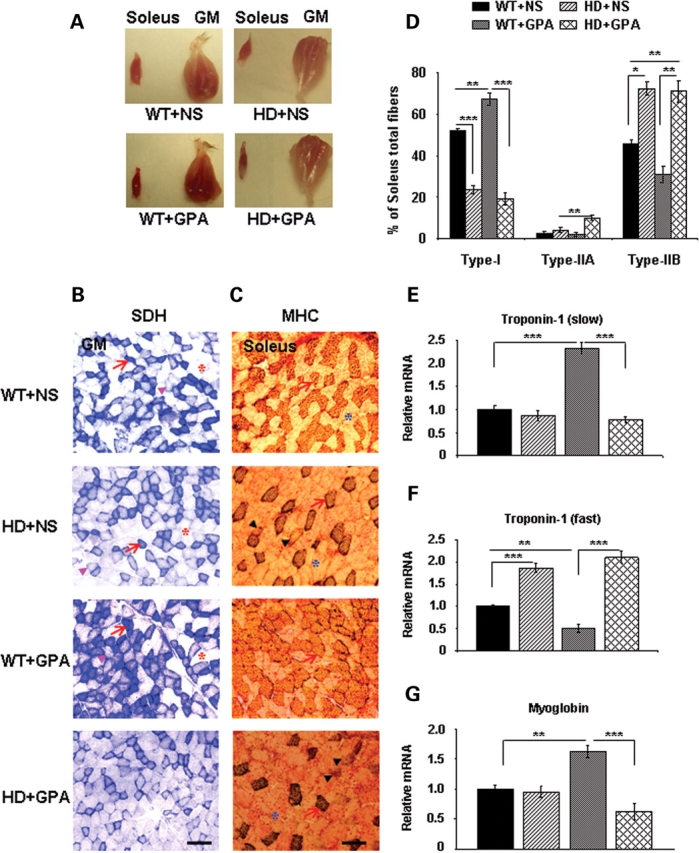
Decreased proportion of oxidative type I fibers and oxidative capacity in soleus and gastrocnemius (GM) muscles from NLS-N171-82Q HD mice under chronic energy deprivation conditions. (A) Gross morphology of soleus and GM from WT and HD mice treated with NS or GPA. Muscles from WT mice showed a distinct red color characteristic of oxidative muscle, whereas HD muscles from HD mice are light red color under baseline conditions. GPA treatment caused a redder appearance in WT mice, but not in HD+GPA mice. (B) Histochemical staining for succinate dehydrogenase (SDH) in muscle sections from WT and HD mice treated with NS or GPA. Arrows indicate dark stained type I oxidative fibers, arrowheads mark to intermediate type IIA oxidative fibers and asterisks point to type IIB glycolytic fibers (Scale bar = 100 µm). (C) Analysis of myosin heavy chain (MHC) isoforms (type I, IIA and IIB) expression in soleus muscle from WT and HD mice using a specific antibody for MHC. Arrows indicate dark stained oxidative type I fibers, arrowheads mark to intermediate oxidative type IIA fibers and asterisks point to glycolytic type IIB fibers (Scale bar = 100 µm). (D) Quantification of MHC isoforms (type I, IIA and IIB) in soleus muscle. Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (n = 5 mice in each group). (E–G) Quantitative real-time PCR analysis of fiber type markers such as troponin 1 (fast), troponin 1 (slow) and myoglobin in soleus muscle of WT and HD mice treated with NS or GPA. Data are expressed as mean ± SEM. **P < 0.01, ***P < 0.001 (n = 5 mice in each group).
We examined SDH activity in GM muscle of HD mice after GPA treatment (Fig. 2B). Histochemical measurement of SDH in GM muscle showed a decreased SDH staining in HD mice when compared with WT (Fig. 2B). SDH activity increased significantly in GPA-treated WT mice, while it was further decreased in GPA-treated HD mice, consistent with a decrease in the total number of mitochondria and/or mitochondrial activity, whereas increased SDH staining in GPA-treated WT mice suggests increased oxidative capacity.
PGC1-α/β plays an important role in mitochondrial biogenesis, respiration, oxidative phosphorylation and muscle fiber type conversion from glycolytic to more oxidative fibers (32,33,40,53). The reduced expression of PGC1-α/β in soleus muscle (an oxidative fibers rich muscle) prompted us to analyze different muscle fiber types in HD mice. The distribution of muscle fibers can be classified on the basis of their content of different myosin heavy chain (MHC) isoforms. To investigate the effect of GPA treatment on muscle fiber type composition, MHC staining was analyzed in soleus muscle sections of WT and HD mice both at baseline and following GPA treatment (Fig. 2C). The relative proportions of type I, IIA and IIB fibers in soleus muscles were determined by immunostaining with specific MHC antisera. Type 1 oxidative fibers being highly enriched with mitochondria stained dark, whereas Type IIB glycolytic fibers are light pink. Type IIA fibers being intermediate fibers stained light brown (Fig. 2C). Immunohistochemical analysis of soleus muscle sections revealed significantly decreased oxidative type I fibers and significantly increased type IIB glycolytic fibers in HD mice under basal conditions, whereas type IIA fibers were unchanged (Fig. 2C and D). We observed a significant up-regulation of type 1 fibers and significant down-regulation of type IIB fibers following GPA treatment in WT mice. On the other hand, following GPA treatment, HD mice showed a significant up-regulation of only intermediate type IIA fibers, while type I and type IIB were unchanged (Fig. 2D).
We further measured the muscle fiber type-specific isoforms of troponin mRNA expression in skeletal muscle. We found no significant changes in mRNA expression of troponin 1 (slow; a marker of oxidative muscle fibers) in soleus muscle of HD mice compared with WT mice (Fig. 2E). GPA treatment significantly increased troponin 1 (slow) mRNA expression by 2.3-fold in WT mice, while there was no change in HD mice (Fig. 2E). The mRNA expression of troponin 1 (fast; a marker of glycolytic muscle fibers) was significantly increased in HD mice when compared with WT mice at baseline (Fig. 2F). Following GPA treatment troponin 1 (fast), mRNA expression decreased significantly in WT mice and was unchanged in HD mice (Fig. 2F). The mRNA expression of another gene myoglobin (enriched in oxidative muscle) was unchanged in HD mice in basal conditions (Fig. 2G). GPA treatment resulted in up-regulation of myoglobin mRNA expression in WT mice, but not in HD mice.
To evaluate more specifically muscle fiber type composition, we measured the mRNA expressions of four different isoforms of MHC in soleus muscle of WT and HD mice (Supplementary Material, Fig. S2). MHC I was significantly down-regulated and MHC-IIB was up-regulated in HD mice under basal conditions, while MHCs-IIA and IIX were unchanged (Supplementary Material, Fig. S2). GPA treatment caused a significant increase of oxidative MHC-I fibers in WT mice at the expense of intermediate MHC-IIA and MHC-IIX and glyocolytic MHC-IIB fibers. In HD mice also GPA treatment resulted in slightly increased MHC-I mRNA expression, however, the degree of up-regulation was more pronounced in WT mice (Supplementary Material, Fig. S2). The mRNA transcript expression of MHC-IIA, MHC-IIX and MHC-IIB fibers did not change significantly following GPA treatment in HD mice. These results suggest muscle fiber type conversion in HD mice with significantly decreased oxidative fibers and increased glycolytic fibers in HD mice when compared with WT mice under chronic energy deprivation conditions.
Altered mitochondrial morphology and number and decreased rotarod performance in HD mice
In order to see ultrastructural morphology and arrangement of skeletal muscle mitochondria in all treatment groups, we performed electron microscopic analysis (Fig. 3A). Qualitative photomicrographs show an altered morphology and alignment of mitochondria along the Z-axis in HD mice under basal conditions when compared with WT mice, where mitochondria are well organized (Fig. 3A, upper). In GPA-treated WT and HD mice, an altered orientation of mitochondria along the Z-axis and varying size of mitochondria were observed. GPA treatment in WT mice caused an increase in numbers of mitochondria, which was absent in HD mice (Fig. 3A upper). Higher magnification (19 000×) photomicrographs depict normal shape and size of mitochondria and the presence of well assembled cristae in mitochondria of soleus muscle of WT mice (Fig. 3A, lower). Mitochondria in GPA-treated and untreated HD mice are slightly larger in size with irregular assembly of cristae. GPA treatment also caused an enlargement of mitochondria in WT mice. Quantitative analysis of electron photomicrographs of the different groups revealed a 20% decrease in mitochondrial number and 10% decrease in mitochondrial area in HD mice when compared with WT mice (Fig. 3B and C). Following GPA treatment in WT mice, the mitochondria number and mitochondrial area significantly increased by 25 and 81%, respectively, whereas there was no effect in HD mice (Fig. 3B and C).
Figure 3.
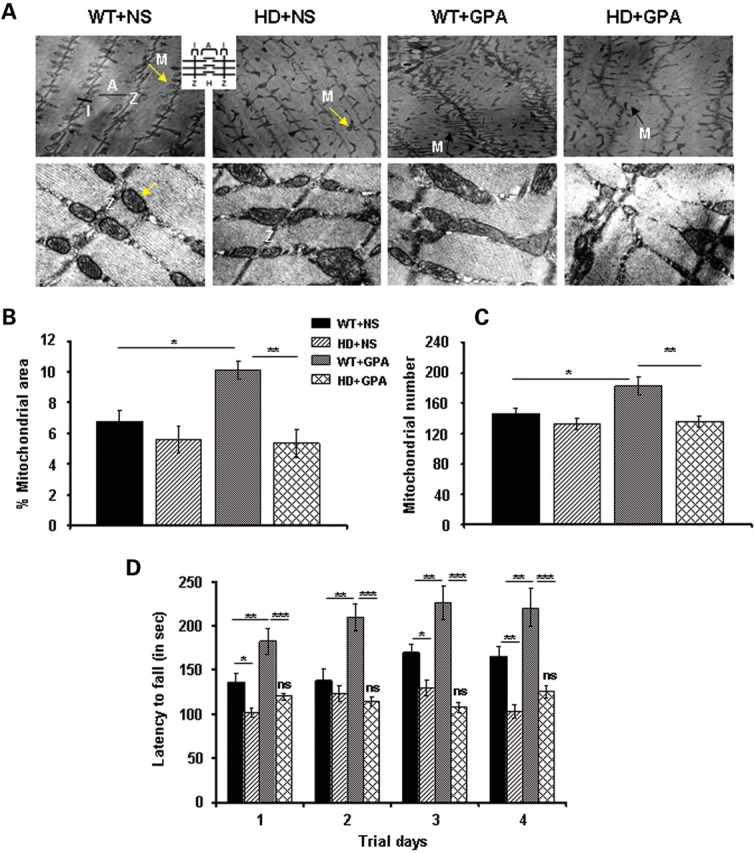
Decreased mitochondrial area/number and rotarod performance in NLS-N171-82Q HD mice under energy deprivation conditions. (A) Transmission electron microscopic analysis of soleus muscle from WT and HD mice treated with NS or GPA. Upper panel shows the mitochondria on lower magnification (4800×) and lower panel shows mitochondria on higher magnification (19 000×). Arrow indicates mitochondria along Z-axis. M, mitochondria; Z, Z-axis; I, I band; A, A band. Inset depicts schematic diagram of muscle fiber anatomy. An altered morphology and alignment of mitochondria along the Z-axis was observed in HD mice under basal conditions when compared with WT mice, where mitochondria are well organized (Fig. 3A, upper). GPA treatment in WT and HD mice caused an altered orientation of mitochondria along the Z-axis, and varying size of mitochondria, while an increase in numbers of mitochondria was observed only in WT mice (Fig. 3A, upper). Higher magnification depicts normal shape and size of mitochondria and the presence of well assembled cristae in mitochondria of soleus muscle of WT mice (Fig. 3A, lower). Mitochondria in GPA-treated and untreated HD mice are slightly larger in size with irregular assembly of cristae. GPA treatment also caused an enlargement of mitochondria in WT mice (Scale bars = 0.2 µm for upper panel and 2 µm for lower panel). (B and C) Corresponding quantification of percentage mitochondria area fraction and mitochondria number from the transmission electron microscopic images of soleus muscles from WT and HD mice. Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01 (n = 8). (D) Rotarod performance was measured in GPA-treated WT and HD mice at the 10th week of GPA treatment. Mice were tested for latency to fall on accelerated rotarod in thrid consecutive trials per day for 4 days. Data are expressed as mean ± SEM. ns = non-significant versus HD + NS. *P < 0.05, **P < 0.01, ***P < 0.001 (n = 10 mice per group).
To investigate the effect of reduced muscle oxidative capacity and altered mitochondrial number and morphology on behavior, we measured motor performance in GPA-treated mice (Fig. 3D). Latency of fall on rotarod was measured at the 10th week of GPA treatment (Supplementary Material, Fig. S7). At basal level, there was a significant decrease in motor performance in HD mice when compared with WT mice. GPA treatment in WT mice caused a significantly improved rotarod performance, whereas there were no significant changes in HD mice following GPA administration (Fig. 3D).
Altered muscle gene expression and metabolic pathways in HD muscle
We performed Affymetrix microarray expression profiling of ∼45 037 genes on RNA isolated from soleus muscle of WT and HD mice. A total number of 1974 (100%) probe sets were significantly (P < 0.05) altered in HD mice when compared with WT mice, of which 1020 (51.7%) were down-regulated and 954 (48.3%) up-regulated. After GPA treatment, the number of significantly altered probe sets was increased to 2517 (100%) in HD mice when compared with WT mice, out of which 1092 (43.4%) were up-regulated and 1425 (56.6%) down-regulated.
In order to see the coordinated expression of genes involved in muscle function, metabolism and plasticity in the treated and non-treated groups, we carried out a gene-set enrichment analysis (GSEA) of a priori-defined groups of genes (55,56). The size of the gene sets was determined on the basis of the number of genes present in each gene set after filtering out those genes not in the expression dataset. The genes were ranked, position of each gene set was identified and maximum enrichment score and nominal P-value were calculated. Gene sets for electron transport chain, oxidative phosphorylation, striated muscle contraction, gluconeogenesis, glycolysis and muscle, fat and connective tissue-specific genes were significantly enriched (increased) in soleus muscle of GPA-treated WT mice, but not in HD mice (Supplementary Material, Table S1). GPA treatment in HD mice caused a significant increase in gene set expression of several metabolic pathways, including the cellto-cell adhesion signaling pathway, ADP-ribosylation factor pathway, apoptosis modulation by HSP-70, GAP junction, cell cycle and inositol phosphate metabolism (Supplementary Material, Table S1). The gene pathways significantly enriched in different treatment groups are shown in Supplementary Material, Table S1. Four representative enrichment score plots of GSEA and their corresponding heat maps for WT+GPA and HD+GPA are presented in Figure 4A. Significantly increased expression of oxidative phosphorylation, electron transport chain and muscle function genes is evident from the enrichment plot and heat map in WT+GPA when compared with HD+GPA (Fig. 4A and B). These results show enrichment of gene expression involved in enhanced muscle oxidative capacity and mitochondrial biogenesis in WT mice following GPA treatment, but not in HD mice.
Figure 4.
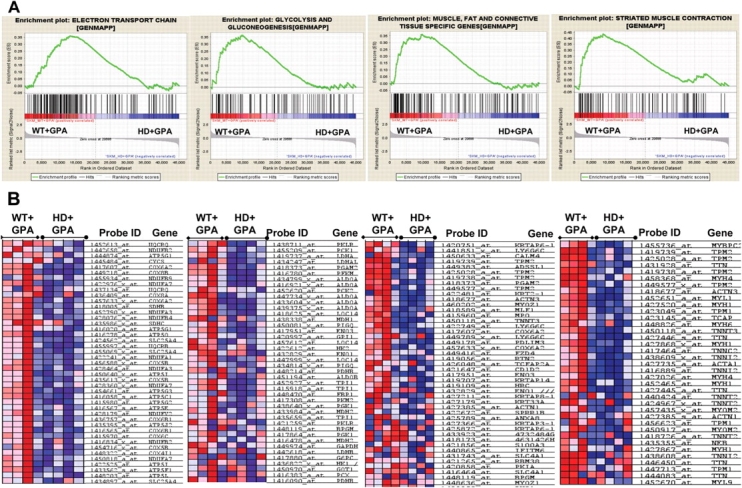
Affymetrix microarray gene expression profile and metabolic studies in soleus muscle revealed enrichment of pathways involved in mitochondrial biogenesis and muscle function in WT mice under chronic energy deprivation conditions. (A) We performed gene set enrichment analysis (GSEA) of gene expression profile in soleus muscles obtained from GPA-treated WT and HD mice. Four representatives gene set enrichment plots for pathways involved in the electron transport chain, glycolysis and gluconeogenesis, muscle fat and connective tissue-specific genes, and striated muscle contraction pathway are shown. These pathways are significantly enriched in WT mice treated with GPA when compared with HD+GPA mice. Upper part of the enrichment plot indicates the enrichment score on the y-axis and corresponding maximum peak. Middle part shows the location of genes in gene set as hits in a ranked list of genes. Middle left part indicates positively correlated genes in WT+GPA group when compared with negatively correlated genes in HD+GPA group. Lower part shows the histogram for the rank of genes in a 45 000 genes rank ordered dataset (n = 4 mice/group). (B) Cluster analysis of genes and corresponding heat map from enrichment plots show the expression levels of a subset of genes in metabolic pathways as described above (red, increased gene expression; blue, reduced gene expression).
Inspection of the PGC-1α pathway shows a significantly decreased expression of downstream PGC-1α target genes in muscle of HD mice under basal conditions (Table 1). GPA treatment caused an increase in expression of oxidative phosphorylation and PGC-1α target genes in WT mice, but not in HD mice (Table 1). Further, analysis of genes involved in TCA cycle, glycolysis, fatty acid metabolism, fatty acid biosynthesis, muscle function, muscle fiber conversion, myogenic differentiation and muscle contractile function showed increased numbers of significantly altered expression of genes in GPA-treated WT mice when compared with GPA-treated HD mice (Supplementary Material, Tables S2–S7).
Table 1.
Affymetrix microarray analysis of oxidative phosphorylation and PGC-1α target genes expression in mouse skeletal muscle
| Gene symbol | Probe set ID | Gene name | HD+NS versus WT+NS |
HD+GPA versus WT+GPA |
||
|---|---|---|---|---|---|---|
| Fold change | P-value | Fold change | P-value | |||
| Ndufa1 | 1422241_a_at | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 1 | 1.06 | 0.671 | −1.19 | 0.0114 |
| Ndufa3 | 1428464_at | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 3 | 1.17 | 0.265 | −1.18 | 0.0496 |
| 1452790_x_at | 1.14 | 0.351 | −1.23 | 0.0339 | ||
| Ndufa5 | 1417286_at | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 5 | −1.04 | 0.721 | −1.13 | 0.0766 |
| Ndufa7 | 1422976_x_at | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 7 | 1.12 | 0.445 | −1.25 | 0.0246 |
| Ndufa11 | 1444154_at | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 11 | −1.65 | 0.413 | 1.35 | 0.453 |
| Ndufa12 | 1433513_x_at | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 12 | 1.03 | 0.807 | −1.23 | 0.0441 |
| Ndufb2 | 1442658_at | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 2 | 1.33 | 0.403 | −1.49 | 0.05 |
| Ndufb4 | 1428076_s_at | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 4 | 1.04 | 0.799 | −1.23 | 0.013 |
| Ndufb8 | 1458404_at | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 8 | 1.01 | 0.983 | −2.38 | 0.103 |
| Ndufs7 | 1451312_at | NADH dehydrogenase (ubiquinone) Fe-S protein 7 | 1.03 | 0.823 | −1.21 | 0.0489 |
| Ndufs4 | 1444464_at | NADH dehydrogenase (ubiquinone) Fe-S protein 4 | 2.44 | 0.038 | −1.8 | 0.0265 |
| Sdha | 1445317_at | Succinate dehydrogenase complex, subunit A | −1.15 | 0.353 | −1.1 | 0.742 |
| 1426688_at | −1.04 | 0.68 | −1.07 | 0.508 | ||
| Sdhb | 1418005_at | Succinate dehydrogenase complex, subunit B, iron sulfur (Ip) | 1.11 | 0.454 | −1.22 | 0.0449 |
| Uqcrb | 1455997_a_at | Ubiquinol-cytochrome c reductase binding protein | 1.11 | 0.457 | −1.2 | 0.0467 |
| Cox4i2 | 1421373_at | Cytochrome c oxidase subunit IV isoform 2 | −1.13 | 0.05 | −1.21 | 0.0184 |
| Cox8b | 1449218_at | Cytochrome c oxidase, subunit VIIIb | 1.1 | 0.487 | −1.26 | 0.0301 |
| Cox6a2 | 1417607_at | Cytochrome c oxidase, subunit VI a, polypeptide 2 | 1.15 | 0.311 | −1.27 | 0.0229 |
| Cox6b2 | 1435275_at | Cytochrome c oxidase subunit VIb polypeptide 2 | −2.46 | 0.01 | −3.06 | 0.0329 |
| Cox15 | 1426693_x_at | COX15 homologue, cytochrome c oxidase assembly | −1.26 | 0.0739 | −1.22 | 0.0441 |
| Cox8c | 1455167_at | Cytochrome c oxidase, subunit VIIIc | −1.34 | 0.006 | −1.21 | 0.237 |
| 1443789_x_at | 3.7 | 0.012 | 1.51 | 0.05 | ||
| Cyc1 | 1416604_at | Cytochrome c-1 | 1.03 | 0.795 | −1.13 | 0.211 |
| Cycs | 1445484_at | Cytochrome c, somatic | −1.25 | 0.0128 | −1.76 | 0.0106 |
| Atp2b3 | 1445679_at | ATPase, Ca++ transporting, plasma membrane 3 | −1.19 | 0.0324 | 1.17 | 0.374 |
| Atp2c1 | 1442742_at | ATPase, Ca++-sequestering | −1.51 | 0.073 | −2.41 | 0.036 |
| Atp8a1 | 1433965_at | ATPase, class I, type 8A, member 1 | −1.52 | 0.0364 | −1.3 | 0.0182 |
| 1454728_s_at | −1.29 | 0.115 | −1.3 | 0.0246 | ||
| Abcc10 | 1428009_a_at | ATP-binding cassette, sub-family C, member 10 | 1.54 | 0.375 | −2.27 | 0.0018 |
| Atp5g1 | 1416020_a_at | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit c | 1.08 | 0.656 | −1.21 | 0.0383 |
| 1444874_at | 1.3 | 0.472 | −1.63 | 0.148 | ||
| Atpaf1 | 1436358_at | ATP synthase mitochondrial F1 complex assembly factor 1 | −1.41 | 0.05 | −1.12 | 0.286 |
| Atpaf2 | 1426474_at | ATP synthase mitochondrial F1 complex assembly factor 2 | −1.02 | 0.864 | −1.17 | 0.0147 |
| Atp5o | 1416278_a_at | ATP synthase, H+ transporting, mitochondrial F1 complex, O subunit | 1.12 | 0.366 | −1.2 | 0.0395 |
| Atp5s | 1459949_at | ATP synthase, H+ transporting, mitochondrial F0 complex, subunits | −1.64 | 0.05 | −1.46 | 0.034 |
| PGC-1α target genes | ||||||
| Ppargc1a | 1460336_at | PEroxisome proliferative activated receptor, gamma, coactivator 1α | −1.26 | 0.032 | −1.4 | 0.0113 |
| Ppargc1b | 1449945_at | Peroxisome proliferative activated receptor, gamma, coactivator 1β | −1.13 | 0.634 | −1.17 | 0.0496 |
| Pparg | 1420715_a_at | Peroxisome proliferator activated receptor gamma | 1.68 | 0.0328 | 2.79 | 0.0013 |
| Ppara | 1449051_at | Peroxisome proliferator activated receptor alpha | −1.33 | 0.257 | −1.41 | 0.396 |
| Ppard | 1439797_at | Peroxisome proliferator activator receptor delta | −1.02 | 0.923 | −1.17 | 0.236 |
| Nrf1 | 1424787_a_at | Nuclear respiratory factor 1 | −1.24 | 0.614 | −1.24 | 0.05 |
| Pparbp | 1450402_at | Peroxisome proliferator activated receptor binding protein | −1.51 | 0.225 | −1.47 | 0.374 |
| 1448708_at | −1.31 | 0.05 | 1.02 | 0.876 | ||
| Esrra | 1460652_at | Estrogen related receptor, alpha | 1.17 | 0.422 | −1.19 | 0.0283 |
| Esrrb | 1422986_at | Estrogen related receptor, beta | 1.67 | 0.281 | −1.27 | 0.353 |
| Pprc1 | 1426381_at | Peroxisome proliferative activated receptor, γ coactivator-related 1 | −1.08 | 0.467 | 1.31 | 0.0171 |
| Tfam | 1456215_at | Transcription factor A, mitochondrial | −1.05 | 0.749 | −1.1 | 0.562 |
| Transcriptional factors upstream to PGC-1α | ||||||
| Taf6 | 1418593_at | TAF6 RNA polymerase II, TATA box binding protein (TBP) factor | −1.09 | 0.401 | −1.11 | 0.0116 |
| Taf2 | 1434238_at | TAF2 RNA polymerase II, TATA box binding protein (TBP) factor | 1.01 | 0.922 | 1.24 | 0.0296 |
| Taf15 | 1419846_at | TAF15 RNA polymerase II, TATA box binding protein (TBP) factor | 1.81 | 0.269 | −4.2 | 0.05 |
| Creb1 | 1423402_at | cAMP responsive element binding protein 1 | −1.81 | 0.0012 | −1.19 | 0.385 |
| Crtc1 | 1434476_at | CREB regulated transcription coactivator 1 | −1.54 | 0.0221 | 1.11 | 0.443 |
| Btaf1 | 1435953s_at | BTAF1 RNA polymerase II, B-TFIID transcription factor-associated | 1.12 | 0.696 | 1.38 | 0.0418 |
P < 0.05 considered significant.
To further evaluate mitochondrial activity in HD and to validate the microarray data, we analyzed the expression of PGC-1α and its downstream target genes and genes involved in mitochondrial biogenesis in soleus muscle by quantitative RT–PCR (Fig. 5A and B). We found significantly reduced PGC-1α expression in HD mice when compared with WT mice. PGC-1β, another transcription factor with homology to PGC-1α, co-activates another set of transcription factors, including nuclear transcription factors and drives the formation of oxidative fibers in muscle (57). PGC-1β was unchanged in HD mice under basal conditions. GPA administration in WT mice resulted in significantly increased PGC-1α and PGC-1β mRNA expression in WT mice, which was abrogated in HD mice (Fig. 5A). GPA treatment in HD mice caused further significant down-regulation of PGC-1β in HD mice when compared with untreated HD mice. PGC-1α plays a prominent role in regulating mitochondrial biogenesis and cellular respiration by modulating the expression of transcription factors such as NRF-1, NRF-2 and estrogen-related receptor-α (ERR-α) (58–68). NRF-2 mRNA expression was significantly decreased in HD mice when compared with WT mice, whereas NRF-1 and ERR-α were not changed significantly (Fig. 5A and B). Following GPA treatment, NRF-1 expression significantly increased in WT mice, whereas NRF-2 was not changed in either group. Interestingly, following GPA administration, HD mice displayed a significantly reduced NRF-1 and ERR-α mRNA expression compared with untreated HD mice.
Figure 5.
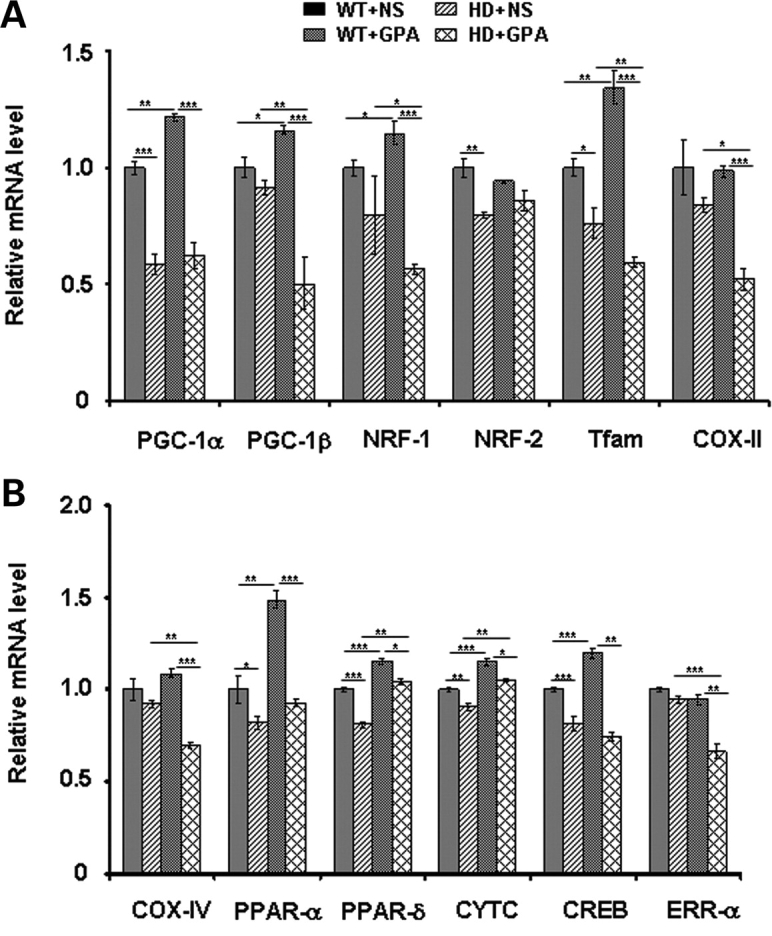
PGC-1α and its downstream target genes transcription in NLS-N171-82Q HD mice under chronic energy deprivation conditions. (A and B) We confirmed microarray gene expression data by quantitative real-time PCR. Relative mRNA expression levels of PGC-1α and its downstream target genes (PGC-1β, NRF-1, NRF-2, PPAR-α, PPAR-δ, CREB and ERR-α), genes involved in mitochondrial function (COX-II, COX-IV and CYTC) and mitochondrial biogenesis (Tfam) was measured in soleus muscles from GPA-treated and untreated WT and HD mice. Relative mRNA expression was normalized to β-actin. Data are expressed as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (n = 5 mice/group).
We analyzed the expression of PPAR-α and PPAR-δ, which co-act with PGC-1α in a positive feedback loop and regulate several metabolic pathways, including glucose metabolism, fatty acid oxidation, mitochondrial biogenesis and muscle function (58,61). As shown in Figure 5, the expression of PPAR-α and PPAR-δ is significantly reduced in muscle of HD mice. GPA treatment resulted in significantly increased PPAR-α and PPAR-δ expression in WT as well as in HD mice, however, the degree of PPAR-α increase was more pronounced in WT mice. CREB enhances the expression of PGC-1α by binding to cAMP response element (CRE) site in the PGC-1α promoter. We found a significant decrease in CREB mRNA expression in HD mice in basal conditions when compared with WT mice. Following GPA treatment, CREB went up in WT mice, while up-regulation of CREB was abrogated in HD mice (Fig. 5).
PGC-1α increases the expression of the transcription factor Tfam, a nuclear encoded mitochondrial transcription factor and a target of NRF-1, which after translocation to mitochondria increases the transcription and replication of the mitochondrial genome leading to mitochondrial biogenesis (38,60,62). Tfam mRNA expression was decreased significantly in HD mice. In WT mice, Tfam significantly increased following GPA treatment, but significantly decreased further in HD mice (Fig. 5A). These results are consistent with the reduced numbers of mitochondria and mitochondrial area observed by electron microscopy in GPA-treated HD mice when compared with WT mice (Fig. 3). Further, we assessed the mRNA expression of genes necessary for mitochondrial function such as cytochrome c oxidase subunits (e.g. COX-II and COX-IV) and Cyt C (Fig. 5A and B). Cyt C mRNA expression was significantly decreased in HD mice under basal conditions, it was up-regulated significantly in both WT and HD mice following GPA treatment. COX-II and COX-IV mRNA expression were unchanged in HD mice. There were no significant decrease in COX-II and COX-IV mRNA expression in WT+GPA mice, but they were significantly decreased in HD+GPA mice (Fig. 5).
Adenoviral-mediated delivery of PGC-1α in the muscle increases muscle oxidative capacity and reverses blunted response for GPA in HD mice
To further evaluate whether PGC-1α may be protective in HD, we have injected PGC-1α overexpressing adenoviral vector directly into the skeletal muscle of WT and transgenic HD mice. We have observed bright green fluorescence in the green fluorescent protein (GFP) adenoviral-transduced muscle fibers of HD mice (Fig. 6A). Following injection of PGC-1α overexpressing adenovirus, we have observed the expression of PGC-1α and MHC-I gene in muscle fibers of GPA-treated and untreated WT and HD mice (Fig. 6B). PGC-1α and MHC-I mRNA expression was significantly decreased in muscle of HD mice when compared with WT mice (Fig. 6B). We observed that PGC-1α overexpression caused a significant increase in PGC-1α and MHC-I mRNA expression in WT and HD mice following GPA treatment. PGC-1α overexpression also reversed the blunted response against GPA in HD mice.
Figure 6.

Adenoviral vector-mediated delivery of PGC-1α increases the muscle oxidative capacity and reverses the blunted response for GPA in transgenic HD mice. (A) Expression of GFP labeled PGC-1α adenoviral vectors in the muscle of HD transgenic mice 4 weeks after injection. A vast majority of adenoviral transduced muscle fibers exhibiting green fluorescence of GFP. (B) Relative mRNA expression of PGC-1α and oxidative muscle fiber marker MHC-I in the muscles from PGC-1α adenoviral injected WT and HD mice. mRNA levels were normalized to β-actin. P < 0.05, *= versus control, # = versus AAV/GFP. (C) Histochemical staining for succinate dehydrogenase (SDH) in muscle sections from PGC-1α adenoviral injected WT and HD mice treated with NS or GPA. Overexpression of PGC-1α resulted in increased number of dark stained type I oxidative fibers in muscle of WT and HD mice. Arrows indicate dark stained type I oxidative fibers, arrowheads mark to intermediate type IIA oxidative fibers and asterisks point to type IIB glycolytic fibers (Scale bar = 100 µm).
Histochemical staining of SDH in muscle tissue showed decreased SDH expression in HD mice when compared with WT (Fig. 6C). SDH activity increased significantly in GPA-treated WT mice, while it was further decreased in GPA-treated HD mice. Overexpression of PGC-1α resulted in increased number of dark stained type I oxidative fibers in muscle of WT and HD mice following GPA treatment (Fig. 6C).
Impaired PGC-1α transcription and function in muscle of HD patients
Impairment in transcription of PGC-1α and downstream target genes and abnormal muscle function in NLS-N171-82Q mice prompted us to ask, whether muscle tissue from HD patients also display these abnormalities. We examined mRNA expression of PGC-1α and target genes in muscle biopsies from symptomatic HD patients. We found a significant decrease of PGC-1α (32%) and PGC-1β (50%) but unchanged NRF-1 and Tfam mRNA expression in HD patients when compared with control subjects (Fig. 7A). We also analyzed mRNA expression of oxidative muscle markers such as troponin 1 (slow) and myoglobin. We found significantly decreased troponin 1 (slow) (24%) and myoglobin (35%) in muscle of HD patients (Fig. 7A). Interestingly, PPAR-α mRNA expression was significantly up-regulated in HD patients, while PPAR-δ, PPAR-γ and COX-IV were unchanged (Supplementary Material, Fig. S3). These results suggest impaired PGC-1α transcription and reduced oxidative capacity in muscles of HD patients.
Figure 7.
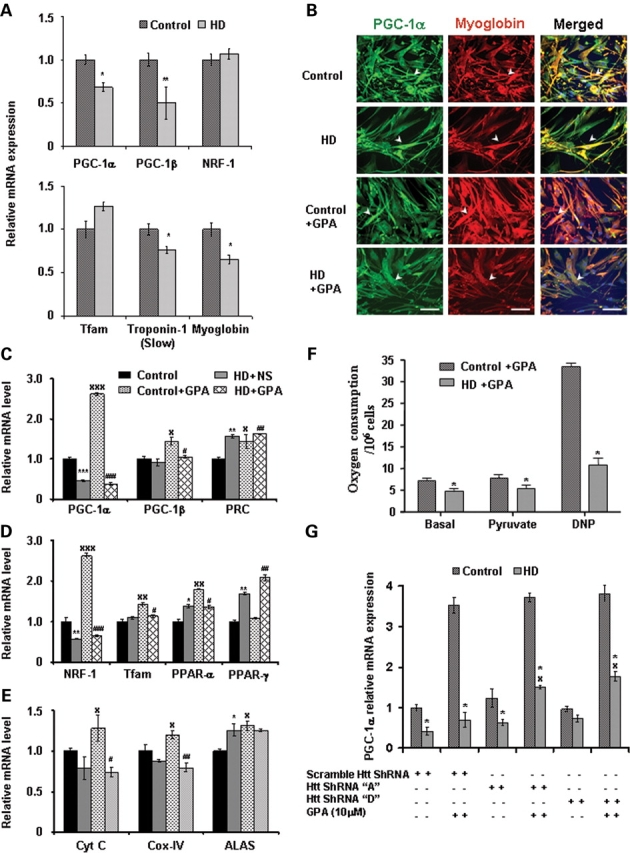
Impaired PGC-1α and target genes transcription, decreased oxidative capacity and cellular respiration in muscle biopsies and myoblasts from HD patients. (A) Quantitative real-time PCR analysis was performed in RNA isolated from muscle biopsies from symptomatic human HD patients and matched control subjects. Relative mRNA expression of PGC-1α and target genes and oxidative muscle markers (myoglobin and troponin I slow) was measured by normalizing values to β-actin. Data are expressed as the mean ± SEM. *P < 0.05, **P < 0.01 (n = 9 control subjects and n = 13 HD patients). (B) We established muscle cell (myoblast) culture from HD patients and control subjects. To create chronic energy deprivation conditions, myoblast cultures were treated with GPA for 7 days. Immunofluorescence analysis of PGC-1α (green fluorescence), oxidative muscle marker myoglobin (red fluorescence) and DAPI (blue fluorescence) in myoblast cultures from HD patients and control subjects. Co-localization of PGC-1α and myoglobin is shown in merged images (yellow fluorescence). Arrowheads indicate immunoreactivity for PGC-1α and myoglobin. In HD myoblasts, decreased immunostaining and co-localization of PGC-1α and myoglobin is observed. Following GPA treatment, PGC-1α and myoglobin expression are increased in control myoblasts, but not in HD myoblasts. Scale bars = 50 µm. (C–E) Quantitative real-time PCR analysis was performed in RNA isolated from GPA-treated myoblast cultures from HD patients and control subjects. Relative mRNA expression of PGC-1α and target genes and mitochondrial function genes was measured by normalizing values to β-actin. Data are expressed as mean ± SEM. * and x = versus WT+NS, # = versus WT+GPA, *P < 0.05, **P < 0.01, ***P < 0.001 (n = 3 control subjects and n = 5 HD patients). (F) Measurement of cellular oxygen consumption in GPA-treated myoblast cultures from HD patients and control subjects. Oxygen consumption was recorded under basal conditions and after addition of pyruvate and the mitochondrial uncoupler dinitrophenol (DNP). Data are expressed as mean ± SEM. *P < 0.05 (n = 3 control subjects and n = 5 HD patients). (G) Increased PGC-1α mRNA expression in HD myoblasts by knockdown of mutant huntingtin. Control and HD myoblasts were transiently transfected with plasmid vector containing scramble and ShRNA target sequences A and D against mutant huntingtin. Knockdown of mutant huntingtin followed by GPA treatment caused a significant increase of PGC-1α mRNA expression in HD myoblasts. Data are expressed as mean ± SEM of two experiments. * = versus Control, x = versus scramble ShRNA. *, xP < 0.05.
We established primary muscle cell (myoblast) cultures from muscle biopsies of control subjects (n = 3) and symptomatic HD patients (n = 5). Both the control subject myoblasts and HD myoblasts showed immunostaining for the muscle-specific marker Desmin (Supplementary Material, Fig. S4). Desmin immunostaining revealed the presence of apoptotic vacuoles in HD myoblasts when compared with control myoblasts (Supplementary Material, Fig. S4). Phase contrast microscopy of myoblast cultures showed that myoblasts from control subjects showed fine morphology and grew in a regular pattern, whereas HD myoblasts showed abnormal morphology and grew in an irregular pattern (Supplementary Material, Fig. S5). Similarly, myotubes differentiated from HD myoblasts also showed an irregular pattern of growth when compared with control myotubes (Supplementary Material, Fig. S6).
To investigate the effect of chronic energy deprivation conditions on PGC-1α transcription and function in myoblasts of control and HD subjects, we chronically treated myoblasts with GPA. Using immunocytochemistry, we observed reduced co-localization of PGC-1α (green) and myoglobin (red) (a protein highly expressed in oxidative muscle fibers) in HD myoblasts when compared with myoblasts of control subjects (Fig. 7B). Treatment with GPA caused a minimal increase in PGC-α and myoglobin immunostaining in HD myoblasts, whereas in control myoblasts pronounced immunostaining of PGC-1α and myoglobin was observed (Fig. 7B). These results show PGC-1α transcriptional interference and reduced oxidative capacity in myoblasts of HD patients under basal as well as energy stressed conditions.
We studied the mRNA expression of PGC-1α and downstream target genes both at baseline and following treatment with GPA in HD myoblasts. At baseline, there was a significant down-regulation of PGC-1α and NRF-1 and significant up-regulation of PRC (PGC-1α-related co-activator), PPAR-α, PPAR-γ and aminolevulinate synthase (ALAS) gene expression in HD myoblasts (Fig. 7C–E). Following treatment of control myoblasts with GPA, there were significant increases in PGC-1α, PGC-1β, PRC, NRF-1, Tfam, PPAR-α, Cyt C, COX IV and ALAS which were absent in the myoblasts from HD subjects. Only PPAR-γ was unchanged in WT mice after GPA treatment, but increased significantly in HD mice. At baseline there was no significant difference in oxygen consumption between control and HD myoblast (data not shown). Following GPA treatment, HD myoblasts showed reduced oxygen consumption at rest, with pyruvate and with the uncoupling compound dinitrophenol (Fig. 7F).
As mutant Htt inhibits PGC-1α transcriptional activity (38), we knocked down mutant Htt expression in cultured myoblasts using a specific target ShRNA sequence against mutant Htt (Fig. 7G). At baseline PGC-1α mRNA expression was significantly decreased in scrambled ShRNA transfected HD myoblasts when compared with control myoblasts (Fig. 7G). We found that knockdown of mutant Htt with ShRNAs A and D in HD myoblast resulted in a 20–25% increase of PGC-1α mRNA expression. GPA treatment further significantly induced PGC-1α mRNA expression up to ∼1.4-fold in mutant Htt knockdown HD myoblasts when compared with scramble sequence transfected HD myoblasts (Fig. 7G). PGC-1α transcript expression was comparable in control myoblasts following GPA treatment in all the conditions. Both ShRNAs targeting sequence A and D showed similar knockdown efficiency.
DISCUSSION
Recent studies showed that impaired functioning of PGC-1α contributes to mitochondrial dysfunction and appears to play an important role in HD pathogenesis (39,40). In striata from postmortem HD patient brains, striata from an HD knock-in mouse model and the STHdhQ111 cultured HD striatal cell line, there was a marked reduction in the expression of PGC-1α mRNA (39). It was suggested that mutant Htt interferes with formation of the CREB/TAF4 complex that regulates transcription of the gene encoding PGC-1α. The STHdhQ111 striatal neuronal line exhibited reduced expression of known mitochondrial gene targets of PGC-1α, including Cyt C and cytochrome oxidase IV. In knock-in HD mice (with 140 CAG repeats inserted into the murine Htt gene), the expression of PGC-1α was reduced several fold in medium spiny neurons, but increased almost 50-fold in nNOS interneurons, consistent with a role of PGC-1α in the selective vulnerability of medium spiny neurons and the resistance of interneurons (which are spared in HD).
Expression of PGC-1α is rapidly induced in response to cold, and the coactivator regulates expression of key components of adaptive thermogenesis including UCP-1, which uncouples respiration resulting in heat production in brown adipose tissue (36). There was marked hypothermia at baseline and following cold exposure in several HD mouse models (40). Following cold exposure, UCP-1 expression did not increase in brown adipose tissue from N171-82Q HD mice relative to WT animals, implicating impaired ability of PGC-1α to induce UCP-1 and activate thermogenesis in these mice. In primary brown adipocytes of N171-82 HD mice, a reduced ATP/ADP ratio and reduced numbers of mitochondria, as well as vacuolated brown adipose tissue, occur similar to the findings in PGC-1α-deficient mice (34). Using microarray expression data from the caudate nucleus of HD patient postmortem brain tissue, the authors reported reduced expression of 24 out of 26 PGC-1α target genes (40).
Other findings are consistent with impaired PGC-1α function in HD. There is substantial evidence for oxidative stress in HD plasma, HD postmortem brain tissue and transgenic mice (63,64). The increase in oxidative damage may be a consequence of a deficiency in PGC-1α and PGC-1β since they are required for induction of many reactive oxygen species detoxifying enzymes such as Cu/Zn superoxide dismutase, manganese superoxide dismutase, glutathione peroxidase and glutathione synthesis (59,65).
There is strong evidence that myopathy may play a role in HD. Polyglutamine inclusions as well as changes in gene expression and calcium regulation are observed in muscle from both HD mice and HD patients (4,25,42,45,46). Myopathy was reported as a first symptom of HD in a marathon runner (43). Cardiac dysfunction with abnormal mitochondria occurs in R6/2 HD transgenic mice, consistent with findings in PGC-1α-deficient mice (33,44,66). In R6/2 HD transgenic mice, the quadriceps and soleus muscles develop normally until 6weeks of age, after which they severely atrophy (4). In another study of muscle in R6/2 mice, there was progressive atrophy and supersensitivity to acetylcholine, as well as morphological abnormalities of neuromuscular junctions (5). This is of interest, since PGC-1α controls the development of neuromuscular junctions (67). The progressive muscle atrophy, which occurs in HD transgenic mice and patients, may be a consequence of impaired PGC-1α function, since it protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription (68).
In the present studies, we investigated whether PGC-1α dysfunction plays a role in muscle abnormalities in HD, by examining changes in an HD transgenic mouse model, myoblast cultures from HD patients and muscle biopsies from HD patients. We utilized the NLS-N171-82Q transgenic mouse model of HD in which the N171-82Q fragment is fused to a nuclear localization signal (69). In these mice, NLS-N171-82Q fragments accumulate in the nucleus and the mice develop progressive loss of weight, decreased motor performance, and loss of spontaneous activity, nuclear aggregates and premature death.
We found significant reductions of PGC-1α and Tfam in soleus muscle (type I fiber-enriched) and of PGC-1α, PGC-1β and Tfam in type IIB fiber-enriched EDL and smaller reductions of PGC-1α and Tfam in GM in which fiber types are mixed. We confirmed the reduced concentration of PGC-1α using western blots in both the soleus and EDL muscles. These results suggest that progressive muscle atrophy and morphological abnormalities of neuromuscular junctions observed in earlier studies could be a consequence of impaired PGC-1α and target gene expression (4,5).
To examine muscle function in HD mice under energy deprivation conditions, we treated HD mice with the creatine analogue GPA for 10 weeks. GPA treatment being a metabolic challenge, in many respects mimics the effects of endurance exercise leading to increased muscle oxidative capacity, increased glutamate transporter content and citrate synthase activities (49,51). Administration of GPA causes energy stressed conditions, depletes intramuscular ATP/AMP ratios leading to activation of muscle AMPK and muscle mitochondrial biogenesis (52). Inactivation of AMPK blocks this, as well as increased expression of PGC-1α. AMPK directly phosphorylates PGC-1α and increases its expression through a feedback loop (53). Thus, GPA treatment induces PGC-1α expression and mitochondrial biogenesis through activation of AMPK in muscle cells.
Following administration of GPA, there was a depletion of ATP and PCr in both WT and HD transgenic mice, which was greater in the HD mice. At baseline, there were reduced AMPK mRNA and protein levels in the muscle of HD mice. Following treatment with GPA, both AMPK mRNA and protein levels and PGC-1α mRNA significantly increased in WT mice, but there was no effect in the HD mice.
Skeletal muscle responds to increased energy demand such as during endurance exercise training, calcium fluxes and electrical stimulation by converting type II glycolytic muscle fibers (fast-twitch or fatigable fibers) into oxidative fibers (slow-twitch or fatigue resistant fibers). Muscle fibers can be classified as ‘slow-twitch’ fatigue resistant fibers which contain numerous mitochondria and use oxidative phosphorylation. These type I and IIA fibers allow for continuous activity with less fatigue. The type I and IIA fibers contain large amounts of myoglobin, are rich in mitochondria and generate ATP oxidatively. In contrast, type IIX and IIB fibers are ‘fast-twitch fatiguable’ fibers which have few mitochondria and which utilize glycolysis to generate ATP (41). PGC-1α plays a key role in the formation of slow-twitch (type 1) muscle fibers. PGC-1α expresses highly in type I fibers enriched muscle such as the soleus muscle and very low expression in type II fibers rich muscle such as EDL and GM (41). Interestingly, overexpression of PGC-1α in type II fibers enriched muscles causes fiber type conversion and increased expression of genes characteristic of type I fibers (41). In cultured muscle cells, PGC-1α activates transcription in cooperation with Mef2 proteins and serves as a target for calcineurin signaling, which has been implicated in slow fiber gene expression (41). In PGC-1α knockout mice, there is a shift from type I and type IIA towards type IIX and IIB muscle fibers which is accompanied by myopathy and exercise intolerance (70).
We, therefore, examined our HD transgenic mice for fiber typing using SDH histochemistry, immunohistochemistry to MHC isoforms and type I fiber-specific genes expression such as troponin and myoglobin. Examination of the soleus and GM showed that the muscle of WT mice were redder (consistent with more type I fibers) than those of the HD mice, and these differences were much more marked following GPA treatment. These observations are therefore consistent with an earlier study, where forced expression of PGC-1α resulted in an increased proportion of type I fibers and increased expression of mitochondrial markers and type I fibers specific contractile proteins, leading to red muscle coloration (41). SDH histochemistry confirmed that type I fibers increased following GPA treatment in WT mice, but they appeared to decrease further in the HD mice. The proportions of type I, type IIA and type IIB fibers were determined quantitatively using immunocytochemistry to MHC isoforms. There was a significant reduction in type I fibers and increase in type IIB fibers in the HD mice. Levels of troponin mRNA and myoglobin mRNA were consistent with these histological observations. These findings are also consistent with changes that occur in mice, which have a muscle-specific knockout of PGC-1α (70).
We carried out further studies to determine effects on mitochondrial area and mitochondrial number using electron microscopy. In WT mice, mitochondria are uniform in size and align regularly along the Z lines, whereas in the HD mice, the mitochondria were irregular and poorly aligned. Following treatment with GPA, there was a significant increase in mitochondrial area and number in WT mice, but no change in the HD mice. Similarly, PGC-1α-deficient mice show fewer and smaller mitochondria in soleus muscle (33).
We also examined Affymetrix gene expression profiling in GPA-treated HD mice (56). Predefined sets of genes were analyzed using GSEA. This analysis showed increased expression of genes involved in oxidative phosphorylation, electron transport chain and muscle function in the GPA-treated WT mice when compared with GPA-treated HD mice. The changes in PGC-1α, PGC-1β, as well as the downstream genes were confirmed using quantitative RT–PCR. In soleus muscle of HD mice, mRNA for PGC-1α, NRF-2, Tfam, PPARα, Cyt C and CREB were all reduced at baseline. Following treatment of WT mice, there were significant increases in PGC-1α, PGC-1β, Tfam, COX IV, PPARα, PPARδ, Cyt C and CREB, whereas there were no alterations of these genes following treatment of HD mice with GPA. Adenoviral-mediated PGC-1α overexpression resulted in increased muscle oxidative capacity and reversal of blunted response for GPA. These results corroborate with earlier studies, where PGC-1α overexpression was found to increase oxidative muscle fibers. (41).
In cultures of myoblasts from HD subjects, there is increased apoptosis, defective cell differentiation and formation of Htt inclusions (71). We studied HD myoblasts for mRNA levels both at baseline and following treatment with GPA. We found reduced levels of PGC-1α and NRF-1 at baseline. Following treatment of control myoblasts with GPA, there were increases in PGC-1α, PGC-1β, NRF-1, Tfam, PPAR-α, Cyt C and COX IV, which were absent in the myoblasts from HD subjects. Induction of NRF-1 and Tfam following GPA treatment is consistent with a previous study, where ectopic expression of PGC-1α in myotubes induced the expression of downstream transcription factors such as NRF-1 and Tfam (60). Immunocytochemical staining showed reduced expression of PGC-1α in HD myoblasts both at baseline and following treatment with GPA. The HD myoblasts showed reduced oxygen consumption at rest, as well as with pyruvate and following treatment with the uncoupling compound dinitrophenol. In muscle biopsies from HD patients and controls, we found significant decreases in PGC-1α and PGC-1β. There were also significant reductions in troponin 1 (slow) and myoglobin, markers of slow oxidative type 1 fibers.
In concert, the present findings, therefore, show that there is both reduced PGC-1α and its downstream genes controlling oxidative metabolism in muscle of HD transgenic mice in myoblasts from HD patients, and in muscle biopsies from HD patients. Both type I fibers and numbers of mitochondria were reduced in HD mice as compared with WT following GPA treatment. Furthermore, the ability to up-regulate PGC-1α and mitochondrial biogenesis in response to an energetic stress was blocked in HD transgenic mice and myoblasts from HD patients.
The finding that PGC-1α expression is impaired in muscle and brain of HD transgenic mice and patients raises the possibility that molecules that activate PGC-1α may be therapeutically useful. Such molecules are in development because reduced PGC-1α expression has been strongly implicated in obesity and type II diabetes (35). The present findings showing impaired PGC-1α function in muscle in HD, as well as in brain of both HD transgenic mice and patients suggest that PGC-1α is a therapeutic target and may play a critical role in HD disease pathogenesis. Furthermore, muscle may provide a readily accessible tissue in which to monitor therapeutic interventions.
MATERIALS AND METHODS
Transgenic animals and treatments
The NLS-N171-82Q mice were obtained from Dr. David Borchett and have the mouse prion promoter driving expression of 82-CAG repeat. In NLS-N171-82Q HD mice, a nuclear localization signal (NLS) derived from atrophin-1 was fused to the N-terminus of an N171-82Q construct (69). These mice develop phenotypes identical to N171-82Q mice. HD offspring were genotyped by PCR assay of DNA obtained from tail tissue. The animals were housed at Weill Medical College animal house and were kept on a 12-h light/dark cycle, with food and water continuously available. Experiments were carried out using procedures that minimized pain and discomfort. All experiments were conducted within National Institutes of Health guidelines for animal research and were approved by the Weill Cornell Medical College Animal Care and Use Committee.
WT littermates and HD male and female mice were divided equally in different treatment groups. WT and HD mice were treated with GPA, which is a creatine analogue that competes for the transport of creatine in the skeletal muscle and inhibit creatine kinase activity. Chronic GPA treatment causes chronic energy depletion by depleting intracellular PCr and ATP concentrations and mimics endurance exercise training conditions. Thus, chronic GPA treatment is a method to pharmacologically create chronic energy deprivation conditions. WT and HD mice at the age of 16 weeks received GPA by single i.p. injection daily for 10 weeks (0.2 ml/mouse; 0.5 m). WT and HD mice which received a daily single injection of saline (0.2 ml/mouse) for the same duration, served as a control as described earlier (52).
Transmission electron microscopy
Transmission electron microscopy was carried out in soleus muscle from different groups. See supplemental experimental procedures for a brief description.
Histochemical staining for SDH
For detail see Supplementary Material, experimental procedure.
MHC immunohistochemistry
Immunohistochemical staining of different isoforms of muscle was performed in soleus muscle of WT and HD mice following earlier published method (72). For detail see supplemental experimental procedure.
Western blot
Tissue samples from the skeletal muscle of WT and HD mouse and myoblast cultures were homogenized. The membranes were incubated overnight at 4°C for PGC-1α (1:2000, kind gift of Dr B.M. Spiegelman), AMPK (1:500, Santa Cruz, CA), pAMPK Thr 172 (1:500, Santa Cruz) and β-actin (1:10 000, Chemicon). For detail see supplemental experimental procedure.
HPLC assay for ATP, PCR and their metabolites
We have reported the method in our previous publications (73,74). Detailed methods are described in the Supplementary Material, Experimental procedure.
Myoblast cell culture and GPA treatment
Myoblasts derived from muscle biopsies were obtained from control subjects and HD patients with informed consent (71). The myoblasts were cultured as previously described (75). Myoblasts were grown in HAM's F10 medium (GIBCO, Invitrogen, San Diego, CA) supplemented with 15% fetal bovine serum (FBS) (Invitrogen), 0.5 mg/ml bovine serum albumin, 4 ng/ml insulin, 10 ng/ml epidermal growth factor, 0.39 mg/ml dexamethasone, 0.1 mg/ml streptomycin and 100 U/ml penicillin. To create chronic energy deprivation conditions, myoblast cultures were treated with 20 µm GPA dissolved in F-10 medium for 7 days.
Myoblast culture transient transfection
For transient transfection myoblasts from control subjects and HD patients were maintained in HAM's F10 medium with 15% FBS in 6-well plate. Transient transfection was performed with 4 µg plasmids per well, at a confluence of ∼65–70% using Lipofectamine™ 2000 as per manufacturer's protocol. For RNA interference of Htt, two different Htt shRNA target sequences (Htt ShRNA A and D) were inserted separately in pSUPER.retro.neo+GFP (8.37 kb) plasmids. pSUPER-control vector was constructed using a nucleotide sequence with no significant homology to any mammalian gene sequence and served as a scramble non-silencing control. HindIII and BglII restriction sites in pSUPER vector were used for insertion of shRNA target sequences. ShRNA target sequences are available upon request.
Measurement of myoblast oxygen consumption
Gene expression analysis by RT–PCR
Total RNA was isolated from frozen WT and HD mouse skeletal muscle and from cultured myoblast using TriZol Reagent as per manufacture's protocol. Primer sequences used for RT–PCR experiments are listed in Supplementary Material, Table S8. For detail see Supplementary Material, Experimental procedure.
Affymetrix gene expression profiling
Total RNA was isolated from muscle using QiaZen RNeasy RNA extraction kit according to the manufacturer's recommended protocol (Valencia, CA). Affymetrix microarray gene expression was carried out by NIH microarray consortium at Yale University, CT. The RNA samples were analyzed for integrity, using RNA 6000 Nano LabChip Kit and 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). The RNA was processed according to the standard Affymetrix protocol and analyzed using the Mouse Genome 430 2.0 Gene Chips. Data analysis was carried out using GeneSpring GX 7.3 software (Agilent Technologies). To test the differential expression of predefined sets of related genes between different treatment groups, GSEA was carried out (56).
Adenoviral injection
PGC-1α adenovirus was a kind gift of Bruce M. Spiegelman (Dana Farber Cancer Research Center, Harvard University). The PGC-1α adenovirus contained the gene for GFP in tandem with the PGC-1α gene. PGC-1α adenovirus were injected into the muscle of WT and HD mice following earlier published method (76).
Statistical analysis
Statistical analysis was performed using GraphPad InStat statistical analysis software version 3.05 for Windows (San Diego, CA). The mean significant difference in the experimental groups was determined using one-way analysis of variance followed by Tukey–Kramer post hoc multiple comparisons test. Values of P < 0.05 were considered to be statistically significant.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work was supported by the NS 39258 and Huntington's disease Society of America Coalition for the Cure.
ACKNOWLEDGEMENTS
We are thankful to Dr B.M Spiegelman for providing huntingtin ShRNA target sequence and PGC-1α adenoviral vector. Dr David Borchett is thanked for providing us with NLS-N171-82Q transgenic HD mice. We are also thankful to Laragen Inc. Los Angels, CA, for mice PCR genotyping. Secretarial assistance of Greta Strong is gratefully acknowledged.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Pratley R.E., Salbe A.D., Ravussin E., Caviness J.N. Higher sedentary energy expenditure in patients with Huntington's disease. Ann. Neurol. 2000;47:64–70. [PubMed] [Google Scholar]
- 2.Goodman A.O., Murgatroyd P.R., Medina-Gomez G., Wood N.I., Finer N., Vidal-Puig A.J., Morton A.J., Barker R.A. The metabolic profile of early Huntington's disease- a combined human and transgenic mouse study. Exp. Neurol. 2008;210:691–698. doi: 10.1016/j.expneurol.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 3.Djousse L., Knowlton B., Cupples L.A., Marder K., Shoulson I., Myers R.H. Weight loss in early stage of Huntington's disease. Neurology. 2002;59:1325–1330. doi: 10.1212/01.wnl.0000031791.10922.cf. [DOI] [PubMed] [Google Scholar]
- 4.Sathasivam K., Hobbs C., Turmaine M., Mangiarini L., Mahal A., Bertaux F., Wanker E.E., Doherty P., Davies S.W., Bates G.P. Formation of polyglutamine inclusions in non-CNS tissue. Hum. Mol. Genet. 1999;8:813–822. doi: 10.1093/hmg/8.5.813. [DOI] [PubMed] [Google Scholar]
- 5.Ribchester R.R., Thomson D., Wood N.I., Hinks T., Gillingwater T.H., Wishart T.M., Court F.A., Morton A.J. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington's disease mutation. Eur. J. Neurosci. 2004;20:3092–3114. doi: 10.1111/j.1460-9568.2004.03783.x. [DOI] [PubMed] [Google Scholar]
- 6.Grafton S.T., Mazziotta J.C., Pahl J.J., St George-Hyslop P., Haines J.L., Gusella J., Hoffman J.M., Baxter L.R., Phelps M.E. Serial changes of cerebral glucose metabolism and caudate size in persons at risk for Huntington's disease. Arch. Neurol. 1992;49:1161–1167. doi: 10.1001/archneur.1992.00530350075022. [DOI] [PubMed] [Google Scholar]
- 7.Feigin A., Leenders K.L., Moeller J.R., Missimer J., Kuenig G., Spetsieris P., Antonini A., Eidelberg D. Metabolic network abnormalities in early Huntington's disease: an [(18)F]FDG PET study. J. Nucl. Med. 2001;42:1591–1595. [PubMed] [Google Scholar]
- 8.Antonini A., Leenders K.L., Spiegel R., Meier D., Vontobel P., Weigell-Weber M., Sanchez-Pernaute R., de Yebenez J.G., Boesiger P., Weindl A., et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington's disease. Brain. 1996;119(Pt 6):2085–2095. doi: 10.1093/brain/119.6.2085. [DOI] [PubMed] [Google Scholar]
- 9.Kuwert T., Lange H.W., Boecker H., Titz H., Herzog H., Aulich A., Wang B.C., Nayak U., Feinendegen L.E. Striatal glucose consumption in chorea-free subjects at risk of Huntington's disease. J. Neurol. 1993;241:31–36. doi: 10.1007/BF00870669. [DOI] [PubMed] [Google Scholar]
- 10.Jenkins B.G., Koroshetz W.J., Beal M.F., Rosen B.R. Evidence for impairment of energy metabolism in vivo in Huntington's disease using localized 1H NMR spectroscopy. Neurology. 1993;43:2689–2695. doi: 10.1212/wnl.43.12.2689. [DOI] [PubMed] [Google Scholar]
- 11.Koroshetz W.J., Jenkins B.G., Rosen B.R., Beal M.F. Energy metabolism defects in Huntington's disease and effects of coenzyme Q10. Ann. Neurol. 1997;41:160–165. doi: 10.1002/ana.410410206. [DOI] [PubMed] [Google Scholar]
- 12.Lodi R., Schapira A.H., Manners D., Styles P., Wood N.W., Taylor D.J., Warner T.T. Abnormal in vivo skeletal muscle energy metabolism in Huntington's disease and dentatorubropallidoluysian atrophy. Ann. Neurol. 2000;48:72–76. [PubMed] [Google Scholar]
- 13.Saft C., Zange J., Andrich J., Muller K., Lindenberg K., Landwehrmeyer B., Vorgerd M., Kraus P.H., Przuntek H., Schols L. Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington's disease. Mov. Disord. 2005;20:674–679. doi: 10.1002/mds.20373. [DOI] [PubMed] [Google Scholar]
- 14.Browne S.E., Bowling A.C., MacGarvey U., Baik M.J., Berger S.C., Muqit M.M., Bird E.D., Beal M.F. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann. Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 15.Gu M., Gash M.T., Mann V.M., Javoy-Agid F., Cooper J.M., Schapira A.H. Mitochondrial defect in Huntington's disease caudate nucleus. Ann. Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 16.Tabrizi S.J., Cleeter M.W., Xuereb J., Taanman J.W., Cooper J.M., Schapira A.H. Biochemical abnormalities and excitotoxicity in Huntington's disease brain. Ann. Neurol. 1999;45:25–32. doi: 10.1002/1531-8249(199901)45:1<25::aid-art6>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 17.Seong I.S., Ivanova E., Lee J.M., Choo Y.S., Fossale E., Anderson M., Gusella J.F., Laramie J.M., Myers R.H., Lesort M., et al. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum. Mol. Genet. 2005;14:2871–2880. doi: 10.1093/hmg/ddi319. [DOI] [PubMed] [Google Scholar]
- 18.Milakovic T., Johnson G.V. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J. Biol. Chem. 2005;280:30773–30782. doi: 10.1074/jbc.M504749200. [DOI] [PubMed] [Google Scholar]
- 19.Beal M.F., Brouillet E., Jenkins B., Henshaw R., Rosen B., Hyman B.T. Age-dependent striatal excitotoxic lesions produced by the endogenous mitochondrial inhibitor malonate. J. Neurochem. 1993;61:1147–1150. doi: 10.1111/j.1471-4159.1993.tb03633.x. [DOI] [PubMed] [Google Scholar]
- 20.Brouillet E., Hantraye P., Ferrante R.J., Dolan R., Leroy-Willig A., Kowall N.W., Beal M.F. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc. Natl Acad. Sci. USA. 1995;92:7105–7109. doi: 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ludolph A.C., He F., Spencer P.S., Hammerstad J., Sabri M. 3-Nitropropionic acid-exogenous animal neurotoxin and possible human striatal toxin. Can. J. Neurol. Sci. 1991;18:492–498. doi: 10.1017/s0317167100032212. [DOI] [PubMed] [Google Scholar]
- 22.Brouillet E., Jenkins B.G., Hyman B.T., Ferrante R.J., Kowall N.W., Srivastava R., Roy D.S., Rosen B.R., Beal M.F. Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J. Neurochem. 1993;60:356–359. doi: 10.1111/j.1471-4159.1993.tb05859.x. [DOI] [PubMed] [Google Scholar]
- 23.Orr A.L., Li S., Wang C.E., Li H., Wang J., Rong J., Xu X., Mastroberardino P.G., Greenamyre J.T., Li X.J. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 2008;28:2783–271492. doi: 10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choo Y.S., Johnson G.V., MacDonald M., Detloff P.J., Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004;13:1407–1420. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- 25.Orth M., Cooper J.M., Bates G.P., Schapira A.H. Inclusion formation in Huntington's disease R6/2 mouse muscle cultures. J. Neurochem. 2003;87:1–6. doi: 10.1046/j.1471-4159.2003.02009.x. [DOI] [PubMed] [Google Scholar]
- 26.Panov A.V., Gutekunst C.A., Leavitt B.R., Hayden M.R., Burke J.R., Strittmatter W.J., Greenamyre J.T. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat. Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 27.Cha J.H. Transcriptional dysregulation in Huntington's disease. Trends Neurosci. 2000;23:387–392. doi: 10.1016/s0166-2236(00)01609-x. [DOI] [PubMed] [Google Scholar]
- 28.Sugars K.L., Rubinsztein D.C. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- 29.Chen-Plotkin A.S., Sadri-Vakili G., Yohrling G.J., Braveman M.W., Benn C.L., Glajch K.E., DiRocco D.P., Farrell L.A., Krainc D., Gines S., et al. Decreased association of the transcription factor Sp1 with genes downregulated in Huntington's disease. Neurobiol. Dis. 2006;22:233–241. doi: 10.1016/j.nbd.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Dunah A.W., Jeong H., Griffin A., Kim Y.M., Standaert D.G., Hersch S.M., Mouradian M.M., Young A.B., Tanese N., Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- 31.Nucifora F.C., Jr, Sasaki M., Peters M.F., Huang H., Cooper J.K., Yamada M., Takahashi H., Tsuji S., Troncoso J., Dawson V.L., et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 32.Steffan J.S., Kazantsev A., Spasic-Boskovic O., Greenwald M., Zhu Y.Z., Gohler H., Wanker E.E., Bates G.P., Housman D.E., Thompson L.M. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl Acad. Sci. USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leone T.C., Lehman J.J., Finck B.N., Schaeffer P.J., Wende A.R., Boudina S., Courtois M., Wozniak D.F., Sambandam N., Bernal-Mizrachi C., et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin J., Wu P.H., Tarr P.T., Lindenberg K.S., St-Pierre J., Zhang C.Y., Mootha V.K., Jager S., Vianna C.R., Reznick R.M., et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 35.Puigserver P., Spiegelman B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 36.Puigserver P., Wu Z., Park C.W., Graves R., Wright M., Spiegelman B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 37.Lin J., Handschin C., Spiegelman B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Kelly D.P., Scarpulla R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 39.Cui L., Jeong H., Borovecki F., Parkhurst C.N., Tanese N., Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 40.Weydt P., Pineda V.V., Torrence A.E., Libby R.T., Satterfield T.F., Lazarowski E.R., Gilbert M.L., Morton G.J., Bammler T.K., Strand A.D., et al. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 41.Lin J., Wu H., Tarr P.T., Zhang C.Y., Wu Z., Boss O., Michael L.F., Puigserver P., Isotani E., Olson E.N., et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 42.Gizatullina Z.Z., Lindenberg K.S., Harjes P., Chen Y., Kosinski C.M., Landwehrmeyer B.G., Ludolph A.C., Striggow F., Zierz S., Gellerich F.N. Low stability of Huntington muscle mitochondria against Ca2+ in R6/2 mice. Ann. Neurol. 2006;59:407–411. doi: 10.1002/ana.20754. [DOI] [PubMed] [Google Scholar]
- 43.Kosinski C.M., Schlangen C., Gellerich F.N., Gizatullina Z., Deschauer M., Schiefer J., Young A.B., Landwehrmeyer G.B., Toyka K.V., Sellhaus B., et al. Myopathy as a first symptom of Huntington's disease in a Marathon runner. Mov. Disord. 2007;22:1637–1640. doi: 10.1002/mds.21550. [DOI] [PubMed] [Google Scholar]
- 44.Mihm M.J., Amann D.M., Schanbacher B.L., Altschuld R.A., Bauer J.A., Hoyt K.R. Cardiac dysfunction in the R6/2 mouse model of Huntington's disease. Neurobiol. Dis. 2007;25:297–308. doi: 10.1016/j.nbd.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strand A.D., Aragaki A.K., Shaw D., Bird T., Holton J., Turner C., Tapscott S.J., Tabrizi S.J., Schapira A.H., Kooperberg C., et al. Gene expression in Huntington's disease skeletal muscle: a potential biomarker. Hum. Mol. Genet. 2005;14:1863–1876. doi: 10.1093/hmg/ddi192. [DOI] [PubMed] [Google Scholar]
- 46.Turner C., Cooper J.M., Schapira A.H. Clinical correlates of mitochondrial function in Huntington's disease muscle. Mov. Disord. 2007;22:1715–1721. doi: 10.1002/mds.21540. [DOI] [PubMed] [Google Scholar]
- 47.Gines S., Seong I.S., Fossale E., Ivanova E., Trettel F., Gusella J.F., Wheeler V.C., Persichetti F., MacDonald M.E. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington's disease knock-in mice. Hum. Mol. Genet. 2003;12:497–508. doi: 10.1093/hmg/ddg046. [DOI] [PubMed] [Google Scholar]
- 48.Fitch C.D., Jellinek M., Mueller E.J. Experimental depletion of creatine and phosphocreatine from skeletal muscle. J. Biol. Chem. 1974;249:1060–1063. [PubMed] [Google Scholar]
- 49.Shoubridge E.A., Challiss R.A., Hayes D.J., Radda G.K. Biochemical adaptation in the skeletal muscle of rats depleted of creatine with the substrate analogue beta-guanidinopropionic acid. Biochem. J. 1985;232:125–131. doi: 10.1042/bj2320125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Gorman E., Beutner G., Wallimann T., Brdiczka D. Differential effects of creatine depletion on the regulation of enzyme activities and on creatine-stimulated mitochondrial respiration in skeletal muscle, heart, and brain. Biochim. Biophys. Acta. 1996;1276:161–170. doi: 10.1016/0005-2728(96)00074-6. [DOI] [PubMed] [Google Scholar]
- 51.Yaspelkis B.B., 3rd, Castle A.L., Farrar R.P., Ivy J.L. Effect of chronic electrical stimulation and beta-GPA diet on GLUT4 protein concentration in rat skeletal muscle. Acta Physiol. Scand. 1998;163:251–259. doi: 10.1046/j.1365-201x.1998.00377.x. [DOI] [PubMed] [Google Scholar]
- 52.Zong H., Ren J.M., Young L.H., Pypaert M., Mu J., Birnbaum M.J., Shulman G.I. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl Acad. Sci. USA. 2002;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jager S., Handschin C., St-Pierre J., Spiegelman B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stein S.C., Woods A., Jones N.A., Davison M.D., Carling D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem. J. 2000;345(Pt 3):437–443. [PMC free article] [PubMed] [Google Scholar]
- 55.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E., et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 56.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arany Z., Lebrasseur N., Morris C., Smith E., Yang W., Ma Y., Chin S., Spiegelman B.M. The transcriptional coactivator PGC-1beta drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab. 2007;5:35–46. doi: 10.1016/j.cmet.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 58.Mootha V.K., Handschin C., Arlow D., Xie X., St Pierre J., Sihag S., Yang W., Altshuler D., Puigserver P., Patterson N., et al. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc. Natl Acad. Sci. USA. 2004;101:6570–6575. doi: 10.1073/pnas.0401401101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.St-Pierre J., Lin J., Krauss S., Tarr P.T., Yang R., Newgard C.B., Spiegelman B.M. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J. Biol. Chem. 2003;278:26597–26603. doi: 10.1074/jbc.M301850200. [DOI] [PubMed] [Google Scholar]
- 60.Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G., Mootha V., Troy A., Cinti S., Lowell B., Scarpulla R.C., et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 61.Huss J.M., Kelly D.P. Nuclear receptor signaling and cardiac energetics. Circ. Res. 2004;95:568–578. doi: 10.1161/01.RES.0000141774.29937.e3. [DOI] [PubMed] [Google Scholar]
- 62.Larsson N.G., Wang J., Wilhelmsson H., Oldfors A., Rustin P., Lewandoski M., Barsh G.S., Clayton D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 63.Browne S.E., Beal M.F. Oxidative damage in Huntington's disease pathogenesis. Antioxid. Redox Signal. 2006;8:2061–2073. doi: 10.1089/ars.2006.8.2061. [DOI] [PubMed] [Google Scholar]
- 64.Hersch S.M., Gevorkian S., Marder K., Moskowitz C., Feigin A., Cox M., Como P., Zimmerman C., Lin M., Zhang L., et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2’dG. Neurology. 2006;66:250–252. doi: 10.1212/01.wnl.0000194318.74946.b6. [DOI] [PubMed] [Google Scholar]
- 65.St-Pierre J., Drori S., Uldry M., Silvaggi J.M., Rhee J., Jager S., Handschin C., Zheng K., Lin J., Yang W., et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 66.Arany Z., He H., Lin J., Hoyer K., Handschin C., Toka O., Ahmad F., Matsui T., Chin S., Wu P.H., et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 67.Handschin C., Kobayashi Y.M., Chin S., Seale P., Campbell K.P., Spiegelman B.M. PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007;21:770–783. doi: 10.1101/gad.1525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sandri M., Lin J., Handschin C., Yang W., Arany Z.P., Lecker S.H., Goldberg A.L., Spiegelman B.M. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl Acad. Sci. USA. 2006;103:16260–16265. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schilling G., Savonenko A.V., Klevytska A., Morton J.L., Tucker S.M., Poirier M., Gale A., Chan N., Gonzales V., Slunt H.H., et al. Nuclear-targeting of mutant huntingtin fragments produces Huntington's disease-like phenotypes in transgenic mice. Hum. Mol. Genet. 2004;13:1599–1610. doi: 10.1093/hmg/ddh175. [DOI] [PubMed] [Google Scholar]
- 70.Handschin C., Chin S., Li P., Liu F., Maratos-Flier E., Lebrasseur N.K., Yan Z., Spiegelman B.M. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J. Biol. Chem. 2007;282:30014–30021. doi: 10.1074/jbc.M704817200. [DOI] [PubMed] [Google Scholar]
- 71.Ciammola A., Sassone J., Alberti L., Meola G., Mancinelli E., Russo M.A., Squitieri F., Silani V. Increased apoptosis, Huntingtin inclusions and altered differentiation in muscle cell cultures from Huntington's disease subjects. Cell Death Differ. 2006;13:2068–2078. doi: 10.1038/sj.cdd.4401967. [DOI] [PubMed] [Google Scholar]
- 72.Behan W.M., Cossar D.W., Madden H.A., McKay I.C. Validation of a simple, rapid, and economical technique for distinguishing type 1 and 2 fibres in fixed and frozen skeletal muscle. J. Clin. Pathol. 2002;55:375–380. doi: 10.1136/jcp.55.5.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Manfredi G., Yang L., Gajewski C.D., Mattiazzi M. Measurements of ATP in mammalian cells. Methods. 2002;26:317–326. doi: 10.1016/S1046-2023(02)00037-3. [DOI] [PubMed] [Google Scholar]
- 74.Matthews R.T., Yang L., Jenkins B.G., Ferrante R.J., Rosen B.R., Kaddurah-Daouk R., Beal M.F. Neuroprotective effects of creatine and cyclocreatine in animal models of Huntington's disease. J. Neurosci. 1998;18:156–163. doi: 10.1523/JNEUROSCI.18-01-00156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blau H.M., Webster C. Isolation and characterization of human muscle cells. Proc. Natl Acad. Sci. USA. 1981;78:5623–5627. doi: 10.1073/pnas.78.9.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martinov V.N., Sefland I., Walaas S.I., Lømo T., Njå A., Hoover F. Targeting functional subtypes of spinal motoneurons and skeletal muscle fibers in vivo by intramuscular injection of adenoviral and adeno-associated viral vectors. Anat. Embryol. 2002;205:215–221. doi: 10.1007/s00429-002-0233-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.