

Abstract
Holoprosencephaly (HPE) is the most common developmental anomaly of the human forebrain; however, the genetics of this heterogeneous and etiologically complex malformation is incompletely understood. Heterozygous mutations in SIX3, a transcription factor gene expressed in the anterior forebrain and eyes during early vertebrate development, have been frequently detected in human HPE cases. However, only a few mutations have been investigated with limited functional studies that would confirm a role in HPE pathogenesis. Here, we report the development of a set of robust and sensitive assays of human SIX3 function in zebrafish and apply these to the analysis of a total of 46 distinct mutations (19 previously published and 27 novel) located throughout the entire SIX3 gene. We can now confirm that 89% of these putative deleterious mutations are significant loss-of-function alleles. Since disease-associated single point mutations in the Groucho-binding eh1-like motif decreases the function in all assays, we can also confirm that this interaction is essential for human SIX3 co-repressor activity; we infer, in turn, that this function is important in HPE causation. We also unexpectedly detected truncated versions with partial function, yet missing a SIX3-encoded homeodomain. Our data indicate that SIX3 is a frequent target in the pathogenesis of HPE and demonstrate how this can inform the genetic counseling of families.
INTRODUCTION
In humans, holoprosencephaly (HPE) encompasses an extremely broad clinical spectrum, with individual examples ranging from a structurally normal brain with only subtle clinical microsigns (such as a single central incisor) to the clinically overt single-eye with proboscis facies, and undivided forebrain, more typical of the extreme forms (1,2). Factors contributing to the cumulatively frequent occurrence of HPE (one in 250 during embryogenesis) are the numerous genes and environmental agents implicated in its causation (reviewed in 1–4). Most of these HPE genes, such as SIX3 (5), participate in developmental processes that are shared among vertebrates and allow for evaluations in model systems to illustrate some of the key aspects of its pathogenesis. Nevertheless, there are often as many dissimilarities as there are congruencies between animal models and humans with respect to many phenotypic details (reviewed in 6). Various models, such as the ‘multiple-hit hypothesis’ (7), have been promoted, which acknowledge these differences and postulate that the underlying genetic composition of sequence changes are likely to be complex (i.e. requiring multiple factors: genetic and/or environmental) in humans and thereby establishing a basis for some of these cross-species differences.
Six3 was originally described as one of the earliest genes to be expressed in the anterior forebrain (8–13). Slightly later in development, it plays a second and distinct role as a constituent component of a complex network of eye field transcription factors that regulate eye and lens development (14). A clue to at least one of the mechanisms and functions of Six3 surfaced when several groups reported the association of the Six3 transcription factor with members of the groucho co-receptor sub-family by yeast two-hybrid and co-immunoprecipitation studies (15–17). It is now generally accepted that this interaction is important for the growth and regional patterning of the vertebrate forebrain territory (13,18). The specification and maintenance of this anterior character depends on the repression of key genes during forebrain development and the pathways affected include Wnts, BMP and Nodal signaling (18–20). In particular, Six3 is described as one of several key factors that maintain the anterior character of the forebrain (21) because conditional elimination of its activity leads to progressive caudalization of the forebrain in both mice (22) and zebrafish (reviewed in 23) (24,25). Some of the best-understood caudalizing agents are the Wnt factors (26,27) whose over-activity can explain many of the observed effects of diminished Six3 function in animals. Indeed, the Wnt1 gene is a direct target of the repressive action of Six3 in vivo (18). Additional effects of Six3 include its interaction with Geminin (28), which influences the cellular decisions between proliferation and differentiation of neuroepithelial cells. Finally, Six3 has also been shown to activate the expression of lens specification genes (14). Thus, Six3 acts at multiple times and places and can involve the activation as well as the repression of key genes. Consequently, it seems apparent that Six3 is a component of multiple transcription factor complexes in a context-specific manner.
Mice heterozygous for null mutations in Six3 are described to be fertile and phenotypically normal (18); however, an essential role for murine Six3 in forebrain and eye development is clearly established from homozygous null embryos (14,22). In contrast, human HPE patients are typically heterozygous for sequence changes within the SIX3 gene (5,29–33). While the complete genetic composition of HPE mutations for any particular HPE case is unknown, the majority of well-documented cases to date have measurably diminished function in only one HPE gene per individual proband (Muenke lab, unpublished observations; 1, 5, 7, 29–33). Indeed, the 46 individual SIX3 mutations studied in this report (Table 1) are typical of the single family-specific change seen in this, and other, HPE cases. While we cannot exclude important genetic interactions between SIX3 and other factors, our understanding of the range of factors that could be functionally important is incomplete.
Table 1.
Summary of mutations
 |
To fully assess the significance of the many SIX3 alleles associated with HPE, it is critical to determine the extent to which their biological activities, inferred from orthologs studied in model organisms, are affected by their specific mutations. Setting the questions of the precise mechanistic consequences and/or hypothetical co-factor(s) aside, we set out to develop a robust and high-throughput functional assay that could be used to directly measure and compare WT and mutant SIX3 activities. Here we show that zebrafish is an adaptable and versatile system to execute this strategy. Using one biosensor assay, we found that 89% of detected SIX3 mutations are loss-of-function alleles. However, while truncating mutations eliminating SIX3’s DNA-binding domain lacked function in this biosensor assay, these mutant alleles retained bioactivity in a second biosensor assay. Mutations in the Groucho-binding eh1-like motif decreased function in both assays.
RESULTS
Evaluation of suitable assays
Since the mechanism(s) underlying HPE causation resulting from putative SIX3 dysfunction is incompletely understood, potentially any quantifiable parameter attributed to zebrafish six3, or its vertebrate orthologs, might be suitable to generate an initial test system. It would still be left to future studies to elaborate which of the many known biological properties attributed to Six3 orthologs were most consistently affected in HPE patients. In addition, our ultimate approach was to seek out assays that would be robust, rapid, and readily scalable to the large numbers of mutations available for study. Hence, previously described assays that quantified aspects of DNA binding or specific target gene activation/repression that we considered (14,18), and that might be clearly relevant to a fraction of mutations, were ultimately set aside in favor of assays that estimated the function of the complete SIX3 protein molecule in vivo. In this regard, zebrafish has distinct advantages over cell-based systems (34) in that large numbers of essentially uniform embryos can be rapidly collected and studied. Therefore, in order to screen for effects of SIX3 mutations on biological activity, we narrowed our focus ultimately to two assays measuring the ability of injected human SIX3 alleles to either rescue the headless phenotype [generated by simultaneous Tcf3 morphilino injection (18,25)] (rescue assay; Fig. 1A–F) or to induce dorsalized phenotypes (35,36) that we noted arising from higher dose SIX3 injections (overexpression assay; Fig. 1G–N), a phenomenon that was independently reported for zebrafish six3 mRNA injections (19). These two assays, respectively, measure the ability of SIX3 to repress wnt1 expression (Fig. 1D–F) or to shift the balance of dorsal–ventral patterning markers in the BMP pathway (Fig. 1K–N). This latter role for SIX3 in BMP dorsal–ventral patterning during gastrulation is a novel finding that is consistent with the reported repression of BMPs by Six3 during brain patterning in Xenopus (20). Here we demonstrate that the human SIX3 gene can alter the dorsal–ventral specification pathway influenced by BMPs in an analogous manner.
Figure 1.
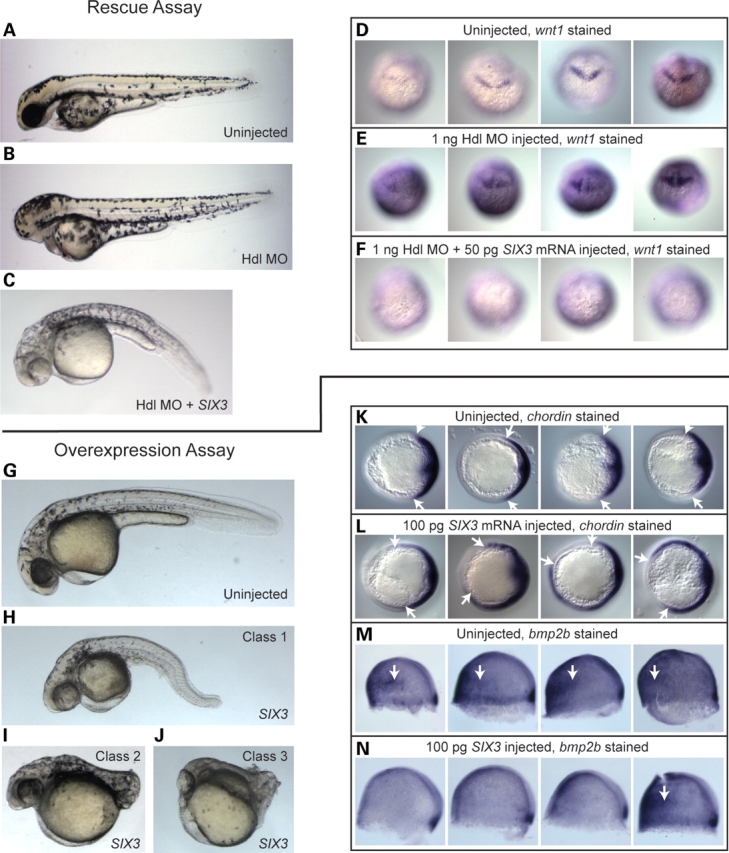
WT human SIX3 is biologically potent in zebrafish embryos. (A–N) Phenotypes, classification criteria and molecular validation underlying a biosensor assay based on rescue of eye formation [rescue assay: (A–F)] and a second biosensor assay based on embryonic dorsalization [overexpression assay: (G–N)]. Embryos were injected with various combinations of an antisense MO targeting translation of the Hdl/Tcf3 protein and/or (WT) SIX3 mRNA and incubated until the mid-gastrula stage for WISH with chordin (43) and bmp2b (41) probes (K–N), at the bud stage for WISH with a wnt1 (42) probe (D–F) or until the second day of development for live scoring (A–C and G–J). Rescue assay (A–F): Hdl MO alone caused an eyeless phenotype (B) while co-injection of 50 pg WT human SIX3 mRNA efficiently restored eye development (C). WISH confirmed that injection of Hdl MO alone (E) expanded wnt1 expression levels relative to uninjected embryos in 63% (N = 30) of the embryos examined (D), although wnt1 staining intensity in both groups displayed some variability. Co-injection of SIX3 mRNA reversed this expansion and completely shut down wnt1 expression in 93% (N = 27) of the embryos examined (F). Overexpression assay (G–N): injection of 50 pg SIX3 alone produced a range of phenotypes (H–J) that we grouped into three classes. These SIX3-induced phenotypes match classical embryonic dorsalized phenotypes seen, for instance, in BMP pathway mutants (35,36). WISH on mid-gastrula-stage embryos injected with 100 pg SIX3 confirmed dorsalization on the molecular level, with clear ventral expansion of the dorsal marker chordin in 65% (N = 26) of SIX3-injected embryo (L) and 0% (N = 24) of control embryos (K). Similarly, ventral reduction of the ventral marker bmp2b was apparent in 73% (N = 26) of SIX3-injected embryos (N), compared with 10% (N = 21) of control embryos (M). Embryos in (D–F) are presented in a dorsal view, in (K and L) are presented in an animal pole view, with dorsal to the right and in (M and N) are presented in a lateral view, with dorsal to the right. White arrows in (K) and (L) point to the ventral limits of chordin stain. White arrows in (M) and (N) point to ventrally situated patches of bmp2b stain, which were lost in the majority of SIX3-injected embryos.
Figure 2 illustrates the informative comparison between these two assays. Both assays reach a plateau level, but with very different dose–response slopes. We noted that the slope of the headless rescue assay was so steep that small differences in bioactivity were quite difficult to replicate. In contrast, the slope of the dose-response curve of the overexpression assay was a far superior discriminator between SIX3 mutations with different degrees of activity.
Figure 2.

Comparison of the two biosensor assays. Various doses of WT SIX3 mRNA were injected into embryos either with (for rescue assay calibration) or without (for overexpression assay calibration) co-injection of 1 ng Hdl MO. The Y-axis represents the fraction of embryos that achieved the theoretical maximum score on two distinct criteria scales. For the rescue assay, the measurable parameter was the presence of rescued eyes with the theoretical maximum of 100%. For the overexpression assay, the measurable parameter was the distribution of phenotypic classes that were seen and the theoretical maximum was a PI score (see Materials and Methods) of 3, which would be achieved if exclusively class 3 embryos were obtained. Note the rapid transition from half of the embryos rescued by 2.5 pg SIX3 mRNA to nearly all rescued by 5.0 pg SIX3 mRNA, explaining the high variability we observed with this assay (Fig. 5).
Systematic comparison of bioactivities of mutant six3 proteins
The reproducibility of the overexpression assay (Fig. 3A–C and Supplementary Material, Table S1) allows us to confidently conclude that each mutation has a characteristic level of activity, which we have color-coded in Figs 3 and 4 and Table 1 as black for apparently normal, orange for hypomorphic and red for apparent complete loss of function. A summary of the activity (based on the most sensitive discriminator: the overexpression assay) and position of the mutations is given in Fig. 4. Although frameshift (asterisk) and truncation mutations (hash) are understandably diminished in measurable activity, it was not obvious at the outset that we would observe differences between missense changes at the same amino acid position, as we indeed did (e.g. compare 29 with 30 or 40 with 41). Similarly, although the general clustering of mutations in the SIX domain and homeodomain had suggested their importance, we can now confirm this with concrete measurements. Interestingly, not all of the missense changes to the highly conserved homeodomain impair function to the same extent, and some variations have little effect (e.g. L226V) (5,37). What clearly emerges from this analysis is the utility of evaluating and comparing a large number of mutations at the same time. The degree of impairment measured varies with the particular mutation and many, but not all, are significantly impaired. For example, mutations 2 and 17 were detected in the same individual. We can now report that only the frameshift allele (i.e. 17*) has a detectable abnormality. Of course, these biological activity estimates are assay-specific and do not necessarily imply dysfunction in other important functions.
Figure 3.

(A–C) Relative strengths of the 46 variant SIX3 alleles tested (see Table 1 for clinical details). Open circles represent the individual experiments. The Y-axis represents normalized outcomes, as explained in Material and Methods. Filled circles represent the average outcome and the vertical bars represent the 95% confidence interval, calculated by simulation using a multinomial model. Raw values are reported in Supplementary Material, Table S1. Alleles are presented 5′–3′. (A) Alleles 1–16 (see Table 1 and Supplementary Material, Table S1 for clinical details), spanning the N-terminus, the poly-glycine domain and N-terminal half of the Six domain. (B) Alleles 17–32, spanning the C-terminal half of the SIX domain and N-terminal half of the homeodomain. (C) Alleles 33–46, spanning the C-terminal half of the homeodomain, the C-terminal motifs and the C-terminus. Data points are color-coded to represent allele strengths in the overexpression assay, as also indicated in Table 1 and Fig. 4: color: red = <50% activity, orange = 50–90% activity and black >90% activity.
Figure 4.

Summary of structural alterations detected in the human SIX3 protein. A virtual translation of the human cDNA sequence used for our functional analysis is depicted with positions of variations according to individual mutation number (Table 1) and color representing activity in the overexpression assay: red = <50% activity, orange = 50–90% activity and black = >90% activity. An N-terminal poly-glycine segment (gray) is the most striking difference between multiple-species alignments and is present in human and rodent sequences but is largely absent in chick, zebrafish and Xenopus (data not shown). An uncommon length variant 205–207dupGGC adds an additional Glycine69 residue and was found in normal controls and can explain minor numbering differences among different SIX3 mutation reports. The canonical SIX domain (blue and underlined) also contains two eh1-like motifs (salmon-colored); note functional mutations were detected in both motifs. The homeodomain (purple and italic) is a target for in-frame deletions (e.g. 34**) and frameshift (e.g. 25*), whereas typical premature termination signals are indicated by the mutation number and the hash symbol. Note two putative C-terminal motifs (green) (38) are also functionally implicated by mutations leading to impaired activity.
Mutational analysis as a tool to dissect the role of important motifs
Using naturally occurring mutations to identify important structural motifs is a classical approach to gene structure–function analysis. Here we exploit this strategy to clarify the role of the poly-glycine stretch in the N-terminus of SIX3 (Fig. 4, shaded gray). The mutations flanking the poly-glycine domain indicate that there is a mild reduction in activity associated with these changes; hence, this domain may well participate in functional interactions not obvious otherwise. Interestingly, the poly-glycine stretch is not present in the zebrafish Six3 ortholog, raising the possibility that there are no natural interaction partners for the poly-glycine domain in our biosensor assay and that the functional requirement represents an essential cis (or homo-meric) interaction. The effect of mutations in the two C-terminal motifs (shaded green) (38) strongly argues that these are additional sites of interaction requiring further analysis. The eh1-like motifs are implicated in direct binding to Groucho co-factors (colored salmon). Our data shows that the mutations 4 and 5 within the first eh1 motif virtually eliminate measurable SIX3 function. Mutation 18 within the putative second eh1 motif has a strong effect as well, though it has little effect on activity in the rescue assay at the dosage used (Fig. 5). These data indicate that the Groucho co-repressor function is a readily measurable parameter of mutated versions of SIX3, and a physiologically important one with respect to HPE.
Figure 5.

Side-by-side comparison of relative signaling strengths for various predicted SIX3 truncation alleles in the two biosensor assays. Open circles represent individual experiments. The Y-axis represents the normalized outcomes, as explained in Material and Methods and Supplementary Material, Table S2. Filled circles represent the average outcome and the vertical bars represent the 95% confidence interval, calculated by simulation using a multinomial model. The two most N-terminal truncations (patients 8 and 11), as well as point mutations in the first eh1-domain (patients 4 and 5), cause loss of function in both assays; however, downstream truncations in which the N-terminal 129 amino acids are intact generally retained activity in the rescue assay (patients 12, 15, 16, 19, 23 and 24, but not 17), as did a point mutation in the second eh1 domain (patient 18).
Truncation mutations without DNA-binding motifs can still participate in the rescue of Tcf3 morphants
An unexpected finding stemming from our attempt to develop the headless rescue assay was the discovery that only a small fraction of these mutations had diminished function. We now understand that at the dose we were employing (50 pg), hypomorphic mutations in key residues would be mischaracterized based on having already reached the plateau activity level (Fig. 2). Although this would lead to exaggerated positive results in the rescue assay, this assay is still informative because negative results should correspond to true negatives and positive results to some level of basal activity. eh1-domain mutants 4 and 5 were clear examples of loss of function in both assay systems (Fig. 5), supporting our conclusion that the Groucho co-repressor function is key. However, we noted that even frameshift and truncation mutants retained the ability to rescue the Tcf3 morphants, despite the lack of an encoded homeodomain, as long as the first 39 amino acids of the Six3 domain were retained (Fig. 5), suggesting at least partial repressive activity for truncated forms. We now view these results (together with the distinct slopes of the dose–response curves) as two lines of evidence for distinct co-repressor complexes. Further, the fact that SIX3 lacking the homeodomain can still rescue loss of Tcf3 phenotypes suggests that SIX3 co-factors available in the zebrafish system can directly participate in DNA binding and site selection. In contrast, the overexpression assay is exquisitely sensitive to structural alterations in the SIX3 homeodomain. We note that these conclusions are based on cooperative interactions between the exogenous human protein and its zebrafish host co-factors, which form the basis of both biosensor assays.
DISCUSSION
The relative success of our approach gives considerable impetus to expanding the use of zebrafish to develop screening assays to determine initial functional properties of putative mutant genes. The speed and versatility of these assays allow for rapid preliminary analysis. Indeed, we would argue that they serve as the most cost-effective bridge between initial screening studies and any more detailed physiological studies in the future. A robust screening method to determine either the types of pathways or degrees of measurable dysfunction is a crucial step prior to embarking on more expensive and labor-intensive investigations. This is particularly true when a large number of mutations require study.
Based on our results articulated in this report, we can envision future experiments, such as replacing the normal murine Six3 gene with selected mutated versions and assessing the effects. Indeed, a report emerging after the completion of our studies showed that a fraction of mice heterozygous for an engineered mutation orthologous to V250A display forebrain anomalies that model human HPE and that additional loss of shh can increase HPE penetrance to 100% in certain backgrounds (39). These same authors also used zebrafish assays similar to ours to measure activity for five mutations we examined, confirming most of our conclusions except for their finding that R257P (patient 41) and 556_557dupGG (patient 24) are not sufficient for rescue of Hdl morphant eyes.
The promise of these kinds of studies will ultimately depend on there being parallel studies leading to a better understanding of the co-morbid factors (e.g. additional genetic variants or environmental influences) required to generate a full HPE phenotype. Such confirmations are also important because relying on a zebrafish response apparatus to measure human gene functionality within specific signaling pathways can certainly lead to a certain number of false-positive and false-negative results that mask the true role of a given allele in HPE pathogenesis. However, similar concerns can be voiced for virtually any functional assay, even those using human cells. These caveats noted, our zebrafish data on SIX3 allow us to make several conclusions.
First, we conclude that the majority of mutations in SIX3 detected in HPE patients affect its BMP repressor function. Second, mutations in the N-terminal Groucho-binding eh1-like motif decreased the ability of SIX3 to repress either BMP or Wnt signaling. Third, truncated alleles bearing N-terminal sequences but lacking the DNA-binding domain retained the ability to repress Wnt signaling, but not BMP signaling.
Therefore, although our biosensor assays are not intended to mimic human physiological events, they do uncover unexpected differences in allele activity that may lead to important new insights into Six3 complex composition. Which of these various roles is most relevant to HPE pathogenesis is not entirely understood. In fact, despite the absence of obvious co-morbid mutations in other HPE genes in this group of patients, we remain convinced that additional genetic or environmental factors must be invoked to help explain not only the generation of HPE phenotypes, but also their wide variability (6,7).
MATERIALS AND METHODS
Study population
We analyzed approximately 600 HPE patients (collectively comprising the entire spectrum of HPE brain malformations and collected prospectively over 15 years) for potential sequence variations in the SIX3 gene under our NHGRI approved brain research protocol. In addition, we studied 125 unrelated individual normal controls obtained as anonymous samples from the Coriell Institute for Medical Research that matched the predominant Northern European ethnicity of our HPE cases. In the cases extracted from literature reports (cited in Table 1), only the nature of the mutation was used to design constructs; whereas the patients and their clinical descriptions are known to us only through these published sources (29–33). Similarly, anonymous instances of mutations in the SIX3 gene were shared with us from prospective studies performed under CLIA standards by GeneDx (Gaithersburg, MD, USA).
Mutation screening, PCR amplification and DNA sequencing
A strategy for screening the SIX3 gene has been described previously (5). Amplification of human genomic DNA was performed in a 30 µl reaction volume, using 60–100 ng DNA template, 50 µm each of deoxynucleotide triphosphate, 0.25 µm of each primer, 3 µl of 10× PCR Amplification buffer (Invitrogen), 1.5 µl 10× Enhancer buffer (Invitrogen) and 0.3 µl Taq polymerase. All reactions were performed using a PTC-255 thermocycler (MJ Research, MA, USA). Typical PCR cycling parameters were: 95°C for 4 min followed by 30 cycles at 95°C, annealing at 62°C, extension at 72°C for 1 min and a final extension step of 72°C for 5 min. One half of the PCR product was used for denaturing high-pressure liquid chromatography analysis (Trangenomics WAVE™) and the remainder was retained for direct DNA sequencing. Amplicons displaying heterozygous profiles were purified using a high pure PCR purification kit (Roche, IN, USA) and bi-directionally sequenced using the BigDye™ version 3.1 terminator cycle sequencing kit according to the manufacturer’s protocol (Applied Biosystems, CA) on an ABI 3100 automated sequencer.
Site-directed mutagenesis and construct design
The cDNA-derived coding region of the SIX3 gene (a kind gift of J. Rommens, University of Toronto) was directionally cloned as an (5′–3′) EcoRI to XbaI fragment into pcDNA3.1 by standard methods. This clone will be available from addgene.org for a nominal fee. Site-directed mutagenesis was performed with a minor modification of the Transformer kit (Promega, WI, USA) by Transponics (York, PA, USA) to introduce the human sequence variations.
Zebrafish biosensor assays
Embryos were obtained from WT adult AB strain zebrafish via natural mating and raised at 28°C in standard media (0.0001% methylene blue, 0.006% sea salt). Human SIX3 alleles in pcDNA3.1 were linearized with XbaI and the sense strand was synthesized using the T7 Message Machine kit (Ambion) followed by the synthetic addition of polyA (Ambion). The first biosensor assay we developed measured the ability of SIX3 alleles to repress hyperactive WNT signaling in embryos that were depleted for the tcf3/headless (hdl) gene via antisense morpholino oligonucleotide (MO) microinjection (18,25). In the absence of exogenous Six3 protein, Headless MO-injected embryos fail to develop eyes and other anterior structures; however, co-injection of wild-type (WT) zebrafish six3 (18) or human SIX3 (our findings) can completely rescue this. Using this system, we measured the relative activity of distinct SIX3 alleles as a function of their ability to induce eyes when 50 pg mRNA was injected along with 1 ng of the Tcf3/Hdl MO. The WT-adjusted activity (WTAA) for these experiments were calculated as follows: WTAA = (% eyeless for SIX3EXP)/(average % eyeless for SIX3WT). This biosensor assay turned out to have a very sharp transition from eyeless to rescued states which made reproducible measurements difficult and so we developed a second biosensor assay that quantified the embryonic dorsalization that arises from injection of SIX3 alone. A total of 100 pg of WT or variant SIX3 mRNAs was microinjected into zebrafish embryos and the embryos were incubated to the pharyngula stage (28–32 h post-fertilization) and scored as WT, dead or belonging to one of the following classes. Class 1 (C1) was the weakest phenotype, ranging from embryos with curved tails and reduced ventral fins to embryos with significantly shortened yolk extensions. In class 2 (C2) embryos, the tail is dramatically shortened and malformed. Class 3 embryos are broadly malformed and shortened, sitting entirely atop the yolk, with small or absent eyes and frequently bursting yolks in the more extreme cases. Activity was quantified as follows: a phenotypic index (PI) was determined for each group of injected embryos by assigning a value to each embryo of each phenotypic class (WT = 0; C1 = 1; C2 = 2; C3 = 3) except for dead embryos which were disregarded, multiplying the number of embryos in each class by the corresponding assigned value and dividing the sum by the total number of embryos. PIs were calculated for WT SIX3, and the WTAA for all other experiments were calculated by dividing the PI of each experiment by the average overall PI for WT SIX3. For each construct, a minimum of two measurements were performed, using two independent syntheses, to avoid false-negative results. To improve statistical certainty, when only two data points were available for a given allele and differed by more than 0.4 units on the WTAA scale or when only three data points were available and differed by more than 0.6 units on the WTAA scale, at least one additional measure was taken; 95% confidence intervals were generated by 10,000 simulations using multinomial distribution as a statistical model. See supplementary methods in the Supplementary Material available online for a complete mathematical description.
Histology and photography
For live photography, embryos were mounted in 2% methyl cellulose and imaged on a Leica MZ16 dissecting microscope with a Zeiss Axiocam HrC whole-mount in situ hybridizations (WISH) were performed as described (40) and embryos were cleared in 2:1 benzyl benzoate/benzyl alcohol, mounted in canada balsam and photographed on a Leica MZ16 dissecting microscope for bright field images of bmp2b (41) stains and on a Zeiss Axioplan 2 compound microscope for differential interference contrast images of wnt1 (42) and chordin (43) stains.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
FUNDING
This work was supported in part by the Division of Intramural Research, NHGRI, NIH, USA.
ACKNOWLEDGEMENTS
We would like to thank the patients who participated in these studies and the continuing support of clinicians around the world in referring such cases.
Conflict of Interest statement. To the best of our knowledge, none of the authors have any financial conflict of interest related to this study.
REFERENCES
- 1.Muenke M., Beachy P.A. Holoprosencephaly. In: Scriver, Beaudet, Valle, Sly, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th edn. Vol. 4. New York, NY: McGraw-Hill; 2000. pp. pp. 6203–6230. [Google Scholar]
- 2.Dubourg C., Bendavid C., Pasquier L., Henry C., Odent S., David V. Holoprosencephaly. Orphanet J. Rare Dis. 2007;2:8. doi: 10.1186/1750-1172-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Monuki E. The morphogen signaling network in forebrain development and holoprosencephaly. J. Neuropathol. Exp. Neurol. 2007;66:566–575. doi: 10.1097/nen.0b013e3180986e1b. [DOI] [PubMed] [Google Scholar]
- 4.Cohen M.M., Jr Holoprosencephaly, clinical, anatomic and molecular dimensions. Birth Defects Res. A Clin. Mol. Teratol. 2006;76:658–673. doi: 10.1002/bdra.20295. [DOI] [PubMed] [Google Scholar]
- 5.Wallis D.E., Roessler E., Hehr U., Nanni L., Wiltshire T., Richiri-Costa A., Gillessen-Kaesbach G., Zackai E.H., Rommens J., Muenke M. Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat. Genet. 1999;22:196–198. doi: 10.1038/9718. [DOI] [PubMed] [Google Scholar]
- 6.Krauss R.S. Holoprosencephaly: new models, new insights (online version) Expert Rev. Mol. Med. 2007;9 doi: 10.1017/S1462399407000440. DOI:1.1017/S1462399407000440. [DOI] [PubMed] [Google Scholar]
- 7.Ming J.E., Muenke M. Multiple hits during early embryonic development: digenic diseases and holoprosencephaly. Am. J. Hum. Genet. 2002;71:1017–1032. doi: 10.1086/344412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oliver G., Mailhos A., Wehr R., Copeland N.G., Jenkins N.A., Gruss P. Six3, a murine homlogue of the sine oculis gene, demarcates the most anterior border of the developing neural plate and is expressed during eye development. Development. 1995;121:4045–4055. doi: 10.1242/dev.121.12.4045. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi M., Toyama R., Takeda H., Dawid I.B., Kawakami K. Overexpression of the forebrain-specific homeobox gene six3 induces rostral forebrain enlargement in zebrafish. Development. 1998;125:2973–2982. doi: 10.1242/dev.125.15.2973. [DOI] [PubMed] [Google Scholar]
- 10.Lagutin O., Zhu C.C., Furuta Y., Rowitch D.H., McMahon A.P., Oliver G. Six3 promotes the formation of ectopic optic vesicle-like structure in mouse embryos. Dev. Dyn. 2001;221:342–349. doi: 10.1002/dvdy.1148. [DOI] [PubMed] [Google Scholar]
- 11.Seo H.C., Curtiss J., Mlodzik M., Fjose A. Six class homeobox genes in Drosophila belong to three distinct families and are involved in head development. Mech. Dev. 1999;83:127–139. doi: 10.1016/s0925-4773(99)00045-3. [DOI] [PubMed] [Google Scholar]
- 12.Loosli F., Winkler S., Wittbrodt J. Six3 overexpression initiates the formation of ectopic retina. Genes Dev. 1999;13:649–654. doi: 10.1101/gad.13.6.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carl M., Loosli F., Wittbrodt J. Six3 inactivation reveals its essential role for the formation and patterning of the vertebrate eye. Development. 2002;129:4057–4063. doi: 10.1242/dev.129.17.4057. [DOI] [PubMed] [Google Scholar]
- 14.Liu W., Lagutin O.V., Mende M., Streit A., Oliver G. Six3 activation of Pax6 expression is essential for mammalian lens induction and specification. EMBO J. 2006;25:5383–5395. doi: 10.1038/sj.emboj.7601398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu C.C., Dyer M.A., Uchikawa M., Kondoh H., Lagutin O.V., Oliver G. Six3-mediated auto repression and eye development requires its interaction with members of the Groucho-related family of co-repressors. Development. 2002;129:2835–2849. doi: 10.1242/dev.129.12.2835. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi M., Nishikawa K., Suzuki T., Yamamoto M. The homeobox protein Six3 interacts with the Groucho corepressor and acts as a transcriptional repressor in eye and forebrain formation. Dev. Biol. 2001;232:315–325. doi: 10.1006/dbio.2001.0185. [DOI] [PubMed] [Google Scholar]
- 17.Lopez-Rios J., Tessmar K., Loosli F., Wittbrodt J., Bovolenta P. Six3 and Six6 activity is modulated by members of the groucho family. Development. 2003;130:185–195. doi: 10.1242/dev.00185. [DOI] [PubMed] [Google Scholar]
- 18.Lagutin O.V., Zhu C.C., Kobayashi D., Topczewski J., Shimamura K., Puelles L., Russell H.R., Mckinnon P.J., Solnica-Krezel L., Oliver G. Six3 repression of Wnt signaling in the anterior neuroectoderm is essential for vertebrate forebrain development. Genes Dev. 2003;17:368–379. doi: 10.1101/gad.1059403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inbal A., Kim S.-H., Shin J., Solnica-Krezel L. Six3 represses Nodal activity to establish early brain asymmetry in zebrafish. Neuron. 2007;55:407–415. doi: 10.1016/j.neuron.2007.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gestri G., Carl M., Appolloni I., Wilson S.W., Barsacchi G., Andreazzoli M. Six3 functions in anterior neural plate specification by promoting cell proliferation and inhibiting Bmp4 expression. Development. 2005;132:2401–2413. doi: 10.1242/dev.01814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Houart C., Caneparo L., Heisenberg C.-P., Barth K.A., Take-Uchi M., Wilson S.W. Establishment of the telencephalon during gastrulation by local antagonisms of Wnt signaling. Neuron. 2002;35:255–265. doi: 10.1016/s0896-6273(02)00751-1. [DOI] [PubMed] [Google Scholar]
- 22.Lavado A., Lagutin O.V., Oliver G. Six3 inactivation causes progressive caudalization and aberrant patterning of the mammalian diencephalon. Development. 2007;135:441–450. doi: 10.1242/dev.010082. [DOI] [PubMed] [Google Scholar]
- 23.Wilson S.W., Houart C. Early steps in the development of the forebrain. Dev. Cell. 2004;6:167–181. doi: 10.1016/s1534-5807(04)00027-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van de Water S., van de Wetering M., Joore J., Esseling J., Bink R., Clevers H., Zivkovic D. Ectopic Wnt signal determines the eyeless phenotype of zebrafish masterblind mutant. Development. 2001;128:3877–3888. doi: 10.1242/dev.128.20.3877. [DOI] [PubMed] [Google Scholar]
- 25.Kim C.-H., Oda T., Itoh M., Jiang D., Artinger K.B., Chandrasekharappa S.C., Driver W., Chitnis A.B. Repressor activity of Headless/Tcf3 is essential for vertebrate head formation. Nature. 2000;407:913–916. doi: 10.1038/35038097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niehrs C. Head in the WNT. Trends Genet. 1999;15:314–319. doi: 10.1016/s0168-9525(99)01767-9. [DOI] [PubMed] [Google Scholar]
- 27.Yamaguchi T.P. Heads or tails: Wnts and anterior–posterior patterning. Curr. Biol. 2001;11:R713–R734. doi: 10.1016/s0960-9822(01)00417-1. [DOI] [PubMed] [Google Scholar]
- 28.Del Bene F., Tessmar-Raible K., Wittbrodt J. Direct interaction of geminin and Six3 in eye development. Nature. 2004;427:745–749. doi: 10.1038/nature02292. [DOI] [PubMed] [Google Scholar]
- 29.Pasquier L., Dubourg C., Blayau M., Lazaro L., Le Marec B., David V., Odent S. A new mutation in the SIX-domain of SIX3 gene causes holoprosencephaly. Eur. J. Hum. Genet. 2000;8:797–800. doi: 10.1038/sj.ejhg.5200540. [DOI] [PubMed] [Google Scholar]
- 30.Dubourg C., Lazaro L., Pasquier L., Bendavid C., Blayay M., Le Duff F., Durou M.-R., Odent S., David V. Molecular screening of SHH, ZIC2, SIX3 and TGIF genes in patients with features of holoprosencphaly spectrum: mutation review and genotype–phenotype correlations. Hum. Mutat. 2004;24:43–51. doi: 10.1002/humu.20056. [DOI] [PubMed] [Google Scholar]
- 31.Pasquier L., Dubourg C., Gonzales M., Lazaro L., Odent S., Encha-Razavi F. First occurrence of aprosencephaly/atelencephaly and holoprosencephaly in a family with a SIX3 gene mutation and phenotype/genotype correlation in our series of SIX3 mutations. J. Med. Genet. 2005;42 doi: 10.1136/jmg.2004.023416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ribeiro L.A., El-Jaick K.B., Muenke M., Richieri-Costa A. SIX3 mutations with holoprosencephaly. Am. J. Med. Genet. 2007;140A:2577–2583. doi: 10.1002/ajmg.a.31377. [DOI] [PubMed] [Google Scholar]
- 33.El-Jaick K.B., Fonesca R.F., Moreira M.A., Ribeiro M.G., Bolognese A.M., Dias S.O., Pereira E.T., Castilla E.E., Orioli I.M. Single median maxillary central incisor: new data and mutation review. Birth Defects Res. A Clin. Mol. Teratol. 2007;79:573–580. doi: 10.1002/bdra.20380. [DOI] [PubMed] [Google Scholar]
- 34.Lengler J., Krausz E., Tomarev S., Prescott A., Quinlan R.A., Graw J. Antagonistic action of Six3 and Prox1 at the γcrystalline promotor. Nuc. Acids Res. 2001;29:515–526. doi: 10.1093/nar/29.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hammerschmidt M., Serbedzija G.N., McMahon A.P. Genetic analysis of dorsoventral pattern formation in the zebrafish: requirement of a BMP-like ventralizing activity and its dorsal repressor. Genes Dev. 1996;10:2452–2461. doi: 10.1101/gad.10.19.2452. [DOI] [PubMed] [Google Scholar]
- 36.Kishimoto Y., Lee K.-H., Zon L., Hammerschmidt M., Schulte-Merker S. The molecular nature of zebrafish swirl: BMP2 function is essential during early dorsoventral patterning. Development. 1997;124:4457–4466. doi: 10.1242/dev.124.22.4457. [DOI] [PubMed] [Google Scholar]
- 37.Laflamme C., Filion C., Labelle Y. Functional characterization of SIX3 homeodomain mutations in holoprosencephaly: interaction with the nuclear receptor NR4A3/NOR1. Hum. Mutat. 2004;24:502–508. doi: 10.1002/humu.20102. [DOI] [PubMed] [Google Scholar]
- 38.Weasner B., Salzer C., Kumar J.P. Sine oculis, a member of the SIX family of transcription factors, directs eye formation. Dev. Biol. 2007;303:756–771. doi: 10.1016/j.ydbio.2006.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geng X., Speirs C., Lagutin O., Inbal A., Solnica-Krezel L., Jeong Y., Epstein D.J., Oliver G. Haploinsufficiency of Six3 fails to activate Sonic hedgehog expression in the ventral forebrain and causes holoprosencephaly. Dev. Cell. 2008;15:236–247. doi: 10.1016/j.devcel.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thisse C., Thisse B. High resolution whole-mount in situ hybridization. Zebrafish Sci. Monit. 1998;5:8–9. [Google Scholar]
- 41.Martinez-Barbera J.P., Toresson H., Da Rocha S., Krauss S. Cloning and expression of three members of the zebrafish Bmp family: Bmp2a, Bmp2b and Bmp4. Gene. 1997;98:53–59. doi: 10.1016/s0378-1119(97)00292-8. [DOI] [PubMed] [Google Scholar]
- 42.Molven A., Njolstad P.R., Fjose A. Genomic structure and restricted neural expression of the zebrafish wnt-1 (int-1) gene. EMBO J. 1991;10:799–807. doi: 10.1002/j.1460-2075.1991.tb08012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller-Bertoglio V.E., Fisher S., Sanchez A., Mullins M.C., Halpern M.E. Differential regulation of chordin expression domains in mutant zebrafish. Dev. Biol. 1997;192:537–550. doi: 10.1006/dbio.1997.8788. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.