

Abstract
Hutchinson–Gilford progeria syndrome (HGPS) is an accelerated aging disorder caused by point mutation in LMNA encoding A-type nuclear lamins. The mutations in LMNA activate a cryptic splice donor site, resulting in expression of a truncated, prenylated prelamin A called progerin. Expression of progerin leads to alterations in nuclear morphology, which may underlie pathology in HGPS. We generated transgenic mice expressing progerin in epidermis under control of a keratin 14 promoter. The mice had severe abnormalities in morphology of skin keratinocyte nuclei, including nuclear envelope lobulation and decreased nuclear circularity not present in transgenic mice expressing wild-type human lamin A. Primary keratinocytes isolated from these mice had a higher frequency of nuclei with abnormal shape compared to those from transgenic mice expressing wild-type human lamin A. Treatment with a farnesyltransferase inhibitor significantly improved nuclear shape abnormalities and induced the formation of intranuclear foci in the primary keratinocytes expressing progerin. Similarly, spontaneous immortalization of progerin-expressing cultured keratinocytes selected for cells with normal nuclear morphology. Despite morphological alterations in keratinocyte nuclei, mice expressing progerin in epidermis had normal hair grown and wound healing. Hair and skin thickness were normal even after crossing to Lmna null mice to reduce or eliminate expression of normal A-type lamins. Although progerin induces significant alterations in keratinocyte nuclear morphology that are reversed by inhibition of farnesyltransferasae, epidermal expression does not lead to alopecia or other skin abnormalities typically seen in human subjects with HGPS.
INTRODUCTION
Hutchinson–Gilford progeria syndrome (HGPS; OMIM no. 176670) is a rare, sporadic genetic disorder with phenotypic features of accelerated aging (1–4). It is caused by de novo dominant mutation in LMNA (5–7). LMNA encodes A-type nuclear lamins, with the predominant somatic cell isoforms lamin A and lamin C arising by alternative RNA splicing (8–10). Lamins are intermediate filament proteins that polymerize to form the nuclear lamina, a meshwork of intermediate filaments associated with the inner membrane of the nuclear envelope (9,11–13). HGPS is one of a spectrum of phenotypically diverse diseases, sometimes referred to as ‘laminopathies,’ caused by mutations in LMNA or genes encoding other proteins of the nuclear envelope (14).
Lamin A is synthesized as a precursor prelamin A, which is farnesylated and carboxymethylated at its carboxyl-terminus (15). To yield lamin A, farnesylated prelamin A is cleaved near its carboxyl-terminus in a reaction catalyzed by ZMPSTE24 endoprotease (16–18). LMNA mutations that cause HGPS create an abnormal splice donor site within exon 11, leading to an mRNA that encodes prelamin A with 50 amino acids deleted from its carboxyl-terminal domain (5,6). This truncated prelamin A has been designated progerin. As progerin lacks the ZMPSTE24 endoproteolytic site, it retains the farnesylated and carboxymethylated cysteine at its carboxyl-terminus (19–21).
Cultured fibroblasts from human subjects with HGPS and mouse models of the disease as well as transfected cells expressing progerin have abnormal nuclear morphology, including lobulation or ‘blebbing’ of the nuclear envelope, increased nuclear surface area, thickening of the nuclear lamina, loss of peripheral heterochromatin and clustering of nuclear pores complexes (5,6,22–26). Chemical inhibitors of farnesyltransferase that block prenylation of progerin, reduction of expression using RNA interference and treatment of cells with morpholino oligonucleotides that correct the aberrant RNA splicing generating progerin reverse these nuclear shape defects (25,27–32). All of these studies of progerin’s effects on nuclear morphology have been in cultured fibroblasts or transfected cultured cells and there are no data showing a cause and effect relationship between altered nuclear structure and tissue pathology in HGPS.
Skin is dramatically affected in human subjects with HGPS, with alopecia a universal feature (1,3,4,33–36). We, therefore, hypothesized that overexpression of progerin in epidermal keratinocytes would lead to alterations in nuclear morphology and concurrent alopecia. To test this hypothesis, we generated lines of transgenic mice expressing progerin or wild-type human lamin A in epidermis using a keratin 14 (K14) promoter and examined keratinocyte nuclear morphology and skin structure and function.
RESULTS
Expression of progerin and wild-type human lamin A in epidermis of transgenic mice
We generated minigenes for expressing progerin and wild-type human lamin A by cloning cDNAs downstream of a human K14 promoter (Fig. 1A). The K14 promoter has been shown to drive transgene expression in the basal layer of the epidermis and the outer root sheath of hair follicles (37–39). We engineered the minigenes so that the expressed proteins contained a FLAG epitope tag at their amino termini to facilitate their detection using antibodies. To confirm that the constructs expressed the encoded proteins prior to generating transgenic mice, we performed confocal immunofluorescence microscopy using anti-FLAG antibodies on transiently transfected immortalized mouse keratinocytes and detected nuclear rim fluorescence (data not shown). Minigenes encoding FLAG-tagged progerin and wild-type human lamin A were microinjected separately into B6/CBA F1 fertilized mouse oocytes that were subsequently transferred to pseudopregnant foster mothers. Analysis of DNA extracted from tail clippings by polymerase chain reaction and Southern blot hybridization showed presence of the minigenes in 12 transgenic founders (data not shown). Backcrosses of three founder mice containing the lamin A minigene and five containing the progerin minigene to B6/CBA F1 mice generated lines containing minigenes.
Figure 1.
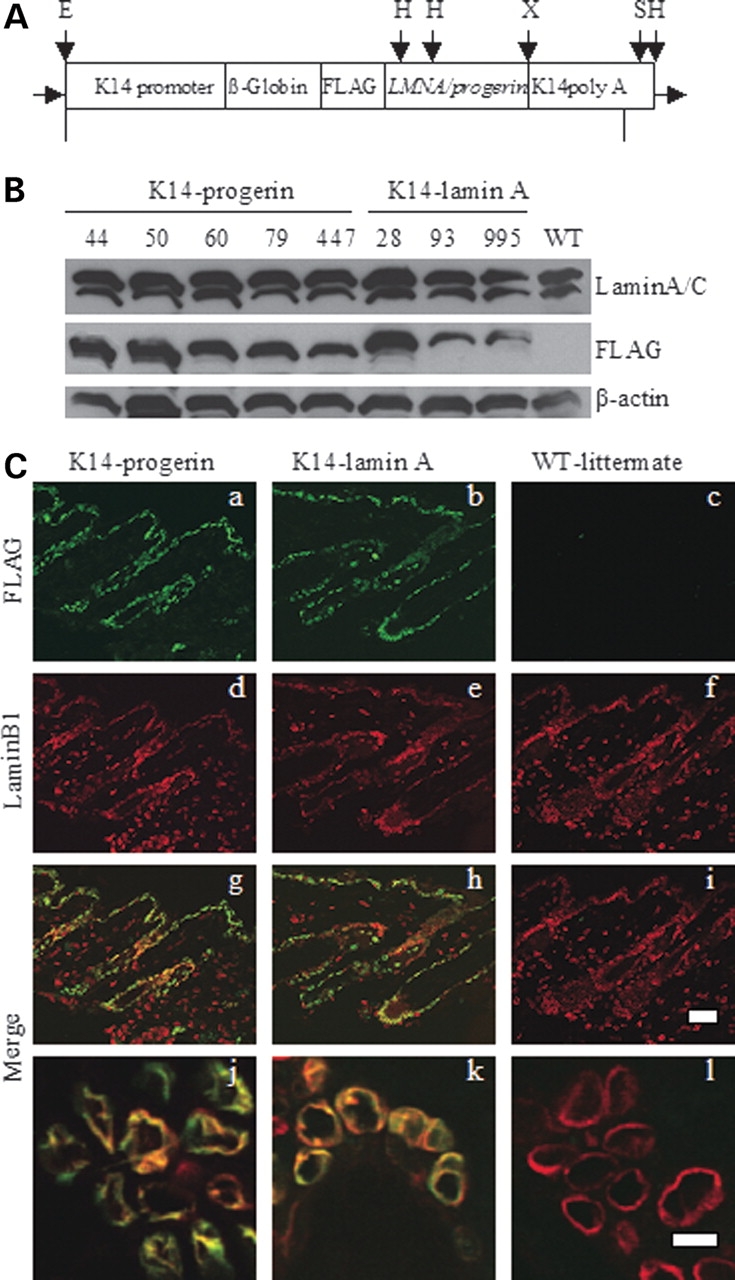
(A) Minigene constructs used for epidermal expression of wild-type human lamin A and progerin. The constructs contained a K14 promoter and ß-globin intron followed by DNA containing an ATG start codon and a FLAG sequence fused in frame to cDNAs encoding either prelamin A (LMNA) or progerin followed by K14 poly A tail. Major restriction endonuclease sites indicated are E: EcoRI, H: HindIII, S: SphI, X: XbaI. Fragments of 5319 and 5170 base pairs (bp), respectively, for wild-type lamin A and progerin were generated by digestion with EcoRI and SphI and microinjected into pronuclei of fertilized mouse oocytes. Diagram not to scale. (B) Immunoblotting demonstrating transgene expression in mouse skin. Blots were probed with mouse anti-lamin A/C (Lamin A/C), mouse anti-FLAG (FLAG) and mouse anti-actin (ß-actin) antibodies. Lanes numbered 44, 50, 60, 79, 447 are from five transgenic mouse lines containing the FLAG-progerin transgene (K14-progerin) and lanes numbered 28, 93, 995 are from three transgenic mouse lines with the FLAG-wild-type human lamin A transgene (K14-lamin A); WT indicates proteins extracted from skin of a non-transgenic littermate. Note faster migration of FLAG-progerin compared to FLAG-wild-type human lamin A. (C). Confocal immunofluorescence micrographs of dorsal skin sections from transgenic expressing FLAG-progerin (K14-progerin), FLAG-wild-type human lamin A transgene (K14-lamin A) and a non-transgenic littermate (WT-littermate). Panels show signals using anti-FLAG antibodies (a–c; green), anti-lamin B1 (d–f; red). Merge (g–i) shows overlay of anti-FLAG and anti-lamin B1 signals of panels immediately above. Lower panels (j–l) show overlay (yellow) of anti-FLAG and anti-lamin B1 signals of other fields at higher magnification. Bars: 50 µm (a–i) and 10 µm (j–l).
To examine the expression of FLAG-tagged progerin and wild-type human lamin A encoded by the transgenes, we performed immunoblotting of protein extracts from various organs of mice at 28–32 days of age. FLAG-lamin A or FLAG-progerin was detected by immunoblotting of proteins extracted from dorsal skin (Fig. 1B) and ear (data not shown) of the respective transgenic mouse lines but not non-transgenic controls. FLAG-tagged proteins encoded by the transgenes were not detected in heart or liver and detected at only minimal levels in tongue of transgenic mice (data not shown). Based on immunoblotting with antibodies against lamins A and C and antibodies against β-actin as a loading control, FLAG-progerin and FLAG-lamin A were estimated to be expressed at levels approximately one and a half to two times endogenous lamin A in different lines (Fig. 1B).
We further examined the expression of progerin and wild-type lamin A in skin of the transgenic mice by immunofluorescence microscopy. FLAG-tagged progerin or wild-type human lamin A was detected in the basal layer of the epidermis and the outer root sheath of hair follicles in skin sections from transgenic mice but not non-transgenic wild-type littermates (Fig. 1Ca and b). The fluorescence signals generated using anti-FLAG antibodies corresponded to those generated by antibodies against endogenous lamin B1 in epidermis, including hair follicles (Fig. 1Cd, e, g and h). No fluorescent signal was detectable in skin sections from non-transgenic littermates with anti-FLAG antibodies in which lamin B1 was present (Fig. 1Cc, f and i). Examination of epidermal cells at higher magnification revealed alterations in nuclear shape in the transgenic mice expressing progerin compared to transgenic mice expressing human lamin A or non-transgenic mice, including a ‘rougher’ and less circular contour of the nuclear periphery (Fig. 1Cj–l).
Electron microscopic analysis of nuclear shape abnormalities in epidermal keratinocytes of transgenic mice expressing progerin
Because the immunofluorescence microscopic analysis suggested that epidermal keratinocytes in transgenic mice expressing progerin had abnormal nuclear morphology, we used the electron microscopy to obtain higher resolution images of keratinocyte nuclei. We used transgenic mice from three lines expressing FLAG-progerin and one line expressing wild-type human lamin A that had a level of expression approximately two times of endogenous lamin A. Examination of keratinocytes in the basal layer of epidermis and the outer root sheath of hair follicles showed marked nuclear shape alterations in the transgenic mice expressing progerin (Fig. 2A). The keratinocyte nuclei appeared less round and to have increased surface area with multiple lobulations or irregular extensions. This abnormal nuclear morphology was observed in 80% of keratinocytes examined (n = 70) in the mice expressing progerin. Keratinocyte nuclei at the same locations in the skin of transgenic mice expressing wild-type human lamin A were much more elliptical and similar in shape to nuclei of non-transgenic littermate controls (Fig. 2A). Of keratinocytes examined in skin of mice expressing wild-type human lamin A (n = 30) and non-transgenic controls (n = 70), less than 5% had nuclei that were not primarily elliptical in shape. Electron dense inclusions or pseudoinclusions were noticed in ∼30% of keratinocyte nuclei in a transgenic mouse line expressing progerin (Fig. 2Aj, arrow) and not present in keratinocytes of mice expressing wild-type human lamin A or non-transgenic controls. While some of the keratinocytes expressing progerin appeared to show electron-dense areas in the cytoplasm, the cells were not more compact (shrunken), as cross-sectional whole cell diameters in electron micrographs were similar to those in keratinocytes from mice expressing wild-type human lamin A and non-transgenic controls.
Figure 2.
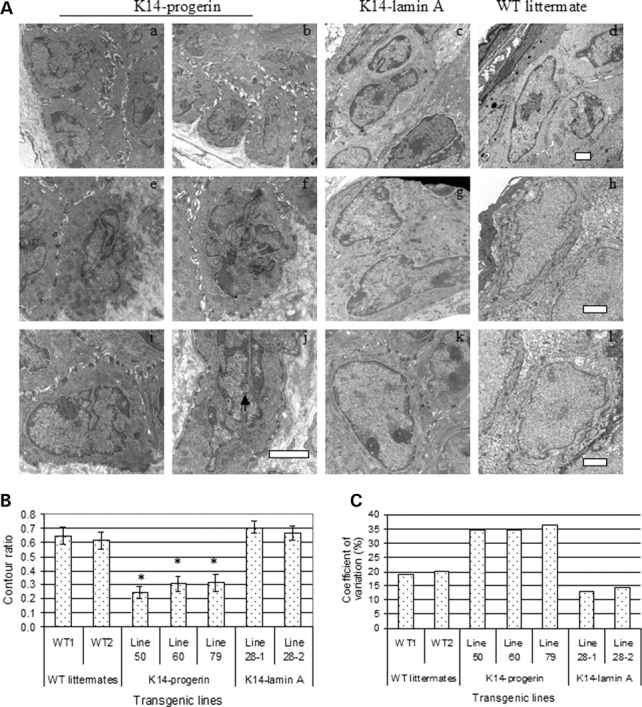
(A) Electron micrographs of epidermal keratinocyte nuclei in skin sections from transgenic mice expressing progerin (K14-progerin; a, b, e, f, i, j) or wild-type human lamin A (K14-lamin A; c, g, k). Sections from non-transgenic controls (WT littermate; d, h, l) are also shown. Arrowhead shows a nuclear pseudoinclusion (j). Bars: 2 µm. (B) Nuclear circularity expressed as contour ratio from different transgenic mouse lines. WT1 and WT2 are results for non-transgenic wild-type littermates; line 50, line 60 and line 79 (K14-progerin) are results for three different transgenic lines expressing progerin; line 28-1 and line 28-2 (K14-lamin A) are results for two different transgenic mice from a line expressing wild-type human lamin A. Values are means ± SD for n = 5–19 nuclei measured. Contour ratios of keratinocyte nuclei in mice expressing progerin were significantly less (*P < 0.01) than those in non-transgenic wild-type littermates or mice expressing wild-type human lamin A. (C) Coefficients of variation for contour ratios of keratinocyte nuclei in B.
We assessed the circularity of keratinocyte nuclei by determining the contour ratio based on electron micrographs with same magnification (n = 5–19). Mean contour ratios of keratinocyte nuclei from three lines of mice expressing FLAG-progerin were significantly lower than those in transgenic mice expressing FLAG-lamin A and non-transgenic controls (Fig. 2B). The coefficient of variation of keratinocyte nuclear shape in mice expressing progerin in epidermis was significantly larger than the variation dispersion of keratinocyte nuclear shape in mice expressing human wild-type lamin A and non-transgenic littermates from each strain (Fig. 2C). Hence, the nuclear shape alterations in epidermal keratinocytes induced by progerin expression are characterized by decreased nuclear circularity, resulting greater nuclear surface area and greater morphological diversity.
Abnormal nuclear morphology of progerin-expressing keratinocytes in primary culture and effects of farnesyltransferase inhibition
Epidermal keratinocytes were isolated from transgenic mice expressing progerin and wild-type human lamin A in epidermis and used to establish primary cultures. Immunoblotting showed expression of FLAG-progerin and FLAG-lamin A in primary keratinocyte cultures derived from four lines of each of these transgenic mice (Fig. 3A). The immunofluorescence microscopic analysis confirmed that the cultured cells were indeed keratinocytes as assessed by detection of keratin 14 and that the progerin-expressing cells had abnormal nuclear morphology (Fig. 3B). Nuclei of cultured progerin-expressing keratinocytes were variable in contour, less circular and had ‘rough-appearing’ nuclear envelopes than nuclei of keratinocytes isolated from transgenic mice expressing wild-type lamin A or non-transgenic controls, in which the nuclei were virtually uniformly elliptical (Fig. 3B). Several of the progerin-expressing keratinocytes also had ‘blebs’ or protrusions of the nuclear envelope.
Figure 3.
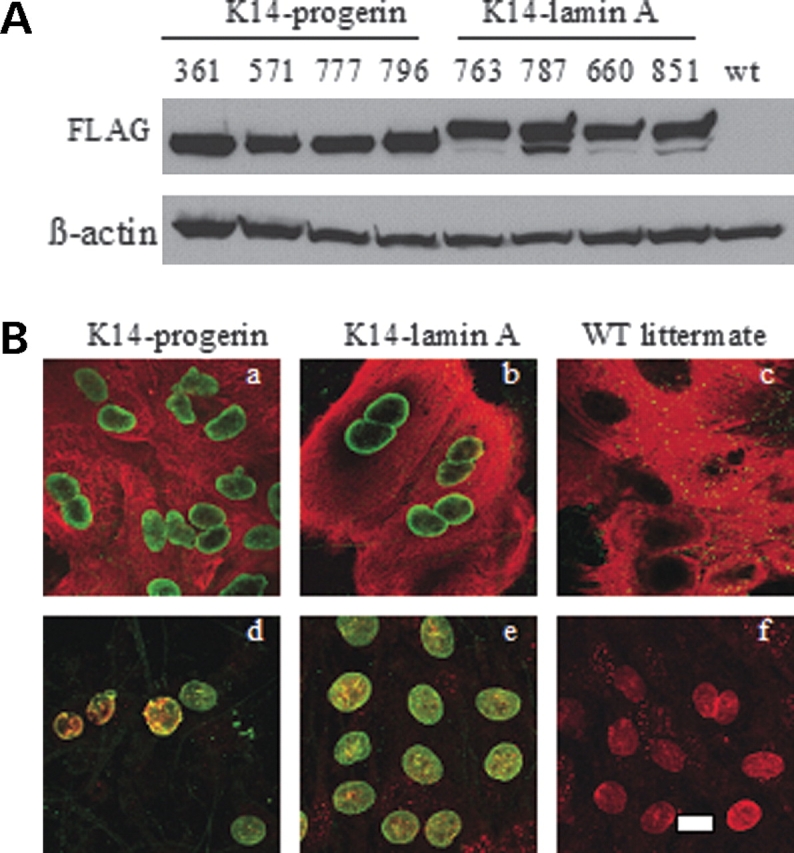
(A) Immunoblotting demonstrating transgene expression in cultured keratinocytes isolated from transgenic mice expressing FLAG-progerin and FLAG-wild-type lamin A. Blots were probed with mouse anti-FLAG (FLAG) and mouse anti-actin (ß-actin) antibodies. Lanes numbered 361, 571, 777 and 796 are from two transgenic mouse lines containing the FLAG-progerin transgene (K14-progerin) and lanes numbered 763, 787, 660 and 851 are from two transgenic mouse lines with the FLAG-wild-type human lamin A transgene; wt indicates proteins extracted from skin of a non-transgenic littermate. (B) Confocal immunofluorescence micrographs of cultured mouse keratinocytes expressing FLAG-progerin (K14-progerin; panels a and d), FLAG-wild-type human lamin A transgene (K14-lamin A; panels b and e) and a non-transgenic littermate (WT littermate; panels c and f). Panels a, b and c show double labeling with anti-FLAG (green) and anti-keratin 14 (red) primary antibodies. For panels d, e and f, cells were labeled with anti-FLAG (green) and anti-lamin A/C (red) antibodies and overlay (yellow) of signals are shown. Bar: 10 µm.
Farnesyltransferase inhibitors have been shown to reverse abnormalities in nuclear morphology in cultured fibroblasts (25,27,28,32) and transfected cultured Hela cells (30) expressing progerin. We, therefore, tested if inhibition of farnesyltransferase would have similar effects on progerin-expressing keratinocytes isolated from the transgenic mice. Cultured mouse keratinocytes expressing progerin or wild-type human lamin A were treated with farnesyltransferase inhibitor BMS-214662 to inhibit protein farnesylation. Treatment of keratinocytes expressing progerin for 48 h reversed the abnormal nuclear envelope morphology as assessed by confocal immunofluorescence microscopy (Fig. 4A). Farnesyltransferase inhibitor treatment also led to the formation of intranuclear foci composed presumably non-prenylated progerin (Fig. 4A). Assessment of nuclear envelope morphology in 200–500 cells treated with either BMS-214662 or vehicle (dimethylsulfoxide) in four different experiments showed that the mean percentage of progerin-expressing cells with abnormal nuclear morphology (less circular and/or ‘blebs) was 28% after farnesyltransferase inhibitor treatment compared to 64% after treatment with vehicle (Fig. 4B). In cultured keratinocytes expressing wild-type lamin A, 23% had abnormal nuclear morphology after treatment with vehicle and 19% after treatment with farnesyltransferase inhibitor (Fig. 4B). Assessment of 200–500 cells in two different experiments showed that the farnesyltransferase inhibitor induced the formation of multiple intranuclear foci in 93% keratinocytes expressing progerin, which was significantly higher than 13% of cells treated with vehicle (Fig. 4C). Only 1% of the keratinocytes expressing wild-type human lamin A treated with vehicle and 6% treated with the farnesyltransferase inhibitor had intranuclear foci (Fig. 4C).
Figure 4.
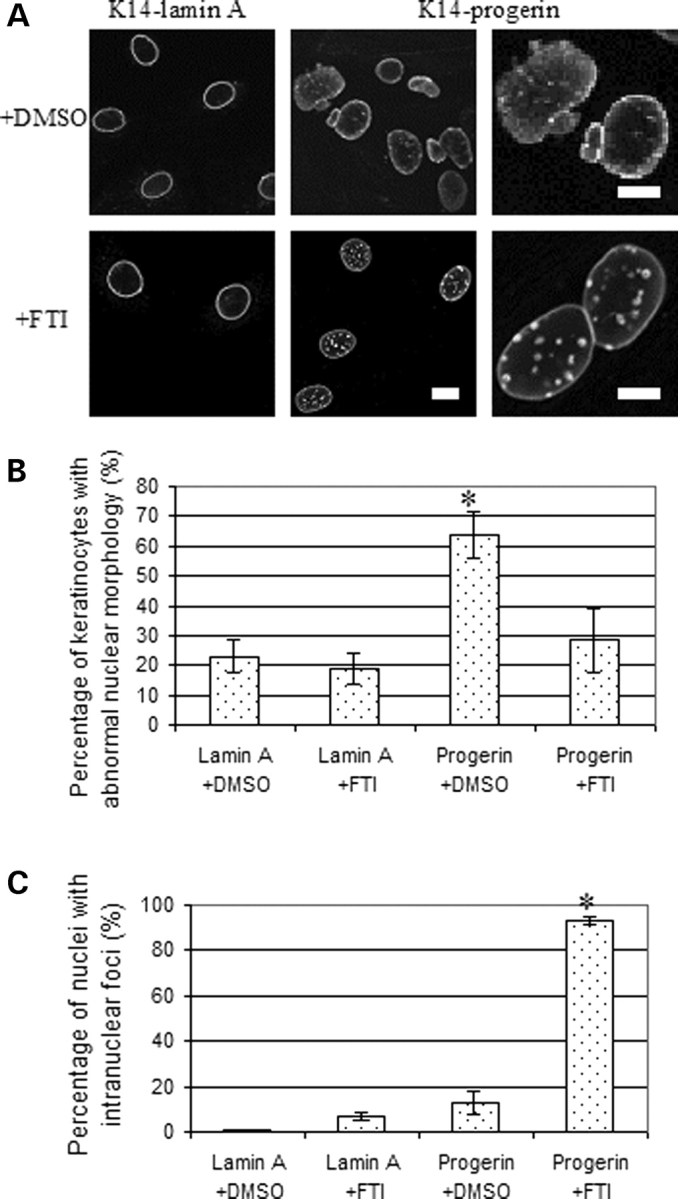
(A) Representative confocal immunofluorescence micrographs of cultured keratinocytes expressing FLAG-wild-type human lamin A (K14-lamin A) and FLAG-progerin (K14-progerin) after treatment with farnesyltransferase inhibitor BMS-214662 or dimethylsulfoxide vehicle. Top panels (+DMSO) show cells after 48 h of treatment with dimethylsulfoxide and lower panels (+FTI) show cells after 48 h of treatment with BMS-214662 labeled with anti-FLAG antibodies. Bar: 10 µm for four left-most panels at left; 5 µm for two panels at right. (B) Percentages of cultured mouse keratinocytes expressing wild-type human lamin A (Lamin A) or progerin (Progerin) with abnormal nuclear morphology after 48 h of treatment with farnesyltransferase inhibitor (+FTI) or dimethylsulfoxide (+DMSO). Nuclei in 200–500 cells in 25 microscopic fields per sample were scored for abnormal morphology (‘rough’ rim fluorescence, nuclear envelope ‘blebs’) versus normal morphology (smooth, mostly circular nuclear rim fluorescence). Values are means ± SD for n = 4 experiments. *P < 0.05 for progerin-expressing cells +FTI compared to +DMSO. (C) Percentages of cultured mouse keratinocytes expressing wild-type human lamin A (Lamin A) or progerin (Progerin) with intranuclear foci after 48 h of treatment with farnesyltransferase inhibitor (+FTI) or dimethylsulfoxide (+DMSO). Nuclei in 200–500 cells in 25 microscopic fields per sample were scored for the presence of fluorescent nuclear foci. Values are means ± SD for n = 2 experiments. *P < 0.05 for progerin-expressing cells +FTI compared to +DMSO.
Effect of spontaneous immortalization on nuclear shape of progerin-expressing keratinocytes
Primary keratinocytes were spontaneously immortalized during continuous subculture and multiple passages. Six cultures of immortalized mouse keratinocytes expressing wild-type human lamin A were established from 19 attempts and six cultures of mouse keratinocytes expressing progerin were established from 16 attempts. When cultures did not immortalize, all cells generally died by the fourth passage. The immunofluorescence microscopic analysis showed that the nuclear morphology of the majority of keratinocytes expressing progerin gradually became normal and after 20 passages was not very different than cultured keratinocytes expressing wild-type human lamin A (Fig. 5A). Assessment of nuclear envelope morphology in 200–500 cells from five cell lines expressing progerin and five expressing wild-type human lamin A showed that at first passage 64% of primary keratinocytes expressing progerin and 23% of primary keratinocytes expressing wild-type human lamin A had abnormal nuclear morphology (Fig. 5B). In contrast, at passage 20, only 8% of spontaneously immortalized keratinocytes expressing progerin and 4% of those expressing wild-type human lamin A had abnormal nuclear morphology (Fig. 5B). Hence, spontaneous immortalization by continuous subculture selected for progerin-expressing keratinocytes with normal nuclear morphology.
Figure 5.

(A) Representative confocal immunofluorescence micrographs of cultured keratinocytes expressing FLAG-progerin (K14-progerin) and FLAG-wild-type human lamin A (K14-lamin A) during spontaneous immortalization by continuous subculture. Top panels show cultured keratinocytes expressing progerin and bottom panels cultured keratinocytes expressing wild-type human lamin A at passage 1 (P1), passage 8 (P8) and passage 20 (P20) labeled with anti-FLAG antibodies. Bar: 10 µm. (B) Percentages of primary cultured mouse keratinocytes at passage 1 expressing wild-type human lamin A (LaminA-p) or progerin (Progerin-p) and spontaneously immortalized keratinocytes at passage 20 expressing wild-type human lamin A (LaminA-I) or progerin (Progerin-I) with abnormal nuclear morphology. Nuclei in 200–500 cells in 25 microscopic fields per sample were scored for abnormal morphology (‘rough’ rim fluorescence, nuclear envelope ‘blebs’) versus normal morphology (smooth, mostly circular nuclear rim fluorescence). Values are means ± SD for n = 5 experiments on different cell lines. *P < 0.05 for progerin-expressing primary keratinocytes at passage 1 (Progerin-p) compared to spontaneously immortalized progerin-expressing keratinocytes at passage 20 (Progerin-I).
Transgenic mice expressing progerin in epidermis have normal hair and wound healing
Growth curves and survival of all lines of transgenic mice expressing progerin or wild-type human lamin A in epidermis were not significantly different from non-transgenic controls. We hypothesized that the mice expressing progerin in epidermis would develop some of the skin abnormalities present in human subjects with HGPS. Despite the pronounced morphological alterations in keratinocyte nuclei, skin and hair in all lines of transgenic mice expressing progerin in epidermis appeared grossly normal even up to 19 months of age.
As proper differentiation, growth and migration of keratinocytes are critical for normal hair growth (40), we expected that the transgenic mice expressing progerin in these cells could have hair abnormalities more subtle than grossly apparent alopecia. The photographic analysis of hair at days 1–3 (hair follicle morphogenesis), day 14 (early catagen), day 21 (telogen) and day 28 (anagen) after birth showed no significant differences between transgenic mice expressing either progerin or wild-type human lamin A and non-transgenic littermates (data not shown). No hair, hair follicle or skin abnormalities were detected by histopathological examination of tissue sections from the 28-day-old mice expressing progerin (data not shown). There were no significant differences in hair density and full skin thickness (epidermis plus dermis) between transgenic mice expressing progerin, transgenic mice expressing wild-type lamin A and non-transgenic littermates.
For a more thorough assessment of hair, we examined the patterns of hair growth after hair cycle-inducing depilation. Male transgenic mice 7–8 weeks of age expressing progerin or wild-type human lamin A in epidermis were anesthetized and depilated by waxing of the dorsal skin. The area was photographed at days 1–3, 7, 10 and 13 after depilation in three mice of each genotype and no visual differences in skin or hair re-growth were observed. Skin appeared pink the first week and then turned grey on day 7 and black on day 10 after depilation (Fig. 6A) suggesting normal progression of hair follicle morphogenesis (41). Hairs fully grew back in mice expressing either progerin or wild-type human lamin A by day 13 (Fig. 6A). This hair growth pattern of mice after depilation was visually similar to that previously reported for normal mice (42). Mice were sacrificed 21 days after depilation and skin sections examined microscopically. FLAG-progerin and FLAG-lamin A were detected by immunohistofluorescence in basal epidermis and the outer root sheaths of hair follicles in cells that also were labeled with antibodies against keratin 14 (Fig. 6B). Antibodies against A-type lamins labeled nuclei at multiple layers of the skin and hair follicles (Fig. 6B). No significant difference was observed in the skin and hair follicles of depilated skin in mice expressing progerin or wild-type human lamin A. The histopathological analysis of sections from the depilated areas demonstrated similar overall skin and hair follicle structure (Fig. 6C). The result showed that the expression of the progerin in epidermis did not cause abnormalities in hair follicle cycling, hair follicle morphology or skin morphology after depilation.
Figure 6.
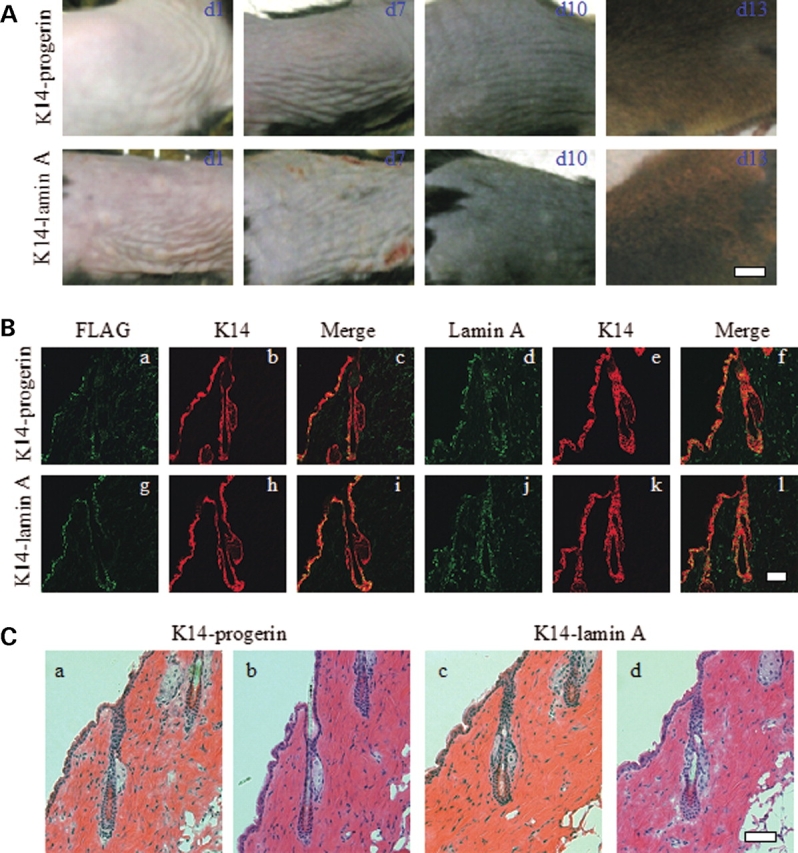
(A) Photographs of sections of dorsal skin from transgenic mice expressing progerin (K14-progerin) or wild-type human lamin A (K14-lamin A) 1 day (d1), 7 days (d7), 10 days (d10) and 13 days (d13) after depilation. Bar: 0.5 cm. (B) Confocal immunohistofluorescence micrographs of dorsal skin sections from transgenic mice expressing progerin (K14-progerin) or wild-type human lamin A (K14-lamin A) 21 days after depilation. Panels show signals using anti-FLAG antibodies (a, g; green), anti-lamin A antibodies (d, j; green), anti-keratin 14 (K14) antibodies (b, h, e, k; red). Merge (c, i, f, l) shows overlay (yellow) of anti-FLAG or anti-lamin A and anti-K14 signals of panels immediately to the left. Bars: 50 µm. (C) Representative sections of skin 21 days after depilation from transgenic mice expressing progerin (K14-progerin) or wild-type human lamin A (K14-lamin A) stained with hematoxylin and eosin showing no significant differences. Bar: 50 µm.
Because directed migration of keratinocytes is critical to wound re-epithelialization (43–45), we next examined wound healing in transgenic mice expressing progerin in the epidermis. Three male mice of similar body weights 7– 8 weeks of age expressing either progerin or wild-type human lamin A in epidermis, along with three non-transgenic controls, were wounded at two dorsal sites. No statistically significant differences in wound closure rates were detectable between the different transgenic lines and the non-transgenic littermates (Fig. 7A). Histological examination of hematoxylin and eosin stained sections of skin 11 days after wounding did not reveal differences in reconstituted epidermis between either of the transgenic mice and the non-transgenic controls (Fig. 7B). All of the healing skins showed thickened epidermis and new hair follicles (Fig. 7B). These experiments showed that the mice with the progerin expression in epidermal keratinocytes had similar wound healing capacity as wild-type mice.
Figure 7.
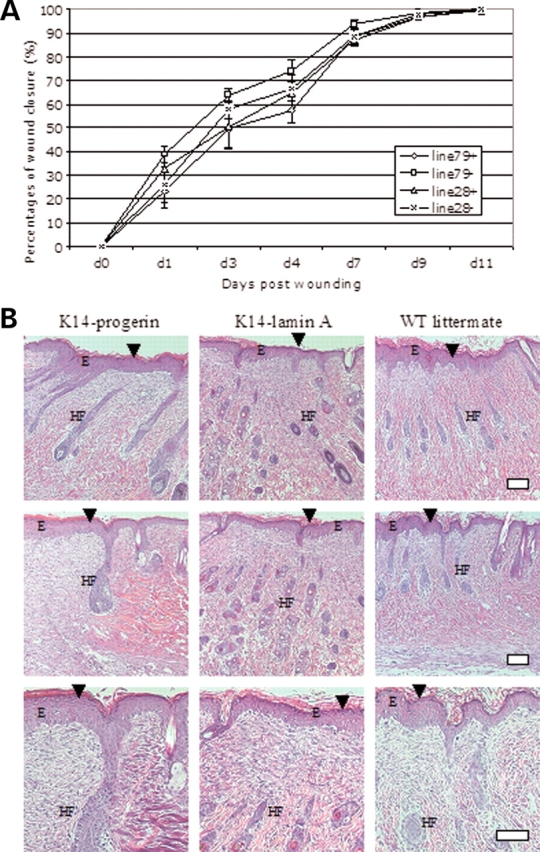
(A) Rates of wound healing in transgenic mice expressing progerin or wild-type human lamin A in epidermis. Graphs show mean percentages of wound closure (y-axis) versus days post wounding (x-axis) for transgenic mice expressing progerin (line 79+), their non-transgenic littermates (line 79−), transgenic mice expressing wild-type human lamin A (line 28+) and their non-transgenic littermates (line 28−). Values are means ± SD for n = 6 wounds in three mice from each group (two wounds per mouse); no differences between groups were significant. (B) Representative sections of skin 11 days after wounding (just prior to complete closure) from transgenic mice expressing progerin (K14-progerin), transgenic mice expressing wild-type human lamin A (K14-lamin A) and a non-transgenic control (WT littermate) stained with hematoxylin and eosin showing no significant differences. Healing skins had thickened epidermis (E) and new hair follicles (HF). Arrowheads show healing wounds. Bar: 50 µm.
Expression of progerin in epidermis has no effects on skin in mice with reduced or no A-type lamins
In cells of human subjects with HGPS, there may be decreased levels of wild-type lamin A (6,46). To determine if progerin had effects on skin or hair in the absence of A-type lamins, we crossed transgenic mice expressing progerin or wild-type human lamin A to Lmna+/− mice (47). Breeding generated F2 mice that expressed FLAG-tagged progerin or wild-type lamin A on Lmna−/−, Lmna+/− or Lmna+/+ backgrounds. Immunohistofluorescence microscopy showed appropriate expression of FLAG-progerin and FLAG-lamin A in epidermis of these mice (data not shown). No significant difference among the different genotypes was observed. Epidermis in skin sections from Lmna−/−, Lmna+/− or Lmna+/+ mice not crossed to the transgenic were not labeled with anti-FLAG antibodies and the Lmna−/− did not have detectable endogenous A-type lamin expression (data not shown). Epidermis appeared thicker in skin sections from Lmna−/− mice compared to the skin of the mice of the other genotypes (data not shown).
Full skin thickness and hair density were graded in skin of male and female mice sacrificed at an age of 28–30 days (Fig. 8). Despite having a thicker epidermis, Lmna−/− mice had a thinner dermis, resulting in decreased full-skin thickness, and decreased hair density compared to Lmna+/− or Lmna+/+ mice. Lmna−/− mice with transgenic epidermal expression of progerin or wild-type lamin A similarly had significantly decreased full-skin thickness and hair density, which was no different than that in Lmna−/− mice. Full-skin thickness and hair density were not significantly different in male or female mice of other Lmna backgrounds whether or not they transgenically expressed progerin or wild-type human lamin A. Progerin did not affect skin thickness or hair density when expressed in epidermis of mice with reduced expression of A-type lamins.
Figure 8.

Hair density (bars filled with dots) and full skin thickness (bars with diagonal lines) in male (M) and female (F) transgenic mice expressing progerin (K14-progerin), transgenic mice expressing wild-type human lamin A (K14-lamin A) and non-transgenic mice (WT) on Lmna−/−, Lmna+/− and Lmna+/+ backgrounds. Hair density and skin thickness were scored on a scale of 1 (lowest) to 5 (highest). Values are means ± SD for n = 5 to 6 mice. Lmna−/− mice expressing either progerin or wild-type lamin A or without a transgene had significantly decreased hair density and full-skin thickness compared to Lmna+/− and Lmna+/+ mice.
DISCUSSION
Our results show that the expression of progerin in epidermal keratinocytes caused nuclear morphological abnormalities. Similar expression of wild-type human lamin A did not lead to these abnormalities. The results were consistent across several transgenic lines, demonstrating that the nuclear-shape alterations were caused by progerin expression and not genetic alterations caused by integration of the transgenes. Nuclear-shape alterations observed in keratinocytes expressing progerin in vivo were similar to those that have been reported previously in cultured cells expressing the protein (5,6,22–32). In a careful and quantitative analysis using light and electron microscopy, Goldman et al. (23) demonstrated increased nuclear lobulation and significantly decreased nuclear contour ratios in cultured fibroblasts expressing progerin compared to normal cells. Similar alterations in nuclear morphology have been reported in fibroblasts from genetically modified mice and human subjects with restrictive dermopathy lacking Zmpste24, the endoprotease responsible for the processing of prelamin A to lamin A (17,32,48–50). Few studies prior to ours have examined nuclear shape in tissues. Navarro et al. (49) published an immunofluorescence micrograph showing one keratinocyte in a frozen section skin biopsy of a human subject with restrictive dermopathy lacking Zmpste24 that showed absent A-type lamins at one nuclear pole and an overall inhomogeneous localization of A-type lamins. Pendás et al. (17) used electron microscopy to demonstrate abnormalities in nuclear architecture of cardiac muscle cells in mice lacking Zmpste24.
K14 promoter-driven expression of abnormal epidermal proteins such as mutant keratins can lead to abnormal skin and hair phenotypes in transgenic mice (51,52). We, therefore, hypothesized that the transgenic expression of progerin in mouse epidermis using a K14 promoter would lead to malfunctioning of epidermal keratinocytes and skin abnormalities. Our results showed that despite inducing alterations in nuclear morphology, progerin expression in epidermis did not lead to any significant skin pathology. This suggests that HGPS-associated skin and hair pathology may be caused not by progerin activity in the epidermis but rather by systemic factors or by abnormalities it may induce in other skin components such as dermis.
At least three other studies characterizing mice expressing progerin have been published. Yang et al. (53) reported decreased subcutaneous fat in mice containing a ‘knock in’ HGPS Lmna mutation. Varga et al. (54) reported no consistent differences in hair loss and skin quality in transgenic mice carrying a human bacterial artificial chromosome containing a mutant LMNA leading to progerin expression. Neither of these studies included a detailed analysis of keratinocyte nuclear morphology, hair growth, hair follicle density or wound healing. Very recently, Sagelius et al. (55) generated transgenic mice on a different genetic background that expressed progerin under the control of a keratin 5 promoter, which should give similar tissue-selective expression as the K14 promoter we used. They reported grossly ‘thin’ hair in some lines of progerin-expressing mice but normal hair follicle density, similar to our results. Depending on progerin expression levels in the transgenic line, these investigators found epidermal hyperplasia, which is not a common feature of HGPS, altered hair follicle structure as well as sub-epidermal abnormalities. One of their transgenic mouse lines had deceased survival secondary to failure to eat but not as a clear result of skin pathology. Sagelius et al. (55) did not provide data on keratinocyte nuclear morphology, wound healing or hair growth after depilation. Further work is required to reconcile the differences between some of the results reported by Sagelius et al. (55) and those in the present study.
The dissociation between abnormal ‘cellular phenotype’ and tissue pathology in mice expressing progerin in epidermis raises questions, especially considering the attention given to nuclear-shape abnormalities in HGPS. Despite the fact that cultured fibroblasts have been used in most studies, many investigators in the field have attempted to associate altered nuclear morphology with pathogenesis in HGPS and other diseases caused by mutations in LMNA. Alterations in cardiomyocyte nuclear morphology are observed in sections of hearts from Lmna−/−, LmnaH222P/H222P and LmnaN195K/N195K mice that develop cardiomyopathy (56–58). Expression of a lamin A variant that causes Emery–Dreifuss muscular dystrophy in hearts of transgenic mice leads to marked nuclear structural alterations and severe myocardial damage (59). In contrast, Lmna−/− mice have altered hepatocyte nuclear morphology but no reported liver abnormalities (47). It is possible that alterations in nuclear morphology induced by abnormalities in A-type lamins lead to pathology only in certain tissues, such as striated muscle, in which cells are continuously subjected to mechanical stress. In contrast, cells in tissues such as liver or epidermis may be able to function mostly normally despite having significant alterations in the nuclear shape.
Previous studies (25,27,28,30,32) and our current results have shown that the nuclear-shape abnormalities in cultured cells expressing progerin are reversed by treatment with farnesyltransferase inhibitors. Much more importantly, systemic administration of farnesyltransferase inhibitors improves the overall phenotypes and survivals of mice with a ‘knock in’ HGPS Lmna mutation or Zmpste24 deficiency (53,60,61). These results have led to an open label clinical trial of a farnesyltransferase inhibitor in human subjects with HGPS (4). It would be interesting to determine if improvements in specific clinical parameters in these mice or human subjects correlate with reversal of nuclear-shape alterations in appropriate tissues. In addition, we also found that continuous subculture of keratinocytes expressing progerin selected for cells with normal nuclear morphology. This suggests that factors other than the prenylation of progerin play an important role in determining nuclear morphology in HGPS.
MATERIALS AND METHODS
Construction of progerin and human lamin A minigenes
To generate a cDNA encoding progerin, total RNA was extracted from cultured fibroblasts from a subject with HGPS (24) and first strand cDNA synthesized by reverse transcription. Primers containing engineered restriction endonuclease sites were then used for polymerase chain reaction to generate a cDNA corresponding to a 3′ portion of progerin. Polymerase chain-reaction products were digested with appropriate restriction endonucleases and ligated into pSVF containing cDNA encoding human prelamin A with a FLAG epitope tag at its amino-terminus, which was similarly treated with appropriate restriction endonucleases. FLAG-prelamin A cDNA had been previously generated in our laboratory (62). To generate the minigenes, cDNAs encoding FLAG-progerin and FLAG-prelamin A were excised from plasmids by restriction endonuclease digestion with EcoRI and SalI, and cloned into the BamH1 site of pG3Z•K14 (37,38) (kindly provided by Dr Elaine Fuchs, Rockefeller University). The cDNAs were cloned downstream of the K14 promoter and β-globin intron and upstream of a K14 polyadenine tail coding sequence (see Fig. 1A). Restriction endonuclease digestion analysis and DNA sequencing using an ABI 3100 capillary sequencer (Applied Biosystems) were carried out to characterize the constructs and ensure proper sequences.
Transfection of cultured keratinocytes for immunofluorescence microscopy
Immortalized mouse keratinocytes were cultured using mitomycin-treated J2-3T3 feeder cells and previously described media and methods (63). The cells were transfected using Lipofectamine PLUS™ following the manufacturer’s instructions (Invitrogen). Cultured cells were fixed and processed for immunofluorescence microscopy as described previously (62,64). Primary antibody used was mouse anti-FLAG M5 (Sigma-Aldrich) diluted at 1:200 and secondary antibody used were fluorescein isothyocyanate-conjugated goat anti-mouse IgG (Jackson ImmunoResearch Laboratories). Immunofluorescence microscopy was performed on a LSM 510 META confocal laser scanning system attached to an Axiovert 200 inverted microscope (Carl Zeiss). Images were processed using Photoshop software (Adobe Systems) on a Macintosh G4 computer (Apple Computer).
Generation of transgenic mice and progeny breeding
Minigenes encoding FLAG tagged human prelamin A and FLAG-tagged progerin were excised from plasmids by restriction endonuclease digestion with EcoRI and Sph I (Fig. 1A). The minigenes were microinjected separately into superovulated B6/CBA F1 fertilized oocytes in vitro and then the oocytes were transferred to pseudopregnant foster mothers to produce transgenic founders. Microinjection and oocyte transfer were performed at the Herbert Irving Comprehensive Cancer Center Transgenic Mouse Facility at Columbia University.
Founder transgenic mice were identified by the polymerase chain reaction analysis of DNA extracted from tail clippings using primer pairs corresponding to sequences in FLAG and human lamin A. Polymerase chain reactions using primers corresponding to sequences in β-globin and β-actin were simultaneously performed as internal controls. The southern blot analysis was performed to confirm the presence of transgenes using DNA extracted from tails and a 595 base-pair probe-containing sequences corresponding to the FLAG coding sequence and the 5′ end of human prelamin A cDNA. Transgenic mice were backcrossed to wild-type B6/CBA F1 to obtain stable transgenic offspring and adequate numbers of individuals for further experiments. All transgenic offspring were genotyped by polymerase chain reaction using tail DNA obtained prior to 2 weeks of age. Mice were fed a chow diet and autoclaved water and housed in a disease-free barrier facility with 12 h/12 h light/dark cycle at 25 ± 2°C. The Institutional Animal Care and Use Committee at Columbia University Medical Center approved the protocols for generation and breeding of transgenic mice.
Heterozygous transgenic mice were crossed to Lmna+/− mice (47) to produce transgenic mice on the Lmna+/− background. Sister–brother matings between these F1 mice were set up to produce mice with transgenes on Lmna+/−, Lmna+/+ and Lmna−/− backgrounds. Mice were genotyped by polymerase chain reaction using primer pairs corresponding to sequences in FLAG and human lamin A.
Protein electrophoresis and immunoblotting
Diced dorsal skin was homogenized in a lysis buffer containing 0.5% (v/v) Triton-X100, 2 mm Tris–HCl (pH 6.8), 2 mm ethylenediaminetetraacetic acid, 2 mm phenylmethanesulphonylfluoride and 1% (v/v) of a protease inhibitor cocktail (Sigma-Aldrich). Proteins were extracted from the insoluble portion in 2% (w/v) SDS, 2 mm Tris–HCl (pH 7.5) and 2 mm ethylenediaminetetraacetic acid. The proteins were separated by 10% SDS-polyacrylamide slab gel electrophoresis at 125 to 200 V for 1.5 to 2.0 h at room temperature. For immunoblotting, proteins were transferred to nitrocellulose membrane by electroblotting at 70 V for 2.5 h at 4°C. Immunoblotting was performed using mouse anti-FLAG M5 antibody at 1:500 dilution, mouse anti-lamin A/C antibody MANLAC1 4A7 (65) (kindly provided by Dr Glenn Morris, Center for Inherited Neuromuscular Diseases) at 1:100 dilution or mouse anti-beta-actin antibody (Santa Cruz Biotechnology) at 1:5000 dilution. ECL-horseradish peroxidase-conjugated anti-mouse NA931 (GE Healthcare) was used at 1:5000 to 1:10 000 dilution to detect primary antibodies. Signals were detected using SuperSignal® West Pice Chemiluminescent Substrate Kit (Pierce) and X-ray film (Kodak).
Immunohistofluorescence microscopy
Dorsal skins of mice were dissected at age 28–30 days and cut parallel to the vertebral line in order to obtain longitudinal hair follicle sections. Frozen skin samples were embedded in Tissue-Tek® O.C.T. compound embedding medium (Fisher Scientific) immediately after harvesting using previously published methods (41). Embedded frozen blocks were sectioned at 6 µm thickness, collected on silane-coated slides and fixed in acetone at −20°C for 10 min after air-dried. Acetone-fixed frozen sections were presoaked and washed three times in phosphate-buffered saline (2 min each wash) and successively treated with 10% normal goat serum and 1% bovine serum albumin in phosphate-buffered saline at room temperature for 30 min in a dark box. Slides then were incubated with primary antibodies diluted in phosphate-buffered saline containing 10% normal goat serum and 1% bovine serum albumin at room temperature for 1.5 h. Primary antibodies were mouse anti-FLAG M5 at 1:200 dilution, mouse anti-lamin A/C (MANLAC1) at 1:30 dilution, rabbit anti-keratin 14 AF64 (Covance Research Products) at 1:1000 dilution and rabbit anti-lamin B1 (66) at 1:1000 to 1:2000 dilution. After incubation with primary antibodies, sections were washed three times with phosphate buffered saline (1 min each wash) and then incubated for 30 min in a dark box with fluorescein isothyocyanate-conjugated goat anti-mouse IgG and Rhodamine Red™- X-conjugated goat anti-rabbit IgG (Invitrogen) diluted 1:200 in phosphate-buffered saline containing 10% normal goat serum and 1% bovine serum albumin. After washing three more times in phosphate-buffered saline, coverslips were mounted with gel/mount™ (Biomeda Corp.) and sealed with clear nail polish. Immunofluorescence microscopy was performed on a LSM 510 META confocal laser scanning system attached to an Axiovert 200 inverted microscope. Images were processed using Photoshop software on a Macintosh G4 computer.
Electron microscopy
Small pieces of full-thickness dorsal skin were fixed in glutaraldehyde in 0.1 M Sorenson’s buffer (pH 7.2) immediately after sacrificing and shaving the mice. Samples then were post-fixed, processed and examined using a JEOL JEM-1200 EXII transmission electron microscope as previously described (58). Pictures were taken using an ORCA-HR digital camera (Hamamatsu) and edited using Photoshop software on a Macintosh G4 computer.
To analyze nuclear roundness or circularity, we calculated contour ratios (4π × area/perimeter2), as described by Goldman et al. (23). The contour ratio for a circle is 1.0 and it approaches 0 as the nucleus becomes more lobulated. Perimeters, areas and circularities of keratinocyte nuclei were measured on electron micrographs by tracing the outline of the nucleus with a freehand selections tool (ImageJ 1.37v, National Institutes of Health). These parameters were measured for nuclei with same magnification in 5–19 cells that appeared to be sectioned close to the center of a nucleus rather than near the surface. Means, standard deviations and significances of differences using two-tailed Student t-test with unequal variance were calculated using Excel software (Microsoft). Variation dispersion of nuclear shape was calculated as coefficient of variation: 100% × SD/mean.
Keratinocyte isolation, culture and farnesyltransferase inhibitor treatment
Mouse epidermal keratinocytes were isolated based on previously described protocols (63,67). Briefly, mice 2–3 months of age were sacrificed by cervical dislocation, shaved and sequentially washed in 0.075% providone solution, autoclaved distilled water, 70% ethanol and autoclaved distilled water. Skin was removed and placed in phosphate-buffered saline containing 200 U/ml penicillin and 200 µg/ml streptomycin. All of the subcutaneous tissue was scraped off and the cutaneous tissue incubated in 0.25% trypsin for 2 h at 37°C. Epidermis was separated from dermis and stirred twice at room temperature for 20 min each time in FAD medium (three parts Dulbecco modified Eagle’s minimal medium and one part Ham’s F12 supplemented with 1.8 × 10−4 M adenine, 100 IU/ml penicillin and 100 µg/ml streptomycin) containing 10% fetal bovine serum. Cells were filtered through a 70 µm Teflon mesh (Fisher Scientific), centrifuged for 10 min at 2000 rpm and resuspended in complete FAD medium [FAD containing final concentrations of 10% fetal bovine serum, 0.5 µg/ml hydrocortisone (MP Biomedicals), 5 µg/ml insulin (Sigma-Aldrich), 10−10 M cholera enterotoxin (Sigma-Aldrich or MP Biomedicals) and 10 ng/ml epidermal growth factor (PeperoTech Inc. or MP Biomedicals)]. Cells were then seeded onto collagen-coated dishes (Fisher Scientific) in the presence of a feeder layer of mitomycin-treated J2-3T3 cells and incubated at 37°C and 5% CO2. Medium was changed every 2–3 days. To generate spontaneously immortalized keratinocytes, the primary cultured keratinocytes were subcultured when the cell density reached 50–70% confluency or after 1 week of culture and subjected to multiple rounds of subculture.
BMS-214662 (kindly provided by Dr Stephen G. Young, University of California Los Angeles) was used to inhibit farnesyltransferase in cultured keratinocytes. Cells were treated with BMS-214662 for 48 h at a dose of 2.5 µM as described previously (32). Cells incubated with vehicle (dimethylsulfoxide) were used as controls. Immunofluorescence microscopy was performed as described above using mouse anti-FLAG M5 at 1:200 dilution, rabbit anti-keratin 14 AF64 at 1:1000 dilution and rabbit anti-lamin A/C (66) at 1:200 as primary antibodies.
Gross and histological analysis of mouse skin and hair
To grossly assess the skin and hair, mice were photographed using a PowerShot A85 or SD1000 digital camera (Cannon). Photos were processed using Photoshop software on a Macintosh G4 computer. For histological analysis, skin samples were fixed in formalin for at least 2 days, embedded in paraffin, sectioned at 6 µm thickness and stained with hematoxylin and eosin. Sections were photographed using an AxioVision digital imaging system (Carl Zeiss) connected to a light microscope (Nikon).
Full-skin thickness and hair density were graded blindly (grader was unaware of mouse genotype) using dorsal skin sections at same location of approximately 2 cm2 from shaved mice at an age of 28–30 days. Sections were spread on aluminum foil and examined by eye. Full-skin thickness was graded on a scale of 1–5, with grade 1 ≤ 0.25 mm, grade 3 about 0.50 mm, grade 5 ≥ 1.0 mm and grades 2 and 4 intermediate skin thickness, respectively, between grades 1 and 3, and 4 and 5. Hair density was also graded on a scale from 1 to 5 with grade 1 corresponding to skin sections with visible hair shafts covering less than one-third of the surface area, grade 3 grossly equal portions of the surface area with and without visible hair shafts and grade 5 surface area completely covered with hair shafts and grades 2 and 4 intermediate hair shaft densities, respectively, between grades 1 and 3 and 4 and 5.
Depilation
Depilation was carried out when mice were 7–8 weeks of age. Male mice were anesthetized by intraperitoneal injection of 150–200 µl of a solution that was three quarters volume 0.9% NaCl and one quarter volume a mixture of 85.8% Ketaset (Fort Dodge Animal Health; 100 mg/ml ketamine HCl) and 14.2% Rompun (Bayer; 2% Xylazine). A melted mixture of 50% bleached beeswax and 50% resin gum (Sigma-Aldrich) was applied on the back of the anesthetized mice and the cooled wax was peeled off with hair when it was hard enough to hold the hair. Depilated areas of mouse skin were photographed 1, 3, 7, 10 and 13 days after the procedure. Mice were sacrificed 21 days after depilation for the histological analysis of the skin and hair follicles.
Wound healing
Male mice 7–8 weeks of age were anesthetized by intraperitoneal injection of 80–100 mg/kg ketamine and 5–10 mg/kg xylazine. The dorsum was shaved and cleaned with 10% w/v povidine iodine solution and 70% ethanol. Two full thickness skin excisions approximately 8 mm × 7 mm were created on the flanks of the animals. A subcutaneous injection of buprenorphine (0.05–0.1 mg/kg) was administered post-operatively for analgesia prior to recovery from anesthesia and a triple antibiotic ointment was used topically afterward to prevent possible infection. Wounds were left to heal by secondary intention and wound areas measured everyday or every other day to monitor the healing progress. Photographs of the wounded areas were taken at days 4, 7, 9 and 11. Mice were sacrificed and skin samples harvested at day 11, when the wounds were almost fully closed, for histological analysis.
FUNDING
This work was supported by a grant to H.J.W. from the US National Institutes of Health (AG025240). K.D. was also supported by a grant from the US National Institutes of Health (AG025302).
ACKNOWLEDGEMENTS
We thank Kristy Brown (Columbia University) for help with electron microscopy, Revekka Boguslavsky (Columbia University) for generating the progerin cDNA, Elaine Fuchs (Rockefeller University) for the K14 promoter construct, Chyuan-sheng ‘Victor’ Lin (Columbia University) for microinjection of mouse oocytes, Glenn E. Morris (Center for Inherited Neuromuscular Diseases) for antibodies and Stephen G. Young (University of California Los Angeles) for insightful discussions and supplying BMS-214662.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Hutchinson J. Case of congenital absence of hair, with atrophic condition of the skin and its appendages, in a boy whose mother had been almost wholly bald from alopecia areata from the age of six. Lancet. 1886;I:923. doi: 10.1177/095952878606900127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilford H. Ateleiosis and progeria: continuous youth and premature old age. Brit. Med. J. 1904;2:914–918. [Google Scholar]
- 3.DeBusk F.L. The Hutchinson–Gilford progeria syndrome. J. Pediatr. 1972;80:697–724. doi: 10.1016/s0022-3476(72)80229-4. [DOI] [PubMed] [Google Scholar]
- 4.Merideth M.A., Gordon L.B., Clauss S., Sachdev V., Smith A.C., Perry M.B., Brewer C.C., Zalewski C., Kim H.J., Solomon B., et al. Phenotype and course of Hutchinson–Gilford progeria syndrome. N. Engl. J. Med. 2008;358:592–604. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eriksson M., Brown W.T., Gordon L.B., Glynn M.W., Singer J., Scott L., Erdos M.R., Robbins C.M., Moses T.Y., Berglund P., et al. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Sandre-Giovannoli A., Bernard R., Cau P., Navarro C., Amiel J., Boccaccio I., Lyonnet S., Stewart C.L., Munnich A., Merrer M.L., et al. Lamin A truncation in Hutchinson–Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 7.Cao H., Hegele R.A. LMNA is mutated in Hutchinson–Gilford progeria (MIM 176670) but not in Wiedemann–Rautenstrauch progeroid syndrome (MIM 264090) J. Hum. Genet. 2003;48:271–274. doi: 10.1007/s10038-003-0025-3. [DOI] [PubMed] [Google Scholar]
- 8.Gerace L., Blobel G. The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell. 1980;19:277–287. doi: 10.1016/0092-8674(80)90409-2. [DOI] [PubMed] [Google Scholar]
- 9.Fisher D.Z., Chaudhary N., Blobel G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl Acad. Sci. USA. 1986;83:6450–6454. doi: 10.1073/pnas.83.17.6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin F., Worman H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993;268:16321–16326. [PubMed] [Google Scholar]
- 11.Aebi U., Cohn J., Buhle L., Gerace L. The nuclear lamina is a meshwork of intermediate-type filaments. Nature. 1986;323:560–564. doi: 10.1038/323560a0. [DOI] [PubMed] [Google Scholar]
- 12.McKeon F.D., Kirschner M.W., Caput D. Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature. 1986;319:463–468. doi: 10.1038/319463a0. [DOI] [PubMed] [Google Scholar]
- 13.Goldman A.E., Maul G., Steinert P.M., Yang H.Y., Goldman R.D. Keratin-like proteins that coisolate with intermediate filaments of BHK-21 cells are nuclear lamins. Proc. Natl Acad. Sci. USA. 1986;83:3839–3843. doi: 10.1073/pnas.83.11.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Worman H.J., Bonne G. ‘Laminopathies’: a wide spectrum of human diseases. Exp. Cell Res. 2007;313:2121–2133. doi: 10.1016/j.yexcr.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beck L.A., Hosick T.J., Sinensky M. Isoprenylation is required for the processing of the lamin A precursor. J. Cell Biol. 1990;110:1489–1499. doi: 10.1083/jcb.110.5.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergo M.O., Gavino B., Ross J., Schmidt W.K., Hong C., Kendall L.V., Mohr A., Meta M., Genant H., Jiang Y., et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl Acad. Sci. USA. 2002;99:13049–13054. doi: 10.1073/pnas.192460799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pendás A.M., Zhou Z., Cadiñanos J., Freije J.M., Wang J., Hultenby K., Astudillo A., Wernerson A., Rodríguez F., Tryggvason K., et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 2002;31:94–99. doi: 10.1038/ng871. [DOI] [PubMed] [Google Scholar]
- 18.Corrigan D.P., Kuszczak D., Rusiñol A.E., Thewke D.P., Hrycyna C.A., Michaelis S., Sinensky M.S. Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24. Biochem. J. 2005;387:129–138. doi: 10.1042/BJ20041359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rusiñol A.E., Sinensky M.S. Farnesylated lamins, progeroid syndromes and farnesyl transferase inhibitors. J. Cell Sci. 2006;119:3265–3272. doi: 10.1242/jcs.03156. [DOI] [PubMed] [Google Scholar]
- 20.Young S.G., Fong L.G., Michaelis S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria–new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J. Lipid Res. 2005;46:2531–2558. doi: 10.1194/jlr.R500011-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Young S.G., Meta M., Yang S.H., Fong L.G. Prelamin A farnesylation and progeroid syndromes. J. Biol. Chem. 2006;281:39741–39745. doi: 10.1074/jbc.R600033200. [DOI] [PubMed] [Google Scholar]
- 22.Bridger J.M., Kill I.R. Aging of Hutchinson–Gilford progeria syndrome fibroblasts is characterised by hyperproliferation and increased apoptosis. Exp. Gerontol. 2004;39:717–724. doi: 10.1016/j.exger.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 23.Goldman R.D., Shumaker D.K., Erdos M.R., Eriksson M., Goldman A.E., Gordon L.B., Gruenbaum Y., Khuon S., Mendez M., Varga R., et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl Acad. Sci. USA. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paradisi M., McClintock D., Boguslavsky R.L., Pedicelli C., Worman H.J., Djabali K. Dermal fibroblasts in Hutchinson–Gilford progeria syndrome with the lamin A G608G mutation have dysmorphic nuclei and are hypersensitive to heat stress. BMC Cell Biol. 2005;6:27. doi: 10.1186/1471-2121-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang S.H., Bergo M.O., Toth J.I., Qiao X., Hu Y., Sandoval S., Meta M., Bendale P., Gelb M.H., Young S.G., et al. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson–Gilford progeria syndrome mutation. Proc. Natl Acad. Sci. USA. 2005;102:10291–10296. doi: 10.1073/pnas.0504641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McClintock D., Gordon L.B., Djabali K. Hutchinson–Gilford progeria mutant lamin A primarily targets human vascular cells as detected by an anti-Lamin A G608G antibody. Proc. Natl Acad. Sci. USA. 2006;103:2154–2159. doi: 10.1073/pnas.0511133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Capell B.C., Erdos M.R., Madigan J.P., Fiordalisi J.J., Varga R., Conneely K.N., Gordon L.B., Der C.J., Cox A.D., Collins F.S. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson–Gilford progeria syndrome. Proc. Natl Acad. Sci. USA. 2005;102:12879–128784. doi: 10.1073/pnas.0506001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glynn M.W., Glover T.W. Incomplete processing of mutant lamin A in Hutchinson–Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum. Mol. Genet. 2005;14:2959–2969. doi: 10.1093/hmg/ddi326. [DOI] [PubMed] [Google Scholar]
- 29.Huang S., Chen L., Libina N., Janes J., Martin G.M., Campisi J., Oshima J. Correction of cellular phenotypes of Hutchinson–Gilford progeria cells by RNA interference. Hum. Genet. 2005;118:444–450. doi: 10.1007/s00439-005-0051-7. [DOI] [PubMed] [Google Scholar]
- 30.Mallampalli M.P., Huyer G., Bendale P., Gelb M.H., Michaelis S. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson–Gilford progeria syndrome. Proc. Natl Acad. Sci. USA. 2005;102:14416–14421. doi: 10.1073/pnas.0503712102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scaffidi P., Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson–Gilford progeria syndrome. Nat. Med. 2005;11:440–445. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toth J.I., Yang S.H., Qiao X., Beigneux A.P., Gelb M.H., Moulson C.L., Miner J.H., Young S.G., Fong L.G. Blocking protein farnesyl transferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc. Natl Acad. Sci. USA. 2005;102:12873–12878. doi: 10.1073/pnas.0505767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fleischmajer R., Nedwich A. Progeria (Hutchinson–Gilford) Arch. Dermatol. 1973;107:253–258. [PubMed] [Google Scholar]
- 34.Badame A.J. Progeria. Arch. Dermatol. 1989;125:540–544. [PubMed] [Google Scholar]
- 35.Gillar P.J., Kaye C.I., McCourt J.W. Progressive early dermatologic changes in Hutchinson–Gilford progeria syndrome. Pediatr. Dermatol. 1991;8:199–206. doi: 10.1111/j.1525-1470.1991.tb00859.x. [DOI] [PubMed] [Google Scholar]
- 36.Jansen T., Romiti R. Progeria infantum (Hutchinson–Gilford syndrome) associated with scleroderma-like lesions and acro-osteolysis: a case report and brief review of the literature. Pediatr. Dermatol. 2000;17:282–285. doi: 10.1046/j.1525-1470.2000.01775.x. [DOI] [PubMed] [Google Scholar]
- 37.Vassar R., Rosenberg M., Ross S., Tyner A., Fuchs E. Tissue-specific and differentiation-specific expression of a human K14 keratin gene in transgenic mice. Proc. Natl Acad. Sci. USA. 1989;86:1563–1567. doi: 10.1073/pnas.86.5.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X., Zinkel S., Polonsky K., Fuchs E. Transgenic studies with a keratin promoter-driven growth hormone transgene: prospects for gene therapy. Proc. Natl Acad. Sci. USA. 1997;94:219–226. doi: 10.1073/pnas.94.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan T., Ghahary A., Dmare J., Yang L., Iwashina T., Scott P.G., Tredget E.E. Development, characterization, and wound healing of the keratin 14 promoted transforming growth factor-beta1 transgenic mouse. Wound Rep. Reg. 2002;10:177–187. doi: 10.1046/j.1524-475x.2002.11101.x. [DOI] [PubMed] [Google Scholar]
- 40.Panteleyev A.A., Jahoda C.A., Christiano A.M. Hair follicle predetermination. J. Cell Sci. 2001;114:3419–3431. doi: 10.1242/jcs.114.19.3419. [DOI] [PubMed] [Google Scholar]
- 41.Paus R., Müller-Röver S., Veen C., Maurer M., Eichmüller S., Ling G., Hofmann U., Foitzik K., Mecklenburg L., Handjiski B. A comprehensive guide for the recognition and classification of distinct stages of hair follicle morphogenesis. J. Invest. Dermatol. 1999;113:523–532. doi: 10.1046/j.1523-1747.1999.00740.x. [DOI] [PubMed] [Google Scholar]
- 42.Müller-Röver S., Handjiski B., Veen C., Eichmüller S., Foitzik K., McKay I.A., Stenn K.S., Paus R. A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J. Invest. Dermatol. 2001;117:3–15. doi: 10.1046/j.0022-202x.2001.01377.x. [DOI] [PubMed] [Google Scholar]
- 43.Coulombe P.A. Towards a molecular definition of keratinocyte activation after acute injury to stratified epithelia. Biochem. Biophys. Res. Commun. 1997;236:231–238. doi: 10.1006/bbrc.1997.6945. [DOI] [PubMed] [Google Scholar]
- 44.Santoro M.M., Gaudino G. Cellular and molecular facets of keratinocyte reepithelization during wound healing. Exp. Cell Res. 2005;304:274–286. doi: 10.1016/j.yexcr.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 45.Sivamani R.K., Garcia M.S., Isseroff R.R. Wound re-epithelialization: modulating keratinocyte migration in wound healing. Front. Biosci. 2007;12:2849–2868. doi: 10.2741/2277. [DOI] [PubMed] [Google Scholar]
- 46.Reddel C.J., Weiss A.S. Lamin A expression levels are unperturbed at the normal and mutant alleles but display partial splice site selection in Hutchinson–Gilford progeria syndrome. J. Med. Genet. 2004;41:715–717. doi: 10.1136/jmg.2004.019323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sullivan T., Escalante-Alcalde D., Bhatt H., Anver M., Bhat N., Nagashima K., Stewart C.L., Burke B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 1999;147:913–921. doi: 10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fong L.G., Ng J.K., Meta M., Coté N., Yang S.H., Stewart C.L., Sullivan T., Burghardt A., Majumdar S., Reue K., et al. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc. Natl Acad. Sci. USA. 2004;101:18111–18116. doi: 10.1073/pnas.0408558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Navarro C.L., De Sandre-Giovannoli A., Bernard R., Boccaccio I., Boyer A., Geneviève D., Hadj-Rabia S., Gaudy-Marqueste C., Smitt H.S., Vabres P., et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum. Mol. Genet. 2004;13:2493–2503. doi: 10.1093/hmg/ddh265. [DOI] [PubMed] [Google Scholar]
- 50.Navarro C.L., Cadiñanos J., De Sandre-Giovannoli A., Bernard R., Courrier S., Boccaccio I., Boyer A., Kleijer W.J., Wagner A., Giuliano F., et al. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum. Mol. Genet. 2005;14:1503–1513. doi: 10.1093/hmg/ddi159. [DOI] [PubMed] [Google Scholar]
- 51.Fuchs E. Keratin genes, epidermal differentiation and animal models for the study of human skin diseases. Biochem. Soc. Trans. 1991;19:1112–1115. doi: 10.1042/bst0191112. [DOI] [PubMed] [Google Scholar]
- 52.Fuchs E., Coulombe P.A. Of mice and men: genetic skin diseases of keratin. Cell. 1992;69:899–902. doi: 10.1016/0092-8674(92)90607-e. [DOI] [PubMed] [Google Scholar]
- 53.Yang S.H., Meta M., Qiao X., Frost D., Bauch J., Coffinier C., Majumdar S., Bergo M.O., Young S.G., Fong L.G. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson–Gilford progeria syndrome mutation. J. Clin. Invest. 2006;116:2115–2121. doi: 10.1172/JCI28968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Varga R., Eriksson M., Erdos M.R., Olive M., Harten I., Kolodgie F., Capell B.C., Cheng J., Faddah D., Perkins S., et al. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson–Gilford progeria syndrome. Proc. Natl Acad. Sci. USA. 2006;103:3250–3255. doi: 10.1073/pnas.0600012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sagelius H., Rosengardten Y., Hanif M., Erdos M.R., Rozell B., Collins F.S., Eriksson M. Targeted transgenic expression of the mutation causing Hutchinson–Gilford progeria syndrome leads to proliferative and degenerative epidermal disease. J. Cell Sci. 2008;121:969–978. doi: 10.1242/jcs.022913. [DOI] [PubMed] [Google Scholar]
- 56.Nikolova V., Leimena C., McMahon A.C., Tan J.C., Chandar S., Jogia D., Kesteven S.H., Michalicek J., Otway R., Verheyen F., et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J. Clin. Invest. 2004;113:357–369. doi: 10.1172/JCI19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arimura T., Helbling-Leclerc A., Massart C., Varnous S., Niel F., Lacène E., Fromes Y., Toussaint M., Mura A.M., Keller D.I., et al. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet. 2005;14:155–169. doi: 10.1093/hmg/ddi017. [DOI] [PubMed] [Google Scholar]
- 58.Mounkes L.C., Kozlov S.V., Rottman J.N., Stewart C.L. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum. Mol. Genet. 2005;14:2167–2180. doi: 10.1093/hmg/ddi221. [DOI] [PubMed] [Google Scholar]
- 59.Wang Y., Herron A.J., Worman H.J. Pathology and nuclear abnormalities in hearts of transgenic mice expressing M371K lamin A encoded by an LMNA mutation causing Emery–Dreifuss muscular dystrophy. Hum. Mol. Genet. 2006;15:2479–2489. doi: 10.1093/hmg/ddl170. [DOI] [PubMed] [Google Scholar]
- 60.Fong L.G., Frost D., Meta M., Qiao X., Yang S.H., Coffinier C., Young S.G. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006;311:1621–1623. doi: 10.1126/science.1124875. [DOI] [PubMed] [Google Scholar]
- 61.Yang S.H., Qiao X., Fong L.G., Young S.G. Treatment with a farnesyltransferase inhibitor improves survival in mice with a Hutchinson–Gilford progeria syndrome mutation. Biochim. Biophys. Acta. 2008;1781:36–39. doi: 10.1016/j.bbalip.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Östlund C., Bonne G., Schwartz K., Worman H.J. Properties of lamin A mutants found in Emery-Dreifuss muscular dystrophy, cardiomyopathy and Dunnigan-type partial lipodystrophy. J. Cell Sci. 2001;114:4435–4445. doi: 10.1242/jcs.114.24.4435. [DOI] [PubMed] [Google Scholar]
- 63.Li E.R., Owens D.M., Djian P., Watt F.M. Expression of involucrin in normal, hyperproliferative and neoplastic mouse keratinocytes. Exp. Dermatol. 2000;9:431–438. doi: 10.1034/j.1600-0625.2000.009006431.x. [DOI] [PubMed] [Google Scholar]
- 64.Östlund C., Ellenberg J., Hallberg E., Lippincott-Scjwartz J., Worman H.J. Intracellular trafficking of emerin, the Emery–Dreifuss muscular dystrophy protein. J. Cell Sci. 1999;112:1709–1179. doi: 10.1242/jcs.112.11.1709. [DOI] [PubMed] [Google Scholar]
- 65.Manilal S., Randles K.N., Aunac C., Nguyen M., Morris G.E. A lamin A/C beta-strand containing the site of lipodystrophy mutations is a major surface epitope for a new panel of monoclonal antibodies. Biochim. Biophys. Acta. 2004;1671:87–92. doi: 10.1016/j.bbagen.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 66.Cance W.G., Chaudhary N., Worman H.J., Blobel G., Cordon-Cardo C. Expression of the nuclear lamins in normal and neoplastic human tissue. J. Exp. Clin. Cancer Res. 1992;11:233–246. [Google Scholar]
- 67.Romero M.R., Carroll J.M., Watt F.M. Analysis of cultured keratinocytes from a transgenic mouse model of psoriasis: effects of suprabasal integrin expression on keratinocyte adhesion, proliferation and terminal differentiation. Exp. Dermatol. 1999;8:53–67. doi: 10.1111/j.1600-0625.1999.tb00348.x. [DOI] [PubMed] [Google Scholar]