

Abstract
Usher syndrome (USH) is a genetically heterogeneous group of autosomal recessive deaf-blinding disorders. Pathophysiology leading to the blinding retinal degeneration in USH is uncertain. There is evidence for involvement of the photoreceptor cilium, photoreceptor synapse, the adjacent retinal pigment epithelium (RPE) cells, and the Crumbs protein complex, the latter implying developmental abnormalities in the retina. Testing hypotheses has been difficult in murine USH models because most do not show a retinal degeneration phenotype. We defined the retinal disease expression in vivo in human USH using optical imaging of the retina and visual function. In MYO7A (USH1B), results from young individuals or those at early stages indicated the photoreceptor was the first detectable site of disease. Later stages showed photoreceptor and RPE cell pathology. Mosaic retinas in Myo7a-deficient shaker1 mice supported the notion that the mutant photoreceptor phenotype was cell autonomous and not secondary to mutant RPE. Humans with PCDH15 (USH1F), USH2A or GPR98 (USH2C) had a similar retinal phenotype to MYO7A (USH1B). There was no evidence of photoreceptor synaptic dysfunction and no dysplastic phenotype as in CRB1 (Crumbs homologue1) retinopathy. The results point to the photoreceptor cell as the therapeutic target for USH treatment trials, such as MYO7A somatic gene replacement therapy.
INTRODUCTION
Usher syndrome (USH), an autosomal recessive deaf-blindness, was historically divided into three clinical types but more recently has emerged as a genetically heterogeneous disease group with at least 12 causative chromosomal loci. The disease-causing genes encode proteins of different classes. Some of the proteins are thought to be integrated into a network (1–3). Although functional evidence for such a network in the retina is lacking, it follows from this suggestion that a defect in any of the components of the USH network could lead to dysfunction in the entire complex with the result being sensorineural degeneration (1,2). It has been proposed that the pathophysiology in the retina may involve the photoreceptor ciliary apparatus or synapse or possibly both. One USH protein, MYO7A, has also been localized to both photoreceptor cilium and adjacent retinal pigment epithelium (RPE) cells (3–5). Further, a link between the USH network and the Crumbs protein complex has been found recently and it was suggested that developmental aberrations in the retina might occur in USH (6,7).
Testing hypotheses about USH pathomechanisms in the mammalian retina has been difficult because most mouse models of human USH genotypes have not shown a retinal degeneration phenotype (3,8). We inquired in human USH about the primary sites of retinal disease expression using optical imaging and visual measurements. Evidence is presented for a common photoreceptor disease phenotype among USH genotypes and this phenotype does not include photoreceptor synaptic dysfunction or hallmarks of Crumbs-like developmental aberrations in the retina. The mutant photoreceptor phenotype in a shaker1 mouse model of MYO7A-USH1B was also clarified to be primary and not secondary to mutant RPE.
RESULTS
MYO7A leads to photoreceptor and then RPE disease in man
An en face view of the ocular fundus with infrared light is shown in a normal subject and an individual with USH1B due to MYO7A mutations (Fig. 1A and B; Table 1). Compared with the normal appearance, the MYO7A-mutant fundus has greater visibility of the choroidal circulation and pigmentary aggregations. These findings, typical of late-stage retinal degenerations (9), suggest depigmentation or loss of the RPE layer underlying the neural retina. Superimposed on the fundus views are cross-sectional optical images along the vertical meridian to illustrate the otherwise-transparent retinal laminar architecture. Topography of the light-sensitive photoreceptor cell layer is also remarkably different in the USH1B subject compared with normal (Fig. 1A and B, insets). The photoreceptor layer (or outer nuclear layer, ONL) of the normal retina peaks centrally and declines with distance from the fovea with more gradually thinning in the superior retina. F6,P1 at age 41 retains only a central island of diminished ONL. The abnormal ONL and RPE layers are accompanied by severely reduced vision (data not shown).
Figure 1.
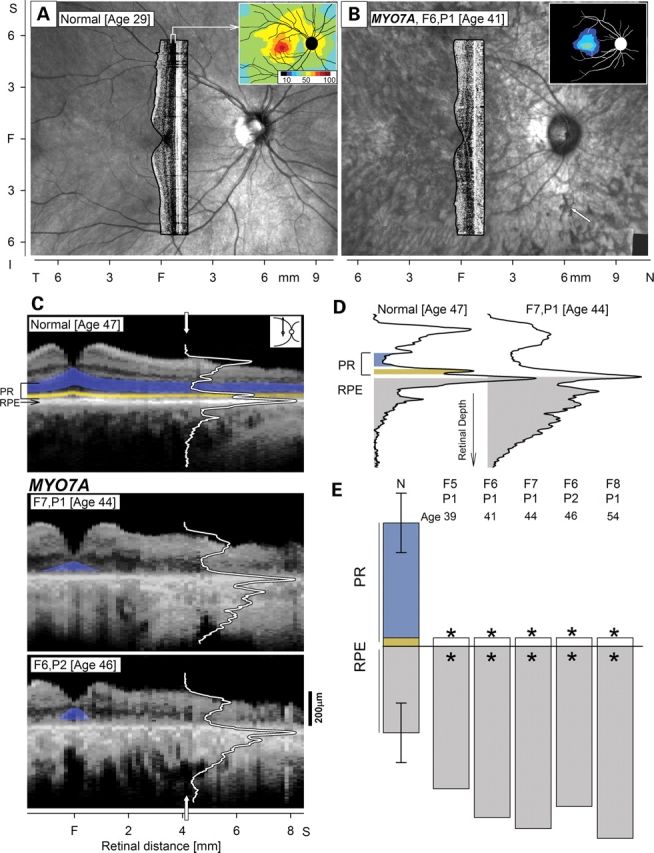
Human MYO7A-mutant retinas analyzed for photoreceptor (PR) and retinal pigment epithelium (RPE) disease. En face images of infrared reflectance in a normal (A) and an USH1B subject (B) with superimposed cross-sectional images and insets (upper right) of PR (outer nuclear layer, ONL) topography. Arrow (B, lower right) points to pigment migration in the USH1B image. (C) Vertical cross-sections of a normal subject and two MYO7A-mutant retinas with advanced disease. Reflectivity profiles (white lines) are overlaid on the section at 4.1 mm in superior retina. Highlighted are ONL (blue) and inner/outer segment (IS/OS) layer (yellow). (D) Reflectivity profiles with signal features representing PR (ONL, blue; IS/OS, yellow) and RPE (sub-RPE backscatter index, gray) are shown. (E) Quantitation of the PR and RPE parameters derived from cross-sections at the superior retinal locus in five MYO7A-mutant retinas at advanced stages compared with results from a normal population (N; n = 26; ages 5–68 years). PR abnormalities in the MYO7A data co-localize with abnormal RPE (asterisk; significantly reduced for PR or increased for RPE). Error bars on normal are ±2SD from mean normal parameters. F, fovea; T, temporal; N, nasal; S, superior.
Table 1.
Mutations in study subjects
| Subtype (gene) | Family | Patient | Gender/age (years) | Amino acid | Referencea |
|---|---|---|---|---|---|
| Usher 1B (MYO7A) | 1 | 1 | M/6 | Tyr333X | This study |
| Tyr333X | |||||
| 2 | M/8 | Tyr333X | This study | ||
| Tyr333X | |||||
| 2 | 1 | M/17 | Glu495X | This study | |
| Cys31X | |||||
| 3 | 1 | F/34 | Arg1240Gln | This study | |
| Arg1240Gln | |||||
| 4 | 1 | M/35 | Arg669X | This study | |
| Arg1591fs | |||||
| 5 | 1 | F/39 | Leu1858Pro | This study | |
| Glu1716X | |||||
| 6 | 1 | F/41 | Ala1288Pro | This study | |
| Arg1743Trp | |||||
| 2 | F/46 | Ala1288Pro | This study | ||
| Arg1743Trp | |||||
| 7 | 1 | M/44 | Gly163Arg | This study | |
| IVS6+1G>T | |||||
| 8 | 1 | F/61 | Gln1798X | This study | |
| Usher 1F (PCDH15) | 1 | 1 | M/10 | Arg245X | This study |
| Arg245X | |||||
| Usher 2A (USH2A) | 1 | 1 | F/18 | Glu767fs | (46) (F2,P4) |
| Glu767fs | |||||
| 2 | F/25 | Glu767fs | (46) (F2,P5) | ||
| Glu767fs | |||||
| 3 | M/26 | Glu767fs | (46) (F2,P1) | ||
| Glu767fs | |||||
| 2 | 1 | F/24 | Cys759Phe | (47) (F12,P1) | |
| Trp3955X | |||||
| 3 | 1 | M/38 | Glu767fs | (46) (F4,P1) | |
| N405fs | |||||
| 4 | 1 | F/41 | Glu767fs | (47) (F14,P1) | |
| 5 | 1 | F/43 | Cys759Phe | (47) (F16,P1) | |
| Val218Glu | |||||
| 6 | 1 | M/44 | Glu767fs | (47) (F13,P1) | |
| 7 | 1 | F/46 | Cys759Phe | (47) (F15,P1) | |
| 8 | 1 | F/56 | Glu767fs | (46) (F5,P1) | |
| Usher 2C (GPR98) | 1 | 1 | F/31 | Gln2301X | (46) (F1,P6); (48) (F1848) |
| Ile2906fs | |||||
| 2 | F/33 | Gln2301X | (46) (F1,P5); (48) (F1848) | ||
| Ile2906fs | |||||
| 3 | F/42 | Gln2301X | (46) (F1,P1); (48) (F1848) | ||
| Ile2906fs | |||||
| CSNB1A (NYX) | 1 | 1 | M/18 | p.Trp350X | (34) (F610) |
| 2 | 1 | M/60 | p.Leu205_Arg207dup | This study | |
| CSNB2A (CACNA1F) | 1 | 1 | M/42 | p.Pro197AlafsX | This study |
| LCA8 (CRB1) | 1 | 1 | F/20 | Cys948Tyr | (11) (P3) |
| Cys1218Phe |
CSNB, congenital stationary night blindness; LCA, Leber congenital amaurosis.
aPrevious reports of genotype and/or phenotype by the authors.
Structural integrity of the ONL and RPE was quantified in optical cross-sections that spanned central and superior retina (Fig. 1C–E). Normal cross-sections show discernible laminae: there are cellular layers of low reflectivity and intervening higher reflectivity laminae representing synaptic and nerve fiber connections. Deep in the retina is a multi-peaked signal that can be subdivided into components representing photoreceptor inner and outer segments (PR IS/OS), RPE and anterior choroid (Fig. 1C) (10,11). MYO7A-mutant retinas in F7,P1 and F6,P2, at ages 44 and 46 respectively, differ from normal in that there is disturbed laminar architecture and increased backscatter signal originating deep to the RPE layer. The deep backscatter in these USH1B subjects corresponds qualitatively to the choroidal visibility secondary to RPE depigmentation from the en face fundus view of F6,P1 (Fig. 1B).
Three structural parameters, two representing photoreceptors (ONL thickness and PR IS/OS) and one for RPE depigmentation, were measured at 4 mm in the superior retina in four MYO7A-mutant retinas with advanced vision loss (Fig. 1D and E). Representative reflectivity profiles at this locus show the photoreceptor and RPE parameters in a normal subject compared with F7,P1 at age 44 (Fig. 1D). The USH1B profile has absence of the ONL and PR IS/OS signals; the RPE depigmentation parameter is also abnormal. Four late-stage MYO7A-mutant retinas were quantified for these parameters and all had severe photoreceptor loss and RPE structural abnormalities (Fig. 1E).
To determine the site of earliest disease expression, we performed similar quantitative analyses in USH1B subjects at younger ages (Fig. 2). Two siblings were studied in their first two decades of life. F1,P1, at age 6 years, had a large expanse of ONL which became undetectable at about 5 mm from the fovea. Both photoreceptor and RPE structural parameters were normal at the 4 mm superior locus. A sibling, F1,P2, studied at ages 8 and 15 years showed disease progression in this 7-year interval. The photoreceptor signal, which was measurable but abnormally reduced, became undetectable; RPE structural integrity, however, was maintained at both ages. In another USH1B family, longitudinal data of F2,P1 showed progression of the photoreceptor abnormality over an 8-year interval (from ages 17 to 25 years), but the RPE parameter was unchanged. These intra- and inter-familial and longitudinal data support the hypothesis that photoreceptor structural abnormalities antedate detectable RPE disturbance in USH1B.
Figure 2.
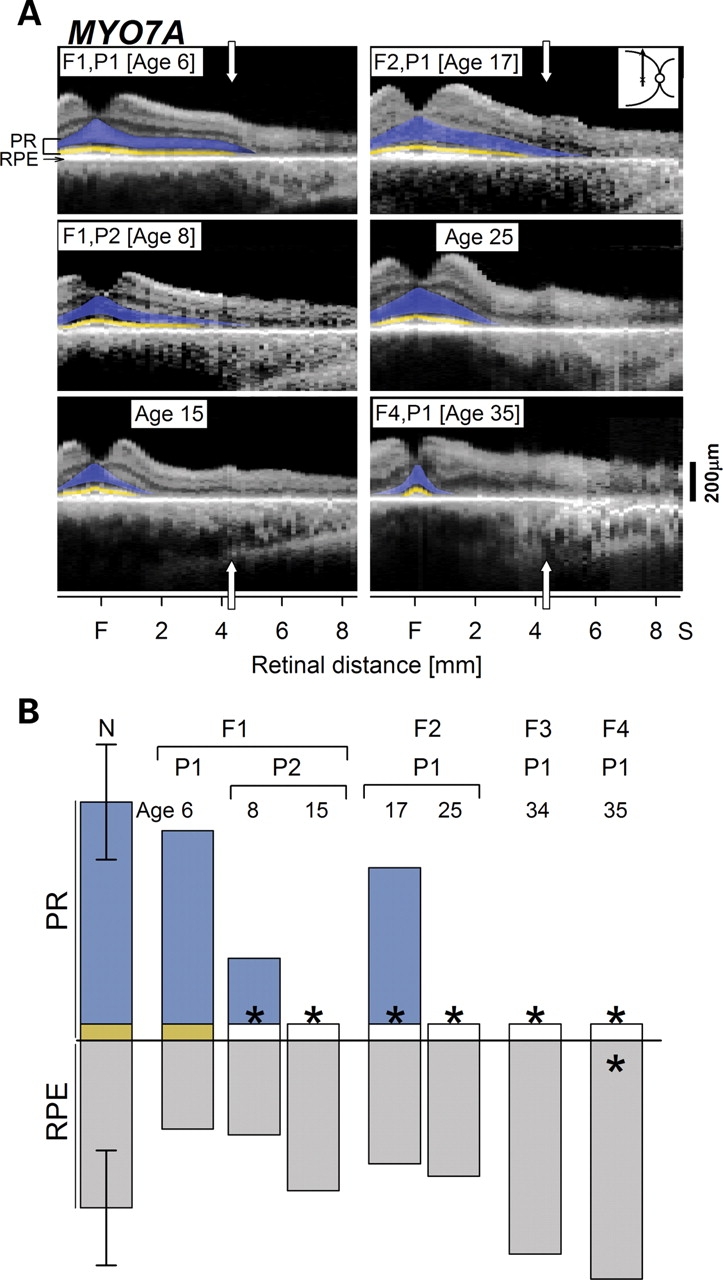
MYO7A-mutant retinas at early ages and stages show photoreceptor (PR) before retinal pigment epithelium (RPE) disease expression. (A) Optical scans of the central-superior retina (inset, right) in three young USH1B subjects show preserved PR lamination; longitudinal data in two of the individuals (F1,P2 over 7 years; F2,P1 over 8 years) show diminution of superior extent. For comparison, the scan from F4,P1, a 35-year-old USH1B subject, is shown. (B) Quantitation of the PR and RPE signals from the scans at 4.1 mm in the superior retina in normal subjects (N; n = 26; ages 5–68) compared with the MYO7A-mutant retinas. PR parameters become significantly reduced (asterisk), while the RPE parameter remains within normal limits except in F4,P1 at age 35 years, in which both parameters are abnormal. F, fovea; S, superior.
Mutant PR phenotype in shaker1 mice is primary, not secondary to RPE defect
None of the alleles of shaker1 mice undergo photoreceptor degeneration (3); however, there is an excessive accumulation of opsin in the photoreceptor connecting cilium and a retarded distal migration of the OS disc membranes (12). These mutant phenotypes in the photoreceptors could result from a MYO7A deficiency in the photoreceptors, or, indirectly from a deficiency in the RPE. Immunolabeling of rodent retinas has shown that the vast majority of MYO7A resides in the RPE (4). Lack of MYO7A affects melanosome localization (13), and results in a slowed degradation of ingested photoreceptor disc membranes by the RPE (14). The second defect could feasibly feedback to slow down disc membrane renewal, and thus generate the photoreceptor phenotypes. We distinguished between these two hypotheses, by studying homozygous shaker14626SB mice (a null allele of Myo7a, which is on chromosome 7). Through bacterial artificial chromosome transgenesis, these mice expressed wild-type (WT) Myo7a from their X-chromosome. Tissues of the transgenic female shaker1 mice thus contain mosaics of MYO7A-null and MYO7A-positive cells due to X-inactivation of the X-chromosome with or without the Myo7a transgene, respectively (15). In the retina, this mosaic pattern is evident by light microscopy of the melanosome distribution within different RPE cells (Fig. 3A). Examination of mosaic retinas, immunogold labeled for opsin, showed that the abnormal accumulation of opsin in the photoreceptor-connecting cilium was independent of whether the overlying RPE cell was mutant or WT. Instead it correlated with the absence of MYO7A in the connecting cilium (Fig. 3B–D), indicating that it arises directly from loss of MYO7A in the photoreceptors, and not indirectly as a result of mutant RPE.
Figure 3.
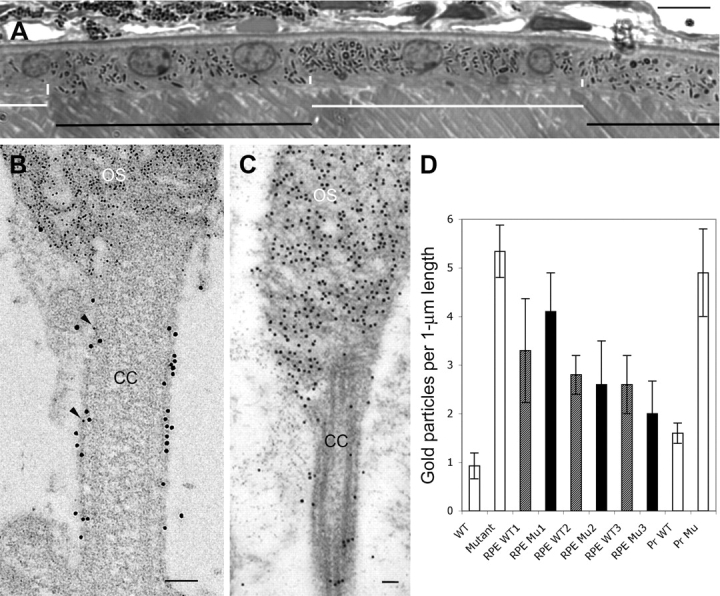
Photoreceptor phenotype in shaker1 mice is associated with lack of photoreceptor (PR) MYO7A, and is not secondary to retinal pigment epithelium (RPE) defect. (A) Light micrograph of a mosaic retina, showing wild-type (WT) (black horizontal bars) and mutant (white horizontal bars) RPE cells, as indicated by the distribution of the melanosomes. Short vertical white bars indicate the apical region, which is devoid of melanosomes in the mutant cells (and contains melanosomes in the WT cells). Scale bar (upper right) = 10 µm. (B) Electron micrograph of the connecting cilium (CC) and basal outer segment (OS) discs of a PR in a mosaic retina. The section was double-labeled with opsin antibodies (identified by goat anti-mouse (GAM) IgG, conjugated to 5 nm gold particles) and MYO7A antibodies (identified by goat anti-rabbit IgG, conjugated to 10 nm gold particles). The presence of MYO7A indicates that this PR cell expressed the Myo7a transgene. The PR was adjacent to a mutant RPE cell (data not shown). The scarcity of opsin label in the CC (only two 5 nm particles are evident, indicated by arrowheads) is consistent with a WT phenotype. (C) Electron micrograph of the CC and basal OS discs of a PR in a Myo7a-null retina. The section was labeled with opsin antibodies (identified by GAM IgG, conjugated to 10 nm gold particles). In the absence of MYO7A, the CC contains significant opsin label, as shown previously (12). Scale bars in B and C = 100 nm. (D) Bar graph showing opsin immunogold particle density measured in the PR connecting cilia of (from left to right): WT retinas; MYO7A-null retinas; three different mosaic retinas, with counts separated for photoreceptors interfacing with normal RPE (WT; shaded bars) and those interfacing with mutant RPE (Mu; filled bars), as determined by the distribution of the melanosomes (13). These data indicate that the genotype of the RPE cell has no significant effect on the quantity of opsin label in the connecting cilia. The last two bars show opsin immunogold particle density according to whether a given CC was also labeled with MYO7A antibodies. The PR cilia that were labeled with MYO7A antibodies (Pr WT) showed opsin labeling that was similar to that in WT retinas (e.g. panel B), whereas the cilia that were not labeled with MYO7A antibodies (Pr Mu) had opsin labeling that was more comparable with that in MYO7A-null retinas. A minimum of 20 PR cells was counted in each retina. Data for the first two and last two bars were obtained from three animals of each genotype. Error bars indicate ±SEM. Student t-test analysis showed no significant difference between counts for photoreceptors interfacing with normal RPE and those interfacing with mutant RPE in mosaic retinas. There was also no significant difference between photoreceptors in WT retinas and MYO7A-labeled photoreceptors in mosaic retinas; and between photoreceptors in MYO7A-null retinas and photoreceptors that were not labeled with MYO7A antibodies in mosaic retinas.
PCDH15, USH2A and GPR98 also manifest photoreceptor before RPE disease
Photoreceptor and RPE analyses were also performed in 14 individuals, representing 10 different molecularly defined families with three other USH genotypes (Fig. 4; Table 1). As in the MYO7A-mutant retinas, measurements were made at 4 mm in the superior retina. An USH1F-PCDH15 patient had longitudinal measurements made between the ages of 10 and 20 years. For clinical perspective, there were stigmata of retinal degeneration (attenuated retinal vessels and peripheral pigmentary retinopathy) noted on all examinations during this decade. Photoreceptor and RPE parameters were normal at ages 10 and 14 years; at ages 17 and 20 years, however, there was loss of photoreceptor structure but no detectable RPE structural abnormality (Fig. 4A). A spectrum of abnormalities was detected in 10 individuals with USH2A. Only one subject, F1,P1 at age 18, had normal photoreceptor and RPE OCT (optical coherence tomography)-based parameters; clinically, there were signs of degenerative change further in the peripheral retina. Four USH2A subjects showed photoreceptor losses but a normal RPE parameter; and the remaining five subjects had both photoreceptor and RPE abnormalities (Fig. 4B). Two of three siblings from a family with USH2C-GPR98 had an abnormal photoreceptor layer without detectable RPE disease while the third had both photoreceptor and RPE structural defects (Fig. 4C). All members of the USH2C-GPR98 family had clinically evident retinal degeneration peripheral to the measurement site at 4 mm. The comparative quantitative analyses of photoreceptor and RPE structural changes in different USH genotypes indicate a shared mechanism of photoreceptor disease preceding RPE abnormality.
Figure 4.

Photoreceptor (PR) structure is lost before retinal pigment epithelium (RPE) disease is structurally apparent in all Usher syndrome (USH) genotypes. (A–C) Optical scans of the central-superior retina in multiple individuals within each genotype (USH2A, GPR98) or longitudinal measurements in the same individual (PCDH15 over a decade). Quantitation of PR and RPE signals shows PR losses without or with RPE abnormalities but no examples of RPE abnormalities preceding PR loss. The spectrum of ages of individuals with USH2A available for study reveals a proposed disease sequence from normal PR and RPE to abnormalities in only PR to disease in both layers. The family with GPR98-mutant retina also reveals this spectrum of changes. F, fovea; S, superior. (D) ONL (outer nuclear layer) thickness is plotted as a function of sub-RPE backscatter index in the four USH genotypes (key) at the superior retinal locus. Data from longitudinal measurements are connected by dashed lines. Normal variability is described by the ellipse encircling 95% confidence interval of a bivariate Gaussian distribution. Unfilled circles, normal subjects (n = 25; ages 5–58 years).
We summarized the association between ONL thickness and RPE depigmentation at the superior retinal locus in all USH genotypes (Fig. 4D). All genotypes had individuals with normal photoreceptor layer and RPE, abnormally reduced photoreceptor and normal RPE, or abnormalities in both of the OCT-based parameters. There were no examples of RPE abnormalities with normal ONL thickness. Longitudinal data in a subset of individuals followed the suggested sequence of photoreceptor loss preceding RPE abnormality. The generalizability of the observation was tested at a temporal retinal locus in the same subjects and the results were similar (data not shown).
No evidence in human USH for photoreceptor synaptic dysfunction or a Crumbs homologue1 (CRB1)-like retinal dysplasia
Localization of USH proteins in the synaptic regions of photoreceptor cells has led to the hypothesis that there may be a role of the USH protein network in photoreceptor synaptic function (7,16). Defective synaptic transmission from photoreceptors to second-order neurons would be predicted to manifest as disproportionately preserved photoreceptor layer thickness for the amount of visual dysfunction. We tested the hypothesis about synaptic abnormality in USH by studying the relationship between ONL structure and visual function at two locations with normally high photoreceptor densities (17). Comparisons were made with normal subjects, a primary photoreceptor retinal degeneration and congenital stationary night blinding (CSNB) diseases with known defective photoreceptor synaptic transmission (Fig. 5A and B).
Figure 5.
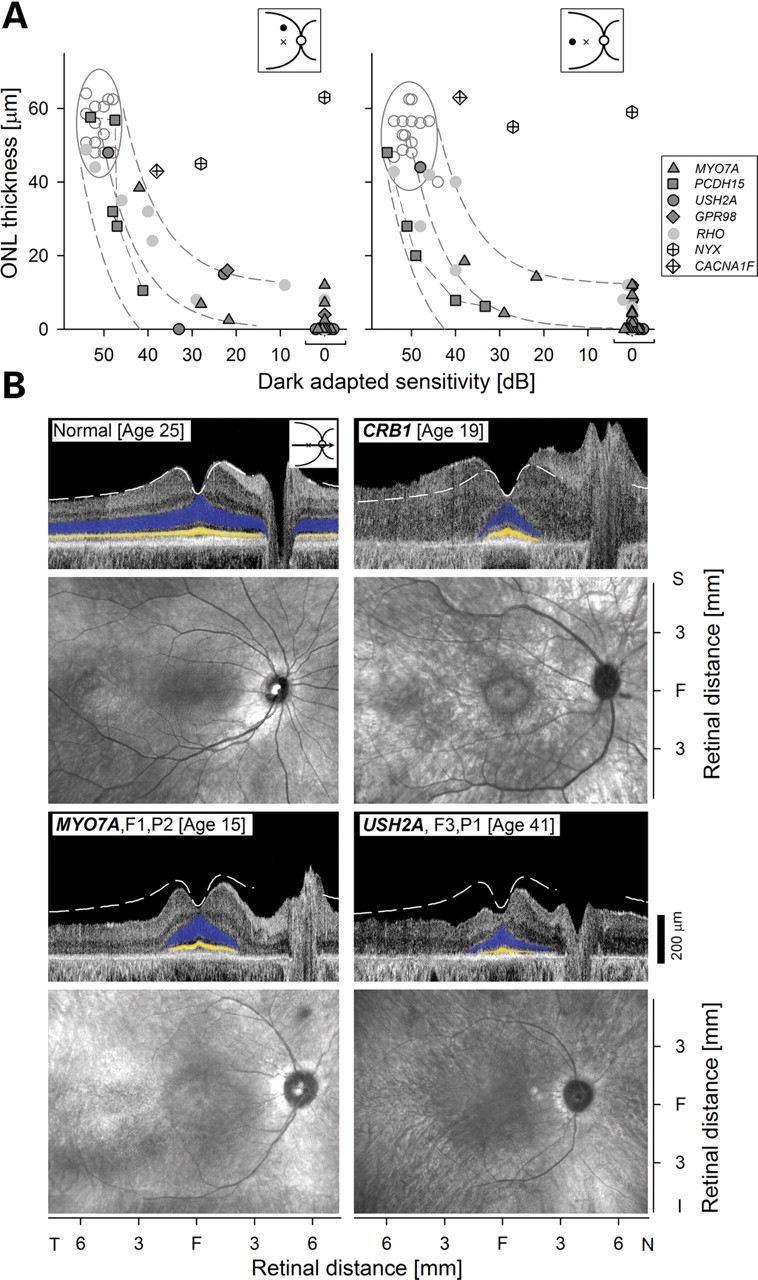
Usher syndrome (USH) genotypes: testing hypotheses about dysfunction at the photoreceptor (PR) synapse and interaction with the Crumbs network. (A) Relationship between PR nuclear layer structure [outer nuclear layer (ONL) thickness] and visual function (dark-adapted sensitivity, 500 nm) at superior (left) and temporal (right) retinal loci (insets at upper right: loci, filled circle; fovea, cross). USH genotypes (filled symbols) are compared with CSNB (congenital stationary night blinding) genotypes (unfilled symbols with ‘plus’), diseases that compromise synaptic transmission from PRs to second-order neurons. Data from 11 individuals with adRP Class B rhodopsin mutations are also shown (gray circles) (18,19). Normal variability is described by ellipses encircling the 95% confidence interval of a bivariate Gaussian distribution. Dashed lines indicate the idealized model of the relationship between structure and function in pure PR degenerations and the region of uncertainty that results by translating the normal variability along the idealized model (20). (B) Cross-sectional OCT (optical coherence tomography) scans along the horizontal meridian (upper) and en face infrared reflectance images (lower) for a normal subject (top left), a subject with a CRB1 mutation (top right), and subjects with MYO7A or USH2A mutations (bottom row). Highlighted are ONL (blue) and inner/outer segment (IS/OS) layer (yellow). Dashed white lines on scans indicate mean normal thickness (n = 27). The dysplastic and thickened CRB1-mutant retina is in contrast to the thinned retinas in USH genotypes.
Normal ONL thickness at the superior retinal locus averages 56 µm (SD = 6 µm; n = 14; age = 19–56 years). In the photoreceptor degeneration due to mutations in the rhodopsin gene (18,19), thinning of the ONL is related predictably to dark-adapted vision loss over a 5 log unit range from normal to severely abnormal (Fig. 5A) (20). This relationship in pure photoreceptor degenerations is based on an idealized model where loss of function is proportional to the square of ONL thinning (20). The relationship of function and structure in all subjects with USH genotypes also followed the idealized model expected from photoreceptor degenerations (20). As positive controls, data are also plotted for three individuals with CSNB where the structure–function relationship does not conform to the idealized model. Two CSNB1 subjects with mutations in the NYX gene (Table 1) encoding nyctalopin, a protein known to be associated with photoreceptor synapses (21), had reductions in dark-adapted sensitivity but relatively preserved ONL thickness. A subject with CSNB2 due to a mutation in CACNA1F gene (Table 1), which encodes for the α1F subunit of the Cav1.4 voltage-dependent calcium channel, has only about 1 log unit of dark-adapted vision disturbance but more ONL than anticipated from the other disease relationships measured. Generalizability of these results was tested at a temporal retinal locus and the findings were as in the superior retina (Fig. 5B). Among the USH subjects examined, there were no examples of negative electroretinograms, waveforms traditionally associated with synaptic transmission abnormalities (such as in CSNB1 and CSNB2) between photoreceptors and the inner retina (22).
A link between the USH and the Crumbs protein networks was recently described (6). There were interaction, colocalization and coexpression patterns leading to the suggestion of an important role of the two networks in retinal development, such as apical–basal polarity and patterning of the retina. It was postulated that these developmental processes might be disrupted in inherited disorders associated with defects in the networks. The only human retinal disease with a Crumbs defect is the early-onset recessive disease caused by mutations in the CRB1 gene (11). The disease is characterized by retinal laminar disruption and severe visual disturbances (Fig. 5B). None of the USH genotypes showed a retinopathy that resembled the dysplastic and thickened retina, which is a feature of CRB1 disease (Fig. 5B).
DISCUSSION
Auditory and vestibular criteria led to early clinical subtyping of USH (reviewed in 23,24) and these subtypes were then studied for differences in severity of the associated eye disease (25–29). Such approaches were helpful in the clinic for counseling purposes; and, in the molecular laboratory, subtyping led to progress in linkage analyses and eventual gene identification. Following the discovery phase of many USH genes, it was recognized that the encoded proteins are diverse in class and function, but at least in heterologous systems, interactions among many of the proteins could result in an integrated protein network. These observations have prompted hypotheses about the pathophysiology of the sensorineural degenerations (1,3,7,16). The lack of murine models with retinal degeneration for most USH genotypes puts an onus on human studies to explore pathological mechanisms in the retina (3,8).
We inquired whether there were differences in human USH retinal disease expression in different genotypes and whether the expression would be consistent with primary photoreceptor cell death (3), with photoreceptor synaptic dysfunction (16), or with abnormal retinal development (6). USH1B from MYO7A mutations, the most common molecular form of USH type 1, was investigated first because there are two proposed cellular sites of retinal disease – photoreceptors and RPE. MYO7A is the only USH protein in the RPE (3). Recent progress toward somatic gene therapy in USH1B has made it important to know the target cell type (3). A gene replacement strategy has been used to correct defects in vitro and by subretinal injection in shaker1 mice (30). A large body of scientific work in the Myo7a-deficient shaker1 mouse retina has identified and characterized an RPE transport defect of phagosomes and melanosomes (3,5,12,13,30,31). Yet, there is also a minor presence of MYO7A in the photoreceptor ciliary region (5), a subcellular site where other USH proteins of the network are thought to cause disease effects (1,3,8). The present results in young humans with USH1B from MYO7A mutations support the concept that the site of disease action is the photoreceptor. Of interest, the pattern of photoreceptor layer thickness loss preceding an RPE structural defect has recently been illustrated using optical imaging in patients with the primary photoreceptor degeneration due to mutations in the rhodopsin gene (Fig. 3A in Ref. 32).
A primary RPE defect due to MYO7A mutation cannot be excluded by our non-invasive studies and could theoretically be additive to a photoreceptor abnormality causing acceleration of the time course of retinal degeneration. Quantitative immunogold labeling of human and mouse retinas has shown that the distribution of MYO7A, in terms of organelle association and relative apical–basal distribution, is similar in human and mouse RPE (33), suggesting comparable MYO7A function in the human and mouse RPE. Studies of shaker1 mice with mosaic retinas indicated that the mutant photoreceptor phenotype was independent of whether the adjacent RPE cell was mutant or WT, thus supporting the concept of a primary rather than secondary photoreceptor disease in human USH1B-MYO7A. Nevertheless, because the retinas of shaker1 mice do not degenerate (3), we cannot test whether the RPE or the photoreceptor phenotype is the harbinger of photoreceptor cell death.
Other USH genotypes also showed the same phenotypic pattern as found in USH1B-MYO7A. This suggests uniformity of retinopathy as it relates to primary cell of disease origin in this molecularly heterogeneous group of disorders. These human results, especially those in the USH2A genotype, are also consistent with the conclusions drawn from usherin-null mice that the disease manifests as a photoreceptor degeneration (8). There was no evidence for synaptic dysfunction, as occurs in retinopathies that specifically affect the photoreceptor synapse (21,34,35). The dramatic retinal dysplasia that we previously characterized in CRB1 mutations and attributed to developmental abnormalities (11) was not present in any of the USH genotypes. The lack of a dysplastic phenotype in USH, however, does not exclude a common molecular network (6).
An underlying shared pathomechanism as the root cause for the shared retinal phenotype remains the most parsimonious hypothesis to explain the results in the different USH genotypes. The details of USH molecular pathogenesis will be complex and, at a subcellular level, there may be very different mechanisms leading to the same cell death pathway. Current knowledge of USH protein localization suggests that dysfunction in the region of the photoreceptor connecting cilium may be the pre-apoptotic event (1,3). Slowing or disruption of the unidirectional or bidirectional transport of molecules through the cilium that connects the photosensitive OS to the cell body would be expected to have major functional and structural consequences on these fragile and metabolically hyperactive neurons (36,37). The many events resulting from impaired ciliary transport that could initiate the cell death pathway include mislocalization of molecules to the IS compartment, misrouting of phototransduction proteins including opsin (38,39), and defective OS membrane formation with perturbation of the rate of renewal mechanisms (36). The USH disease group has been discussed as one of the ciliopathies associated with retinal degeneration and it will be important to determine if there is commonality of retinal phenotype between USH and these other diseases, such as the Bardet–Biedl syndrome and RPGR/RPGRIP1 retinopathies. Cell and molecular biological studies are obviously required to elucidate details of the interplay between USH network molecules and between USH and other protein networks involving the cilium (1,3,40).
METHODS
Human subjects
Twenty-four patients (ages 6–61 years) with known USH genotypes were included (Table 1). Also included were three patients with CSNB, one patient with LCA8, 11 patients with adRP caused by rhodopsin mutations and normal subjects. Informed consent was obtained; procedures followed the Declaration of Helsinki and had institutional review board approval.
In vivo microscopy of human retina
Cross-sectional images of the retina were obtained with commercially available OCT instruments (OCT1 and OCT3; Carl Zeiss Meditec, Dublin, CA, USA). Overlapping 4.5 mm-length scans were used to study a 9 mm extent along the vertical and horizontal meridia through the fovea. Each scan, formed by a series of longitudinal reflectivity profiles, was analyzed with custom-developed software (MatLab ver. 6.5; The Math Works, Natick, MA, USA) and published methods (10,11). The PR ONL was defined as published (11,20,41,42). Topographical analysis of the ONL was performed and analyzed as described (41). The axial extent of propagation of the near infrared (INR) illumination light of OCT was estimated from the normalized partial integral of the backscattering signal over retinal depth from the RPE signal peak toward the scleral direction (42). The partial integral was divided by the signal intensity at the RPE peak in order to normalize for the pre-RPE attenuation of the light intensity. We refer to the resulting measure as sub-RPE backscattering index (sRBI). Under the simplifying assumption of dominant backscattering and absorption of the NIR illumination occurring at or near the normal RPE, sRBI may be expected to be proportional to the amount of NIR illumination reaching choroidal layers. This expectation is consistent with experimental results in animals with varying RPE and choroidal pigmentation and in patients with molecularly defined choroideremia and depigmentation of the RPE (42).
Visual function
Psychophysical thresholds were measured (1.7° diameter, 200 ms duration stimuli spaced at 2° intervals) in the same retinal regions as the OCT scans. Rod function was determined using two-color dark-adapted perimetry (500, 650 nm stimuli). Long/middle wavelength cone function was determined with 650 nm stimuli in the dark-adapted state (and compared with normal data determined during the cone plateau phase of dark adaptation) and under light-adapted (600 nm) conditions. Details of the techniques and analysis methods have been published (41–44).
Microscopy of mouse retinas
Studies conformed to UK Home Office regulations, the Association for Research in Vision and Ophthalmology Statement for Use of Animals in Ophthalmic and Vision Research and had institutional approval. Mice were genotyped as described previously (15). Eyes were fixed in 0.1% glutaraldehyde, 2–3% paraformaldehyde in 0.1 M phosphate buffer, and processed for embedment in LR White resin. Semithin sections along the dorso-ventral axis of the retinas were stained with toluidine blue for light microscopy, and ultrathin sections were immunogold labeled with opsin and MYO7A antibodies. Double immunogold labeling was performed as described previously (45), using the same rabbit-generated MYO7A antibody, but a different opsin mouse monoclonal antibody, mAb1D4. Photoreceptor and RPE cells within 2 mm of the optic nerve head were examined, so that data were obtained from all cells in the dorso-ventral axis, except the most peripheral ones. Opsin immunogold labeling in the CC was quantified in sections containing negligible background label by counting particles along a 1 µm length of the axoneme, as described (13).
FUNDING
This research was supported by grants from the National Neuroscience Research Institute, Foundation Fighting Blindness, Hope for Vision, National Eye Institute/National Institutes of Health, Macula Vision Research Foundation, Macular Disease Foundation, The Chatlos Foundation, Research to Prevent Blindness, Ruth and Milton Steinbach Fund, Alcon Research Institute, the Medical Research Council and the Wellcome Trust. D.S.W. is a Jules and Doris Stein RPB Professor.
ACKNOWLEDGEMENTS
The authors thank Elaine Smilko and Malgorzata Swider for their critical help with patient studies; Rose Tobias for mutation analysis of CSNB patients; and Roz Lacey and Kornnika Khanobdee for their assistance in processing the mosaic mouse retinas for microscopy.
Conflict of Interest statement. There are no conflicts of interest.
REFERENCES
- 1.Maerker T., van Wijk E., Overlack N., Kersten F.F., McGee J., Goldmann T., Sehn E., Roepman R., Walsh E.J., Kremer H., et al. A novel Usher protein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum. Mol. Genet. 2008;17:71–86. doi: 10.1093/hmg/ddm285. [DOI] [PubMed] [Google Scholar]
- 2.Lefèvre G., Michel V., Weil D., Lepelletier L., Bizard E., Wolfrum U., Hardelin J.P., Petit C. A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development. 2008;135:1427–1437. doi: 10.1242/dev.012922. [DOI] [PubMed] [Google Scholar]
- 3.Williams D.S. Usher syndrome: animal models, retinal function of Usher proteins, and prospects for gene therapy. Vision Res. 2008;48:433–441. doi: 10.1016/j.visres.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hasson T., Hintzelman M.B., Santos-Sacchi J., Corey D.P., Mooseker M.S. Expression in cochlea and retina of myosin VIIa, the gene product defective in Usher syndrome type 1B. Proc. Natl Acad. Sci. USA. 1995;92:9815–9819. doi: 10.1073/pnas.92.21.9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu X., Vansant G., Udovichenko I.P., Wolfrum U., Williams D.S. Myosin VIIa, the product of the Usher 1B syndrome gene, is concentrated in the connecting cilia of photoreceptor cells. Cell Motil. Cytoskeleton. 1997;37:240–252. doi: 10.1002/(SICI)1097-0169(1997)37:3<240::AID-CM6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 6.Gosens I., van Wijk E., Kersten F.F., Krieger E., van der Zwaag B., Märker T., Letteboer S.J., Dusseljee S., Peters T., Spierenburg H.A., et al. MPP1 links the Usher protein network and the Crumbs protein complex in the retina. Hum. Mol. Genet. 2007;16:1993–2003. doi: 10.1093/hmg/ddm147. [DOI] [PubMed] [Google Scholar]
- 7.Overlack N., Maerker T., Latz M., Nagel-Wolfrum K., Wolfrum U. SANS (USH1G) expression in developing and mature mammalian retina. Vision Res. 2008;48:400–412. doi: 10.1016/j.visres.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 8.Liu X., Bulgakov O.V., Darrow K.N., Pawlyk B., Adamian M., Liberman M.C., Li T. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc. Natl Acad. Sci. USA. 2007;104:4413–4418. doi: 10.1073/pnas.0610950104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milam A.H., Li Z.Y., Fariss R.N. Histopathology of the human retina in retinitis pigmentosa. Prog. Retin. Eye Res. 1998;17:175–205. doi: 10.1016/s1350-9462(97)00012-8. [DOI] [PubMed] [Google Scholar]
- 10.Huang Y., Cideciyan A.V., Papastergiou G.I., Banin E., Semple-Rowland S.L., Milam A.H., Jacobson S.G. Relation of optical coherence tomography to microanatomy in normal and rd chickens. Invest. Ophthalmol. Vis. Sci. 1998;39:2405–2416. [PubMed] [Google Scholar]
- 11.Jacobson S.G., Cideciyan A.V., Aleman T.S., Pianta M.J., Sumaroka A., Schwartz S.B., Smilko E.E., Milam A.H., Sheffield V.C., Stone E.M. Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum. Mol. Genet. 2003;12:1073–1078. doi: 10.1093/hmg/ddg117. [DOI] [PubMed] [Google Scholar]
- 12.Liu X., Udovichenko I.P., Brown S.D.M., Steel K.P., Williams D.S. Myosin VIIa participates in opsin transport through the photoreceptor cilium. J. Neurosci. 1999;19:6267–6274. doi: 10.1523/JNEUROSCI.19-15-06267.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X., Ondek B., Williams D.S. Mutant myosin VIIa causes defective melanosome distribution in the RPE of shaker-1 mice. Nat. Genet. 1998;19:117–118. doi: 10.1038/470. [DOI] [PubMed] [Google Scholar]
- 14.Gibbs D., Kitamoto J., Williams D.S. Abnormal phagocytosis by retinal pigment epithelium that lacks myosin VIIa, the Usher syndrome 1B protein. Proc. Natl Acad. Sci. USA. 2003;100:6481–6486. doi: 10.1073/pnas.1130432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prosser H.M., Rzadzinska A.K., Steel K.P., Bradley A. Mosaic complementation demonstrates a regulatory role for myosin VIIa in stereocilia actin dynamics. Mol. Cell Biol. 2008;28:1702–1712. doi: 10.1128/MCB.01282-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reiners J., Nagel-Wolfrum K., Jurgens K., Marker T., Wolfrum U. Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp. Eye Res. 2006;83:97–119. doi: 10.1016/j.exer.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 17.Curcio C.A., Sloan K.R., Kalina R.E., Hendrickson A.E. Human photoreceptor topography. J. Comp. Neurol. 1990;292:497–523. doi: 10.1002/cne.902920402. [DOI] [PubMed] [Google Scholar]
- 18.Cideciyan A.V., Hood D.C., Huang Y., Banin E., Li Z.Y., Stone E.M., Milam A.H., Jacobson S.G. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc. Natl Acad. Sci. USA. 1998;95:7103–7108. doi: 10.1073/pnas.95.12.7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aleman T.S., Cideciyan A.V., Sumaroka A., Windsor E.A.M., Herrera W., White D.A., Kaushal S., Naidu A., Roman A.J., Schwartz S.B., et al. Retinal laminar architecture in human retinitis pigmentosa caused by rhodopsin gene mutations. Invest. Ophthalmol. Vis. Sci. 2008;49:1580–1590. doi: 10.1167/iovs.07-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobson S.G., Aleman T.S., Cideciyan A.V., Sumaroka A., Schwartz S.B., Windsor E.A., Traboulsi E.I., Heon E., Pittler S.J., Milam A.H., et al. Identifying photoreceptors in blind eyes caused by RPE65 mutations: prerequisite for human gene therapy success. Proc. Natl Acad. Sci. USA. 2005;102:6177–6182. doi: 10.1073/pnas.0500646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgans C.W., Ren G., Akileswaran L. Localization of nyctalopin in the mammalian retina. Eur. J. Neurosci. 2006;23:1163–1171. doi: 10.1111/j.1460-9568.2006.04647.x. [DOI] [PubMed] [Google Scholar]
- 22.Cideciyan A.V., Jacobson S.G. Negative electroretinograms in retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 1993;34:3253–3263. [PubMed] [Google Scholar]
- 23.Kremer H., van Wijk E., Marker T., Wolfrum U., Roepman R. Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum. Mol. Genet. 2006;15:R262–R270. doi: 10.1093/hmg/ddl205. [DOI] [PubMed] [Google Scholar]
- 24.Cohen M., Bitner-Glindzicz M., Luxon L. The changing face of Usher syndrome: clinical implications. Int. J. Audiol. 2007;46:82–93. doi: 10.1080/14992020600975279. [DOI] [PubMed] [Google Scholar]
- 25.Fishman G.A., Kumar A., Joseph M.E., Torok N., Anderson R.J. Usher’s syndrome. Ophthalmic and neuro-otologic findings suggesting genetic heterogeneity. Arch. Ophthalmol. 1983;101:1367–1374. doi: 10.1001/archopht.1983.01040020369005. [DOI] [PubMed] [Google Scholar]
- 26.Piazza L., Fishman G.A., Farber M., Derlacki D., Anderson R.J. Visual acuity loss in patients with Usher’s syndrome. Arch. Ophthalmol. 1986;104:1336–1339. doi: 10.1001/archopht.1986.01050210090031. [DOI] [PubMed] [Google Scholar]
- 27.Smith R.J., Berlin C.I., Hejtmancik J.F., Keats B.J., Kimberling W.J., Lewis R.A., Möller C.G., Pelias M.Z., Tranebjaerg L. Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. Am. J. Med. Genet. 1994;50:32–38. doi: 10.1002/ajmg.1320500107. [DOI] [PubMed] [Google Scholar]
- 28.Hope C.I., Bundey S., Proops D., Fielder A.R. Usher syndrome in the city of Birmingham – prevalence and clinical classification. Br. J. Ophthalmol. 1997;81:46–53. doi: 10.1136/bjo.81.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsilou E.T., Rubin B.I., Caruso R.C., Reed G.F., Pikus A., Hejtmancik J.F., Iwata F., Redman J.B., Kaiser-Kupfer M.I. Usher syndrome clinical types I and II: could ocular symptoms and signs differentiate between the two types? Acta. Ophthalmol. Scand. 2002;80:196–201. doi: 10.1034/j.1600-0420.2002.800215.x. [DOI] [PubMed] [Google Scholar]
- 30.Hashimoto T., Gibbs D., Lillo C., Azarian S.M., Legacki E., Zhang X.M., Yang X.J., Williams D.S. Lentiviral gene replacement therapy of retinas in a mouse model for Usher syndrome type 1B. Gene Ther. 2007;14:584–594. doi: 10.1038/sj.gt.3302897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibson F., Walsh J., Mburu P., Varela A., Brown K.A., Antonio M., Beisel K.W., Steel K.P., Brown S.D. A type VII myosin encoded by the mouse deafness gene shaker-1. Nature. 1995;374:62–64. doi: 10.1038/374062a0. [DOI] [PubMed] [Google Scholar]
- 32.Aleman T.S., Cideciyan A.V., Sumaroka A., Windsor E.A., Herrera W., White D.A., Kaushal S., Naidu A., Roman A.J., Schwartz S.B., Stone E.M., Jacobson S.G. Retinal laminar architecture in human retinitis pigmentosa caused by rhodopsin gene mutations. Invest. Ophthalmol. Vis. Sci. 2008;49:1580–1590. doi: 10.1167/iovs.07-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibbs D., Azarian S.M., Lillo C., Kitamoto J., Klomp A.E., Steel K.P., Libby R.T., Williams D.S. Role of myosin VIIa and Rab27a in the motility and localization of RPE melanosomes. J. Cell Sci. 2004;117:6473–6483. doi: 10.1242/jcs.01580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bech-Hansen N.T., Naylor M.J., Maybaum T.A., Sparkes R.L., Koop B., Birch D.G., Bergen A.A., Prinsen C.F., Polomeno R.C., Gal A., et al. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat. Genet. 2000;26:319–323. doi: 10.1038/81619. [DOI] [PubMed] [Google Scholar]
- 35.Allen L.E., Zito I., Bradshaw K., Patel R.J., Bird A.C., Fitzke F., Yates J.R., Trump D., Hardcastle A.J., Moore A.T. Genotype–phenotype correlation in British families with X-linked congenital stationary night blindness. Br. J. Ophthalmol. 2003;87:1413–1420. doi: 10.1136/bjo.87.11.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kennan A., Aherne A., Humphries P. Light in retinitis pigmentosa. Trends Genet. 2005;21:103–110. doi: 10.1016/j.tig.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Stone J., Maslim J., Valter-Kocsi K., Mervin K., Bowers F., Chu Y., Barnett N., Provis J., Lewis G., Fisher S.K., et al. Mechanisms of photoreceptor death and survival in mammalian retina. Prog. Retin. Eye Res. 1999;18:689–735. doi: 10.1016/s1350-9462(98)00032-9. [DOI] [PubMed] [Google Scholar]
- 38.Marszalek J.R., Liu X., Roberts E.A., Chui D., Marth J.D., Williams D.S., Goldstein L.S. Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell. 2000;21:175–187. doi: 10.1016/s0092-8674(00)00023-4. [DOI] [PubMed] [Google Scholar]
- 39.Chen J., Shi G., Concepcion F.A., Xie G., Oprian D., Chen J. Stable rhodopsin/arrestin complex leads to retinal degeneration in a transgenic mouse model of autosomal dominant retinitis pigmentosa. J. Neurosci. 2006;26:11929–11937. doi: 10.1523/JNEUROSCI.3212-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roepman R., Wolfrum U. Protein networks and complexes in photoreceptor cilia. Subcell. Biochem. 2007;43:209–235. doi: 10.1007/978-1-4020-5943-8_10. [DOI] [PubMed] [Google Scholar]
- 41.Aleman T.S., Cideciyan A.V., Sumaroka A., Schwartz S.B., Roman A.J., Windsor E.A., Steinberg J.D., Branham K., Othman M., Swaroop A., et al. Inner retinal abnormalities in X-linked retinitis pigmentosa with RPGR mutations. Invest. Ophthalmol. Vis. Sci. 2007;48:4759–4765. doi: 10.1167/iovs.07-0453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacobson S.G., Cideciyan A.V., Sumaroka A., Aleman T.S., Schwartz S.B., Windsor E.A., Roman A.J., Stone E.M., MacDonald I.M. Remodeling of the human retina in choroideremia: rab escort protein 1 (REP-1) mutations. Invest. Ophthalmol. Vis. Sci. 2006;47:4113–4120. doi: 10.1167/iovs.06-0424. [DOI] [PubMed] [Google Scholar]
- 43.Jacobson S.G., Voigt W.J., Parel J.M., Apáthy P.P., Nghiem-Phu L., Myers S.W., Patella V.M. Automated light- and dark-adapted perimetry for evaluating retinitis pigmentosa. Ophthalmology. 1986;93:1604–1611. doi: 10.1016/s0161-6420(86)33522-x. [DOI] [PubMed] [Google Scholar]
- 44.Roman A.J., Schwartz S.B., Aleman T.S., Cideciyan A.V., Chico J.D., Windsor E.A., Gardner L.M., Ying G.S., Smilko E.E., Maguire M.G., et al. Quantifying rod photoreceptor-mediated vision in retinal degenerations: dark-adapted thresholds as outcome measures. Exp. Eye Res. 2005;80:259–272. doi: 10.1016/j.exer.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 45.Liu X., Williams D.S. Coincident onset of expression of myosin VIIa and opsin in the cilium of the developing photoreceptor cell. Exp. Eye Res. 2001;72:351–355. doi: 10.1006/exer.2000.0963. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz S.B., Aleman T.S., Cideciyan A.V., Windsor E.A., Sumaroka A., Roman A.J., Rane T., Smilko E.E., Bennett J., Stone E.M., et al. Disease expression in Usher syndrome caused by VLGR1 gene mutation (USH2C) and comparison with USH2A phenotype. Invest. Ophthalmol. Vis. Sci. 2005;46:734–743. doi: 10.1167/iovs.04-1136. [DOI] [PubMed] [Google Scholar]
- 47.Herrera W., Aleman T., Cideciyan A.V., Roman A.J., Banin E., Ben-Yosef T., Gardner L.M., Sumaroka A., Windsor E.A., Schwartz S.B., et al. Retinal disease in Usher syndrome III caused by mutations in the Clarin-1 gene. Invest. Ophthalmol. Vis. Sci. 2008 doi: 10.1167/iovs.07-1505. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 48.Weston M.D., Luijendijk M.W., Humphrey K.D., Möller C., Kimberling W.J. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am. J. Hum. Genet. 2004;74:357–366. doi: 10.1086/381685. [DOI] [PMC free article] [PubMed] [Google Scholar]