

Abstract
Mitochondrial alteration has been long proposed to play a major role in tumorigenesis. Recently, mitochondrial DNA (mtDNA) mutations have been found in a variety of cancer cells. In this study, we examined the contribution of mtDNA mutation and mitochondrial dysfunction in tumorigenesis first using human cell lines carrying a frame-shift at NADH dehydrogenase (respiratory complex I) subunit 5 gene (ND5); the same homoplasmic mutation was also identified in a human colorectal cancer cell line earlier. With increasing mutant ND5 mtDNA content, respiratory function including oxygen consumption and ATP generation through oxidative phosphorylation declined progressively, while lactate production and dependence on glucose increased. Interestingly, the reactive oxygen species (ROS) levels and apoptosis exhibited antagonistic pleiotropy associated with mitochondrial defects. Furthermore, the anchorage-dependence phenotype and tumor-forming capacity of cells carrying wild-type and mutant mtDNA were tested by growth assay in soft agar and subcutaneous implantation of the cells in nude mice. Surprisingly, the cell line carrying the heteroplasmic ND5 mtDNA mutation showed significantly enhanced tumor growth, while cells with homoplasmic form of the same mutation inhibited tumor formation. Similar results were obtained from the analysis of a series of mouse cell lines carrying a nonsense mutation at ND5 gene. Our results indicate that the mtDNA mutations might play an important role in the early stage of cancer development, possibly through alteration of ROS generation and apoptosis.
INTRODUCTION
In 1956, Warburg proposed that one of the important contributing factors to carcinogenesis was the alteration of oxidative phosphorylation owing to injured mitochondria (1). Since then, changes in the number, shape and function of mitochondria have been reported in various cancers (2). The switch of ATP production from mitochondrial oxidative phosphorylation to glycolysis has been suggested to be a marker of tumor development (3). Furthermore, mitochondrial dysfunction has been shown to initiate critical signaling pathways that regulate cell growth (4,5), and nuclear-encoded subunits of mitochondrial complex II have been reported as tumor-suppressor genes involved in hereditary paraganglioma (6,7). Recently, mtDNA mutations have been reported in various tumors (8–11). However, the functional significance of these mutations on tumorigenesis is still under debate (10,12).
Based on the resistance to a respiration inhibitor, rotenone, an efficient approach to isolate mtDNA mutations resulting in defective mitochondrial oxidative phosphorylation has been developed (13,14). In such selective conditions, cells are adapted to rely on the glycolysis for ATP production (15,16), which creates a bioenergetic condition similar to cancer development. Analyzing these rotenone-resistant clones has led to the isolation and characterization of cell lines carrying homoplasmic and heteroplasmic mtDNA mutations with defective oxidative phosphorylation activity (14,17). Among them, C8T and C9T cells carried a frame-shift mutation in the human ND5 gene at heteroplasmic (72% mutant) and near homoplasmic level (14). This mutation disrupted the synthesis of ND5 subunit. The NADH dehydrogenase activity and the assembly of the mtDNA-encoded subunits of complex I were also disrupted in these mutant cells (14). Interestingly, this mutation is identical to one found in colorectal cancer cell lines (18). Similarly, a series of mouse cell lines derived from a mutant isolated in this approach have also been obtained, carrying different content of a ND5 nonsense mutation (16).
In this study, we utilized both the human and mouse cells isolated in vitro to test the influence of heteroplasmic and homoplasmic ND5 mutations on tumorigenesis with in vitro colony forming in soft agar assay and in vivo tumor forming assay with nude mice. We also analyzed the underlying molecular mechanisms by studying the reactive oxygen species (ROS) generation and the apoptotic reaction associated with the altered mitochondrial function. Our data suggest that mtDNA mutations might play an important role in cancer development.
RESULTS
To explore the role of mtDNA mutations in tumorigenesis, we used C8T and C9T cybrids along with a human osteosarcoma cell line 143B as control. Originally, human VA2B cells were exposed to increasing concentrations of complex I inhibitor rotenone over a period of 2 months. Some derivatives of VA2B cells exhibited resistance to rotenone at a concentration as high as 1.2 µm (14). Analyzing these rotenone-resistant clones has led to the isolation of cell lines with defective oxidative phosphorylation activity. Mitochondria from those cells were then repopulated to mtDNA-less (ρ°) 143B.206 cells generating trans-mitochondrial cells (cybrids) carrying hetero- or homo-plasmic mtDNA in the constant 143B nuclear background (19). Among them, C8T and C9T cells carry an insertion in the 5′ end—proximal portion of the ND5 gene of mtDNA. A stretch of eight A′s, starting at position 12417 of human mtDNA, was found to be extended to nine A′s (14). This insertion causes a frame shift, and induces a premature termination product of approximately 6.9 kDa. By primer extension assay, it was determined that C8T carries approximately 72% of mutant mtDNA and C9T contains near-homoplasmic mutant mtDNA (14). Interestingly, this mutation is identical to the one found in colorectal cancer cells (18).
Tumorigenicity of C8T and C9T cells
To investigate the influence of this ND5 mutation on tumor formation, an anchorage-dependence growth test was performed. One thousand cells were seeded in 0.4% soft agar medium. To our surprise, heteroplasmic C8T cells formed more colonies in soft agar than the control 143B cells, which carried the wild-type mtDNA, while homoplasmic C9T cells produced the least colonies (Fig. 1A and B).
Figure 1.
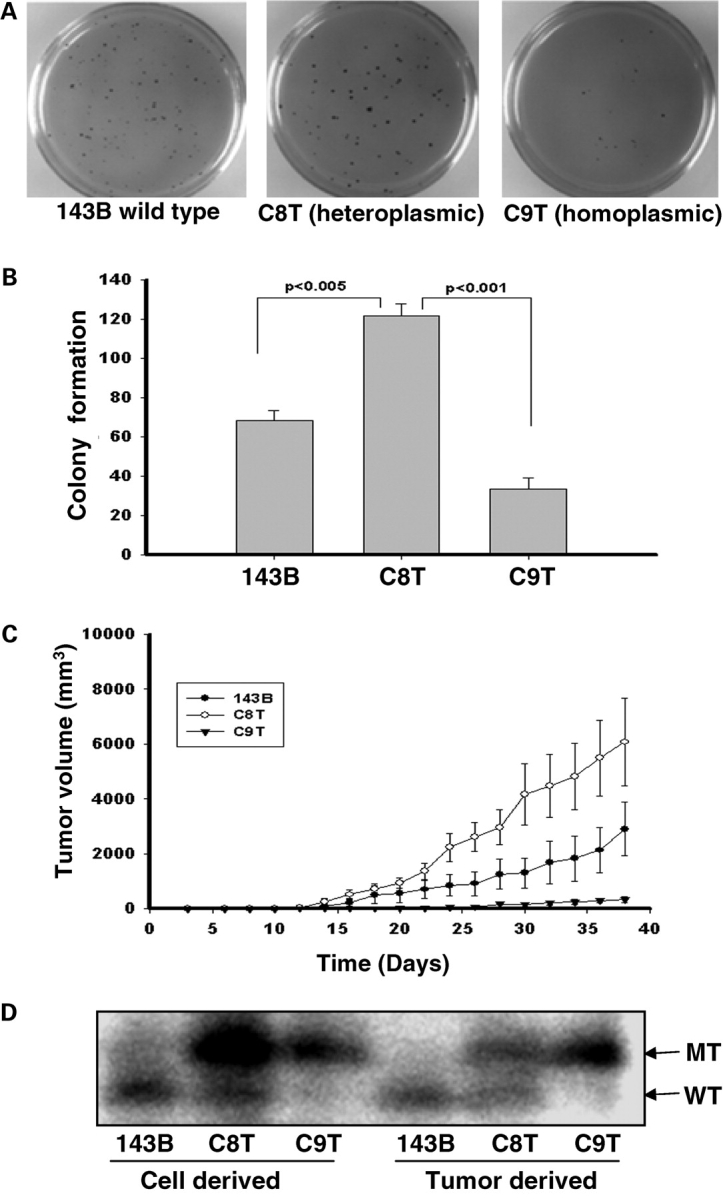
Colony formation in soft agar and tumor growth in the nude mice. (A) 1 × 103 cells were seeded on 60 mm dish contained 0.4% soid agar, and the colonies were stained with p-iodonitortetrazolium violet after 3 weeks. (B) The numbers of colonies were scored with Nucleotech's Gel Expert colony-counting software. (C) For each line 5 × 105 cells were injected into the shoulders of nude mice. Tumor size was measured every 2 days after injection, and was calculated using the formula, a × b2/2, where ‘a’ is the larger and ‘b’ is the smaller of the two dimensions. Three independent experiments with five mice in each group were used in each experiment. The error bars indicate two times of the standard errors of the mean. (D) The quantification of the insertions in the ND5 genes in 143B, C8T and C9T cells along with tumors derived from them was carried out using allele-specific termination of primer extension. The products of analysis were separated on a 20% polyacrylamide 6 m urea-sequencing gel. The upper bands represent the mutant mtDNA with insertion, and the lower bands indicate the wild-type.
To confirm that the cells carrying heteroplasmic ND5 mtDNA exhibited enhanced tumorigenicity and homoplasmic ND5 mtDNA had an inhibitory effect, we injected 5 × 105 of 143B, C8T and C9T cells subcutaneously into nu/nu nude mice. Three separate experiments were carried out, and in each experiment five mice were used in each group. Tumor volume was measured every other day. As shown in Figure 1C, tumors derived from heteroplasmic C8T cells grew faster and were larger in volume than those from wild-type mtDNA carrying 143B cells. No significant tumor growth was observed in mice injected with C9T cells, which contained near-homoplasmic mutant ND5 mtDNA.
These results indicated that heteroplasmic ND5 mutant mtDNA has an enhancing effect while its homoplasmic form exerts inhibition on cell growth in vitro.
To exclude the contribution from other mutations in mtDNA, we sequenced the whole mitochondrial genomes of 143B, C8T and C9T cells utilized in this study. No new mutations other than the one in ND5 gene were found. To further confirm the heteroplasmy of mtDNA in C8T and C9T cells, and to determine if there is a switch in the mtDNA genotype after the tumor growth, an assay based on allele-specific termination of primer extension (14) was carried out on DNA from the cells before injection and the tumors derived from the injected cells. As shown in Figure 1D, 143B cell contains 100% wild-type mtDNA and C9T carries almost 100% mutant ND5. Interestingly, there is a switch of the mtDNA genotype of the C8T cells during the tumor formation, from around 76–36% of mutant mtDNA.
Characterization of C8T and C9T cells
To analyze the effect of decreased wild-type ND5 gene content on mitochondrial functions, the endogenous respiration of 143B, C8T and C9T cells was investigated. We found that basal oxygen consumption in C8T and C9T cells was about 73% and 56% of the level of the control 143B, respectively (Fig. 2A). In the presence of the chemical uncoupler Carbonylcyanide-4-(trifluoromethoxy)-phenylhydrazone (FCCP), which completely uncoupled mitochondrial respiration and raised the respiratory capacity to a maximal level, oxygen consumption in C8T and C9T remained 53% and 17% that of control cells (Fig. 2A). No significant difference was detected with oxygen consumption measured in the presence of ATP synthase (complex V) inhibitor, oligomycin.
Figure 2.
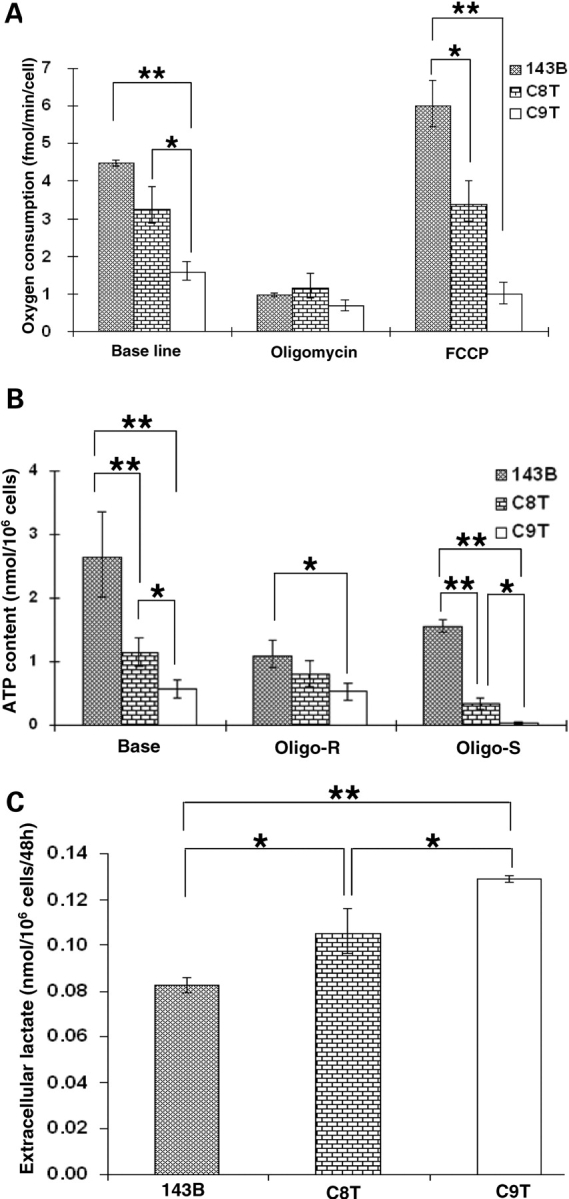
Characterization of 143B, C8T and C9T cells. (A) Oxygen consumption measurements; endogenous respiration was measured in intact cells and after treatment with ATP synthase inhibitor oligomycin and uncoupler FCCP. (B) The total cellular ATP contents were measured in 1 × 106 cells with a luciferase detection kit. The oligomycin-resistant (Oligo-R) ATP contents were determined after cells were incubated with oligomycin for 20 min at 37°C at a concentration of 15 µg/ml. The oligomycin sensitive (Oligo-S) ATP production was calculated by subtracting the oligomycin-resistant portion from the total ATP content. Four measurements were carried out, and the error bars indicate two times of the standard errors of the mean. (C) Extracellular lactate level was determined in the media from 1 × 106 cells and calculated after 48 h of culturing. The error bars indicate the standard errors of the mean (*P < 0.05; **P < 0.01).
ATP synthesis was then measured by luminometry in boiled cells. We found that the ATP contents decreased by 57% and 78% in C8T and C9T cells, respectively (Fig. 2B). We further investigated the mitochondrial oxidative phosphorylation contribution on ATP production with the addition of oligomycin. As shown in Figure 2B, calculated ATP synthesis sensitive to oligomycin (which assumed to be contributed by mitochondrial ATP synthase) further decreased by 79% and 98% in C8T and C9T cells compared with control 143B.
As lactate is the product of anaerobic glycolysis, mitochondrial dysfunction is usually associated with hyperlactatemia in normal aerobic conditions (20). We also measured extracellular lactate levels in 143B, C8T and C9T cells. As shown in Figure 2C, lactate measurements raised by 28% and 56% in C8T and C9T cells, indicating an increasing adaptation on glycolysis with increasing mutant ND5 mtDNA. This trend was then further verified by analysis of the dependence on glucose of cell growth.
Cell viability was investigated in medium containing galactose with increasing glucose. Since galactose is not used efficiently by mammalian cells as a glycolytic substrate, limiting glucose in the medium will have bigger impact on cells with impaired mitochondrial activity (17). As shown in Figure 3A, 143B cells grew well in the galactose medium when supplemented with 0.562 mg/ml [1/8 of that in normal Dulbecco's modified Eagle's medium (DMEM)]. However, in C8T and C9T lines, most cells could not survive when glucose content decreased to 0.562 and 1.125 mg/ml, respectively (Fig. 3B and C).
Figure 3.
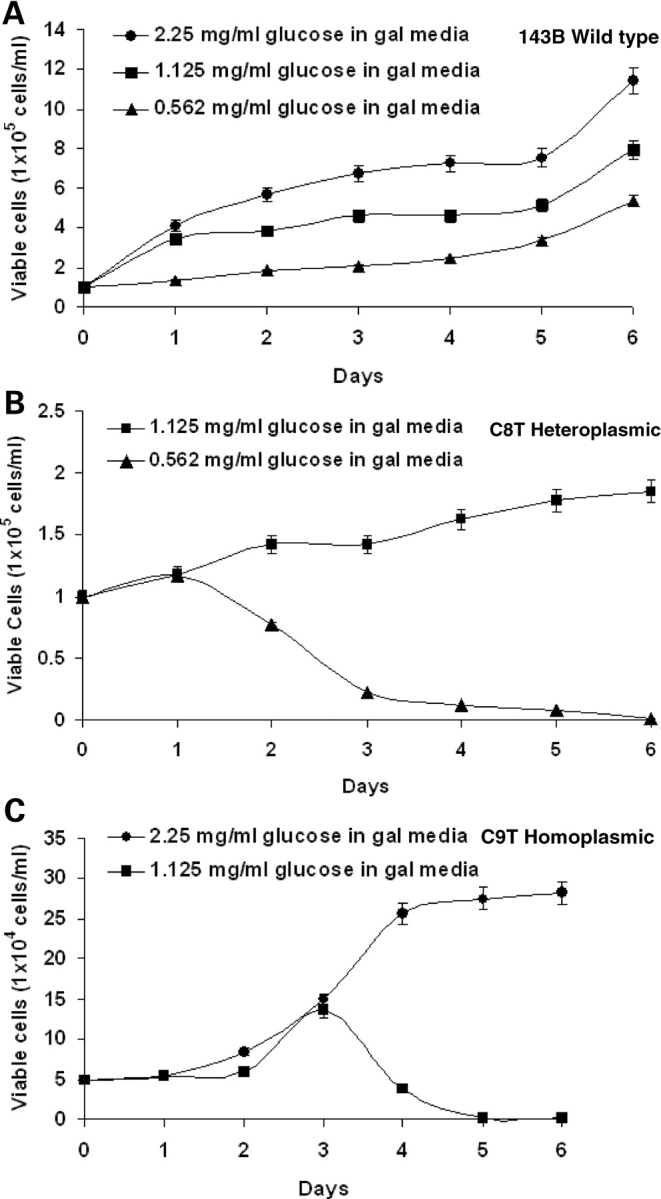
Cell growth analysis of wild and mutant cell lines in galactose media supplemented with different amounts of glucose for different days. (A) Growth curve for wild-type 143B cells in galactose media supplemented with 2.25 mg/ml, 1.125 mg/ml and 0.562 mg/ml glucose. (B) Growth curve of C8T cells in galactose media supplemented with 1.125 mg/ml and 0.562 mg/ml glucose. (C) Growth curve of C9T cells in galactose media supplemented with 2.25 mg/ml and 1.125 mg/ml glucose.
This progressive decline in mitochondrial functions with the increasing mutant ND5 content in the cybrids, together with the fact that there was no other mtDNA mutations detected in C8T and C9T cells, increased our confidence that the mutation in ND5 gene was the major if not the only difference among 143B, C8T and C9T cells.
Mitochondrial reactive oxygen species production in cells with hetero- and homo-plasmic ND5 mutation
To investigate the underlying molecular mechanism mediated by the signal from mitochondrial dysfunction associated with mtDNA mutation to tumorigenesis, we first analyzed the redox signaling by measuring the ROS production. Mitochondrial-specific generation of superoxide was detected by Mitosox, which was rapidly and selectively targeted to the mitochondria. Once in mitochondria, Mitosox is preferentially oxidized by superoxide. As shown in Figure 4A, significant increases of ROS in C8T and C9T were recorded. We then analyzed intracellular ROS levels with fluorescent probe 6-carboxy 2′7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA). Carboxy-H2DCFDA diffuses into cells, and is hydrolyzed to 2′7′-dichlorofluorescein (DCFH). DCFH is trapped within the cells, and could be converted to 2′7′-dichlorofluorescein (DCF) by ROS-mediated oxidation (21). Interestingly, as shown in Figure 4B, compared with 143B cells, while more ROS were still detected in C9T cells, the cellular peroxide level in C8T cells appeared normal. These results suggested an upregulation of antioxidant enzyme activities in C8T cells.
Figure 4.

Measurement of reactive oxygen species (ROS). The levels of ROS at mitochondrial and cytoplasmic compartments were assessed by (A) MitoSOX™ red mitochondrial superoxide indicator and (B) Carboxy-H2DCFDA probe, respectively. After treatment, cells were detached by trypsin–EDTA and immediately analyzed by flow cytometry. Ten thousand individual data points were collected for each sample (*P < 0.05). (C) Steady-state levels of antioxidant proteins and β-tubulin were analyzed by western blot with specific antibodies after SDS–PAGE. (D) Quantitative analysis of protein levels of catalase, Gpx4, SOD1 and SOD2 and in 143B, C8T and C9T cells. Values are mean ± SEM of two gels (*P<0.05, **P<0.01).
To confirm the notion that the difference we observed on the levels of mitochondrial superoxide and cytosolic hydrogen peroxide was because of the upregulation of anti-oxidative enzymes in C8T cells, we then carried out western blot analysis to examine the steady state levels of catalase, glutathione peroxidase 4 (Gpx4), Cu-Zn superoxide dismutase (SOD1) and Mn superoxide dismutase (SOD2), with β-tubulin as loading control.
In C8T cells, the levels of catalase and SOD1 were significantly higher than those in 143B (Fig. 4C and D). This result was consistent with the observation of higher mitochondrial superoxide but relatively low cytosolic hydrogen peroxide level in C8T cells (Fig. 4A and B). The upregulation of mitochondrial SOD2 in C9T cells might be able to explain the relatively lower level of mitochondrial ROS in C9T cells than expected (Fig. 4A).
Combined with the tumorigenesis phenotype observed, it appears that the upregulation of superoxide in mitochondria enhanced the tumor growth, while the high level of cytosolic peroxide could contribute the increased cell death associated with C9T cells in nude mice.
Cell death and survival properties of C8T and C9T cells
To investigate the possible role that apoptosis plays in the antagonistic pleiotropy observed, we analyzed apoptotic properties in 143B, C8T and C9T cells. The apoptotic potency of mitochondria from these cells was first examined using a newly developed assay that demonstrated the contribution of mitochondria in apoptosis (22).
This assay is based on a published procedure which demonstrated that isolated liver nuclei can be induced to undergo morphological changes comparable with apoptosis in Xenopus egg extract (23). Changes in nuclear morphology only occurred when mitochondria are included with the extract (Fig. 5A). Mitochondria from each of the cell lines were isolated and reconstituted with the purified Xenopus oocytes extract. Our results indicate that C9T mitochondria, which carry the homoplasmic ND5 mutation, are more apoptotic compared with the heteroplasmic C8T and the wild-type 143B mitochondria (Fig. 5B).
Figure 5.
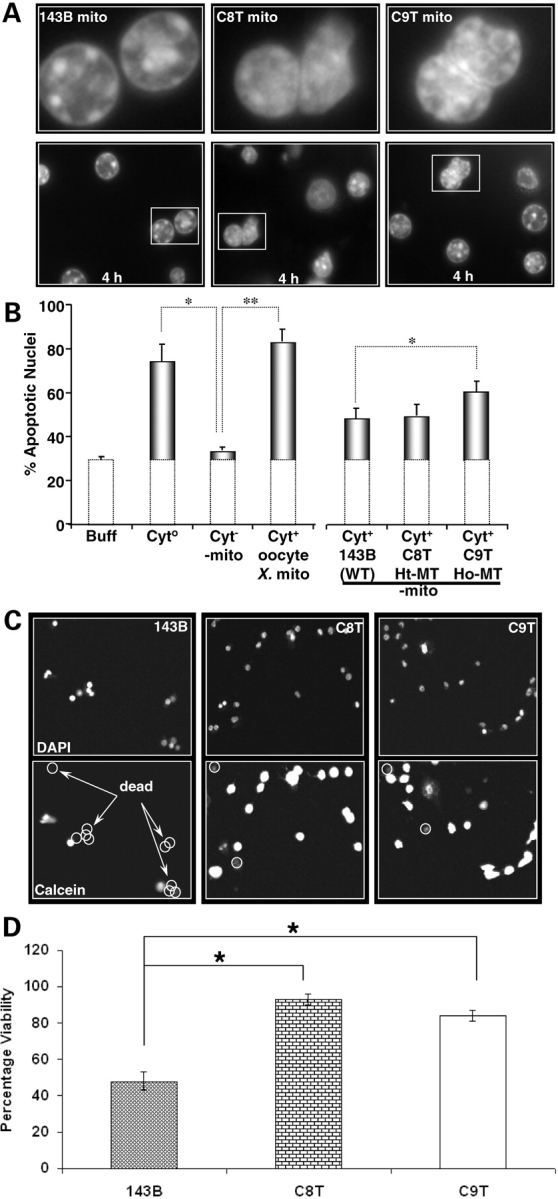
Apoptotic potency of mitochondria. (A) Images of Hoechst-stained nuclei exposed to mitochondria isolated from 143B cell (143B mito), C8T cell (C8T mito) and C9T cell (C9T mito) for 4 h. White framed regions in the lower panels are shown at higher magnification in the upper panels. (B) Percent of apoptotic nuclei at 4 h incubated with buffer alone (Buff), extract from Xenopus oocytes (Cyto), extract from Xenopus oocytes minus mitochondria (Cyt–mito), Cyt–mito plus mitochondria obtained from Xenopus oocytes, wild-type (WT) 143B, heteroplasmic mutant (Ht-MT) C8T and homoplasmic mutant (Ho-MT) C9T. Atleast three measurements were carried out. The error bars indicate the standard errors of the mean (*P < 0.05; **P < 0.01). Sensitivity of cells to oxidative stress. (C) Cells were incubated with tert-butyl hydrogen peroxide (t-BuOOH, 100 µm) for 4.5 h. The top images show DAPI-stained nuclei. The images shown below represent the same field of cells stained with calcein (Cal), which only stains the cytoplasm of cells that are alive. DAPI-labeled nuclei that are calcein-negative are circled and counted as cell deaths. Images were acquired on Olympus FV-500 Laser Scanning Confocal Microscopy. (D) Quantitative analysis of the percentage of cells alive that were exposed to oxidative stress (100 µm; t-BuOOH; 4.5 h). The error bars indicate the SEM (*P < 0.05).
We then assessed the sensitivity of cells to oxidative stress by treating the cells with 100 µm tert-butyl hydrogen peroxide (t-BuOOH) for 4.5 h. Cells were then stained with DAPI to label all cell nuclei and calcein to label viable cells. Cell viability was quantified by counting the number of nuclei that co-localize with calcein-stained cell, which should only be observed in live cells that have maintained their plasma membrane integrity. We found that compared with the control 143B cells, both C8T and C9T cells exhibited resistance to oxidative stress-induced cell death and the effect was more pronounced in the heteroplasmic C8T (Fig. 5C and D).
Combined with the tumorigenesis phenotype and ROS levels observed, our results suggested that upregulation of mitochondrial ROS might lead to cell survival under stress, which in turn contributed to the enhanced tumorigenicity in the heteroplasmic C8T cells. In homoplasmic C9T cells, the very limited ATP production and the increased apoptotic potency might contribute to the inhibited tumorigenicity.
Analysis of mouse cell lines with heteroplasmic and homoplasmic ND5 mutation
To further investigate the role of mtDNA heteroplasmy, we utilized a series of mouse cybrids carrying different levels of a nonsense mutation in ND5 gene (C12081A). These cybrids were generated by transferring mitochondria from a mouse A9 rotenone-resistant clone 3A to a LL/2 mtDNA-less line (16). While the C-to-A mutation at position 12081 changes the arginine codon CGA to a mitochondrial stop codon AGA, it also destroyed a ClaI restriction site. A total of eight cell lines with different ND5 mutation content were selected, and the ND5 heteroplasmy was verified by analysis of ClaI digestion pattern. As shown in Figure 6Aa, in 3A3 and 3A31, mutant mtDNA was hardly detectable above a very weak background signal observed also in the control A9, while, in 3A20-4, the mutant mtDNA was near homoplasmy. A wide range of heteroplasmy at ND5 gene was also verified in 3A33, 3A13, 3A6, 3A25 and 3A27 cells at around 88%, 62%, 52%, 33% and 16%, respectively (Fig. 6Ab). The whole genomes of these clones have been sequenced previously to exclude contribution from other mtDNA mutations.
Figure 6.
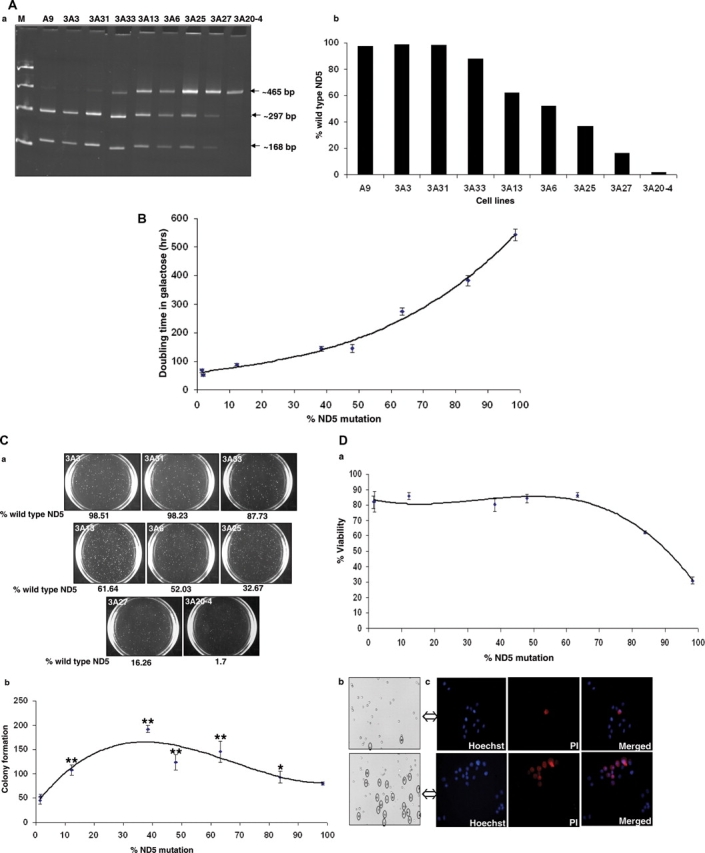
Analysis of mouse cells with varying heteroplasmy of ND5 nonsense mutation. (A) Quantification of the ND5 (A12081C) mutation, by ClaI digestion of a polymerase chain reaction-amplified ND5 fragment (465 bp). (B) Growth capacity in galactose medium represented as doubling time versus %ND5 mutation in various cells. The doubling time was determined at exponential growth phase by (T2 − T1) × log 2/log (C2/C1) where T = time and C = total viable cell count. Values shown are the mean ± SEM of three independent experiments. (C) Colony forming assay in soft agar. (a) 1 × 103 cells were seeded on 60 mm dish contained 0.4% solid agar, and the colonies were analyzed after 3 weeks. Representative pictures of one of the three experiments were shown. (b) The colonies (>0.1 mm) were scored with Nucleotech's Gel Expert colony-counting software. Data represents mean ± SEM of three independent experiments (four replicates for each line). P-values were calculated using one-way analysis of variance comparing control (3A3) versus others using Prism software (*P < 0.05, **P < 0.01). (D) Sensitivity to the oxidative stress. Cells were treated with 200µm tert-butyl hydrogen peroxide for 5 h and their viabilities were plotted against % ND5 mutation (a), and representative pictures of 3A6 and 3A20-4 were shown (b), as dead cells were stained and shown as circled. 3A6 and 3A20-4 cells were also stained by Hoechst and PI for the total and dead cells (c).
Effect of variable ND5 mutation content on mitochondrial function was analyzed previously, and a progressive decline of complex I-dependent respiration was recorded with the increase of ND5 mutant level (16). To confirm, we assessed the mitochondrial function by measuring the doubling time of the cells in galactose media where cells preferentially rely on mitochondrial oxidative phosphorylation for ATP production. As expected, the increase in the ND5 mutation content was accompanied with longer doubling time in galactose medium (Fig. 6B).
We then carried out an anchorage-independent growth test to investigate the effect of ND5 heteroplasmy on tumorigenicity. Equal number of cells (1000 cells/plate) was seeded in soft agar and cells were grown for 3 weeks to form colonies. As shown in Figure 6C, maximum number of colonies was observed in cell lines carrying heteroplasmic ND5 mutation around 40%, and relatively less number of colonies was formed with cells carrying nearly wild-type (3A3 and 3A31) or homoplasmic mutant (3A20-4) lines. This result is consistent with what we observed with human cells, and further supports the notion that the cells carrying certain level of heteroplasmic ND5 mtDNA exhibited enhanced tumorigenicity.
To further investigate the underlying mechanism responsible for the different tumorigenicity between cells carrying hetero- and homoplasmic ND5 mutation, the cell viability under oxidative stress was tested. Cells were treated with t-BuOOH, which is a known ROS-mediated inducer for apoptosis (24). We then analyzed the cell viability by trypan blue exclusion method, and found that the cells became more sensitive to cell death upon induction when ND5 mutation level reached beyond 60% (Fig. 6Da). Representative pictures of cells carrying hetero- (3A6) and homoplasmic (3A20-4) ND5 mutation after t-BuOOH treatment are shown in Figure 6Db. In a complementary experiment, 3A6 and 3A20-4 cells were stained with both Hoechst, a fluorescent dye (Ex 350 nm and Em 460 nm) for total nuclei, and Propidium Iodide (PI, Ex 535 nm and Em 617 nm), which was only taken by apoptotic cell. Again, high percentage of cell death was observed with the homoplasmic 3A20-4 cells (Fig. 6Dc).
DISCUSSION
Mitochondrial dysfunctions have long been reported and were also hypothesized to contribute in cancer development. The switch from mitochondrial oxidative phosphorylation to glycolysis has been suggested to be the bioenergetic signature of cancer cells (3). Yet, the role of mitochondria and particularly the mtDNA mutations, which have been found to be associated with various cancers, in tumorigenesis remains largely unclear. This is mainly because of the fact that most of the mtDNA mutations identified have been found in tumors that are not adapted to growth in the laboratory. Consequently, they are not accessible for sophisticated molecular and biochemical analyses. To overcome this limitation, we took advantage of an established approach to isolate mtDNA mutations in cultured cells (3,14,17) where the conditions mimic the bioenergetic switch from mitochondrial oxidative phosphorylation to glycolysis and thus the derivatives become more like tumor cells. Cell lines carrying mtDNA mutations were obtained and, in particular, trans-mitochondrial cybrids carrying a mtDNA mutation at ND5 gene, which was identical to a natural homoplasmic mutant found in colorectal cancer were generated and were chosen to study the relationship among the accumulation of mtDNA mutation, mitochondrial dysfunction associated with mtDNA mutation and tumorigenesis.
Heteroplasmy is one of the unique features of mitochondrial genetics (25). With increasing mutant ND5 mtDNA, both endogenous respiration and ATP synthesis declined progressively. At the same time, lactate level and dependence on glucose increased, indicating a switch from mitochondrial oxidative phosphorylation to glycolysis from ATP production.
Surprisingly, we found that when the ND5 mutation was heteroplasmic, tumor growth was enhanced. On the other hand, tumor formation was relatively less or inhibited when mutation reached nearly 100%. Hence, there seems to exist a situation of antagonistic pleiotropy that results from mitochondrial dysfunction.
Mitochondria-mediated transformation, by which both C8T and C9T cells were isolated, paved a way to focus on the contribution of mtDNA (19). However, it is possible that changes in the nuclear genome have been accumulated during selection of different cybrids, which could also contribute the phenotypes we observed. To address this concern, advantage was taken of the availability of another series of cell lines carrying multiple heteroplasmy at a mouse ND5 nonsense mutation. The complementary observations from another system with more cybrids containing varying degrees of heteroplasmy strengthened the conclusions significantly.
As for the underling mechanism from mtDNA mutation to tumorigenicity, we focused on three core functions of mitochondria: ATP production through oxidative phosphorylation, generation of ROS which serve as important signaling molecules, and apoptosis regulation (26). On the one hand, disturbed mitochondrial function by some mtDNA mutations has been shown to enhance ROS production (27,28). Furthermore, it has been shown that ROS are involved in many proliferating signaling pathways associated with tumor promotion (29,30). On the other hand, when mitochondrial function is severely damaged, as in the case of some homoplasmic lethal mtDNA mutations, cell growth is inhibited by reduced ATP and/or activation of apoptosis. Specifically, increased ROS level could drive cell proliferation while limited ATP inhibits cell growth. Therefore, for mtDNA mutations to play a role in tumorigenesis, the optimal solution is likely to be heteroplasmy, as we observed with the frame-shift mutation in ND5 gene in our studies.
This interpretation is consistent with a previous report on renal adenocarcinoma where a 50% heteroplasmic deletion in mitochondrial ND1 gene was found in a cancer patient. Interestingly, the mutant mtDNA disappeared in metastatic tumors (31). Two heteroplasmic transitions (T2275C and A8601G) were identified in early-stage breast cancer (32). Somatic mtDNA alterations were observed in preneoplastic lesions of the gastrointestinal tract even in the absence of histopathological evidence of dysplasia (33). It is also interesting to note that previously two pathogenic homoplasmic mtDNA mutations (T8993G and T9176C associated with human mitochondrial encephalomyopathy in the subunit 6 gene of ATP synthase) have been tested for tumorigenesis (34,35). All the tumors derived from the mutant cybrids grew faster and were larger in volume than those from wild-type cybrids in the nude mice assays. Functionally more close to heteroplasmic ND5 mutation investigated in this study, both mutations are relatively mild missense mutation. Furthermore, T8993G was associated with increased ROS production (34), and both cybrids were shown resistance to apoptosis (35).
Oxidation–reduction (redox) reactions that generate ROS have been identified as important mediators in the regulation of signal transduction processes involved in cell growth, differentiation and cell death (36,37). On the other hand, it is generally believed that ROS are a relevant class of carcinogens (38,39), and it has been shown that ROS can stimulate cancer development at all three stages: initiation [the induction of DNA mutations in a somatic cell (40)]; promotion [the stimulation of tumorigenic expansion of the cell clone (41)]; and progression [the malignant conversion of the tumor to cancer (42)]. Interestingly, in some systems, ROS mediate both pro- and anti-apoptosis effects, depending on the ROS concentration (43).
The notion that tumorigenicity exhibited by cells carrying mtDNA mutations was mediated by alteration in ROS and apoptosis was also supported by our findings that mitochondrial ROS was elevated in both C8T and C9T cells and resistance to oxidative stress in C8T cell was significantly increased. The discrepancy between the measurements of mitochondrial superoxide by Mitosox and cytosolic hydrogen peroxide by Carboxy-H2DCFDA might partly be explained by the differential levels of the antioxidant enzymes. It is interesting to note the difference between the apoptotic potency determined by apoptosis initiated by the isolated mitochondria and resistance to oxidative stress measured by cell survival after treatment of t-BuOOH. Although the ND5 homoplasmic C9T showed some resistance to cell death under oxidative stress induced by t-BuOOH, the very limited production of ATP and increased apoptotic potency probably contributed to the inhibited tumorigenicity revealed by both nude mice assay and anchorage-dependency test.
The overexpression of catalase and SOD1 is intriguing. Previously serum levels of SOD1 were reported to be significantly elevated in gastric cancer patients (44), and erythrocyte SOD and catalase activities were shown to be higher in early and advanced stages of non-small-cell lung cancer and small-cell lung cancer (45). Interestingly, apoptosis in cancer cell lines was reduced by catalase through inhibition of caspase 8 activity (46).
Another common concern over the studies on the biological significance of particular mtDNA mutation in cybrids is whether there are contributions from other sources. We have sequenced the whole genome of mtDNA of all cell lines and no additional mutations were found. Other concerns were the shift of mtDNA heteroplasmy and the accumulation of other changes during the passage of the cybrids. These concerns were carefully addressed by using the same earlier passage lines in different experiments and by always carrying out the sequencing analysis before and after the experiments to make sure of the mtDNA genotypes under investigation.
Based on this study and previous investigation from other laboratories, we propose that the mtDNA mutations in cancer development could function as follows: in the initial stage, cancer cells are very mutagenic either because of a carcinogenic insult or the compromised repair mechanism (47,48) and mtDNA is more likely to be mutated at this stage. Because of replicative advantage of the mutant mtDNA molecules, such as previously described for mtDNA carrying the mutation associated with the mitochondrial encephalomyopathy (49), mtDNA mutations enriched to a certain level of heteroplasmy, which would enhance tumor progression owing to either the elevated ROS generation which in turn activates the oncogenic pathways or to the increase in genome instability or to both. After transformation, it may become more important to have a functional respiratory chain than an inhibited one to sustain rapid cell proliferation. In some cases, as for the patient with renal adenocarcinoma reported by Wallace's group, the mutant mtDNAs causing severe mitochondrial defect are selected against and diluted out (31). In other cases, residual mutant mtDNA might escape the selection. In late stages of cancer, the cells are progressively adapted to a glycolytic metabolism because of the hypoxic environment. This may lead to the selection of cells in which the mutations make them mitochondrial function-independent and therefore cells with homoplasmic mtDNA mutation may become predominant in tumors. This could be the reason why homoplasmic mtDNA mutations have been found in late-stage tumors as the same ND5 mutation identified in colorectal cancer lines (18). However, when a homoplasmic status of some mtDNA mutations is reached, the host cells could be converted into non-tumorigenic cells because of the loss of ATP production.
In conclusion, our data suggested that mtDNA mutations could play an important role in certain stage of tumorigenesis. It is interesting to note that it is possible that mtDNA mutations might be much more prevalent than currently believed on the basis of mutations detected in late-stage cancers.
MATERIALS AND METHODS
Cell culture
The 143B (ATCC CRL 8303) is a human osteosarcoma-derived cell line. The C8T and C9T cell lines used in this study were generated as described previously (14). Mouse cell lines carrying different level of a ND5 nonsense mutation were obtained and characterized earlier (16). All the cell lines used in the present work were grown as monolayer in DMEM (Cellgro-Meditech, Inc., Herdon, VA, USA) supplemented with 10% fetal bovine serum (FBS). Early passage lines and frequent checking of the mtDNA genotypes were utilized to avoid the possible accumulation of changes in cell lines involved.
Soft agar assay
One thousand cells were plated in 60 mm diameter dishes prepared with a lower layer of 0.4% agar in DMEM with 10% FBS and overlaid with a 0.27% agar in the same medium. Colonies were analyzed 3 weeks later by staining with p-iodonitortetrazolium violet (INT; Sigma, St Louis, USA). Colonies were scored by Nucleotech's Gel Expert colony-counting software (Westport, CN). Colonies larger than 0.1 mm in diameter were scored as positive.
In vivo tumorigenesis assays
Around 5 × 105 cells of each line were injected subcutaneously into nu/nu male nude mice (6 weeks of age, Charles River Laboratories, Wilmington, MA, USA). Cell viability was determined by trypan blue exclusion assay immediately before injections. Tumor volume was measured every other day.
Mitochondrial DNA analysis
Total DNA was extracted from cells with extraction kit (Promega, Madison, WI, USA). Whole mitochondrial genomes were amplified with three pairs of primers which generate three overlapping fragments covering the whole genome. Each fragment was purified and then sequenced by the chain termination method. The sequence information of the primers will be provided upon request.
Quantification of the mtDNA frame-shift mutations was carried out by allele-specific termination of primer extension method as previously described (14). Briefly, a 359-bp segment of mtDNA between positions 12282 and 12641, containing the ND5 mutation (an A insertion in a row of eight A residues at positions 12417–12424), was amplified. The products of polymerase chain reaction (PCR) were purified with Gel elution kit (QIAGEN Inc., Valencia, CA, USA). Solutions in Sequenase buffer that contained the amplified DNA fragment and a corresponding 32P-labeled primer (positions 12398–12427) in a 1:1 molar ratio were prepared. Nucleotide concentrations were 60 mm dATP and 250 mm ddCTP. The mixtures were heated to 95°C for 3 min and chilled on ice. After addition of 1 ml of diluted Sequenase (USB Corp., Cleveland, OH, USA), they were incubated for 5 min at 45°C. The products were denatured and separated on a 20% polyacrylamide 6 m urea gel. Quantification of the intensity of the bands was done using a PhosphorImager and the IMAGE-QUANT program (Molecular Dynamics Inc., Sunnyvale, CA).
For mouse lines, quantification of the mtDNA mutation (a C-to-A point mutation, which destroys a ClaI site) was carried out by analysis of the products of a restriction digestion reaction. For this purpose, a 465-bp fragment of the ND5 gene was amplified by PCR with the primers ND5-5′-2 and ND5-3′-4 to a 50 µl volume. PCR products were purified and quantified from A260nm. An amount of 2 µg of purified product was subjected to complete digestion with ClaI at 37°C overnight. Under these conditions, the wild-type ND5 fragment was cut into two small fragments of 297 bp and 168 bp, while the mutant ND5 fragment remained intact. An aliquot from the above described reaction mixture was subsequently electrophoresed on a 6% polyacrylamide gel. Quantification of the intensity of the bands was done by PhosphorImager analysis. The sequence of primers used in this study were as follows: ND5-5′-2 (positions 11785–11802), CCCCAATCCTAATTTCAA; ND5-3′-4 (positions 12248–12231), TGCTTGTAGGGCTGCAGT.
Oxygen consumption measurement
The measurements were carried out with a YSI Model 5300 Biological Oxygen Monitor as described earlier (50). Briefly, determination of the O2 consumption rate was carried out in 5 × 106 cells in respiration buffer (20 mm HEPES, pH 7.1, 250 mm sucrose, 2 mm Kpi, 10 mm MgCl2 and 1.0 mm ADP). After recording the base respiration, 2.5 µg/ml oligomycin was added to measure the uncoupled respiration, and then 0.5 µm FCCP was added to measure the maximal respiration.
Adenosine triphosphate determination
ATP measurements were carried out using ATP Determination Kit (Invitrogen, Carlsbad, CA, USA). For luciferase assays, cells were grown in 60 mm dish to approximately 80% confluence, incubated in DMEM containing the following combinations of substrates and inhibitors: glucose (4.5 mg/ml) plus 110 mg/l pyruvate; glucose plus 15 µg/ml of the ATP synthase inhibitor oligomycin. To measure ATP synthesis in the presence of oligomycin, cells were incubated with 15 µg/ml of oligomycin for 20 min before harvesting. Cells were then collected by centrifugation, and resuspended in 100 µl buffer containing 25 mm Tris, pH 7.4 and 150 mm ethylenediamine tetraacetic acid. The above solution was boiled at 100°C for 5 min and centrifuged. The luciferin–luciferase mixture (Invitrogen) was added to the cell suspension (final concentration, 2 mm), and light emission was measured in a Synergy™ HT Multi-Detection Microplate Reader (Bio-Tek Instruments Inc., Winooski, VT, USA). A standard ATP/luminescence curve was constructed by measuring luminescence derived from ATP solutions containing 0, 0.001, 0.01, 0.1, 1 and 10 µm ATP in double-distilled water and luciferin–luciferase mixture.
Lactate measurement
Extracellular lactate level was determined with Lactate Assay Kit (University at Buffalo, NY, USA). Briefly, 2 days before the experiment, cells were seeded in 10 cm dishes. At the time of measurement, cell density was about 60–70% confluent. Media from the dishes were collected and diluted to 50-fold with double-distilled water. Simultaneously, cells were counted by trypan blue exclusion method. Standard graph was generated using different concentrations of lactate in 20 µl of volume and 50 µl of lactate assay solution in 96-well plates followed by incubation in a humidified chamber at 37°C for 30 min. Similarly, different dilutions of media were used to measure extracellular lactate in the linear range. Reaction was stopped by adding 50 µl of 3% acetic acid and absorbance was measured at 490 nm using Synergy HT Microplate Reader.
Growth dependence on glucose
Multiple identical samples of 1–5 × 105 cells were plated on 6-well plates in 2 ml of medium and cells were cultured at 37°C for 24 h. The medium was changed the next day with galactose media (containing 0.9 mg of galactose/ml and 0.5 mg of pyruvate/ml, with 10% dialyzed FBS) supplemented with different concentrations of glucose (4.5, 2.25, 1.125, 0.562 mg/ml). For mouse lines, galactose media were used. Fresh media were supplemented on every second day. Trypan blue exclusion method was then used to count cell numbers. Cells were counted on a daily basis for 6 days. Live cell images were taken using inverted microscope (Nikon Eclipse TE200; Nikon Instruments Inc., Melville, NY).
Reactive oxygen species measurement
The production of ROS at mitochondrial and cytoplasmic level was assessed by MitoSOX™ red mitochondrial superoxide indicator and Carboxy-H2DCFDA probe (all from Invitrogen), respectively. In the 6-well plates, for each analysis 1 × 106 cells were used. Briefly, after the addition of MitoSOX (2.5 µm) or Carboxy-H2DCFDA (20 µm), cells were incubated for 30 min at 37°C in the dark. Cells were washed and harvested in Hank's buffered salt solution (HBSS) and analyzed immediately using a BD FACScan flow cytometer (Becton Dickinson, San Jose, CA) with excitation at 488 nm. Forward and side-scatter were used to gate the viable population of cells. Carboxy-H2DCFDA emit at 530 nm (FL-1) channel, whereas MitoSOX™ Red emit at 590 nm (FL-2 channel). A minimum of 10 000 events was collected. Data were analyzed as single parameter frequency histogram using cell Quest Alias software (Becton Dickinson, San Jose, CA). Results are presented as mean fluorescence intensity.
Western blot analysis
Total cell lysates were separated through an SDS-12% polyacrylamide gel. After electrophoresis, proteins were blotted onto polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membrane was blocked with 5% fish gelatin solution in phosphate buffered saline (PBS) and then incubated with primary antibodies against SOD1, SOD2 (Stressgen, CA, USA), Catalase and GPX4 (gift from Dr Qitao Ran) at 4°C for 2 h. Membranes washed with 0.2% Tween-20 in phosphate-buffered saline (PBST) was incubated with goat anti-mouse and anti-rabbit secondary antibodies labeled with IRDye™ infrared dye 38 (800 nm, Rockland Immunochemicals; Gilbertsville, PA, USA) or with Alexa Fluor® 680 (700 nm, Molecular Probes, OR, USA). Infrared fluorescence signal was detected and analyzed with Odyssay Li-cor bioscience machine according to the manufacturer's guidelines (Li-cor Biosciences, Lincoln, NE).
Apoptosis assay
The cell-free Xenopus apoptotic assay was carried out as recently reported (22): the mitochondrial fraction was isolated as previously described (50) and mixed with the purified Xenopus oocytes extract at the same protein concentration measured for the endogenous Xenopus oocytes. These mixtures were then incubated with liver nuclear fraction for 4 h. Nuclei were stained with 100 µg/µl Hoechst, and scored for the percentage of apoptotic nuclei out of the total nuclei.
Sensitivity of cells to oxidative stress was assessed by treating cultures with 100 µm tert-butyl hydrogen peroxide (t-BuOOH; Sigma) for 4.5 h. Cells were then stained with DAPI and calcein AM (2 µm; Invitrogen). Cell death was quantified by counting the number of nuclei that did not colocalize with calcein-stained cells. Images were acquired on Olympus FV-500 Laser Scanning Confocal Microscopy (Optical Imaging Facility Core at UTHSCSA).
For mouse lines, cells were treated with 200 µm t-BuOOH for 5 h, and cell viability was then analyzed with Vi-Cell cell viability analyzer (Beckman Coulter). In a parallel experiment, above treated cells were rinsed with HBSS and incubated with Hoechst and PI at 10µg/ml and 25 µg/ml, respectively at room temperature for 30 min, and then fixed in 4% paraformaldehyde and images were acquired using the fluorescence microscope (Nikon eclipse Ti-S; Nikon Instruments Inc., Melville, NY) at 400× magnification.
Statistical analysis
Probability (P) values were calculated using the analysis of variance test contained in the Minitab (Minitab Inc., State College, PA)/Prism (Graph Pad Software, Inc., San Diego, CA) programs.
FUNDING
NIH (R01 AG025223 to Y.B. and P30 CA54174 to S.N.) and Wendy Will Case Cancer Fund (to Y.B.).
ACKNOWLEDGEMENTS
We thank Dr Giuseppe Attardi and Dr Anne Chomyn for providing 143B, C8T and C9T cells, Qitao Ran for antibodies against Catalase and Gpx4, and Yuji Ikeno for help with mouse pathology. We also thank Dr Meenakshi Tiwari, Peiqing Hu and Youfen Li for their assistance.
Conflict of Interest statement: None declared.
REFERENCES
- 1.Warburg O. On the origin of cancer cell. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 2.Pedersen P.L. Tumor mitochondria and the bioenergetics of cancer cells. Proc. Exp. Tumor Res. 1978;22:190–274. doi: 10.1159/000401202. [DOI] [PubMed] [Google Scholar]
- 3.Cuezva J.M., Krajewska M., de Heredia M.L., Krajewski S., Santamaria G., Kim H., Zapata J.M., Marusawa H., Chamorro M., Reed J.C. The bioenergetic signature of cancer: a marker of tumor progression. Cancer Res. 2002;62:6674–6681. [PubMed] [Google Scholar]
- 4.Rustin P. Mitochondria, from cell death to proliferation. Nat. Genet. 2002;30:352–353. doi: 10.1038/ng0402-352. [DOI] [PubMed] [Google Scholar]
- 5.Arnould T., Vankoningsloo S., Renard P., Houbion A., Ninane N., Demazy C., Remacle J., Raes M. CREB activation induced by mitochondrial dysfunction is a new signaling pathway that impairs cell proliferation. EMBO J. 2002;21:53–63. doi: 10.1093/emboj/21.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baysal B.E., Willett-Brozick J.E., Lawrence E.C., Drovdlic C.M., Savul S.A., McLeod D.R., Yee H.A., Brackmann D.E., Slattery W.H., III, Myers E.N., et al. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J. Med. Genet. 2002;39:178–183. doi: 10.1136/jmg.39.3.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baysal B.E., Ferrell R.E., Willett-Brozick J.E., Lawrence E.C., Myssiorek D., Bosch A., van der Mey A., Taschner P.E., Rubinstein W.S., Myers E.N., et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 8.Hochhauser D. Relevance of mitochondrial DNA in cancer. Lancet. 2000;356:181–182. doi: 10.1016/S0140-6736(00)02475-2. [DOI] [PubMed] [Google Scholar]
- 9.Penta J.S., Johnson F.M., Wachsman J.T., Copeland W.C. Mitochondrial DNA in human malignancy. Mutat. Res. 2001;488:119–133. doi: 10.1016/s1383-5742(01)00053-9. [DOI] [PubMed] [Google Scholar]
- 10.Brandon M., Baldi P., Wallace D.C. Mitochondrial mutations in cancer. Oncogene. 2006;25:4647–4662. doi: 10.1038/sj.onc.1209607. [DOI] [PubMed] [Google Scholar]
- 11.Chatterjee A., Mambo E., Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006;25:4663–4674. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- 12.Khaidakov M., Reis R.J. Possibility of selection against mtDNA mutations in tumors. Mol. Cancer. 2005;4:36. doi: 10.1186/1476-4598-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao H., Li R., Wang Q., Yan Q., Deng J.H., Han D., Bai Y., Young W.Y., Guan M.X. Maternally inherited aminoglycoside-induced and nonsyndromic deafness is associated with the novel C1494T mutation in the mitochondrial 12S rRNA gene in a large Chinese family. Am. J. Hum. Genet. 2004;74:139–152. doi: 10.1086/381133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hofhaus G., Attardi G. Efficient selection and characterization of mutants of a human cell line which are defective in mitochondrial DNA-encoded subunits of respiratory NADH dehydrogenase. Mol. Cell. Biol. 1995;15:964–974. doi: 10.1128/mcb.15.2.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai Y., Attardi G. The mtDNA-encoded ND6 subunit of mitochondrial NADH dehydrogenase is essential for the assembly of the membrane arm and the respiratory function of the enzyme. EMBO J. 1998;17:4848–4858. doi: 10.1093/emboj/17.16.4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bai Y., Shakeley R.M., Attardi G. Tight control of respiration by NADH dehydrogenase ND5 subunit gene expression in mouse mitochondria. Mol. Cell. Biol. 2000;20:805–815. doi: 10.1128/mcb.20.3.805-815.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bai Y., Hu P., Park J.S., Deng J.H., Song X., Chomyn A., Yagi T., Attardi G. Genetic and functional analysis of mitochondrial DNA-encoded complex I genes. Ann. NY Acad. Sci. 2004;1011:272–283. doi: 10.1007/978-3-662-41088-2_26. [DOI] [PubMed] [Google Scholar]
- 18.Polyak K., Li Y., Zhu H., Lengauer C., Willson J.K., Markowitz S.D., Trush M.A., Kinzler K.W., Vogelstein B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat. Genet. 1998;20:291–293. doi: 10.1038/3108. [DOI] [PubMed] [Google Scholar]
- 19.King M.P., Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 20.Stacpoole P.W. Lactic acidosis and other mitochondrial disorders. Metabolism. 1997;46:306–321. doi: 10.1016/s0026-0495(97)90259-6. [DOI] [PubMed] [Google Scholar]
- 21.Arrigo A.P., Firdaus W.J., Mellier G., Moulin M., Paul C., Diaz-latoud C., Kretz-remy C. Cytotoxic effects induced by oxidative stress in cultured mammalian cells and protection provided by Hsp27 expression. Methods. 2005;35:126–138. doi: 10.1016/j.ymeth.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 22.Saelim N., Holstein D., Chocron E.S., Camacho P., Lechleiter J.D. Inhibition of apoptotic potency by ligand stimulated thyroid hormone receptors located in mitochondria. Apoptosis. 2007;12:1781–1794. doi: 10.1007/s10495-007-0109-1. [DOI] [PubMed] [Google Scholar]
- 23.Newmeyer D.D., Farschon D.M., Reed J.C. Cell-free apoptosis in Xenopus egg extracts: inhibition by Bcl-2 and requirement for an organelle fraction enriched in mitochondria. Cell. 1994;79:353–364. doi: 10.1016/0092-8674(94)90203-8. [DOI] [PubMed] [Google Scholar]
- 24.Pias E.K., Aw T.Y. Early redox imbalance mediates hydroperoxide-induced apoptosis in mitotic competent undifferentiated PC-12 cells. Cell Death Differ. 2002;9:1007–1016. doi: 10.1038/sj.cdd.4401064. [DOI] [PubMed] [Google Scholar]
- 25.Schon E.A. Mitochondrial genetics and disease. Trends Biochem. Sci. 2000;25:555–560. doi: 10.1016/s0968-0004(00)01688-1. [DOI] [PubMed] [Google Scholar]
- 26.Wallace D.C., Ruiz-Pesini E., Mishmar D. mtDNA variation, climatic adaptation, degenerative diseases, and longevity. Cold Spring Harb. Symp. Quant. Biol. 2003;68:479–486. doi: 10.1101/sqb.2003.68.471. [DOI] [PubMed] [Google Scholar]
- 27.Baracca A., Sgarbi G., Mattiazzi M., Casalena G., Pagnotta E., Valentino M.L., Moggio M., Lenaz G., Carelli V., Solaini G. Biochemical phenotypes associated with the mitochondrial ATP6 gene mutations at nt8993. Biochim. Biophys. Acta. 2007;1767:913–919. doi: 10.1016/j.bbabio.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 28.Vives-Bauza C., Gonzalo R., Manfredi G., Garcia-Arumi E., Andreu A.L. Enhanced ROS production and antioxidant defenses in cybrids harbouring mutations in mtDNA. Neurosci. Lett. 2006;391:136–141. doi: 10.1016/j.neulet.2005.08.049. [DOI] [PubMed] [Google Scholar]
- 29.Burdon R.H. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic. Biol. Med. 1995;18:775–794. doi: 10.1016/0891-5849(94)00198-s. [DOI] [PubMed] [Google Scholar]
- 30.Lander H.M. An essential role for free radicals and derived species in signal transduction. FASEB J. 1997;11:118–124. [PubMed] [Google Scholar]
- 31.Horton T.M., Petros J.A., Heddi A., Shoffner J., Kaufman A.E., Graham S.D., Jr, Gramlich T., Wallace D.C. Novel mitochondrial DNA deletion found in a renal cell carcinoma. Genes Chrom. Cancer. 1996;15:95–101. doi: 10.1002/(SICI)1098-2264(199602)15:2<95::AID-GCC3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 32.Wang C.Y., Wang H.W., Yao Y.G., Kong Q.P., Zhang Y.P. Somatic mutations of mitochondrial genome in early stage breast cancer. Int. J. Cancer. 2007;121:1253–1256. doi: 10.1002/ijc.22822. [DOI] [PubMed] [Google Scholar]
- 33.Sui G., Zhou S., Wang J., Canto M., Lee E.E., Eshleman J.R., Montgomery E.A., Sidransky D., Califano J.A., Maitra A. Mitochondrial DNA mutations in preneoplastic lesions of the gastrointestinal tract: a biomarker for the early detection of cancer. Mol. Cancer. 2006;5:73. doi: 10.1186/1476-4598-5-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petros J.A., Baumann A.K., Ruiz-Pesini E., Amin M.B., Sun C.Q., Hall J., Lim S., Issa M.M., Flanders W.D., Hosseini S.H., et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl Acad. Sci. USA. 2005;102:719–724. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shidara Y., Yamagata K., Kanamori T., Nakano K., Kwong J.Q., Manfredi G., Oda H., Ohta S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005;65:1655–1663. doi: 10.1158/0008-5472.CAN-04-2012. [DOI] [PubMed] [Google Scholar]
- 36.Finkel T. Redox-dependent signal transduction. FEBS Lett. 2000;476:52–54. doi: 10.1016/s0014-5793(00)01669-0. [DOI] [PubMed] [Google Scholar]
- 37.Finkel T. Oxidant signals and oxidative stress. Curr. Opin. Cell. Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 38.Feig D.I., Reid T.M., Loeb L.A. Reactive oxygen species in tumorigenesis. Cancer Res. 1994;54:1890s–1894s. [PubMed] [Google Scholar]
- 39.Ames B.N. Dietary carcinogens and anticarcinogens. Oxygen radicals and degenerative diseases. Science. 1983;221:1256–1264. doi: 10.1126/science.6351251. [DOI] [PubMed] [Google Scholar]
- 40.Hussain S.P., Aguilar F., Amstad P., Cerutti P. Oxy-radical induced mutagenesis of hotspot codons 248 and 249 of the human p53 gene. Oncogene. 1994;9:2277–2281. [PubMed] [Google Scholar]
- 41.Nakamura Y., Gindhart T.D., Winterstein D., Tomita I., Seed J.L., Colburn N.H. Early superoxide dismutase-sensitive event promotes neoplastic transformation in mouse epidermal JB6 cells. Carcinogenesis. 1988;9:203–207. doi: 10.1093/carcin/9.2.203. [DOI] [PubMed] [Google Scholar]
- 42.Salim A.S. The permissive role of oxygen-derived free radicals in the development of colonic cancer in the rat. A new theory for carcinogenesis. Int. J. Cancer. 1993;53:1031–1035. doi: 10.1002/ijc.2910530629. [DOI] [PubMed] [Google Scholar]
- 43.Shen Y.H., Wang X.L., Wilcken D.E. Nitric oxide induces and inhibits apoptosis through different pathways. FEBS Lett. 1998;433:125–131. doi: 10.1016/s0014-5793(98)00844-8. [DOI] [PubMed] [Google Scholar]
- 44.Lin Y., Kikuchi S., Obata Y., Yagyu K. Serum copper/zinc superoxide dismutase (Cu/Zn SOD) and gastric cancer risk: a case-control study. Jpn. J. Cancer Res. 2002;93:1071–1075. doi: 10.1111/j.1349-7006.2002.tb01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaynar H., Meral M., Turhan H., Keles M., Celik G., Akcay F. Glutathione peroxidase, glutathione-S-transferase, catalase, xanthine oxidase, Cu-Zn superoxide dismutase activities, total glutathione, nitric oxide, and malondialdehyde levels in erythrocytes of patients with small cell and non-small cell lung cancer. Cancer Lett. 2005;227:133–139. doi: 10.1016/j.canlet.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 46.Perez-Cruz I., Carcamo J.M., Golde D.W. Caspase-8 dependent TRAIL-induced apoptosis in cancer cell lines is inhibited by vitamin C and catalase. Apoptosis. 2007;12:225–234. doi: 10.1007/s10495-006-0475-0. [DOI] [PubMed] [Google Scholar]
- 47.Ames B.N., Durston W.E., Yamasaki E., Lee F.D. Carcinogens are mutagens: a simple test system combining liver homogenates for activation and bacteria for detection. Proc. Natl Acad. Sci. USA. 1973;70:2281–2285. doi: 10.1073/pnas.70.8.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng K.C., Loeb L.A. Genomic stability and instability: a working paradigm. Curr. Top. Microbiol. Immunol. 1997;221:5–18. doi: 10.1007/978-3-642-60505-5_2. [DOI] [PubMed] [Google Scholar]
- 49.Yoneda M., Chomyn A., Martinuzzi A., Hurko O., Attardi G. Marked replicative advantage of human mtDNA carrying a point mutation that causes the MELAS encephalomyopathy. Proc. Natl Acad. Sci. USA. 1992;89:11164–11168. doi: 10.1073/pnas.89.23.11164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deng J.H., Li Y., Park J.S., Wu J., Hu P., Lechleiter J., Bai Y. Nuclear suppression of mitochondrial defects in cells without the ND6 subunit. Mol. Cell. Biol. 2006;26:1077–1086. doi: 10.1128/MCB.26.3.1077-1086.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]