

Abstract
Deletion or duplication of the human chromosome 22q11.2 is associated with many behavioral traits and neuropsychiatric disorders, including autism spectrum disorders and schizophrenia. However, why phenotypes vary widely among individuals with identical deletions or duplications of 22q11.2 and which specific 22q11.2 genes contribute to these phenotypes are still poorly understood. Previous studies have identified a ∼200 kb 22q11.2 region that contributes to behavioral phenotypes in mice. We tested the role of Septin 5 (Sept5), a gene encoded in the ∼200 kb region, in affective behaviors, cognitive capacities and motor activity. To evaluate the impact of genetic backgrounds on behavioral phenotypes of Sept5 deficiency, we used mice on two genetic backgrounds. Our data show that Sept5 deficiency decreased affiliative active social interaction, but this phenotypic expression was influenced by genetic backgrounds. In contrast, Sept5 deficiency decreased anxiety-related behavior, increased prepulse inhibition and delayed acquisition of rewarded goal approach, independent of genetic background. These data suggest that Sept5 deficiency exerts pleiotropic effects on a select set of affective behaviors and cognitive processes and that genetic backgrounds could provide an epistatic influence on phenotypic expression.
INTRODUCTION
The role of constitutive genetic composition in shaping behavior is one of the fundamental issues of understanding individual variations in behavior. Twin studies have amply suggested a robust genetic influence on various behavioral traits and neuropsychiatric disorders in humans. Chromosomal duplications and deletions provide a unique opportunity to further narrow down the chromosomal regions and genes underlying behavioral variations.
Children and adolescents with hemizygosity of a 1.5 or 3 Mb region of chromosome 22q11.2 exhibit altered neuropsychological function, including social interaction deficits, impulsivity, heightened anxiety, a prepulse inhibition (PPI) deficit, cognitive and intellectual impairments and delayed motor development (1–6). Individuals with duplications of this same chromosomal region variably exhibit some of these behavioral variations (7–21). Moreover, defective social thinking, high levels of anxiety and impaired cognitive processing permeate to adulthood (22) and are premorbid, comorbid or even symptomatic elements of schizophrenia and autism spectrum disorders seen at high rates in individuals with 22q11.2 deletions (5,23–30).
Although 22q11.2 likely contains a gene or genes that contribute to a wide range of behavioral phenotypes, identification of specific single 22q11.2 genes remains a formidable task. Because large segments are affected in 22q11.2 deletions and duplications cases, it is not feasible to ascertain the impact of the hemizygosity of individual genes on various phenotypes in humans. Genetically engineered mice have been used to identify small chromosomal segments and individual genes that might contribute to behavioral variations. We previously identified a ∼200 kb human chromosomal segment whose overexpression results in behavioral sensitization in mice; sensitization was blocked by chronic treatment with the antipsychotic drug clozapine (31). Subsequently, it was shown that larger, partly overlapping deletions in the mouse synteny chromosome 16 caused a PPI deficit when part of the deletions encompassed this ∼200 kb region in mice (32,33).
Sept5, a septin family member formerly called cell division control related protein 1 (CDCrel), is encoded in the 200 kb region and is abundantly expressed in the developing and adult brains of mice and humans (34–36). Although its constitutive deletion does not cause detectible structural or developmental abnormalities in the mouse brain (36), Sept5 has been implicated in subtle neuronal and behavioral functions. Sept5 is expressed in presynaptic terminals (35) and exerts negative regulatory control over neurotransmitter release by binding to SNARE-protein (37,38), which has been implicated in neuropsychiatric disorders (39). Moreover, environmental manipulations affect protein levels of this gene; social isolation reduces Sept5 protein levels in the rat striatum (40).
It remains unclear why identical 22q11.2 deletions are associated with a wide range of behavioral phenotypes. As one hypothetical extreme, one of the 22q11.2 genes could cause all behavioral phenotypes. Another extreme possibility is that many 22q11.2 genes could contribute to wide-ranging behavioral phenotypes. Each one of multiple genes could contribute to a single phenotype or multiple phenotypes (i.e. pleiotropy). To delineate the extent to which Sept5 deficiency influences behaviors, we tested Sept5 knockout (KO), heterozygous (HT) and wild-type (WT) mice in a battery of behavioral assays that evaluate affective behaviors (social interaction, elevated plus maze, thigmotaxis, tail suspension and startle), cognitive functions (PPI, rewarded approach, spontaneous alternation and rewarded alternation) and a motor function (spontaneous locomotor activity). Moreover, individuals with identical 22q11.2 deletions exhibit highly variable phenotypes. It is generally assumed that an individual’s genetic background contributes to these variations, but this assumption has not been demonstrated experimentally to be the case. We examined how non-identical genetic backgrounds impact the phenotypes of Sept5 deficiency. Our data show that Sept5 deficiency results in a select set of behavioral phenotypes and that genetic background could alter some phenotypes in mice.
RESULTS
We used two lines of mice with non-identical genetic backgrounds (see Materials and Methods, Mice). One line had a mixed genetic background, referred to as M mice. A second mouse line was created by backcrossing M HT mice to 129X1/SvJ mice, here referred to as 129 enriched (129E) mice.
Social interaction
The Sept5 genotype (i.e. KO, HT and their two WT pair groups) affected active social interaction (F3,94 = 13.96, P < 0.01) and the genotype effect had a significant interaction with genetic backgrounds (F3,94 = 3.05, P < 0.05) (Fig. 1A). Newman–Keuls comparisons confirmed that M KO mice exhibited lower levels of active social interaction than M WT mice, but this effect failed to reach significance in 129E mice. 129E mice generally exhibited higher levels of active social interaction than M mice (F1,94 = 199.94, P < 0.01). Under our experimental condition, mice showed low, almost negligible levels of passive social interaction (Fig. 1B). Although M mice had higher levels of passive social interaction than 129E mice (F1,94 = 4.18, P < 0.05), the four genotype groups did not differ (F3,94 = 1.61, n.s.), even when genetic background was taken into account (F3,94 = 0.47, n.s.).
Figure 1.
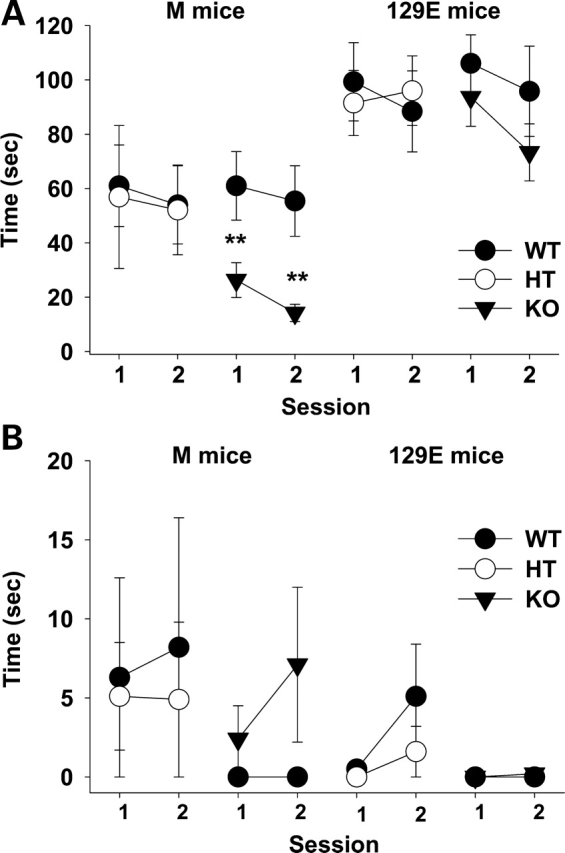
Active (A) and passive (B) social interactions. Interaction time (mean ± SEM) is shown in two successive 5 min sessions. Asterisks indicate statistically significant differences from WT mice at 1% (**) levels, as determined by Newman–Keuls comparisons. M group: WT–HT, n = 9 pairs; WT–KO, n = 16 pairs. 129E group: WT–HT, n = 14 pairs; WT–KO, n = 12 pairs.
To further explore the nature of defective active social interaction in M KO mice, we divided active social interaction into aggressive behaviors and non-aggressive, affiliative social behaviors. Because our experimental condition was designed to optimally evaluate non-aggressive, affiliative social interaction, it was much more common than sporadic and negligible aggressive behaviors (F1,30 =37.55, P < 0.01; data not shown). M WT mice showed significantly more affiliative active social interaction than M KO mice (F1,30 = 10.22, P < 0.01, data not shown), but the two groups did not differ in the amount of aggressive behaviors (F1,30 = 2.74, n.s., data not shown).
Olfactory investigation was the predominant affiliative active social interaction in M WT and M KO mice. However, a deficit in affiliative active social interaction seen in M KO mice does not seem to reflect a generalized deficit in olfactory sensing, as M WT and M KO mice did not differ in avoidance of a compartment containing a non-social olfactory stimulus (2-methylbutyric acid, 1.7 × 10−4 mol) [t(10) = 0.23, n.s., data not shown].
Elevated plus maze
In the elevated plus maze, Sept5 deficient mice spent more time in normally avoided open arms than WT littermates (F2,121 = 3.32, P < 0.05), regardless of genetic background (Genotype×Genetic background interaction, F2,121 = 0.13, n.s.) (Fig. 2A). The significant genotype effect was mainly due to higher levels of time spent in the open arms in HT mice. The 129E mice spent more time in the open arms than M mice (F1,121 = 7.63, P < 0.01). The number of entries to open arms was indistinguishable among genotypes (F2,121 = 2.19, n.s.).
Figure 2.

Anxiety- and affect-related traits. (A) Elevated plus maze. The relative amount of time (% mean ± SEM) spent in the open arms is shown. Because the homogeneity of variance was violated (Hartley’s F-max = 57.87, P < 0.01), data were transformed using a square root. M group: WT, n = 15; HT, n = 20; KO, n = 22. 129E group: WT, n = 20; HT, n = 26; KO, n = 24. (B) Thigmotaxis. Time (mean ± SEM) spent in the marginal area was measured each 5 min during a 30 min period. M group: WT, n = 18; HT, n = 19; KO, n = 7. 129E group: WT, n = 28; HT, n = 27; KO, n = 25. (C) Immobility in a tail suspension test. Time (mean ± SEM) mice remained immobile is shown. WT, wild-type mice; KO, Sept5 knockout mice. M group: WT, n = 25; HT, n = 33; KO, n = 26. 129E group: WT, n = 28; HT, n = 27; KO, n = 25.
Thigmotaxis
Mice placed in an inescapable open field tend to stay in the vicinity of the walls. This tendency or thigmotaxis is interpreted as reflecting, in part, an anxiety- or stress-related trait. Thigmotaxis was not affected by genotype (F2,118 = 1.01, n.s.) or genetic background (F1,118 = 0.79, n.s.) (Fig. 2B). Genotype had no interaction with genetic background (F2,118 = 2.03, n.s.), time (F10,590 = 1.57, n.s.) or genetic background and time (F10,590 = 1.42, n.s.).
Tail suspension
When rodents are held upside-down by the tail, they exhibit mobility in an apparent attempt to escape from the situation. This behavior, or its absence (i.e. immobility), is thought to reflect a basal affective state and a stress response (41). Immobility was not affected by genotype (F2,158 = 1.04, n.s.), genetic background (F1,158 =v3.39, n.s.) or interaction between genetic background and genotype (F2,158 = 2.23, n.s.) (Fig. 2C).
Startle
A startle response was examined against pulses ranging from 75 to 120 dB. Mice exhibited a startle response at high stimulus levels (F9,1215 = 156.12, P < 0.01) (Fig. 3A). Although the intensity of startle was not affected by genotype (F2,135 = 0.02, n.s.) or by interaction between genotype and genetic background (F2,135 = 1.72, n.s.), M mice showed basally higher startle responses than 129E mice at high pulse levels (genetic background, F1,135 = 30.77, P < 0.01; pulse×genetic background, F9,1215 = 29.28, P < 0.01).
Figure 3.

Startle (A) and PPI (B). The magnitude of a startle response and the percentage of PPI are plotted against pulse stimuli (75–120 dB) and prepulse stimuli at 4, 8, 12 and 16 dB (69, 73, 77 and 81 dB presented with the 65 dB background noise), respectively. Data are presented as mean ± SEM. M group: WT, n = 20; HT, n = 25; KO, n = 16. 129E group: WT, n = 28; HT, n = 27; KO, n = 25.
Prepulse inhibition
In rodents and humans, a startle response evoked by a loud sound is reduced by the presentation of a preceding non-startle stimulus, a process termed PPI. PPI increased as the prepulse intensity was increased (F3,405 = 51.53, P < 0.01) (Fig. 3B). KO mice showed higher levels of PPI than WT or HT mice (F2,135 = 4.27, P < 0.05). Higher levels of basal PPI were found in M mice than in 129E mice (F1,135 = 8.78, P < 0.01). M mice showed a higher rate of PPI increase across prepulse levels than did 129E mice (F3,405 = 2.64, P < 0.05).
Rewarded approach
When mice were trained to run on an L-maze to reach a food-baited goal compartment, they gradually learned to reach the goal faster (Fig. 4A) (F5,4310 = 51.03, P < 0.01). Because the three-way interaction was significant (F10,4310 = 2.50, P < 0.01), we applied exploratory two-way analysis of variances (ANOVAs) to the two genetic backgrounds separately. In M mice, Sept5 deficiency delayed, in a gene-dose dependent manner, acquisition of this behavior evenly across sessions (Genotype, F2,405 = 6.26, P < 0.01; genotype×session, F10,2025 = 1.73, n.s.). In contrast, Sept5 deficiency in 129E mice delayed and facilitated this behavior in KO mice and HT mice, respectively, at early sessions only (genotype, F2,457 = 1.22, n.s.; genotype×session, F10,2285 = 3.64, P < 0.01).
Figure 4.

(A) Rewarded approach. Time (mean ± SEM) spent to reach a rewarded goal in the L-maze is shown. M group: WT, n = 19; HT, n = 32; KO, n = 17. 129E group: WT, n = 23; HT, n = 36; KO, n = 18. (B) Spontaneous alternation. All genotypes show a tendency to alternate an arm to visit upon repeated testing (i.e. >50%). M group: WT, n = 22; HT, n = 23; KO, n = 22. 129E group: WT, n = 28; HT, n = 27; KO, n = 25. (C) Rewarded alternation. M group: WT, n = 13; HT, n = 19; KO, n = 9. 129E group: WT, n = 15; HT, n = 27; KO, n = 16.
Spontaneous alternation
In a T-maze, mice have a tendency to visit an unvisited arm rather than a previously visited arm. This phenomenon or spontaneous alternation reflects the flexibility of an animal to alternate their behavior based on the memory of a previously visited arm. Spontaneous alternation was not influenced by genetic background (F1,141 = 0.01, n.s.), genotype (F2,141 = 1.04, n.s.) or interaction (F2,141 = 2.25, n.s.) (Fig. 4B).
Rewarded alternation
When mice are rewarded in one of the two goal arms of the T-maze, their tendency to subsequently alternate behavior is enhanced. This reward-based memory or rewarded alternation is an objective measure to evaluate how a reward experience subsequently influences spontaneous alternation (Fig. 4C). Sept5 deficiency had no effect on rewarded alternation by itself (F2,93 = 0.07, n.s.), with genetic background (F2,93 = 2.09, n.s.) or with genetic background and session (F8,372 = 1.22, n.s.). 129E mice exhibited higher levels of rewarded alternation than M mice (F1,93 = 8.47, P < 0.01).
Spontaneous motor activity
Many of the affective and cognitive behaviors could be confounded by altered basal levels of motor activity. We examined spontaneous locomotor activity in an inescapable open field. Mice exhibited a normal rate of habituation regardless of genetic background (time interval, F5,590 = 113.87, P < 0.01; genetic background×time interval, F5,590 = 1.78, n.s.) (Fig. 5). Sept5 deficiency had no effect on motor activity at any time point, regardless of genetic background (genotype, F2,118 = 1.83, n.s; genotype×genetic background, F2,118 = 1.33, n.s.; genotype×time interval, F10,590 = 0.36, n.s.; genotype×genetic background×time interval, F10,590 = 1.11, n.s.). Overall, M mice exhibited higher levels of basal motor activity than 129E mice (genetic background, F1,118 = 20.53, P < 0.01).
Figure 5.

Spontaneous locomotor activity in an inescapable open field. Distance traveled (mean ± SEM) was measured for 30 min. Each tick on the x-axis represents a 5 min bin. WT, wild-type mice; KO, Sept5 knockout mice. M group: WT, n = 18; HT, n = 19; KO, n = 7. 129E group: WT, n = 28; HT, n = 27; KO, n = 25.
DISCUSSION
Our observations demonstrate several ways Sept5 deficiency influences behavior in mice. First, Sept5 KO mice exhibited a deficit in affiliative active social interaction, but this phenotype depended on the genetic background. Secondly, Sept5 deficiency, regardless of genetic background, decreased an anxiety-related behavior on the elevated plus maze, potentiated PPI and delayed acquisition of rewarded approach. Thirdly, Sept5 deficiency caused no apparent phenotype in thigmotaxis, tail suspension, startle response, spontaneous and rewarded alternation or spontaneous motor activity.
Consistent with the hypothesis that genetic backgrounds could provide an epistatic influence on the phenotypic expression of 22q11.2 deletion, even one generation of shift in genetic background toward 129X1/SvJ masked social interaction deficits in Sept5 deficient mice. This result suggests that genetic background is one of the reasons why identical 22q11.2 deletions cause highly variable phenotypes. In contrast, this level of shift in the genetic background did not affect phenotypes in an anxiety-related behavior in the elevated plus maze, PPI or rewarded approach. It should be noted that these phenotypes still could be affected by alleles that are not present in these two lines of mice. It is estimated that the flanking loci of Sept5 contain alleles originating from 129X1/SvJ and 129S1 in M KO mice and alleles from CD1 in M WT mice. If 129X1/SvJ and 129S1 alleles reduced social interaction in M KO mice, 129E mice, regardless of genotype, would have exhibited social interaction at or below levels of M KO mice. Because 129E mice showed basally higher levels of active social interaction than M mice, our finding is inconsistent with the possibility that lower levels of active social interaction in M KO mice, compared with M WT mice, was caused by alleles in the genetic backgrounds of 129X1/SvJ and 129S1. Nevertheless, currently available breeding strategies, including congenic and other post hoc breeding strategies, do not unequivocally differentiate between the impact of a constitutionally deleted gene and the modifying effect of alleles in the genetic background on a phenotype (42,43). Our data should be interpreted as indicating how a phenotype manifests itself under two non-identical genetic backgrounds in which Sept5 deficiency is embedded.
A previous study reported that Sept5 HT mice were normal in PPI in non-congenic Sept5 HT mice (32). Our data are consistent with this report; both M and 129E Sept5 HT mice showed normal PPI. We extended this observation by demonstrating that homozygous (i.e. KO) mice exhibited an increased PPI. Because an increase in PPI is achieved by antipsychotic drugs (44) and a PPI defect is associated with the genetic susceptibility to many neuropsychiatric disorders (45), the molecular cascades of Sept5 could be exploited to understand the mechanisms of PPI in relation to neuropsychiatric disorders. Such an understanding could guide the development of effective therapies.
In translating phenotypes in mice to humans, our findings raise several important issues. The increased PPI and decreased anxiety-related behavior in the elevated plus maze in Sept5 deficient mice are a sharp contrast to a lower level of PPI and higher levels of anxiety in individuals with 22q11.2 deletions (5,6,26,46). Our finding suggests that individual 22q11.2 genes could exaggerate or attenuate PPI and anxiety-related behaviors, but the final phenotypic expression is determined by summation and/or complex interactions among these intermediate phenotypic elements. Deletions of other 22q11.2 genes are likely to reduce PPI and increase anxiety-related behaviors to the extent that they override the opposing effects of Sept5 deficiency on these behaviors. For example, deletion of Tbx1, another gene encoded in the 200 kb region, has been reported to reduce PPI in mice (32), although another report did not find such a deficit (33). Homozygosity and heterozygosity of catechol-O-methyl transferase (Comt), another 22q11.2 gene, are associated with heightened levels of anxiety-related behaviors in mice (47,48). A future challenge is to characterize the precise ways various 22q11.2 genes individually and collectively affect behaviors.
Although some of the tasks used in this study are thought to commonly reflect certain behavioral traits, Sept5 deficiency did not equally affect behaviors in these tasks. Sept5 heterozygosity reduced avoidance of open arms on the elevated plus maze, but Sept5 deficiency had no effect on thigmotaxis in an inescapable open field or a startle response evoked by a loud sound, although these three tasks are thought to measure anxiety- and fear-related behaviors. It might be premature to assume that ‘anxiety’ can be modeled as a singular process in multiple behavioral tasks, because each of these tasks is thought to contain many distinct components, each having a distinct genetic basis (49). Similarly, Sept5 KO mice were impaired in one type of memory task (i.e. rewarded approach) but not others (i.e. spontaneous alternation and rewarded alternation). In the rewarded approach task, mice run from the start compartment to the only baited, open arm during six trials per day for 6 days. The right or left arm was pseudorandomly chosen as the only open goal arm by the experimenter and the same arm was not chosen as the goal more than twice in a row. Thus, the memory of a visited arm on the previous trial provides no information for the next trial. Unlike spontaneous and rewarded alternation, what is acquired in this task is to ignore or suppress the memory of where the goal arm was located on a previous trial and reach the goal faster. Sept5 might selectively contribute to this type of memory.
Our KO mice, but not HT mice, were impaired in affiliative social interaction and rewarded approach. Although individuals with 22q11.2 deletions show social interaction deficits and cognitive impairments, they are afflicted with 22q11.2 hemizygosity, not homozygosity. However, caution is needed in equating heterozygosity in mice to hemizygosity in humans. First, it is not known whether a gene-dose manifests itself similarly in mice and humans. Secondly, if individuals with 22q11.2 hemizygosity carry low-activity alleles in the remaining copy of SEPT5, such conditions could be functionally closer to homozygosity than heterozygosity. Consistent with such a possibility, a low-activity allele of COMT on the remaining 22q11.2 copy worsens phenotypic expression in cognition and neuropsychiatric disorders in human 22q11.2 hemizygous cases (50–52).
Sept5 deficiency exerted pleiotropic effects on multiple behavioral phenotypes, and phenotypic expression of affiliative active social interaction was altered by genetic background. Together with the observations that Sept5 deficiency does not cause defects in heart or neuronal development in mice (36,53), our findings suggest a polygenic origin of the diverse physical and behavioral phenotypes seen in individuals with 22q11.2 deletions. Our findings provide a glimpse of the extraordinarily complex ways multiple 22q11.2 genes affect the symptomatology of individuals with 22q11.2 haploinsufficiency.
MATERIALS AND METHODS
Mice
Male Sept5 KO, HT and WT littermates were tested, unless otherwise specified. Mice were maintained in a group of up to five in a cage. Mice were given access to food and water ad libitum, unless otherwise specified. The Sept5 KO mouse was originally developed using R1 embryonic stem (ES) cells and bred with CD1 mice (36), designated here as a mixed genetic background mouse or M mice. The R1 mouse ES cell line is a hybrid that contains genetic backgrounds of 129X1/SvJ mice and 129S1/Sv-p+ Tyr+ Kitl SI-J/+ (i.e. 129S1) (54). We used 129X1/SvJ mice for backcrossing of M HT mice, because (i) the allelic composition of the R1 ES cell line is closer to that of 129X1/SvJ mice than to 129S1 mice (55), (ii) CD1 mice are outbred and could introduce more stochastic allelic variations and (iii) the flanking regions of a deleted gene can be made homogenous between WT and KO mice more readily by backcrossing to the strain from which ES cells are derived (42,43). M HT mice were backcrossed to 129X1/SvJ mice for one generation. The resulting HT offspring were inbred to generate an F2 generation, here referred to as 129E mice. The genotype of mice was determined by PCR amplification. When PCR genotypes were ambiguous, Sept5 protein levels were determined by western blotting after behavioral analysis.
Behavioral analysis
Social interaction
Because previous interactions between littermates in their home cages cannot be controlled and may affect subsequent social interactions during testing, non-littermate Sept5 WT and KO pairs and WT and HT pairs were age-matched and tested at the age of 1.5–4 months. Our experimental condition was designed so that non-aggressive, affiliative social interaction was optimally evaluated. Specifically, our experimental procedure did not include prolonged isolation in home cages or establish a resident versus an intruder in the experimental chamber, as both mice were placed in a novel home cage simultaneously. Each mouse was singly placed in a new home cage in the experimental room for 30 min before a pair was placed in a new, third home cage in two 5 min sessions, with a 30 min interval. Social interaction included active and passive forms. Data for the two forms were analyzed separately. Active social interactions included aggressive behaviors (i.e. tail rattle, bites, kicks, sideway offense, boxing, wrestling and pursuit) and non-aggressive, affiliative behaviors (i.e. mounting as part of allogrooming, olfactory investigation and allogroom). Passive interactions included escape, leap, side by side position and submissive posture. Social interaction was recorded and rated by observers blinded to genotype (inter-rater correlation, r = 0.974, P < 0.01). Time spent in active and passive social behaviors was analyzed.
Odorant-induced place avoidance
The apparatus was composed of two home cages (28 × 17 × 12 cm), connected side-by-side, as described (56). A gate (10 × 11 cm) was placed between the two cages. We used M WT mice (n = 6) and M KO mice (n = 6) at the age of 2–4 months. A filter paper scented with water or 2-methylbutyric acid (1.7 × 10−4 mol) was placed in one of the cages, and the amount of time the mouse spent in the other cage was measured for 3 min and used as an index of odor avoidance (57).
Elevated plus maze
The apparatus and procedure were identical to those described in a previous study (41). Sept5 WT, HT and KO mice were age-matched and tested at the age of 3–6 months. Briefly, mice had been brought into a room adjacent to the testing room at least 1 h before testing began. Each mouse was tested on this task only once. Individual mice were placed on the center platform, facing one of the open arms, and tested for 5 min. Behavior was videotaped from an adjacent room through an observation window. An observer blinded to the genotype of mice rated the behavior. We determined the percentage of time spent in and entrances into the open arms with respect to the total time spent in and number of entrances into the open and closed arms, respectively.
Locomotor activity
Spontaneous locomotor activity was tested in four sets of open field apparatus (Truscan, Coulbourn Instruments, PA, USA), as described previously (41). Sept5 WT, HT and KO mice were age-matched and tested at the age of 3–5 months. Horizontal activity was recorded for 30 min. Distance traveled (cm) was used as an index of horizontal locomotor activity. Time(s) spent in the marginal area along the walls was used as an index of thigmotaxis.
Tail suspension
The apparatus and the procedure were identical to those described elsewhere (41). Sept5 WT, HT and KO mice were age-matched and tested at the age of 3–6 months. Briefly, mice were suspended for 6 min by the tail from a metal bar 30 cm above the platform. Behaviors were recorded by a video camera and a rater blinded to genotype recorded the length of time each mouse remained immobile.
Startle and PPI
Mice were tested for startle response and PPI in a startle chamber (S-R LAB, San Diego Instruments, San Diego, CA, USA). Sept5 WT, HT and KO mice were age-matched and tested at the age of 3–5 months. After mice were placed in the startle chamber, a 65 dB background noise level was presented for a 10 min acclimation period and continued throughout the test session. A PPI test consisted of startle trials (PULSE-ALONE, a 120 dB acoustic stimulus, 40 ms), prepulse trials (PREPULSE + PULSE) and no-stimulus trials (NOSTIM). The PREPULSE + PULSE trial consisted of a prepulse (69, 73, 77 and 81 dB with a 65 dB background, 20 ms) followed 100 ms later by the 120 dB pulse. The NOSTIM trial consisted of background noise only (65 dB). Each PPI test began and ended with five presentations of the PULSE-ALONE trial. Each trial type was presented 10 times in a pseudorandom order between these PULSE-ALONE trials. The time between trials was set to average 15 s (12–30 s). The amount of PPI was calculated as a percentage score for a prepulse trial: %PPI = 100 − [[(startle response for PREPULSE + PULSE)/(startle response for PULSE-ALONE)] × 100]. Startle magnitude was calculated as the average response to all of the PULSE-ALONE trials, excluding the first and last blocks of five PULSE-ALONE trials. Each session included acclimation for 5 min, followed by 24 blocks of five consecutive presentations of the pulse. The startle magnitude and PPI% were analyzed.
Spontaneous alternation
Spontaneous alternation was tested on a black Plexiglas T-maze. The three arms were identical in size (30 cm long × 10 cm wide × 20 cm high wall for each arm) and were connected by a central area (10 × 10 × 20 cm). Sept5 WT, HT and KO mice were age-matched and tested at the age of 3–6 months. Mice were placed in the start compartment facing away from the goal arms and confined there for 5 s before the door was opened. Both goal arms were open. Once mice chose and entered one of the goal arms, a door was gently lowered to confine mice in that arm, and 30 s later, they were removed from the arm. The mice were brought back to the start compartment and the door was opened 5 s later. They were allowed to choose either the goal arm just visited or the opposite goal arm. If a mouse did not make a choice within 2 min on the first trial, the mouse was returned to the start compartment and left there for 30 s before resuming a new trial. If a mouse did not enter the goal arms within 2 min on subsequent trials, it was placed in the correctly alternated arm and confined there for 30 s. Such trials were not used for analysis. A total of 10 trials were given. The percentage of alternated choices was used as an index of spontaneous alternation for analysis.
Rewarded approach
The apparatus used for spontaneous alternation was used. Sept5 KO, HT and WT littermates were age-matched and tested at the age of 3–7 months. Food deprivation started 2 days prior to testing and reduced body weights up to 86–96% of their free-feeding body. Although the three-way interaction among genetic background, genotype and day was significant (F14,973 = 2.30, P < 0.01), exploratory ANOVAs showed that food deprivation equally reduced body weights in the three genotype groups in the two genetic backgrounds (M mice, genotype, F2,65 = 1.94, n.s.; day, F7,455 = 36.72, P < 0.01; genotype×day, F14,455 = 0.84, n.s.; 129E mice, genotype, F2,74 = 0.26, n.s.; day, F7,518 = 158.81, P < 0.01; genotype×day, F14,518 = 1.68, n.s). Littermates were first introduced together to the T-maze and allowed to explore all the arms for four 4 min sessions, with 10 min between-session intervals. At least 2 h after these habituation sessions, individual mice were allowed to run from the start compartment to one goal arm with the other goal arm blocked by a door (i.e. L-maze) during six trials per day for 6 days. Mice were allowed to spend up to 10 min to complete each trial. The goal arm was baited with sweetened, condensed milk (diluted 50/50 with water) placed in the food well at the end of the arm. Mice were picked up from the goal compartment immediately after it consumed the condensed milk. Each arm was pseudorandomly designated as the goal arm 50% of the cases for each day; the other arm was blocked by a door. The same arm was not chosen as the goal for more than twice in a row. The inter-trial interval was set within 10 min. This learning reflects an animal’s ability to reach the only available goal faster irrespectively of its spatial location. Latency to the first lick of milk was recorded and used for analysis.
Rewarded alternation
After completion of the 6 days training in rewarded approach, mice were trained for rewarded alternation on six trials per day for 5 days. Food deprivation was effective in reducing body weights during rewarded alternation and mice maintained 88–93% of their free-feeding body. The three-way interaction among genetic background, genotype and day was not significant (F8,372 = 0.89, n.s.) and the two genetic background groups or the three genotype groups did not differ in body weights (genetic background, F1,93 = 0.97, n.s.; genotype, F2,93 = 1.51, n.s.). Both goal arms were baited, but only one arm was pseudorandomly chosen as an open arm and the other goal arm was blocked. Equal numbers of left and right goals were given. During each trial, each mouse was placed in the start compartment and left there for 5 s with a closed door. Each mouse was then allowed to run to the goal arm (i.e. sampling run). After the mouse had been left in the goal arm for approximately 11 s to consume the milk, it was brought back to the start compartment. After the mouse was confined to the start compartment for 5 s, a choice run started. During the choice run, both arms were open, but the previously unvisited arm remained baited and the previously visited arm was left unbaited. Upon choosing one of the goal arms, the mouse was confined there for ∼11 s before the next trial. The inter-trial interval was set within 10 min. The absence of a choice was dealt with in the same way as spontaneous alternation. The numbers of total arm visits and correct visits were recorded. The percentage of correct arm visits (i.e. rewarded alternation) was used for analysis.
Statistical analysis
All data are presented as the mean ± standard error of the mean. Statistical significance was determined by ANOVA followed by Newman–Keuls post hoc comparisons. The Student’s t-test was used when only two groups were compared. The threshold for significance was set at P < 0.05.
FUNDING
This work was supported by a (NIH) grant (HD05311) and a Maltz NARSAD Independent Investigator Award to N.H., a CIHR grant (FRN# 13465) to W.S.T. and the State of New York to S.A.
ACKNOWLEDGEMENTS
We thank Mr Eli Neustadter for building a T-maze and Dr M. Kinoshita for providing Sept5 antibodies. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NICHD or the NIH.
Conflict of Interest statement. N.H. served as a consultant for the IntraCellular Therapies, Inc. in 2008. M.A.G. holds an equity interest in San Diego Instruments, Inc.
REFERENCES
- 1.Goldberg R., Motzkin B., Marion R., Scambler P.J., Shprintzen R.J. Velo-cardio-facial syndrome: a review of 120 patients. Am. J. Med. Genet. 1993;45:313–319. doi: 10.1002/ajmg.1320450307. [DOI] [PubMed] [Google Scholar]
- 2.Swillen A., Devriendt K., Legius E., Eyskens B., Dumoulin M., Gewillig M., Fryns J.P. Intelligence and psychosocial adjustment in velocardiofacial syndrome: a study of 37 children and adolescents with VCFS. J. Med. Genet. 1997;34:453–458. doi: 10.1136/jmg.34.6.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moss E.M., Batshaw M.L., Solot C.B., Gerdes M., Donald-McGinn D.M., Driscoll D.A., Emanuel B.S., Zackai E.H., Wang P.P. Psychoeducational profile of the 22q11.2 microdeletion: a complex pattern. J. Pediatr. 1999;134:193–198. doi: 10.1016/s0022-3476(99)70415-4. [DOI] [PubMed] [Google Scholar]
- 4.Gerdes M., Solot C., Wang P.P., Moss E., LaRossa D., Randall P., Goldmuntz E., Clark B.J., III, Driscoll D.A., Jawad A., et al. Cognitive and behavior profile of preschool children with chromosome 22q11.2 deletion. Am. J. Med. Genet. 1999;85:127–133. [PubMed] [Google Scholar]
- 5.Niklasson L., Rasmussen P., Oskarsdottir S., Gillberg C. Chromosome 22q11 deletion syndrome (CATCH 22): neuropsychiatric and neuropsychological aspects. Dev. Med. Child Neurol. 2002;44:44–50. doi: 10.1017/s0012162201001645. [DOI] [PubMed] [Google Scholar]
- 6.Sobin C., Kiley-Brabeck K., Karayiorgou M. Associations between prepulse inhibition and executive visual attention in children with the 22q11 deletion syndrome. Mol. Psychiatry. 2005;10:553–562. doi: 10.1038/sj.mp.4001609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edelmann L., Pandita R.K., Spiteri E., Funke B., Goldberg R., Palanisamy N., Chaganti R.S., Magenis E., Shprintzen R.J., Morrow B.E. A common molecular basis for rearrangement disorders on chromosome 22q11. Hum. Mol. Genet. 1999;8:1157–1167. doi: 10.1093/hmg/8.7.1157. [DOI] [PubMed] [Google Scholar]
- 8.Ensenauer R.E., Adeyinka A., Flynn H.C., Michels V.V., Lindor N.M., Dawson D.B., Thorland E.C., Lorentz C.P., Goldstein J.L., McDonald M.T., et al. Microduplication 22q11.2, an emerging syndrome: clinical, cytogenetic, and molecular analysis of thirteen patients. Am. J. Human Genet. 2003;73:1027–1040. doi: 10.1086/378818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hassed S.J., Hopcus-Niccum D., Zhang L., Li S., Mulvihill J.J. A new genomic duplication syndrome complementary to the velocardiofacial (22q11 deletion) syndrome. Clin. Genet. 2004;65:400–404. doi: 10.1111/j.0009-9163.2004.0212.x. [DOI] [PubMed] [Google Scholar]
- 10.Portnoi M.F., Lebas F., Gruchy N., Ardalan A., Biran-Mucignat V., Malan V., Finkel L., Roger G., Ducrocq S., Gold F., et al. 22q11.2 duplication syndrome: two new familial cases with some overlapping features with DiGeorge/velocardiofacial syndromes. Am. J. Med. Genet. A. 2005;137:47–51. doi: 10.1002/ajmg.a.30847. [DOI] [PubMed] [Google Scholar]
- 11.Sparkes R., Chernos J., Dicke F. Duplication of the 22q11.2 region associated with congenital cardiac disease. Cardiol. Young. 2005;15:229–231. doi: 10.1017/S1047951105000466. [DOI] [PubMed] [Google Scholar]
- 12.Yobb T.M., Somerville M.J., Willatt L., Firth H.V., Harrison K., MacKenzie J., Gallo N., Morrow B.E., Shaffer L.G., Babcock M., et al. Microduplication and triplication of 22q11.2: a highly variable syndrome. Am. J. Hum. Genet. 2005;76:865–876. doi: 10.1086/429841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de La Rochebrochard C., Joly-Helas G., Goldenberg A., Durand I., Laquerriere A., Ickowicz V., Saugier-Veber P., Eurin D., Moirot H., Diguet A., et al. The intrafamilial variability of the 22q11.2 microduplication encompasses a spectrum from minor cognitive deficits to severe congenital anomalies. Am. J. Med. Genet. A. 2006;140:1608–1613. doi: 10.1002/ajmg.a.31227. [DOI] [PubMed] [Google Scholar]
- 14.Alberti A., Romano C., Falco M., Cali F., Schinocca P., Galesi O., Spalletta A., Di B.D., Fichera M. 1.5 Mb de novo 22q11.21 microduplication in a patient with cognitive deficits and dysmorphic facial features. Clin. Genet. 2007;71:177–182. doi: 10.1111/j.1399-0004.2007.00750.x. [DOI] [PubMed] [Google Scholar]
- 15.Engels H., Brockschmidt A., Hoischen A., Landwehr C., Bosse K., Walldorf C., Toedt G., Radlwimmer B., Propping P., Lichter P., Weber R.G. DNA microarray analysis identifies candidate regions and genes in unexplained mental retardation. Neurology. 2007;68:743–750. doi: 10.1212/01.wnl.0000256367.70365.e0. [DOI] [PubMed] [Google Scholar]
- 16.Mukaddes N.M., Herguner S. Autistic disorder and 22q11.2 duplication. World J. Biol. Psychiatry. 2007;8:127–130. doi: 10.1080/15622970601026701. [DOI] [PubMed] [Google Scholar]
- 17.Cai G., Edelmann L., Goldsmith J.E., Cohen N., Nakamine A., Reichert J.G., Hoffman E.J., Zurawiecki D.M., Silverman J.M., Hollander E., et al. Multiplex ligation-dependent probe amplification for genetic screening in autism spectrum disorders: efficient identification of known microduplications and identification of a novel microduplication in ASMT. BMC Med. Genomics. 2008;1:50. doi: 10.1186/1755-8794-1-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ou Z., Berg J.S., Yonath H., Enciso V.B., Miller D.T., Picker J., Lenzi T., Keegan C.E., Sutton V.R., Belmont J., et al. Microduplications of 22q11.2 are frequently inherited and are associated with variable phenotypes. Genet. Med. 2008;10:267–277. doi: 10.1097/GIM.0b013e31816b64c2. [DOI] [PubMed] [Google Scholar]
- 19.Ramelli G.P., Silacci C., Ferrarini A., Cattaneo C., Visconti P., Pescia G. Microduplication 22q11.2 in a child with autism spectrum disorder: clinical and genetic study. Dev. Med. Child Neurol. 2008;50:953–955. doi: 10.1111/j.1469-8749.2008.03048.x. [DOI] [PubMed] [Google Scholar]
- 20.Wentzel C., Fernstrom M., Ohrner Y., Anneren G., Thuresson A.C. Clinical variability of the 22q11.2 duplication syndrome. Eur. J. Med. Genet. 2008;51:501–510. doi: 10.1016/j.ejmg.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 21.Yu S., Cox K., Friend K., Smith S., Buchheim R., Bain S., Liebelt J., Thompson E., Bratkovic D. Familial 22q11.2 duplication: a three-generation family with a 3-Mb duplication and a familial 1.5-Mb duplication. Clin. Genet. 2008;73:160–164. doi: 10.1111/j.1399-0004.2007.00938.x. [DOI] [PubMed] [Google Scholar]
- 22.Henry J.C., van Amelsvoort T., Morris R.G., Owen M.J., Murphy D.G., Murphy K.C. An investigation of the neuropsychological profile in adults with velo-cardio-facial syndrome (VCFS) Neuropsychologia. 2002;40:471–478. doi: 10.1016/s0028-3932(01)00136-1. [DOI] [PubMed] [Google Scholar]
- 23.Bassett A.S., Hodgkinson K., Chow E.W., Correia S., Scutt L.E., Weksberg R. 22q11 deletion syndrome in adults with schizophrenia. Am. J. Med. Genet. 1998;81:328–337. [PMC free article] [PubMed] [Google Scholar]
- 24.van Amelsvoort T., Henry J., Morris R., Owen M., Linszen D., Murphy K., Murphy D. Cognitive deficits associated with schizophrenia in velo-cardio-facial syndrome. Schizophr. Res. 2004;70:223–232. doi: 10.1016/j.schres.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Chow E.W., Watson M., Young D.A., Bassett A.S. Neurocognitive profile in 22q11 deletion syndrome and schizophrenia. Schizophr. Res. 2006;87:270–278. doi: 10.1016/j.schres.2006.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gothelf D., Feinstein C., Thompson T., Gu E., Penniman L., Van S.E., Kwon H., Eliez S., Reiss A.L. Risk factors for the emergence of psychotic disorders in adolescents with 22q11.2 deletion syndrome. Am. J. Psychiatry. 2007;164:663–669. doi: 10.1176/ajp.2007.164.4.663. [DOI] [PubMed] [Google Scholar]
- 27.Debbane M., Glaser B., David M.K., Feinstein C., Eliez S. Psychotic symptoms in children and adolescents with 22q11.2 deletion syndrome: neuropsychological and behavioral implications. Schizophr. Res. 2006;84:187–193. doi: 10.1016/j.schres.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 28.Antshel K.M., Aneja A., Strunge L., Peebles J., Fremont W.P., Stallone K., AbdulSabur N., Higgins A.M., Shprintzen R.J., Kates W.R. Autistic spectrum disorders in velo-cardio facial syndrome (22q11.2 deletion) J. Autism Dev. Disord. 2007;37:1776–1786. doi: 10.1007/s10803-006-0308-6. [DOI] [PubMed] [Google Scholar]
- 29.Fine S.E., Weissman A., Gerdes M., Pinto-Martin J., Zackai E.H., Donald-McGinn D.M., Emanuel B.S. Autism spectrum disorders and symptoms in children with molecularly confirmed 22q11.2 deletion syndrome. J. Autism Dev. Disord. 2005;35:461–470. doi: 10.1007/s10803-005-5036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kates W.R., Antshel K.M., Fremont W.P., Shprintzen R.J., Strunge L.A., Burnette C.P., Higgins A.M. Comparing phenotypes in patients with idiopathic autism to patients with velocardiofacial syndrome (22q11 DS) with and without autism. Am. J. Med. Genet. A. 2007;143A:2642–2650. doi: 10.1002/ajmg.a.32012. [DOI] [PubMed] [Google Scholar]
- 31.Hiroi N., Zhu H., Lee M., Funke B., Arai M., Itokawa M., Kucherlapati R., Morrow B., Sawamura T., Agatsuma S. A 200-kb region of human chromosome 22q11.2 confers antipsychotic-responsive behavioral abnormalities in mice. Proc. Natl Acad. Sci. USA. 2005;102:19132–19137. doi: 10.1073/pnas.0509635102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paylor R., Glaser B., Mupo A., Ataliotis P., Spencer C., Sobotka A., Sparks C., Choi C.H., Oghalai J., Curran S., et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc. Natl Acad. Sci. USA. 2006;103:7729–7734. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Long J.M., Laporte P., Merscher S., Funke B., Saint-Jore B., Puech A., Kucherlapati R., Morrow B.E., Skoultchi A.I., Wynshaw-Boris A. Behavior of mice with mutations in the conserved region deleted in velocardiofacial/DiGeorge syndrome. Neurogenetics. 2006;7:247–257. doi: 10.1007/s10048-006-0054-0. [DOI] [PubMed] [Google Scholar]
- 34.Caltagarone J., Rhodes J., Honer W.G., Bowser R. Localization of a novel septin protein, hCDCrel-1, in neurons of human brain. Neuroreport. 1998;9:2907–2912. doi: 10.1097/00001756-199808240-00042. [DOI] [PubMed] [Google Scholar]
- 35.Kinoshita A., Noda M., Kinoshita M. Differential localization of septins in the mouse brain. J. Comp. Neurol. 2000;428:223–239. doi: 10.1002/1096-9861(20001211)428:2<223::aid-cne3>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 36.Peng X.R., Jia Z., Zhang Y., Ware J., Trimble W.S. The septin CDCrel-1 is dispensable for normal development and neurotransmitter release. Mol. Cell Biol. 2002;22:378–387. doi: 10.1128/MCB.22.1.378-387.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beites C.L., Xie H., Bowser R., Trimble W.S. The septin CDCrel-1 binds syntaxin and inhibits exocytosis. Nat. Neurosci. 1999;2:434–439. doi: 10.1038/8100. [DOI] [PubMed] [Google Scholar]
- 38.Dong Z., Ferger B., Paterna J.C., Vogel D., Furler S., Osinde M., Feldon J., Bueler H. Dopamine-dependent neurodegeneration in rats induced by viral vector-mediated overexpression of the parkin target protein, CDCrel-1. Proc. Natl Acad. Sci. USA. 2003;100:12438–12443. doi: 10.1073/pnas.2132992100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Honer W.G., Falkai P., Bayer T.A., Xie J., Hu L., Li H.Y., Arango V., Mann J.J., Dwork A.J., Trimble W.S. Abnormalities of SNARE mechanism proteins in anterior frontal cortex in severe mental illness. Cereb. Cortex. 2002;12:349–356. doi: 10.1093/cercor/12.4.349. [DOI] [PubMed] [Google Scholar]
- 40.Barr A.M., Young C.E., Sawada K., Trimble W.S., Phillips A.G., Honer W.G. Abnormalities of presynaptic protein CDCrel-1 in striatum of rats reared in social isolation: relevance to neural connectivity in schizophrenia. Eur. J. Neurosci. 2004;20:303–307. doi: 10.1111/j.0953-816X.2004.03457.x. [DOI] [PubMed] [Google Scholar]
- 41.Zhu H., Lee M., Agatsuma S., Hiroi N. Pleiotropic impact of constitutive fosB inactivation on nicotine-induced behavioral alterations and stress-related traits in mice. Hum. Mol. Genet. 2007;16:820–836. doi: 10.1093/hmg/ddm027. [DOI] [PubMed] [Google Scholar]
- 42.Wolfer D.P., Crusio W.E., Lipp H.P. Knockout mice: simple solutions to the problems of genetic background and flanking genes. Trends Neurosci. 2002;25:336–340. doi: 10.1016/s0166-2236(02)02192-6. [DOI] [PubMed] [Google Scholar]
- 43.Flaherty L., Bolivar V. In: Neurobehavioral Genetics. Jones B.C., Mormede P., editors. New York: Taylor & Francis; 2007. pp. 115–127. [Google Scholar]
- 44.Geyer M.A., McIlwain K.L., Paylor R. Mouse genetic models for prepulse inhibition: an early review. Mol. Psychiatry. 2002;7:1039–1053. doi: 10.1038/sj.mp.4001159. [DOI] [PubMed] [Google Scholar]
- 45.Braff D.L., Geyer M.A., Swerdlow N.R. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology. 2001;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- 46.Baker K.D., Skuse D.H. Adolescents and young adults with 22q11 deletion syndrome: psychopathology in an at-risk group. Br. J. Psychiatry. 2005;186:115–120. doi: 10.1192/bjp.186.2.115. [DOI] [PubMed] [Google Scholar]
- 47.Gogos J.A., Morgan M., Luine V., Santha M., Ogawa S., Pfaff D., Karayiorgou M. Catechol-O-methyltransferase-deficient mice exhibit sexually dimorphic changes in catecholamine levels and behavior. Proc. Natl Acad. Sci. USA. 1998;95:9991–9996. doi: 10.1073/pnas.95.17.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Papaleo F., Crawley J.N., Song J., Lipska B.K., Pickel J., Weinberger D.R., Chen J. Genetic dissection of the role of catechol-O-methyltransferase in cognition and stress reactivity in mice. J. Neurosci. 2008;28:8709–8723. doi: 10.1523/JNEUROSCI.2077-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Henderson N.D., Turri M.G., DeFries J.C., Flint J. QTL analysis of multiple behavioral measures of anxiety in mice. Behav. Genet. 2004;34:267–293. doi: 10.1023/B:BEGE.0000017872.25069.44. [DOI] [PubMed] [Google Scholar]
- 50.Bassett A.S., Caluseriu O., Weksberg R., Young D.A., Chow E.W. Catechol-O-methyl transferase and expression of schizophrenia in 73 adults with 22q11 deletion syndrome. Biol. Psychiatry. 2007;61:1135–1140. doi: 10.1016/j.biopsych.2006.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gothelf D., Eliez S., Thompson T., Hinard C., Penniman L., Feinstein C., Kwon H., Jin S., Jo B., Antonarakis S.E., et al. COMT genotype predicts longitudinal cognitive decline and psychosis in 22q11.2 deletion syndrome. Nat. Neurosci. 2005;8:1500–1502. doi: 10.1038/nn1572. [DOI] [PubMed] [Google Scholar]
- 52.Gothelf D., Michaelovsky E., Frisch A., Zohar A.H., Presburger G., Burg M., viram-Goldring A., Frydman M., Yeshaya J., Shohat M., et al. Association of the low-activity COMT 158Met allele with ADHD and OCD in subjects with velocardiofacial syndrome. Int. J. Neuropsychopharmacol. 2007;10:301–308. doi: 10.1017/S1461145706006699. [DOI] [PubMed] [Google Scholar]
- 53.Lindsay E.A., Vitelli F., Su H., Morishima M., Huynh T., Pramparo T., Jurecic V., Ogunrinu G., Sutherland H.F., Scambler P.J., et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- 54.Nagy A., Rossant J., Nagy R., bramow-Newerly W., Roder J.C. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc. Natl Acad. Sci. USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simpson E.M., Linder C.C., Sargent E.E., Davisson M.T., Mobraaten L.E., Sharp J.J. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat. Genet. 1997;16:19–27. doi: 10.1038/ng0597-19. [DOI] [PubMed] [Google Scholar]
- 56.Agatsuma S., Lee M., Zhu H., Chen K., Shih J.C., Seif I., Hiroi N. Monoamine oxidase A knockout mice exhibit impaired nicotine preference but normal responses to novel stimuli. Hum. Mol. Genet. 2006;15:2721–2731. doi: 10.1093/hmg/ddl206. [DOI] [PubMed] [Google Scholar]
- 57.Kobayakawa K., Kobayakawa R., Matsumoto H., Oka Y., Imai T., Ikawa M., Okabe M., Ikeda T., Itohara S., Kikusui T., et al. Innate versus learned odour processing in the mouse olfactory bulb. Nature. 2007;450:503–508. doi: 10.1038/nature06281. [DOI] [PubMed] [Google Scholar]