

Abstract
BACKGROUND
Trophoblast invasion is a temporally and spatially regulated scheme of events that can dictate pregnancy outcome. Evidence suggests that the potent mitogen epidermal growth factor (EGF) regulates cytotrophoblast (CTB) differentiation and invasion during early pregnancy.
METHODS AND RESULTS
In the present study, the first trimester extravillous CTB cell line SGHPL-4 was used to investigate the signalling pathways involved in the motile component of EGF-mediated CTB migration/invasion. EGF induced the phosphorylation of the phosphatidylinositol 3-kinase (PI3-K)-dependent proteins, Akt and GSK-3β as well as both p42/44 MAPK and p38 mitogen-activated protein kinases (MAPK). EGF-stimulated motility was significantly reduced following the inhibition of PI3-K (P < 0.001), Akt (P < 0.01) and both p42/44 MAPK (P < 0.001) and p38 MAPKs (P < 0.001) but not the inhibition of GSK-3β. Further analysis indicated that the p38 MAPK inhibitor SB 203580 inhibited EGF-stimulated phosphorylation of Akt on serine 473, which may be responsible for the effect SB 203580 has on CTB motility. Although Akt activation leads to GSK-3β phosphorylation and the subsequent expression of β-catenin, activation of this pathway by 1-azakenpaullone was insufficient to stimulate the motile phenotype.
CONCLUSION
We demonstrate a role for PI3-K, p42/44 MAPK and p38 MAPK in the stimulation of CTB cell motility by EGF, however activation of β-catenin alone was insufficient to stimulate cell motility.
Keywords: epidermal growth factor, extravillous cytotrophoblast, motility, PI3-K, p38 mitogen-activated protein kinases
Introduction
Cytotrophoblasts (CTBs) play a pivotal role in the development and maintenance of a successful pregnancy. Extravillous CTBs invade the underlying decidua, surround and migrate into the wall of the uterine spiral arteries through mechanisms that involve trophoblast-mediated apoptosis of endothelial and smooth muscle cells of the vessel wall (Ashton et al., 2005; Harris et al., 2006). Remodelling of the maternal uterine arteries transforms them from small diameter, high resistance vessels into large diameter, low resistance vessels (Robertson et al., 1973). The resulting increase in maternal blood flow is required to meet the increased demands of the developing feto-placental unit. Failure of the extravillous trophoblasts to adequately invade the uterine wall results in poor vessel remodelling and a reduction in placental perfusion, characteristic of common pregnancy complications such as pre-eclampsia, intrauterine growth restriction and pre-term labour (Brosens et al., 1972). Conversely, uncontrolled invasion is indicative of choriocarcinoma.
Human CTB invasion is controlled both temporally and spatially and involves the coordination of a number of distinct processes, including the breakdown of extracellular components through the synthesis and release of matrix metalloproteinases (MMPs), as well as migration towards the maternal spiral arteries. An important component of the migratory processes is cellular motility. Motility can be considered as a cycle of events starting with the extension of cellular processes, frontal adhesion, transcellular contraction and finally rear extension release. Orchestration of these complex events can be mediated through integrin/matrix interactions and/or stimulation of growth factor receptor kinases by a number of external signals (Wells et al., 2002). We have previously reported that CTB motility is stimulated by hepatocyte growth factor (HGF) (Cartwright et al., 1999, 2002; Tse et al., 2002). More recently, we have established a role for epidermal growth factor (EGF) in regulating CTB migration (Barber et al., 2005; LaMarca et al., 2005, 2006).
EGF is a multifunctional growth factor that regulates a variety of fundamental cell properties, such as growth, differentiation, invasion and apoptosis in numerous cell types. Ligand binding induces receptor homo- or heterodimerization that is essential for the phosphorylation of multiple tyrosine (Tyr) residues and the activation of Tyr kinases. These Tyr residues provide docking sites for specific Src homology 2-containing proteins, such as phospholipase Cγ (Di Fiore et al., 1990; Olayioye et al., 2000; Yarden, 2001; Yarden et al., 2001). These interactions launch a variety of intracellular pathways that can involve mitogen-activated protein kinases (MAPKs) or the activation of phosphatidylinositol 3-kinase (PI3-K) (Olayioye et al., 2000; Yarden, 2001; Yarden and Sliwkowski, 2001). Phosphoinositides generated by PI3-K activity trigger activation of Akt kinases through direct binding to the pleckstrin homology domain and the subsequent phosphorylation of Akt at two conserved residues, serine473 (Ser473) and threonine308 (Thr308).
The aim of this study was to examine in more detail the EGF-mediated signalling events responsible for the motile component involved in CTB migration and invasion.
Materials and Methods
Cells and reagents
The human extravillous CTB cell line SGHPL-4 was derived from first trimester chorionic villous tissues. These cells are well characterized and share many characteristics with isolated primary cells, including the expression of cytokeratin-7, HLA class I antigen, HLA-G, BC-1 and CD9 (Cartwright et al., 1999; Cartwright and Balarajah, 2005). Cells were cultured in Ham's F10 Nutrient Mix (Invitrogen Life Technologies, Carlsbad, CA, USA) supplemented with 10% foetal bovine serum (FBS), penicillin G (100 U/ml), streptomycin (100 mg/ml) and 2 mM L-glutamine at 37°C in 5% CO2. Recombinant human EGF was purchased from R&D Systems, Inc. (Minneapolis, MN, USA). PD 98059, LY 294002, SB 203580, Akt inhibitor IV and 1-azakenpaullone were obtained from EMD Biosciences (Madison, WI, USA). The TCF β-catenin reporter plasmid, TOPFLASH (Kim et al., 2000) was obtained from Europa Bioproducts Ltd, Ely, UK.
Boyden chamber chemotaxis assay
A modified Boyden chamber assay was performed as previously described to examine cell migration (Lamarca et al., 2006). Briefly, 10 ng/ml of EGF diluted in serum-free medium was added to the lower compartment of the Boyden chamber (Neuro Probe, Inc., Gaithersburg, MD, USA), and SGHPL-4 cells were serum-starved in medium containing 0.5% FBS for 48 h. The cells were then pre-treated with dimethylsulphoxide (DMSO) (vehicle), 10 µM LY 294002 (specific inhibitor to PI3-K) or 50 µM PD 98059 (specific inhibitor to p42/44 MAPK) for 30 min, trypsinized and subsequently added to the upper compartment of the Boyden chamber at a density of 5.6 × 104cells/well. After a 6 h migration period at 37°C, the cells on the upper side of the membrane were wiped off and the migrated cells were visualized by staining the cells that extended across the porous membrane (Diff-Quik Stain Set, Dade Behring, Inc., Newark, DE, USA). The stained membranes were visualized using a Nikon TE300 inverted epifluorescent microscope (DP Controller v1.2.1.108, Olympus Optical Company, LTD; Nikon USA, Lewisville, TX) and migration was quantified by counting the nuclei that passed through the filter. Stained nuclei from six fields of view (100×) for each experimental condition were counted and the data were expressed as the average number of migrated cells.
Invasion assay
Cellular invasion of SGHPL-4 cells in response to various inhibitors was assessed using the quantitative FluoroBlok invasion assay (BD Discovery Labware, Bedford, MA, USA) according to the manufacturer's instructions. SGHPL-4 cells were serum-starved for 24 h, washed with 1× phosphate-buffered saline (PBS) and pre-treated with DMSO (vehicle), 10 µM LY 294002 or 50 µM PD 98059 for 30 min at 37°C. The cells were trypsinized, resuspended in serum-free medium and seeded onto Matrigel-coated 8 µm FluoroBlok porous membrane inserts at a density of 2.5 × 105 cells per insert. The inserts were lowered into individual wells of a 24-well plate that contained 10 ng/ml of EGF and the plates were incubated at 37°C for 20 h to allow for cell invasion. Then, the invading cells were fluorescently labelled with Calcein AM (Molecular Probes, Eugene, OR, USA) as described previously (Lamarca et al., 2006), and relative fluorescence units were obtained with a fluorescent microtiter plate reader (FLUOstar optima, BMG Labtech, Durham, NC) from four replicates per experimental condition.
Western blotting
SGHPL-4 cells were serum-starved in 0.5% FBS/HamF10 for 24 h at 37°C. Cells were then treated in the absence of serum with 10 ng/ml of EGF or vehicle control (PBS) for 0, 5, 15, 30 or 60 min. Where indicated, cells were pre-treated with DMSO or the appropriate inhibitor for 30 min at 37°C prior to the addition of EGF. Cells were harvested in 1× RIPA (radioimmunoprecipitation assay) buffer containing a protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN, USA) and 1 mM phenylmethylsulphonyl fluoride (Sigma, St Louis, MO, USA), lysates were sheared 3× with a 27G1/2 needle and cell debris was removed by centrifugation at 18 000 g at 4°C. Total protein concentrations were estimated with the Micro BCA™ Protein Assay Kit (Pierce, Milwaukee, WI, USA) and an equal amount of total protein in each well was resolved on 8–10% sodium dodecyl sulphate (SDS)–polyacrylamide gels before transfer to polyvinylidene diflouride membranes (Amersham Biosciences, Piscataway, NJ, USA). Non-specific reactivity was blocked with 5% non-fat dried milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 h at room temperature. Blots were incubated overnight with the following antibodies: rabbit anti-p42/44-Thr202/Thr204, rabbit anti-p42-total, rabbit anti-p38-Thr180/Tyr182, rabbit anti-phospho-Akt-Ser473 or rabbit anti-GSK-3β-Ser9 (1:1000; Cell Signalling Technology, Beverly, MA, USA). The blots were then washed in TBST and incubated with goat or donkey anti-rabbit IgG conjugated to horseradish peroxidase (1:5000; Sigma or Amersham Biosciences, respectively) for 1 h at room temperature, and antigen–antibody complexes were detected using an enhanced chemiluminescence system (Amersham Biosciences). Blots were subsequently stripped in buffer containing 62.5 mM Tris pH 6.7, 2% SDS and 100 mM β-mercaptoethanol and tested for immunoreactivity to a rabbit polyclonal antibody to human β-actin (Sigma). Where indicated, western blots were scanned and the integrated intensity of each band determined using Scion Image for Windows version Beta 4.02. Results were expressed as a ratio to β-actin in the same sample.
Motility assays
Time-lapse digital image microscopy using an Olympus IX70 inverted microscope equipped with a Hamamatsu C4742-95 digital camera was performed to examine cell motility (Cartwright et al., 1999). The microscope and stage were enclosed within a heated (37°C) humidified atmosphere of 5% CO2 (Solent Scientific, UK). SGHPL-4 cells were incubated in medium containing 0.5% FBS for 24 h, at which time they were transferred to the microscope chamber in fresh medium containing vehicle (PBS) or 10 ng/ml of EGF. Where appropriate, cells were pre-treated with the indicated inhibitors for 30 min at 37°C or the vehicle (DMSO) and subsequently transferred to the microscope chamber in the presence or absence of EGF. In accordance with previous studies, 10 ng/ml was found to be the optimal concentration of EGF to stimulate motility (data not shown) and was therefore used for the remainder of the study. Images were captured every 15 min over a period of 6 h and motility was quantified using Image Pro-Plus software (Media Cybernetics, USA). The distance moved was determined by randomly analysing at least 60 cells per treatment, and the experiment was performed a total of three times. Visualizing the cells by this method ensured that only viable cells were analysed throughout the 6 h period.
Transfections and luciferase assays
Transient transfections of SGHPL-4 cells with the β-catenin reporter plasmid TOPFLASH were performed in 6-well plates in the presence of 5% FBS using poly-l-ornithine (15 000 MW) from Sigma mixed with DNA at a ratio of 0.9:1 (w/w). Cells were incubated with the poly-l-ornithine/DNA mixture at a DNA concentration of 2.5 µg/ml at 37°C for 5 h. DMSO (30% in RPMI media) was added for 1 min, washed twice with PBS and fresh media (Hams F10, 10% FBS) was added. Analysis of the cells was carried out 24 h post-transfection. Luciferase assays were performed according to the manufacturer's instructions (BD Bioscience).
Statistical analyses
Data are presented as the mean + SEM. Data from treated groups were compared with vehicle control groups and significant differences were determined by one-way analysis of variance followed by Tukey's post hoc t-test (GraphPad Prism© Home, San Diego, CA, USA).
Results
PI3-k and p42/44 activation are required for EGF-mediated chemotaxis and invasion
We have previously reported that EGF induces in vitro CTB migration and invasion in a dose-dependent fashion (Barber et al., 2005; Lamarca et al., 2006). EGF was a potent chemotactic factor when used at 10 ng/ml in the Boyden chamber, and likewise stimulated the invasion of CTBs through an extracellular matrix. In the present study, these assays were used to investigate the regulatory mechanisms involving EGF and CTB invasiveness. EGF-stimulated SGHPL-4 cell migration and invasion (Fig. 1) was significantly inhibited by pre-treatment with specific inhibitors to p42/44 MAPK (PD 98059) or PI3-K (LY 294002), as compared with vehicle-treated cells. Chemotaxis was inhibited >5-fold in PD 98059- and LY 294002-treated cells when measured in the Boyden chamber (P < 0.001; Fig. 1a). Similarly, invasion through Matrigel was also significantly inhibited (P < 0.001) in SGHPL-4 cells pre-treated with these inhibitors as compared with vehicle-treated cells (Fig. 1b). These data implicate the involvement of both p42/44 MAPK and PI3-K signalling in EGF-regulated CTB chemotaxis and invasion and establish the validity of the model for subsequent studies of the motile mechanisms involved.
Figure 1:
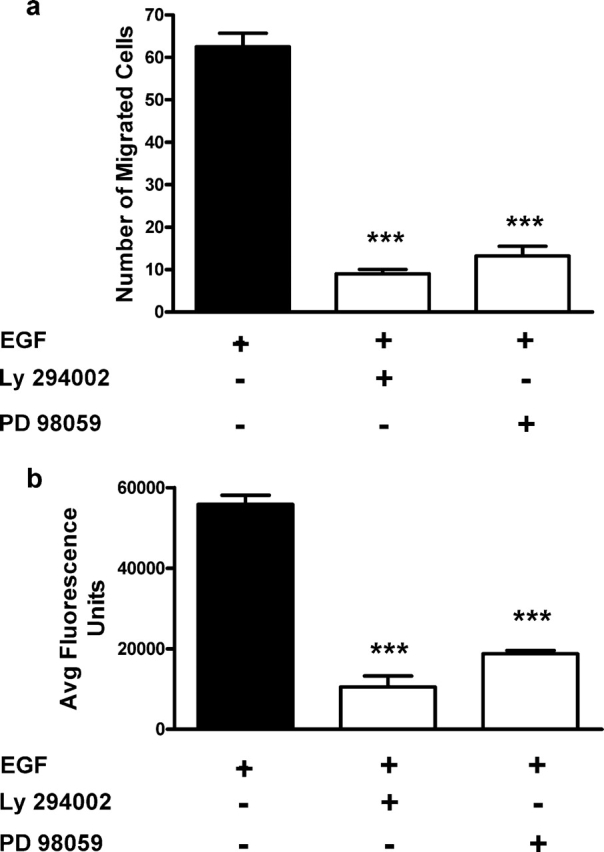
Involvement of mitogen-activated protein kinases (MAPK) and phosphatidylinositol 3-kinase (PI3-K) signalling in cytotrophoblast (CTB) chemotaxis and invasion.
SGHPL-4 cells were pre-treated with 10 µM LY 294002-(specific inhibitor to PI3-K) or 50 µM PD 98059 (specific inhibitor to p42/44 MAPK) for 30 min, and cell migration and invasion assays were performed. Treatment with either LY 294002 or PD 98059 significantly inhibited epidermal growth factor (EGF)-induced (a) chemotaxis in the Boyden chamber and (b) invasion through Matrigel (***P < 0.001). The results presented are the mean + SEM from a representative experiment and each experiment was performed a total of four times.
EGF stimulates CTB motility
An important component of CTB migration and invasion is cellular motility. We therefore determined whether PI3-K and or the MAPK pathways were involved in this aspect of the invasive process. We have previously reported that HGF stimulates not only invasion, but CTB motility through a mechanism involving PI3-K and MAPK activation (Cartwright et al., 1999, 2002). To determine whether EGF shared these characteristics with HGF, time-lapse microscopy was used to examine the motility of SGHPL-4 cells in both the presence and the absence of EGF.
To investigate whether either PI3-K or p42/44 MAPK were involved in SGHPL-4 motility, time-lapse microscopy was performed after pre-treatment with specific inhibitors to these pathways. Inhibition of either PI3-K or p42/44 MAPK significantly reduced the motility of SGHPL-4 cells in a dose-dependent manner (Fig. 2a and b). In addition, treatment with SB 203580, a specific inhibitor of p38 MAPK, inhibited CTB motility in a dose-dependent manner with statistical significance achieved at 10 µM, the lowest dose tested (Fig. 2c).
Figure 2:
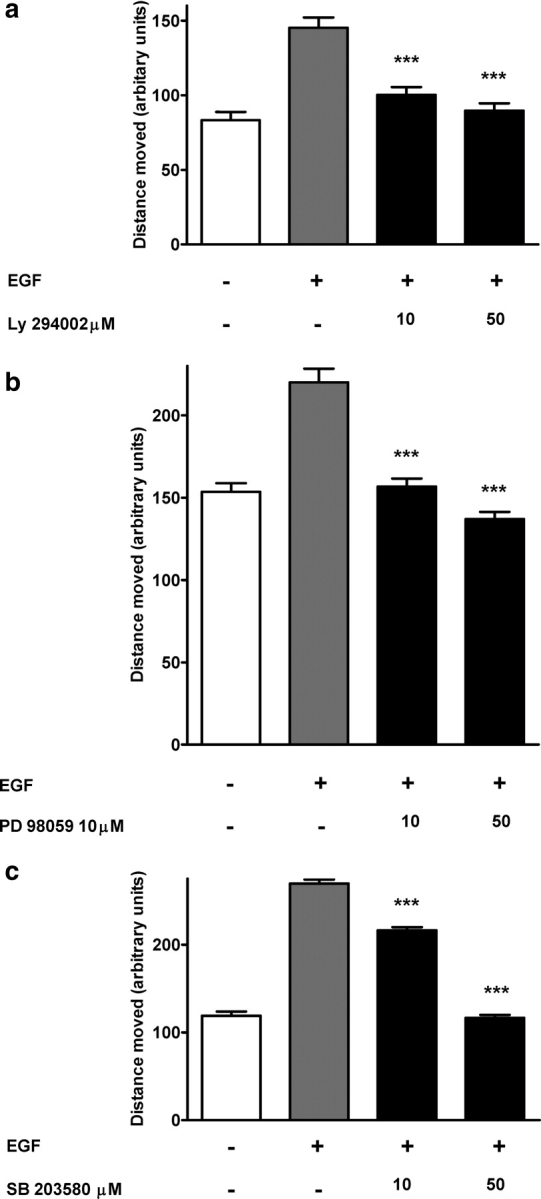
Regulation of CTB motility by PI3-K, p42/44 MAPK and p38 MAPK signalling pathways.
Time-lapse digital microscopy was performed on cells that were pre-treated for 30 min with the indicated doses of either vehicle (dimethylsulphoxide, DMSO) or (a) LY 294002, (b) PD 98059 or (c) SB 203580 (p38 MAPK inhibitor). Cells were then stimulated with 10 ng/ml EGF and motility was monitored over a 6 h period. The data presented in each panel are the mean distance moved + SEM from three independent experiments. The effect of each inhibitor was assessed independently and therefore was compared with contemporaneous control and EGF wells (***P < 0.001).
EGF induces the phosphorylation of MAPKs and PI3-K-regulated proteins
Our functional studies established a role for the PI3-K, p38 and p42/44 MAPK pathways in the regulation of EGF-stimulated cell motility. In order to dissect these pathways and identify possible points of interaction, we studied the time course of activation for each pathway. In these studies, phosphorylation of both p42/44 MAPK and downstream targets of PI3-K, Akt and GSK-3β were activated within 5 min of stimulation (Fig. 3). Although phosphorylation of Akt (Ser473) and p38 (Thr180/Tyr182) MAPK remained greater than the control for up to 60 min, there was a rapid decline after ∼10 min of stimulation (Fig. 3a and b). In contrast, the phosphorylation state of both p42/44 MAPK and GSK-3β remained high throughout the experimental period (Fig. 3c and d).
Figure 3:

PI3-K-mediated p42/44 MAPK activation.
SGHPL-4 cells treated with 10 ng/ml of EGF or vehicle (PBS) for 0, 5, 15, 30 or 60 min. Cell lysates were collected and equal amounts of total protein were subjected to western-blot analyses for (a) phospho-p42/44, (b) phospho-Akt, (c) phospho-p38 MAPK, (d) phospho-GSK-3β. Human β-actin was used as an internal loading control. The results presented are representative of at least three independent experiments.
Akt involvement in EGF-stimulated trophoblast motility
To further explore the role of the PI3-K/Akt pathway in EGF-mediated cell motility, a specific Akt inhibitor was used. Pre-treatment with Akt inhibitor IV resulted in dose-dependent inhibition of trophoblast motility (Fig. 4). Notably, significant trophoblast motility remained when treated with higher concentrations of the Akt inhibitor. However, increasing the concentration of Akt inhibitor IV beyond 2.5 µM had a significant effect on CTB viability.
Figure 4:

Regulation of CTB motility by Akt.
Time-lapse digital microscopy was performed on cells pre-treated with the indicated doses of either (a) Akt inhibitor IV or vehicle (DMSO) for 30 min and then stimulated with 10 ng/ml of EGF or PBS. Motility was monitored for 6 h. Data are presented as the mean + SEM from three independent experiments. Data from treated groups were compared with vehicle control groups and significant differences were determined by one-way analysis of variance followed by Tukey's post hoc t-test (*P < 0.05, **P < 0.01).
Inhibition of GSK-3β does not affect EGF-stimulated trophoblast motility
Activation of Akt has been implicated in the activation of a number of cellular targets including GSK-3β, a protein kinase that phosphorylates a number of downstream targets including the multifunctional transcription factor β-catenin (Frame et al., 2001). In the active state (unphosphorylated), GSK-3β phosphorylates β-catenin, which targets this transcription factor for proteasomal degradation (Frame and Cohen, 2001). In the inactive phosphorylated form, GSK-3β is unable to phosphorylate β-catenin, which allows for the stabilization of β-catenin and the transcriptional activation of downstream target genes (Frame and Cohen, 2001). Our results demonstrate that GSK-3β was rapidly phosphorylated, and therefore inactivated, following treatment with EGF (Fig. 3d). Furthermore, Fig. 5 shows that GSK-3β phosphorylation was inhibited by the PI3-K inhibitor LY 294002.
Figure 5:

EGF-mediated GSK-3β.
SGHPL-4 cells were pre-treated with 50 µM LY 294002 for 30 min. Cells were subsequently treated with 10 ng/ml of EGF or vehicle (PBS) for 30 min. Cell lysates were collected and equal amounts of total protein per well were subjected to western-blot analyses for phospho-GSK-3β. Human β-actin was used as an internal control to verify equal sample loading. The top panel is the integrated density of the individual bands corrected using actin and is the mean + SEM of three separate experiments. The lower panel shows a representative blot of both GSK-3β and actin.
To establish whether phosphorylation of GSK-3β resulted in β-catenin activation, we used the luciferase reporter construct containing the β-catenin/Tcf binding site (TOPFLASH). Stimulation of SGHPL-4 cells with EGF led to a 1.75-fold increase in reporter activity (Fig. 6a). As a result of inhibiting basal GSK-3β activity using 1-azakenpaullone, there was a 2-fold increase in reporter activity at least equivalent to that obtained with 10 ng/ml EGF. Inhibition of Akt using Akt inhibitor IV resulted in a significant dose-dependent inhibition of reporter activity at concentrations previously shown to significantly inhibit trophoblast motility. However, activation of β-catenin alone using 1-azakenpaullone at different concentrations was unable to stimulate trophoblast cell motility (Fig. 6b).
Figure 6:

EGF-mediated β-catenin activation.
(a) SGHPL-4 cells were transiently transfected with the β-catenin reporter plasmid TOPFLASH. Twenty-four hours post-transfection, cells were treated with EGF (6 h) in the presence or absence of the Akt inhibitor IV (1.25 and 2.5 µM). Independent activation of β-catenin was demonstrated using the GSK-3β inhibitor 1-azakenpaullone (20 and 50 nM). (b) The effect of β-catenin activation on SGHPL-4 cell motility was determined following inhibition of GSK-3β with 1-azakenpaullone. Values shown are the mean + SEM of triplicate experiments *P < 0.05, ***P < 0.001.
EGF activates p42/44 MAPK in a PI3-K-independent fashion
Our studies indicate that MAPK (both p42/44 and p38) and PI3-K (Akt) signalling are important regulators of CTB motility. To address whether the MAPKs and PI3-K pathways interact with each other, western-blot analysis was performed for phospho-Akt (Ser473) (Fig. 7a and b) and phospho-p42/44 MAPK (Fig. 7c). Inhibition of PI3-K with LY 294002 inhibited the phosphorylation of Akt (Ser473), whereas the p42/44 MAPK inhibitor PD 98059 had no effect (Fig. 7b and c). Interestingly inhibition of p38 MAPK by SB 203580 had a significant inhibitory effect on Akt activation and the phosphorylation of ser473 in the absence of EGF. The addition of DMSO had no effect on either basal or EGF-stimulated phosphorylation of either Akt or p42/44 MAPK. Basal phosphorylation of p42/44 MAPK was unaffected by inhibition with PD 98059; however, inhibition of PI3-K with LY 294002 resulted in a 50% reduction in the phosphorylation of Akt. This result is consistent with our previous finding that LY 294002 inhibits basal trophoblast motility (Cartwright et al., 2002). A similar but smaller effect on Akt phosphorylation was also observed following the inhibition of p38 MAPK.
Figure 7:
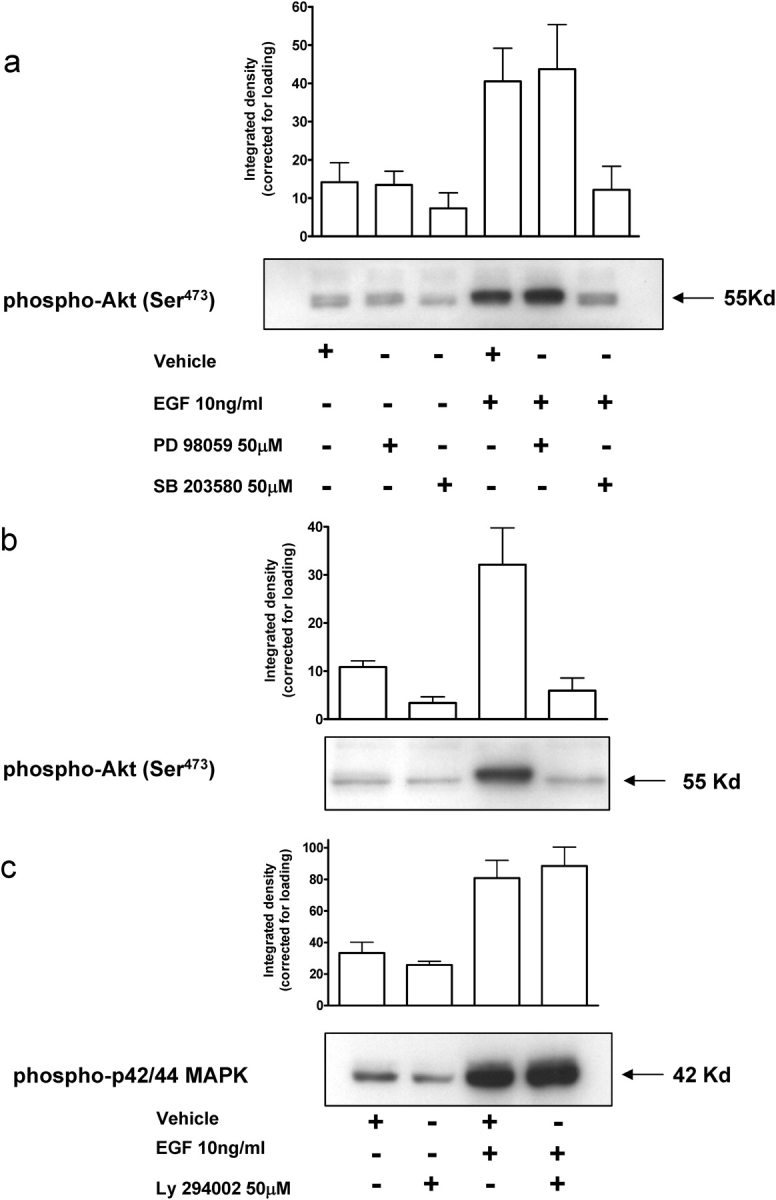
Phosphorylation of MAPKs and PI3-K-regulated proteins by EGF.
SGHPL-4 cells were treated with either the appropriate inhibitor or the vehicle control (DMSO) for 60 min. Cells were then stimulated with 10 ng/ml of EGF for 30 min in the presence or absence of (a) 50 µM PD 98059 or SB 203580 (b and c) 50 µmM LY 2984002. Cell lysates were collected and equal amounts of total protein were subjected to western-blot analyses. The bar chart represents the integrated density of the individual bands corrected for actin and is the mean + SEM of three separate experiments. The figure shows a representative example for each inhibitor.
Discussion
Invasion is a complex biological process involving the coordinated regulation of extracellular matrix remodelling through the secretion and activation of MMPs and stimulation of cellular motility. Although there have been studies examining the signal transduction mechanisms involved in EGF-stimulated trophoblast migration and invasion, very little is known about the motile component of these processes. Using time-lapse microscopy, pharmacological inhibitors and western-blot analysis, we have identified two independent pathways responsible for the regulation of trophoblast motility by EGF. The first results in the activation of p42/44 MAPK; the second is mediated through the phosphorylation of Akt and is independently activated by both PI3-K and more interestingly p38 MAPK.
EGF stimulates the migration and invasion of first and second trimester human CTBs as well as the secretion of MMP-2 and -9 (Bass et al., 1994; Staun-Ram et al., 2004; Anteby et al., 2004). In agreement with these studies and those of Qiu et al. (2004), who used the transformed trophoblast cell line HTR8neo, we were able to demonstrate EGF-stimulated migration and invasion using the first trimester extravillous CTB cell line SGHPL-4. Examination of the pathways involved using the PI3-K inhibitor LY 294002 and the p42/44 MAPK inhibitor PD 98059 corroborated the findings in previous studies (Qiu et al., 2004).
Having established a role for EGF in the regulation of migration and invasion and thereby validating our model, we proceeded to examine these processes in more detail by identifying post-receptor events responsible for the motile component of CTB invasiveness. Using time-lapse microscopy, we assessed cell movement over time. As we previously demonstrated, EGF induced CTB motility (Barber et al., 2005), and using molecular and pharmacological probes, we demonstrate a role for PI3-K, p42/44 MAPK and p38 MAPK in this process. Subsequent analysis indicated PI3-K dependent phosphorylation of Akt (Ser473), and the involvement of Akt in EGF-regulated motility was confirmed using Akt inhibitor IV. To further explore the consequences of Akt activation, we investigated the involvement of the downstream target GSK-3β. GSK-3β is a multi-functional enzyme involved in the regulation of glycogen and protein synthesis and is known to be regulated by EGF in fibroblasts and MCF-7 cells. It is a well-established component of the Wnt signalling pathway and is also thought to play an important role in many other cellular processes, such as proliferation, differentiation and motility (Frame and Cohen, 2001). Most recently, it has been shown to regulate HGF-mediated trophoblast cell survival and invasive differentiation in trophoblasts (Dash et al., 2005; Pollheimer et al., 2006). Unlike most protein kinases, GSK-3β activity is inhibited following phosphorylation and consequently, a number of transcription factors are activated including β-catenin (Frame and Cohen, 2001). β-catenin is a multi-functional protein that is important for cell adhesion, but also mediates cytoplasmic effects leading to transcriptional activation. In the absence of specific activating signals, β-catenin is located either at adherin junctions or bound in a high molecular weight multi-protein complex that includes GSK-3β. In the absence of external stimuli, β-catenin is phosphorylated by GSK-3β, and this retains the β-catenin within the cytoplasm where it undergoes ubiquitination and subsequent degradation (Kimelman and Xu, 2006). Inhibition of GSK-3β kinase activity by its phosphorylation prevents the phosphorylation of β-catenin that is then able to translocate to the nucleus and associate with transcription factors of the Lef/Tcf family to initiate gene expression (Frame and Cohen, 2001).
In the present study, we demonstrate that in trophoblasts EGF phosphorylates GSK-3β, and using a luciferase reporter construct containing the β-catenin/Tcf binding site (TOPFLASH), stimulates β-catenin gene expression. Further analysis showed that this process was mediated through the activation of the PI3-K/Akt pathway. However, when this pathway was activated independently of EGF receptor by inhibiting GSK-3β, there was no effect on trophoblast motility. These results would indicate that either inactivation of GSK-3β and the subsequent increase in gene transcription by β-catenin was not involved in the regulation of trophoblast cell motility or that activation alone is insufficient to stimulate motility. Whether parallel activation signalling pathways, such as the MAPKs, are required warrants further investigation.
Another known signalling molecule responsive to EGF is p38 MAPK. This protein kinase is a member of the MAPK family and has largely been associated with cellular stress responses and apoptosis. In the placenta, activation of p38 MAPK by EGF may play a role in the differentiation of third trimester CTBs (Johnstone et al., 2005). However, its involvement in cell migration and particularly trophoblast cell motility is not well established. In this study, we demonstrate that inhibition of p38 MAPK by SB 203580, the p38α and p38β isoform-specific MAPK inhibitor, had a significant inhibitory effect on trophoblast cell motility. These results were contrary to our previous findings involving HGF stimulation where similar doses had no significant effect. In vascular smooth muscle cells, p38 MAPK exists in a complex with Akt, and activation of p38 MAPK leads to recruitment of MAPKAPK-2 to the p38 MAPK–Akt complex, its phosphorylation, and thus activation. MAPKAPK-2 then phosphorylates Akt on Ser473, leading to full Akt activation (Taniyama et al., 2004). A summary of our results are presented in Fig. 8.
Figure 8:

A schematic depicting the regulation of trophoblast cell motility by EGF.
Stimulation of CTB with EGF via the EGF receptor (EGFR) results in the activation of PI3-K and p42/44 MAPK. Inhibition of either pathway results in inhibition of EGF-stimulated motility. Akt is phosphorylated and therefore activated by EGF through the activation of PI3-K but not p42/44 MAPK. Inhibition of p38 MAPK inhibits both phosphorylation of Akt and CTB motility. Although EGF activates β-catenin, direct activation of β-catenin had no effect on motility.
In conclusion, EGF stimulates extravillous trophoblast cell motility. Following stimulation with EGF, there was an independent activation of PI3-K and p42/44 MAPK, and inhibition of both of these pathways had a significant inhibitory effect on trophoblast cell motility. Inhibition of PI3-K but not p42/44 MAPK led to inhibition of Akt phosphorylation. Interestingly, inhibition of p38 MAPK also resulted in the inhibition of EGF-induced trophoblast motility and Akt phosphorylation, indicating convergence of the PI3-K and p38 MAPK pathways in the control of trophoblast cell motility. As inhibition of β-catenin had no effect on EGF-stimulated CTB motility, the targets for Akt activation under these circumstances remain to be defined.
Funding
This work was supported by a competitive award from Boehringer Ingelheim (HLL), the National Institutes of Health (HD045768; CAM) and the British Heart Foundation (PD).
Acknowledgements
We would like to thank Dr Judith Cartwright for helpful discussions during the course of this study.
References
- Anteby EY, Greenfield C, Natanson-Yaron S, Goldman-Wohl D, Hamani Y, Khudyak V, Ariel I, Yagel S. Vascular endothelial growth factor, epidermal growth factor and fibroblast growth factor-4 and -10 stimulate trophoblast plasminogen activator system and metalloproteinase-9. Mol Hum Reprod. 2004;10:229–235. doi: 10.1093/molehr/gah031. [DOI] [PubMed] [Google Scholar]
- Ashton SV, Whitley GS, Dash PR, Wareing M, Crocker IP, Baker PN, Cartwright JE. Uterine spiral artery remodeling involves endothelial apoptosis induced by extravillous trophoblasts through Fas/FasL interactions. Arterioscler Thromb Vasc Biol. 2005;25:102–108. doi: 10.1161/01.ATV.0000148547.70187.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber KJ, Franklyn JA, McCabe CJ, Khanim FL, Bulmer JN, Whitley GS, Kilby MD. The in vitro effects of triiodothyronine on epidermal growth factor-induced trophoblast function. J Clin Endocrinol Metab. 2005;90:1655–1661. doi: 10.1210/jc.2004-0785. [DOI] [PubMed] [Google Scholar]
- Bass KE, Morrish D, Roth I, Bhardwaj D, Taylor R, Zhou Y, Fisher SJ. Human cytotrophoblast invasion is up-regulated by epidermal growth factor: evidence that paracrine factors modify this process. Dev Biol. 1994;164:550–561. doi: 10.1006/dbio.1994.1223. [DOI] [PubMed] [Google Scholar]
- Brosens IA, Robertson WB, Dixon HG. The role of the spiral arteries in the pathogenesis of preeclampsia. Obstet Gynecol Annu. 1972;1:177–191. [PubMed] [Google Scholar]
- Cartwright JE, Balarajah G. Trophoblast interactions with endothelial cells are increased by interleukin-1beta and tumour necrosis factor alpha and involve vascular cell adhesion molecule-1 and alpha4beta1. Exp Cell Res. 2005;304:328–336. doi: 10.1016/j.yexcr.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Cartwright JE, Holden DP, Whitley GS. Hepatocyte growth factor regulates human trophoblast motility and invasion: a role for nitric oxide. Br J Pharmacol. 1999;128:181–189. doi: 10.1038/sj.bjp.0702757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright JE, Tse WK, Whitley GS. Hepatocyte growth factor induced human trophoblast motility involves phosphatidylinositol-3-kinase, mitogen-activated protein kinase, and inducible nitric oxide synthase. Exp Cell Res. 2002;279:219–226. doi: 10.1006/excr.2002.5616. [DOI] [PubMed] [Google Scholar]
- Dash PR, Whitley GS, Ayling LJ, Johnstone AP, Cartwright JE. Trophoblast apoptosis is inhibited by hepatocyte growth factor through the Akt and beta-catenin mediated up-regulation of inducible nitric oxide synthase. Cell Signal. 2005;17:571–580. doi: 10.1016/j.cellsig.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Di Fiore PP, Segatto O, Taylor WG, Aaronson SA, Pierce JH. EGF receptor and erbB-2 tyrosine kinase domains confer cell specificity for mitogenic signaling. Science. 1990;248:79–83. doi: 10.1126/science.2181668. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LK, Keogh RJ, Wareing M, Baker PN, Cartwright JE, Aplin JD, Whitley GS. Invasive trophoblasts stimulate vascular smooth muscle cell apoptosis by a fas ligand-dependent mechanism. Am J Pathol. 2006;169:1863–1874. doi: 10.2353/ajpath.2006.060265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone ED, Sibley CP, Lowen B, Guilbert LJ. Epidermal growth factor stimulation of trophoblast differentiation requires MAPK11/14 (p38 MAP kinase) activation. Biol Reprod. 2005;73:1282–1288. doi: 10.1095/biolreprod.105.044206. [DOI] [PubMed] [Google Scholar]
- Kim K, Pang KM, Evans M, Hay ED. Overexpression of beta-catenin induces apoptosis independent of its transactivation function with LEF-1 or the involvement of major G1 cell cycle regulators. Mol Biol Cell. 2000;11:3509–3523. doi: 10.1091/mbc.11.10.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelman D, Xu W. Beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene. 2006;25:7482–7491. doi: 10.1038/sj.onc.1210055. [DOI] [PubMed] [Google Scholar]
- LaMarca HL, Ott CM, Honer Zu Bentrup K, Leblanc CL, Pierson DL, Nelson AB, Scandurro AB, Whitley GS, Nickerson CA, Morris CA. Three-dimensional growth of extravillous cytotrophoblasts promotes differentiation and invasion. Placenta. 2005;26:709–720. doi: 10.1016/j.placenta.2004.11.003. [DOI] [PubMed] [Google Scholar]
- LaMarca HL, Nelson AB, Scandurro AB, Whitley GS, Morris CA. Human cytomegalovirus-induced inhibition of cytotrophoblast invasion in a first trimester extravillous cytotrophoblast cell line. Placenta. 2006;27:137–147. doi: 10.1016/j.placenta.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollheimer J, Loregger T, Sonderegger S, Saleh L, Bauer S, Bilban M, Czerwenka K, Husslein P, Knofler M. Activation of the canonical wingless/t-cell factor signaling pathway promotes invasive differentiation of human trophoblast. Am J Pathol. 2006;168:1134–1147. doi: 10.2353/ajpath.2006.050686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Q, Yang M, Tsang BK, Gruslin A. Both mitogen-activated protein kinase and phosphatidylinositol 3-kinase signalling are required in epidermal growth factor-induced human trophoblast migration. Mol Hum Reprod. 2004;10:677–684. doi: 10.1093/molehr/gah088. [DOI] [PubMed] [Google Scholar]
- Robertson WB, Brosens IA, Dixon HG. Placental bed vessels. Am J Obstet Gynecol. 1973;117:294–295. doi: 10.1016/0002-9378(73)90655-8. [DOI] [PubMed] [Google Scholar]
- Staun-Ram E, Goldman S, Gabarin D, Shalev E. Expression and importance of matrix metalloproteinase 2 and 9 (MMP-2 and -9) in human trophoblast invasion. Reprod Biol Endocrinol. 2004;2:59. doi: 10.1186/1477-7827-2-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniyama Y, Ushio-Fukai M, Hitomi H, Rocic P, Kingsley MJ, Pfahnl C, Weber DS, Alexander RW, Griendling KK. Role of p38 MAPK and MAPKAPK-2 in angiotensin II-induced Akt activation in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2004;287:C494–C499. doi: 10.1152/ajpcell.00439.2003. [DOI] [PubMed] [Google Scholar]
- Tse WK, Whitley GS, Cartwright JE. Transforming growth factor-beta1 regulates hepatocyte growth factor-induced trophoblast motility and invasion. Placenta. 2002;23:699–705. doi: 10.1016/s0143-4004(02)90866-0. [DOI] [PubMed] [Google Scholar]
- Wells A, Kassis J, Solava J, Turner T, Lauffenburger DA. Growth factor-induced cell motility in tumor invasion. Acta Oncol. 2002;41:124–130. doi: 10.1080/028418602753669481. [DOI] [PubMed] [Google Scholar]
- Yarden Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37(Suppl 4):S3–S8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]