

Abstract
Experimental evidence from mutant or genetically altered mice indicates that the formation of barrels and the proper maturation of thalamocortical (TC) synapses in the primary somatosensory (barrel) cortex depend on mechanisms mediated by neural activity. Type 1 adenylyl cyclase (AC1), which catalyzes the formation of cAMP, is stimulated by increases in intracellular Ca2+ levels in an activity-dependent manner. The AC1 mutant mouse, barrelless (brl), lacks typical barrel cytoarchitecture, and displays presynaptic and postsynaptic functional defects at TC synapses. However, because AC1 is expressed throughout the trigeminal pathway, the barrel cortex phenotype of brl mice may be a consequence of AC1 disruption in cortical or subcortical regions. To examine the role of cortical AC1 in the development of morphological barrels and TC synapses, we generated cortex-specific AC1 knock-out (CxAC1KO) mice. We found that neurons in layer IV form grossly normal barrels and TC axons fill barrel hollows in CxAC1KO mice. In addition, whisker lesion-induced critical period plasticity was not impaired in these mice. However, we found quantitative reductions in the quality of cortical barrel cytoarchitecture and dendritic asymmetry of layer IV barrel neurons in CxAC1KO mice. Electrophysiologically, CxAC1KO mice have deficits in the postsynaptic but not in the presynaptic maturation of TC synapses. These results suggest that activity-dependent postsynaptic AC1–cAMP signaling is required for functional maturation of TC synapses and the development of normal barrel cortex cytoarchitecture. They also suggest that the formation of the gross morphological features of barrels is independent of postsynaptic AC1 in the barrel cortex.
Keywords: somatosensory cortex, activity dependent, AC1, barrel, thalamocortical, conditional knock-out
Introduction
Since the discovery of “barrels” in 1970 (Woolsey and Van der Loos, 1970), the somatosensory (barrel) cortex of rodents has emerged as a useful model system to examine the mechanism underlying development of complex neural circuits typical of the mammalian brain. In rodents, the pattern of facial whiskers on the snout is recapitulated throughout the ascending trigeminal pathway in the form of “barellettes” in the brainstem, “barreloids” in the ventrobasal (VB) thalamus, and “barrels” in layer IV of the somatosensory cortex. Each mature cortical barrel is composed of a barrel wall and a barrel hollow. During the first postnatal week of development, thalamocortical (TC) axons cluster within the barrel hollow, whereas layer IV neurons arrange their cell bodies to form the barrel wall and acquire an asymmetric morphology by preferentially orienting their dendrites toward the barrel hollow to synapse with TC axons.
Maturation of TC synapses takes place early in postnatal life, when barrels are just forming. One of the proposed mechanisms for synapse maturation during map formation is by Hebbian-based synaptic plasticity mechanisms, such as long-term potentiation (LTP), which is thought to selectively strengthen topographically appropriate synapses through the addition of AMPA receptors at the postsynaptic density (Malinow and Malenka, 2002). Consistent with this model, LTP can be reliably induced at immature TC synapses, and the ratio of AMPA to NMDA receptor-mediated currents (AMPA/NMDA ratio) increase with age (Crair and Malenka, 1995).
Barrelless (brl) mice, a spontaneous mutant mouse line that lacks a barrel map, have a mutation in the adenylyl cyclase 1 (adcy1 or AC1) gene (Welker et al., 1996; Abdel-Majid et al., 1998). AC1 is one of two adenylyl cyclases that are activated by increases in Ca2+ (Wang and Storm, 2003), which at dendritic spines takes place mostly by stimulation of NMDA receptors during neuronal activity (Kovalchuk et al., 2000). AC1 activation leads to an increase in cAMP production, a second messenger whose primary target is cAMP-dependent protein kinase (PKA). In addition to brl mice, knock-out (KO) mice that lack NMDA receptor function only in cortical excitatory neurons (Cx-NR1KO mice) and KO mice that lack the type IIβ regulatory subunit of PKA (PKARIIβ KO mice) have barrel map defects (Iwasato et al., 2000; Inan et al., 2006; Watson et al., 2006). These reports suggest that the function of signaling molecules immediately upstream and downstream of AC1 activation is necessary for barrel map formation.
Both brl and PKARIIβ KO mice have postsynaptic deficits at TC synapses in which LTP induction and the developmental increase in AMPA receptor response at TC synapses are impaired (Lu et al., 2003; Inan et al., 2006). Presynaptic defects are also found at brl TC synapses in which neurotransmitter release efficacy is reduced (Lu et al., 2006). Together, this evidence suggests that barrel map formation relies on proper TC synaptic maturation and that the AC1–PKA signaling pathway regulates the structural and functional maturation of the barrel cortex layer IV neurons. However, because AC1 is present throughout the trigeminal pathway during early postnatal development (Matsuoka et al., 1997; Nicol et al., 2005), it remains unclear whether lack of cortical AC1, subcortical AC1, or both is the source of the defects observed in brl mice.
To investigate the role of cortical AC1 in the development of barrel cytoarchitecture and the functional maturation of TC synapses, we generated cortex-specific KO mice in which AC1 is absent in cortical excitatory neurons (CxAC1KO) but not in any subcortical somatosensory centers. We found that the loss of AC1 in layer IV neurons is sufficient to reproduce the postsynaptic, but not presynaptic, electrophysiological deficits observed in brl TC synapses. Morphologically, while grossly normal barrels are formed, layer IV neurons display reduced dendritic asymmetry in CxAC1KOs compared with controls and layer IV neuron barrel cytoarchitecture is degraded in CxAC1KO mice. Thus, cortical AC1 has specific roles in the functional and morphological maturation of barrel cortex layer IV.
Materials and Methods
Animals.
Emx1-Cre (knock-in ΔNeo, KΔN) mice in which the pgk-neo selection marker gene had been deleted from the locus have been described previously (Iwasato et al., 2004). The AC1-flox allele, in which AC1 exon1 containing transcriptional and translational initiation sites was flanked by a pair of loxPs, was generated by using homologous recombination in E14 embryonic stem cells and deleting a pgk-neo by flp/FRT recombination. AC1-null allele was generated by expressing Cre recombinase in germline of AC1-flox mice. CxAC1KO (Emx-Cre; AC1flox/−) and control (Emx-Cre; AC1+/−, AC1flox/−, AC1flox/+, and AC1+/−) mice were obtained by crossing Emx1-Cre; AC1−/− or Emx1-Cre; AC1+/− male and AC1flox/flox or AC1flox/+ females. In a few histological analyses, Emx1-Cre Tg3 mice (Iwasato et al., 2004) were also used to generate CxAC1KO. In both Emx1-Cre KΔN and Emx1-Cre Tg3 mice, Cre-mediated recombination is restricted to the dorsal telencephalon (Iwasato et al., 2004). All physiological experiments and the majority of histological experiments were performed using mice backcrossed onto C57BL/6 genetic background more than eight generations.
Mice were genotyped by PCR with the AC1-WND primers (5′-CAT GCC CTC TTG GGT ACT GTC TGT C-3′, 5′-CTC CCT TCA GAC CCT GTC ACC TCT G-3′, 5′-CTT GGG TAC TGT CTG TCT AGC CAT C-3′, and 5′-AGG GAC CAA GAT CTG GCC TCT CAT C-3′: 277, 157, and 111 bp for flox, null, and wild-type alleles, respectively) and Cre primers (Iwasato et al., 2004). Primers for the NR1 gene (Iwasato et al., 1997) or IL2 gene (5′-CTAGGCCACAGAATTGAAAGATCT-3′, 5′-GTAGGTGGAAATTCTAGCATCATCC-3′) were run together with Cre primers as a positive control. PCR products were resolved on a 2–3% agarose gel and detected with ethidium bromide.
The experimental procedures and housing conditions for animals were approved by the institutes' Animal Experimental Committees and all animals were cared for and treated humanely in accordance with the institutional Guidelines for Experiments Using Animals.
Reverse transcriptase-PCR.
Quantitative reverse transcriptase-PCR (qRT-PCR) was performed as described previously (Iwasato et al., 2007). In brief, total RNAs were isolated from the barrel cortex of CxAC1KO (n = 4) and AC1flox/− (n = 5) mice at postnatal day 3 (P3) and CxAC1KO (n = 4) and AC1flox/− (n = 4) mice at P7 using RNeasy Mini kit (Qiagen). All samples were digested on-column with RNase-free DNase. cDNA was synthesized from total RNA using Superscript III two-step qRT-PCR kit (Invitrogen). Real-time qPCR was performed using Platinum SYBR green qRT-PCR Super mix UDG with ROX (Invitrogen) by ABI Prism 7700 (ABI). The PCR conditions were 50°C for 2 min, 95°C for 2 min, 45 cycles of 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s. At the end of each program, a melt-curve analysis was performed. All qRT-PCR were performed in triplicate for each cDNA sample. The primer set used was 5′-ccttttggtcaccttcgtgt-3′ and 5′-gctgtgaccagtaagtgcga-3′. Diluted linearized plasmid pS18 (for AC1 exon1 genomic DNA) was used as a standard for absolute cDNA quantification.
cDNA samples were prepared from brain total RNA derived from two each of 6-month-old AC1flox/flox mice and AC1Δ/Δ mice using Superscript II RT (Invitrogen) and oligo-dT primers. PCRs were performed using forward primers (f1, 5′-CCC TGC TCT TCT TTG GTG TG-3′; f2, 5′-GGT TGC TCA TGA GCC TCA TG-3′) and reverse primers (r4, GTT CCT CTC ATG GCC ATG AC; r9, 5′-ATG ACA TTG GCC AGG GTC AC-3′). The PCR conditions were 94°C for 2 min, 30 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 1 min, and 72°C for 7 min.
In situ hybridization.
Using PCR primers (5′-CTTTTCCTGGCGCTGTTCGTG-3′ and 5′-TGTGACCAGTAAGTGCGAGG-3′), a 297 bp fragment within AC1 exon1 was amplified from pS27 cone derived from genomic DNA of 129 strain. This PCR product was purified by electrophoresis and QIAquick Gel Extraction kit (Qiagen) and subcloned into pGEM-T Easy vector (Promega) to generate the pS60 clone. The pS60 clone verified by nucleotide sequencing was used as a template to generate the antisense probe using the Riboprobe System-SP6 (Promega). In situ hybridization was done as described previously (Iwasato et al., 1997, 2000) with minor modifications. Briefly, frozen brains were cut at a thickness of 12 μm in the sagittal or coronal planes on a cryostat. Sections were postfixed in 4% paraformaldehyde (PFA) in PBS, pH 7.2, before pretreatment. Hybridization was done at 58°C for 18 h in hybridization buffer with a 33P-labeled RNA probe. Sections were washed in a series of SSC buffers of increasing stringency after RNaseA treatment. The final wash was done in 0.1× SSC at 60°C for 30 min. The sections were exposed to β-max film (GE Healthcare) for 9–12 d and dipped in NTB3 emulsion (Kodak) followed by exposure for >1 month. After development, sections were stained with cresyl violet. Expression patterns and levels were ensured by two sets of reactions using three each of CxAC1KO and AC1flox/− mice at P7 and two each of CxAC1KO and AC1flox/− mice at P1. Brains of AC1−/− mice at P1 and P7 were used as negative controls.
Cytochrome oxidase staining.
Barrel cortex was removed following the methods described by Strominger and Woolsey (1987), fixed and flattened for 2–4 h in 4% PFA at room temperature, and cut tangentially (parallel to layer IV) on a vibratome (Leica VT1000S or Dosaka Zero1) into 100 μm sections and subject to cytochrome oxidase (CO) staining (Wong-Riley et al., 1978). Sections were incubated with CO reaction solution free-floating for 2 h at room temperature or 10–14 h at 4°C. After visual detection of stain, sections were washed with PBS 3 times and mounted with Aqua Polymount (Polysciences).
Nissl staining.
Nissl stain, which marks cell bodies, was used to reveal cortical “barrel” cytoarchitecture. Briefly, animals were perfused transcardially with ice-cold PBS followed by 4% PFA in PBS. Barrel cortex was cut tangentially on a vibratome (Leica VT1000S or Dosaka Zero1) into 50-μm-thick sections. Sections were then mounted and dried for a day on a slide warmer at 37°C. Slides were dehydrated and rehydrated in graded alcohol, and then fixed in 10% Formalin (Sigma-Aldrich), and stained with 2% cresyl violet solution for 15 min. After dehydration with graded alcohol and xylene, slides were mounted with Cytoseal (Richard-Allen Scientific). The density of neurons in barrel walls and hollows was determined in three representative barrels (b2, c2, and d2) by marking the cells within a closed fixed contour drawn using Neurolucida Software (MicroBrightField). The average cell density of these three barrels was calculated for each animal and compared between genotypes. All counts were done blind to the genotype.
Golgi staining and analysis of dendrites and spines.
Golgi-Cox staining was done as described previously (Datwani et al., 2002a). One hundred micrometer serial coronal sections were cut using a Microslicer (Dosaka). Layer IV spiny stellate neurons whose cell bodies were located at the midlevels of each section and whose dendrites were isolated from those of neighboring neurons were randomly selected from the posteromedial barrel subfield under a light microscope at 40× (or 20×). Dendrites were reconstructed in three dimensions at 100× (or 40×) using Neurolucida software (MicroBrightField). Wedge analysis, which represents the length of dendritic processes binned every 30°, was performed for each neuron using Neuroexplorer (MicroBrightField). Dendritic asymmetry was evaluated by calculating the dendritic length in the hemisphere (180°) with the greatest length of dendrites relative to the total dendritic length in a given neuron. The selection of neurons and the dendritic reconstructions were repeated independently by different investigators to verify the results. Spine analysis was performed on the longest dendrites from each neuron at 100× with an oil-immersion lens. All of these experiments were done blind to genotypes.
Antibodies.
Primary antibodies were as follows: anti-NeuN (1:500) and anti-vesicular glutamate transporter 2 (vGlut2) (1:500) from Millipore Bioscience Research Reagents, and anti-serotonin transporter (5HTT) (1:10,000) from DiaSorin. Secondary antibodies were as follows: mouse IgG-Alexa 488 (1:500), guinea pig IgG-Alexa 488 (1:500) from Invitrogen, and goat IgG (BA-1000; 1:200) from Vector Laboratories.
Immunofluorescence.
Mice were perfused, barrel cortex was flattened, and 100 μm tangential sections were cut as explained above. Sections remained free-floating for all incubations and washes. Briefly, tissue was permeabilized with 0.7% Triton X-100 in PBS for 20 min, incubated with 0.1 m glycine for 30 min, and then blocked with 1% normal goat serum (NGS) and 2 mg/ml BSA in PBS containing 0.01% Triton X-100 (PBST). Sections were then transferred to the primary antibody for incubation overnight at 4°C with appropriate dilution in PBST with 1% NGS. After washing six times in PBST, sections were incubated in the secondary antibody for 2 h at room temperature and then washed again for three times followed by a postfix of the sections with 4% PFA, and then mounting with Fluoromount-G (EMS). Immunolabeling was visualized with a fluorescence microscope and images were collected with a Leica DM confocal scanning microscope using a 10× objective with the channels for Cy3 (red) and Alexa 488 (green) used sequentially.
Whisker lesion experiments.
Whisker lesion experiments were done as previously described (Datwani et al., 2002b). Briefly, pups were anesthetized on ice and whiskers on the left side of the snout were visualized under a light microscope. The C (center) row of whiskers and whisker follicles from P1 and P5 pups were cauterized with a lesion generator (Muromachi Kikai). After cauterization, the pups were revived in a warmed chamber and returned to their mother. The animals were then killed at P7–P8 (for P1 lesions) or P9–P10 (for P5 lesions). For 5HTT immunohistochemistry, 100 μm tangential sections of the cerebral cortex were used. For quantitative analysis, 5HTT-immunostained tangential cortex sections were examined under a light microscope (Leica DMR), and images of the barrel field were acquired using a DC300F digital camera (Leica). Measurements of the entire large whisker representation areas and areas devoted to row C and row D were made using Image-Pro Plus software (Nippon Roper). Whisker pads were removed from the snout and fixed with 4% PFA overnight at 4°C. PFA (4%) was exchanged to 30% sucrose for cryoprotection for 2 d at 4°C, cryoprotected in 30% sucrose, and sectioned at a 60 μm in the tangential plane. These sections were stained with hematoxylin–eosin to confirm the extent and precision of row C follicle lesions. Animals with lesions that spread beyond row C, or did not include at least the first two whiskers in row C, were excluded from analysis. These experiments were performed blind to genotypes.
Slice electrophysiology.
Acute TC slices were prepared as described previously (Lu et al., 2001). The artificial CSF (ACSF) (124 mm NaCl, 5 mm KCl, 1.25 mm NaH2PO4, 1.3 mm MgSO4, 2 mm CaCl2, 26 mm NaHCO3, and 11 mm glucose, pH 7.2 and 290–300 mOsm) was saturated with 95% O2 and 5% CO2. The whole-cell recording solution contained 99 mm cesium gluconate, 17.5 mm CsCl, 8 mm NaCl, 10 mm HEPES, 0.2 mm EGTA, 4 mm Mg-ATP, 0.3 mm GTP, 7 mm phosphocreatine, and 10 mm BAPTA. BAPTA was included in the pipette to prevent inadvertent potentiation of the postsynaptic neuron.
Stimuli were applied to the VB thalamus through bipolar sharpened and insulated stainless-steel microelectrodes (FHC). Data were collected and analyzed on-line using a computer-driven acquisition system (National Instruments) and software that was written under the Igor (WaveMetrics) programming environment.
EPSCs were measured in voltage-clamp mode using in vitro whole-cell voltage-clamp recording techniques following published protocols (Lu et al., 2001). To confirm that excitatory layer IV neurons were recorded, cells were filled with 2% biocytin and the slices were stained using Vectastain Elite ABC kit (Vector Laboratories). To evaluate and monitor the health of the cell, input and series resistances were continuously monitored, with cells that had <300 MΩ input resistance or drifted >20% discarded. Only responses that exhibited short and constant latencies that did not change with increasing stimulus intensity were considered monosynaptic. Because the responses in young cells tend to drift down if the stimulation intensity or the stimulation frequency is high, one-half saturating stimulation strength with a relatively low stimulation frequency (10–15 ms interval) was used to evoke stable EPSCs. Before any experimental manipulation, 10–15 min of stable baseline response was acquired. EPSC amplitudes were calculated by subtracting the mean current during a fixed 3–4 ms window before the stimulus artifact from the mean current during a similar window at the peak of the EPSC.
To measure the AMPA/NMDA current ratio, which is a gross measure of the relative contribution of AMPA and NMDA receptor mediated currents across a population of synapses, we first isolated the AMPA response by voltage clamping the cell at hyperpolarized membrane potentials (−70 mV) while stimulating the thalamus. We then depolarized the cell to +40 mV to relieve the Mg2+ block of the NMDA receptor and added NBQX (2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide) (10 μm) (Tocris) to the perfusate to block AMPA and kainate receptors, leaving a pure NMDA receptor response. These experiments were done at “normal” (half saturating) stimulus strength as a gross measure of the relative amplitude of the AMPA and NMDA receptor currents (Crair and Malenka, 1995; Lu et al., 2001, 2003) in the presence of GABAergic antagonists (10 μm bicuculine, cesium ions in the whole-cell solution block GABAB responses).
For analysis of AMPA “evoked miniature” events, stable whole-cell voltage-clamp recordings were established at −70 mV holding potential and the Ca2+ in the ACSF was exchanged for Sr2+. Sr2+-based ACSF desynchronizes neurotransmitter release, allowing isolated evoked miniature currents to be analyzed (Xu-Friedman and Regehr, 1999). Evoked miniature events were recorded in 1 s epochs every 2 s in Igor Pro using ACSF (2 mm Sr2+) containing 100 μm picrotoxin and 50 μm d-AP5 [d-([amino])-2-amino-5 phosphopentanoic acid] (Tocris) to eliminate inhibitory currents and possible NMDA receptor current contamination, respectively. Data were imported into Mini Analysis (Synaptosoft), amplitude thresholds were set at 2.5 times root mean square noise, and at least 80 events were identified and used for analysis in each cell. Temporal windows were chosen to represent both the fast and slow components of miniature event decay times. A significant advantage of measuring “evoked minis” is that the population is dominated by TC synapses, as opposed to measuring spontaneous minis in tetrodotoxin, which is a random sample of all synapses on the cell.
LTP was induced using a pairing protocol, in which postsynaptic depolarization is “paired” with presynaptic stimulation in whole-cell voltage-clamp mode (Lu et al., 2001). For these experiments, BAPTA was excluded from the pipette solution, and EPSCs from somatosensory cortex layer IV neurons elicited with thalamic stimulation were recorded at −70 mV until a stable baseline of 10 min was obtained. Pairing was then initiated by switching to a holding potential of −10 mV and stimulating at 1 Hz for 100 s. The holding potential was then switched back to −70 mV after pairing. The percentage EPSC change was calculated as the mean EPSC amplitude of 20 sweeps at 35 min after pairing minus the mean EPSC amplitude of 20 sweeps right before pairing (baseline amplitude) divided by the baseline amplitude.
Paired-pulse measurements were done by isolating the NMDA receptor mediated current as described above, and then eliciting pairs of stimuli at interstimulus intervals (ISIs) of 50, 75, 100, and 500 ms. Each condition was presented in pseudorandom order. Ratios were then computed as the mean amplitude response to the second stimulus over the response to the first stimulus.
For (+)-5-methyl-10,11-dihydro-5H-dibenzo(a,d)cyclohepten-5,10-imine maleate (MK801) experiments, NMDA current was isolated and only cells responding with a significant amount of NMDA current (>25 pA) were used. After acquiring a stable baseline response (10–15 min), the stimulation was stopped and ACSF containing (+)-MK801 (10 μm; Tocris Cookson) was perfused onto the slice (1 ml/min flow rate). After 10 min (to allow enough time for MK801 to equilibrate in the recording chamber), stimulation was resumed and response was recorded until no NMDA current could be detected for >5 min. The time course of MK801-dependent blockade was analyzed by normalizing all of the responses to the amplitude of the first NMDA response after MK801 was applied, and the blockade rates were estimated by fitting a double-exponential function (A1e−t/τ1 + A2e−t/τ2) to the decay and calculating a weighted time constant, τ weighted = τ1 × A1/(A1 + A2) + τ2 × A2/(A1 + A2), from the fit parameters.
Results
Generation of CxAC1KO mice
As the first step to generate CxAC1KO mice, we examined whether our floxed AC1 (AC1-flox) allele, in which the putative promoter and exon 1 of AC1 are flanked by two loxP sites (Fig. 1A), is functional. To confirm that Cre-mediated recombination of the floxed allele produces a null allele, we expressed the Cre recombinase protein in the germline of AC1flox/+ mice and obtained progeny heterozygous for the AC1-Δ allele, in which the floxed DNA sequences were deleted by Cre-mediated recombination (Fig. 1A). Then, we intercrossed AC1Δ/+ mice to generate AC1Δ/Δ mice. RT-PCR analyses using primer sets for exons downstream of the floxed sequence detected AC1 transcripts in the AC1flox/flox but not the AC1Δ/Δ brains (Fig. 1B). These results clearly show that the AC1-Δ allele is a null allele of AC1.
Figure 1.

Cre-mediated recombination on the AC1-flox allele produces AC1-null allele. A, Schematics of AC1flox and AC1Δ (= null or −) alleles. AC1Δ allele was generated by expressing Cre recombinase in the germline of AC1flox/+ mice, in which exon 1 and the putative promoter of the AC1 gene are flanked by two loxP sites. B, RT-PCR using primers specific for exons downstream of the floxed region (shown in A) detects AC1 mRNA expression in AC1flox/flox control brains (3, 4) but not in AC1Δ/Δ brains (1, 2), demonstrating that the AC1-flox allele is functional. m, Size markers. C–F, Flattened cortices stained for CO (C, D) or Nissl (E, F) demonstrate that AC1Δ/Δ mice (D, F) completely lack the cortical barrel pattern, unlike control (wild-type) mice (C, E). G, H, Barreloids in VB thalamus of AC1Δ/Δ mice (H) are not as distinct as those of (AC1+/Δ) control mice (G). I, J, Both control (AC1+/Δ) and AC1Δ/Δ mice have normal barrelettes in the brainstem principal nucleus. The whisker-related patterns in AC1Δ/Δ mice throughout the trigeminal pathway are similar to those reported in barrelless (brl) and AC1−/− mice (Welker et al., 1996; Abdel-Majid et al., 1998), further confirming that the AC1flox allele is functional. Scale bar: C–F, 500 μm; G, H, 250 μm; I, J, 125 μm.
It is known that global disruption of the AC1 gene in barrelless (brl) mice completely abolishes barrel patterning and partially impairs barreloids in the VB thalamus (Welker et al., 1996; Abdel-Majid et al., 1998). We examined whether AC1Δ/Δ mice demonstrate phenotypes similar to global AC1-deficient mice. We analyzed patterning of the morphological body map in barrel cortex of AC1Δ/Δ and wild-type mice by CO histochemistry and Nissl staining. CO-dense patches reflect the gross organization of both TC afferent terminals and target neurons, and Nissl staining reveals the layer IV cytoarchitecture. We found that barrel map patterning was completely disrupted in AC1Δ/Δ mice but normal in wild-type controls (Fig. 1C–F). In addition, the pattern of VB thalamic barreloids in AC1Δ/Δ mice was not as distinct as those seen in control (AC1/+/Δ) mice (Fig. 1G,H). Finally, both control (AC1/+/Δ) and AC1Δ/Δ mice had normal barrelettes in the somatosensory brainstem (Fig. 1I,J). These phenotypes were essentially identical with those observed in brl mice, and further confirm that the AC1-flox allele produces an AC1-null allele on Cre-mediated recombination. Hereafter we refer to the AC1Δ allele as the AC1null (or AC1−) allele.
We previously generated Emx1-Cre mice, in which Cre-mediated recombination is first detectable in the dorsal telencephalon at embryonic day 10 (E10) and is restricted to cortical excitatory neurons throughout the animal's life (Fig. 2A) (Iwasato et al., 2000, 2004). In this study, we used the knock-in-ΔNeo (KΔN) line for most experiments and the Tg3 line or both in a few cases (Iwasato et al., 2000, 2004). We crossed these Cre mice with AC1+/− mice to obtain double heterozygous mice, and further crossed these with AC1−/− mice to obtain Emx1-Cre:AC1−/− mice. These mice were then crossed with AC1flox/flox or AC1flox/+ mice to obtain Emx1-Cre:AC1flox/− mice and their littermate controls (Emx1-Cre:AC1+/−, AC1flox/−, and AC1+/−). Emx1-Cre:AC1flox/− mice constitute CxAC1KO mice.
Figure 2.
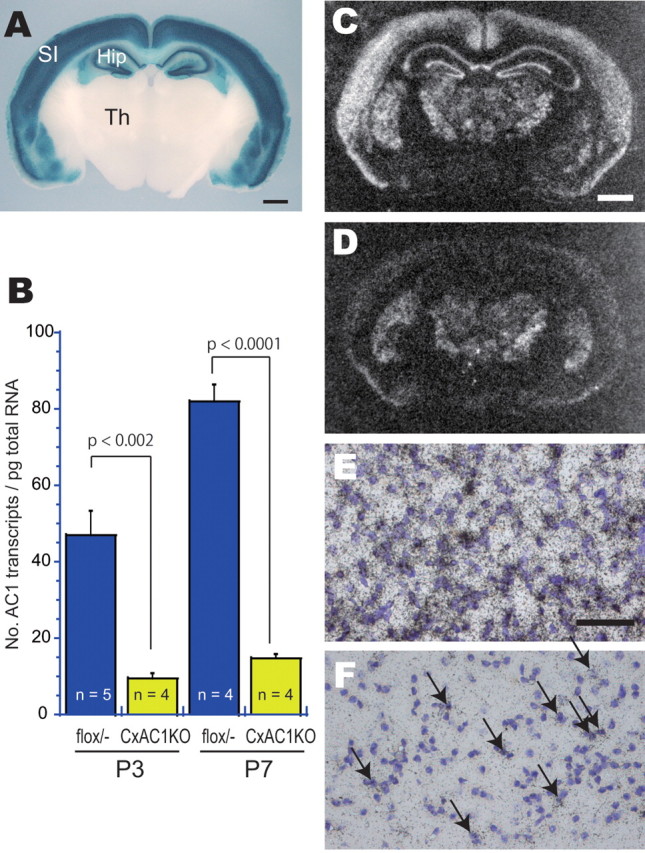
CxAC1KO mice generated using Cre expression driven by Emx1 promoter. A, LacZ staining of 400-μm-thick coronal sections from progeny of Emx1-Cre (KΔN) mice crossed with CAG-CAT-Z reporter mice demonstrates that Cre-mediated recombination is specific to neocortex and hippocampus from embryonic stages (data not shown) to adulthood. For more information, see Iwasato et al. (2000, 2004). SI, Barrel cortex; Hip, hippocampus; Th, thalamus. B, Quantitative RT-PCR of AC1flox/− and CxAC1KO mice barrel cortex at P3 and P7. Number of AC1 transcripts per picogram of total RNA for P1 flox/− (n = 5), P1 CxAC1KO (n = 4), P7 flox/− (n = 4), and P7 CxAC1KO (n = 4) are as follows (mean ± SEM): 46.86 ± 6.37, 9.36 ± 1.42, 81.96 ± 4.43, and 14.83 ± 1.09; Student's t test. C, D, In situ hybridization of AC1 mRNA in coronal sections of P7 brains of AC1flox/− (C, E) and CxAC1KO (D, F) mice at P7. A few AC1-expressing neurons (F, arrows), presumably GABAergic interneurons, are observed in CxAC1KO cortex. Scale bars: A, C, D, 1 mm; E, F, 50 μm.
We compared levels of AC1 expression in the barrel cortex of CxAC1KO and AC1flox/− mice at P3 and P7 by quantitative RT-PCR. At P3 and P7, the level of AC1 mRNA present in the barrel cortex of CxAC1KO mice was reduced to 20.0 and 18.1%, respectively, of the levels in barrel cortex of age-matched AC1flox/− control mice (Fig. 2B). These ratios are similar to the proportion of GABAergic neurons in the barrel cortex (Lin et al., 1985; Beaulieu, 1993). To further confirm the cortex-restricted AC1 disruption in CxAC1KO mice, we examined the distribution of AC1 mRNA by in situ hybridization (Fig. 2C–F). We found that the amount of AC1 mRNA in the neocortex and hippocampus of CxAC1KO mice was much lower than that of control (AC1flox/−) mice both at P1 (data not shown) and P7 (Fig. 2C,D). Other brain regions exhibited similar levels of AC1 mRNA in CxAC1KO and control (AC1flox/−) mice. We detected AC1 expression in only a few cells of the CxAC1KO cortex (Fig. 2F). These AC1-expressing cells are presumably inhibitory neurons, because we and others previously demonstrated that the Emx1 lineage does not give rise to the GABAergic neurons of the neocortex (Iwasato et al., 2000, 2004; Chan et al., 2001; Gorski et al., 2002).
Barrel wall formation, but not the gross barrel pattern, is disturbed in CxAC1KO mice
Intriguingly, analyses of the gross barrel patterning in CxAC1KO somatosensory cortex, using CO staining (n = 6 for both genotypes) as well as presynaptic TC axonal clustering by immunostaining for a TC axon marker, vGlut2 (n = 3 for both genotypes), did not reveal any notable differences from control mice (Fig. 3). Layer IV neuronal organization into barrel walls was examined with Nissl staining (n = 8 for controls and n = 11 for CxAC1KO mice) (Fig. 3C,D) and immunostaining for NeuN, which labels neuronal cell bodies (n = 3 for both genotypes) (Fig. 3E,F). Both staining methods revealed no qualitative difference between CxAC1KO and control mice. We also examined the thalamic barreloid pattern using CO staining in CxAC1KO mice and found a pattern similar to control mice (data not shown). These phenotypes, which were strikingly different from those observed in global AC1 KO mice (Fig. 1C–H), indicate that AC1 function in cortical excitatory neurons is not essential for the gross patterning of cortical barrels and VB thalamic barreloids. However, quantitative analysis of Nissl-stained sections showed that there is a small but statistically significant decrease in the wall-to-hollow ratio of neuronal densities in barrels of CxAC1KO mice (1.28 ± 0.032; n = 11) compared with those of AC1flox/− control mice (1.47 ± 0.035; n = 8; p < 0.01; Student's t test). Therefore, cortical AC1 appears to play a role in the emergence of fine features of layer IV barrel cytoarchitecture.
Figure 3.
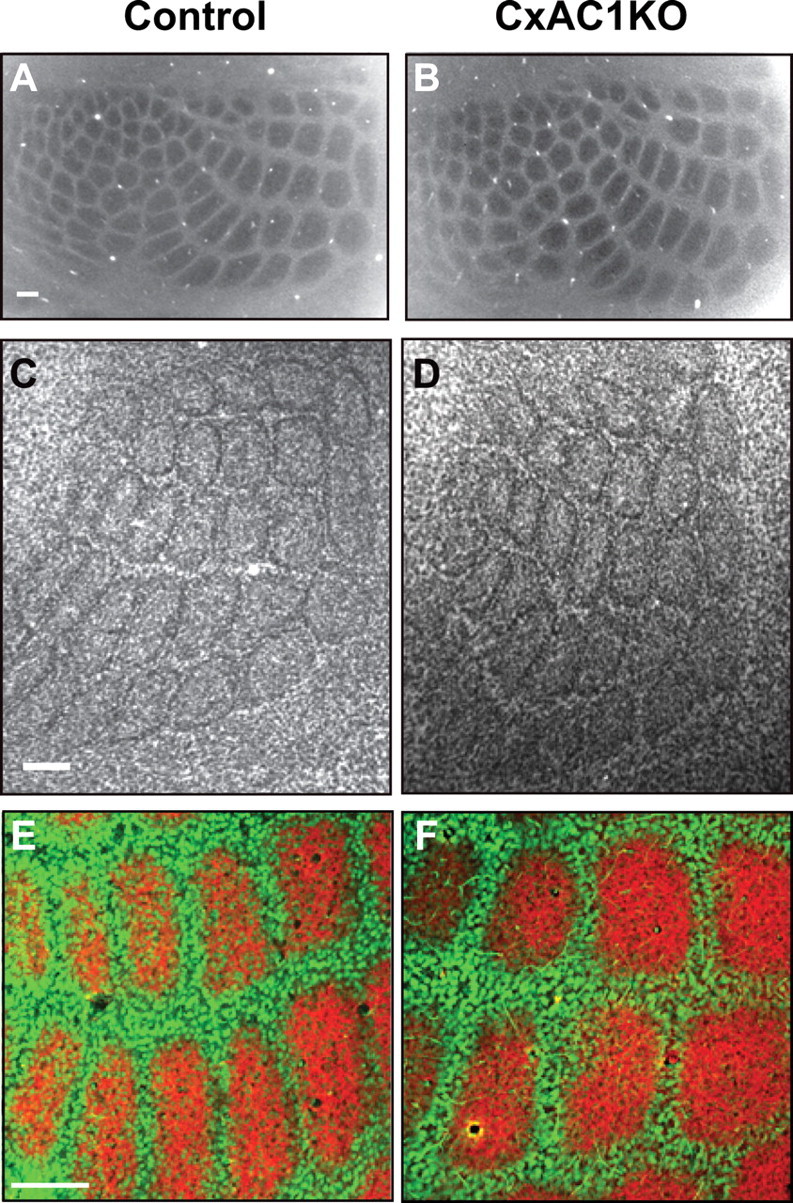
Grossly normal cortical barrel map in CxAC1KO mice. A, B, Tangential sections from P29 control (AC1flox/−) (A) and CxAC1KO (B) mice stained with CO. Whisker barrel patterns are indistinguishable between genotypes. C, D, Nissl-stained tangential sections from P24 control (AC1flox/−) (C) and CxAC1KO (D) mice. As in control mice, layer IV neurons in CxAC1KO mice organize into a grossly normal barrel “wall” and “hollow” pattern characteristic of rodent somatosensory cortex. E, F, Tangential sections from P10 control (AC1flox/−) (E) and CxAC1KO (F) mice immunostained with anti-vGlut2 antibody in red and anti-NeuN in green, which selectively label TC afferents and neuronal cell bodies, respectively. The barrel pattern is qualitatively similar in the two genotypes, showing that the gross presynaptic and postsynaptic patterning of barrels in CxAC1KO mice is not impaired. However, a quantitative analysis of the patterning of layer IV neurons into barrel walls and hollows reveals a small but statistically significant decrease in the wall-to-hollow ratio in the CxAC1KO mice (1.28 ± 0.032; n = 11) compared with AC1flox/− control mice (1.47 ± 0.035; n = 8; p < 0.01, Student's t test). Scale bars, 100 μm.
Dendritic orientation of layer IV neurons is impaired in the barrel cortex of CxAC1KO mice
A prominent feature of barrel cortex cytoarchitecture is the asymmetric orientation of layer IV neuron dendrites toward the barrel hollow. To determine whether layer IV neurons require cortical AC1 function to acquire this asymmetric dendritic morphology, we performed Golgi staining and reconstructed the dendrites of spiny stellate neurons in layer IV barrel cortex of CxAC1KO and control (AC1flox/−) mice (Fig. 4A–D). A total of 60 neurons were analyzed, with 24 control neurons from six animals and 36 CxAC1KO neurons from nine animals. Dendritic asymmetry was determined by calculating the dendritic length in the hemisphere (180°) with the greatest length of dendrites relative to the total dendritic length (360°) in a given neuron (for more details, see Materials and Methods). This analysis showed that the asymmetry of CxAC1KO layer IV neuron dendrites was reduced compared with that of control mice (0.95 ± 0.011 for controls and 0.86 ± 0.017 for CxAC1KOs; p < 0.01, Student's t test) (Fig. 4E). Dendritic span, defined as the largest distance between the most distal dendrite tips of the cell, was also significantly larger in CxAC1KO mice than that of control mice (96.2 ± 5.9 μm for controls and 117.5 ± 6.3 μm for CxAC1KOs; p < 0.05, Student's t test), consistent with the reduced asymmetry of these neurons (Fig. 4F).
Figure 4.
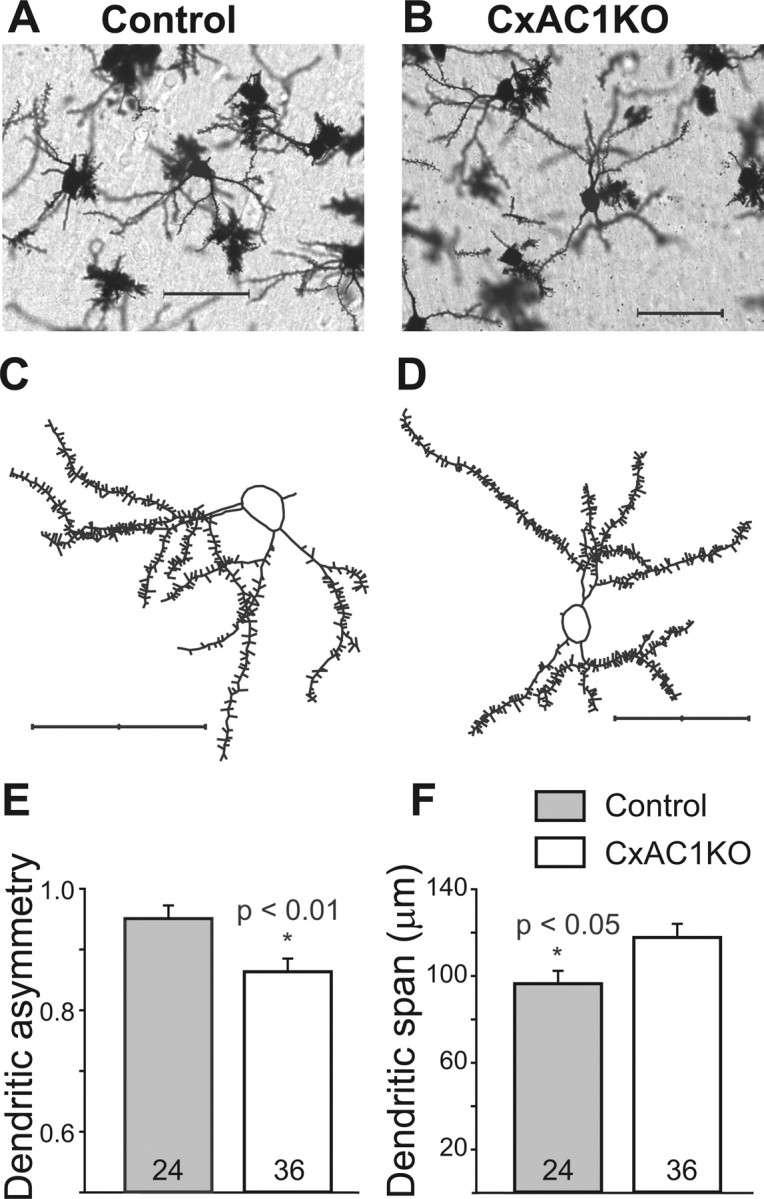
Dendritic asymmetry of layer IV spiny stellate neurons in CxAC1KO mice is reduced compared with littermate controls. A–D, Examples of Golgi-stained layer IV spiny stellate neurons in the barrel cortex (A, B) and their computer-aided reconstructions (C, D) in control (AC1flox/−) (A, C) and CxAC1KO (B, D) mice at P29. E, CxAC1KO mice have significantly lower (*p < 0.01, Student's t test) dendritic asymmetry compared with littermate controls. Dendritic asymmetry, which must be ≥0.5, is defined as the ratio of the dendritic length in the hemisphere with the greatest density of dendrites relative to the total dendritic length. Error bars indicate SEM. F, Dendritic field span, the greatest distance between the most distal dendrite tips of a particular layer IV spiny stellate neuron, is significantly greater (*p < 0.05, Student's t test) in CxAC1KO mice compared with littermate control mice. Scale bars, 50 μm.
To verify these results, the analysis was repeated on the same sections by a different investigator, who reconstructed a total of 37 neurons, 12 neurons from 7 control mice and 25 neurons from 10 CxAC1KO mice. In the end, only three neurons (two CxAC1KO and one control neuron) from the randomly chosen samples were selected for reconstruction by both investigators. This second analysis confirmed that the asymmetry of CxAC1KO layer IV neurons is reduced on average compared with control mice (0.94 ± 0.023 for controls and 0.85 ± 0.023 for CxAC1KOs; p < 0.05, Student's t test). Dendritic span was also found to be relatively larger in CxAC1KO mice compared with controls (105.7 ± 10.1 μm for controls and 128.3 ± 7.6 μm for CxAC1KOs; p = 0.09, Student's t test). In contrast, analyses of the total dendritic length (421.7 ± 55.9 μm for controls and 466.4 ± 43.4 μm for CxAC1KOs; p = 0.61, Student's t test) and spine density (0.69 ± 0.04 μm−1 for controls and 0.74 ± 0.03 μm−1 for CxAC1KOs; p = 0.38, Student's t test) revealed no difference between genotypes. These results indicate that AC1 function in the cortex is required for layer IV neurons in barrel cortex to acquire their typical asymmetric dendritic morphology.
Lesion-induced plasticity of the TC axonal pattern is not changed in CxAC1KO mice
The area of cortex devoted to a particular peripheral sense organ changes if the pattern of sensory input changes. For example, if a row of whiskers and whisker follicles is damaged neonatally, the area of cortex devoted to the damaged row shrinks and the neighboring rows expand (Van der Loos and Woolsey, 1973). In mouse barrel cortex, the critical period for this type of plasticity ends by P4 (Woolsey and Wann, 1976; Durham and Woolsey, 1984; Datwani et al., 2002b; Rebsam et al., 2005).
We examined lesion-induced map plasticity of TC axon clustering in CxAC1KO and control mice by electrocautery of the center (C) row of whiskers and whisker follicles at P1 or P5. Mice were analyzed at P7–P8 (for P1-lesion) or P9–P10 (for P5-lesion) by immunostaining for 5HTT, which is specifically expressed in TC axon terminals (Lebrand et al., 1998; Rebsam et al., 2002) (Fig. 5A–D). Quantification of map plasticity was performed by normalizing each barrel area with respect to the area of the entire large barrel field and then calculating the ratio of D row to C row area. We found that CxAC1KO mice in which whiskers were cauterized at P1 had a similar degree of map plasticity as littermate control mice (D/C ratio for control mice: 2.67 ± 0.24 (mean ± SEM), n = 7; and for CxAC1KO mice: 3.55 ± 0.69; n = 9; p = 0.300, t test) (Fig. 5E–G) (for details of the quantification, see Materials and Methods). Whisker cauterization at P5 did not induce map plasticity in either CxAC1KO or control mice (Fig. 5C–G). Similar results were obtained when examining lesion-induced map plasticity of layer IV cytoarchitecture with Nissl-stained sections (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These results indicate that cortical AC1 function does not play a role in lesion-induced plasticity and in the closure of the critical period for this plasticity.
Figure 5.
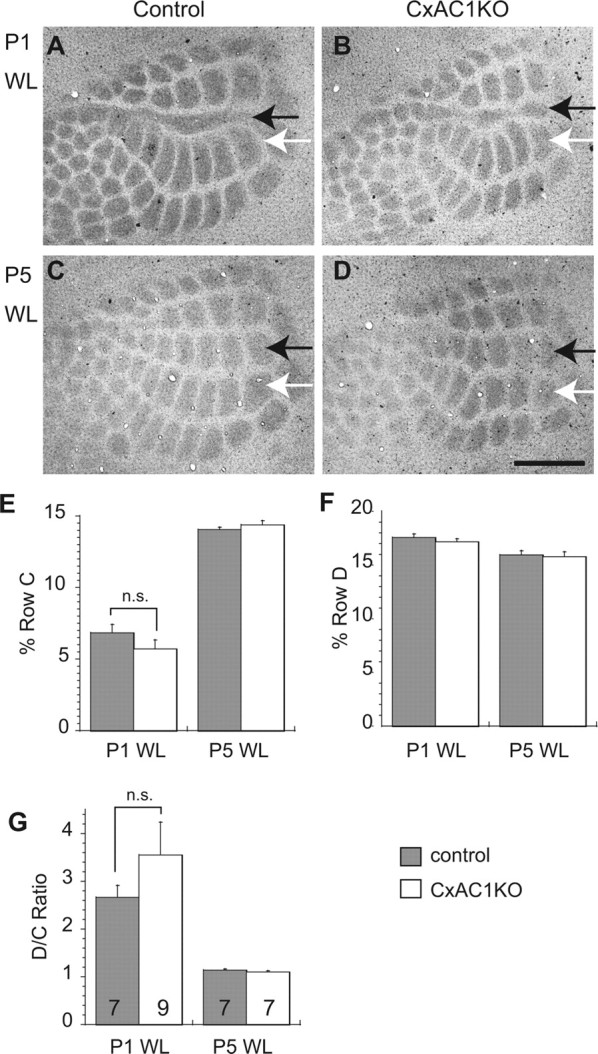
Lesion-induced plasticity is not altered in CxAC1KO mice. A, B, 5HTT immunohistochemistry of tangential sections from barrel cortex shows that C row whisker lesioned at P1 (P1WL) induces robust shrinkage of row C (black arrows) and expansion of neighboring rows (row D, white arrows) in TC axons of both CxAC1KO (B) and their littermate control (AC1flox/−) mice (A). C, D, The critical period for this map plasticity ends by P5 in both CxAC1KO (D) and littermate control mice (C). E–G, Quantification of lesion-induced plasticity in control and CxAC1KO mice. Analyses of percentage row C (E) and row D (F) areas normalized with respect to area of the entire large whisker representation are similar between control and CxAC1KO mice. The D/C ratio calculated by dividing the row D area by the row C area provides a plasticity index (Schlaggar et al., 1993; Datwani et al., 2002b). The D/C ratio analysis reveals no difference in TC axon map plasticity between genotypes (G). Error bars indicate SEM; Student's t test. Scale bar, 500 μm.
CxAC1KO mice have lower AMPA/NMDA ratio and fewer AMPA receptors at TC synapses
Like many glutamatergic synapses, the AMPA/NMDA ratio increases at TC synapses during maturation, likely through the insertion of AMPA receptors at the synapse via LTP-like mechanisms (Malinow and Malenka, 2002). We previously demonstrated that brl mutants have fewer functional AMPA receptors and the developmental increase in AMPA/NMDA ratio is absent at TC synapses of brl mice (Lu et al., 2003). To clarify whether these postsynaptic functional deficits in brl mice are specifically attributable to the absence of cortical AC1 function, we examined AMPA/NMDA ratios (Fig. 6) and AMPA receptor-evoked miniature events (Fig. 7) in CxAC1KOs and littermate control mice.
Figure 6.
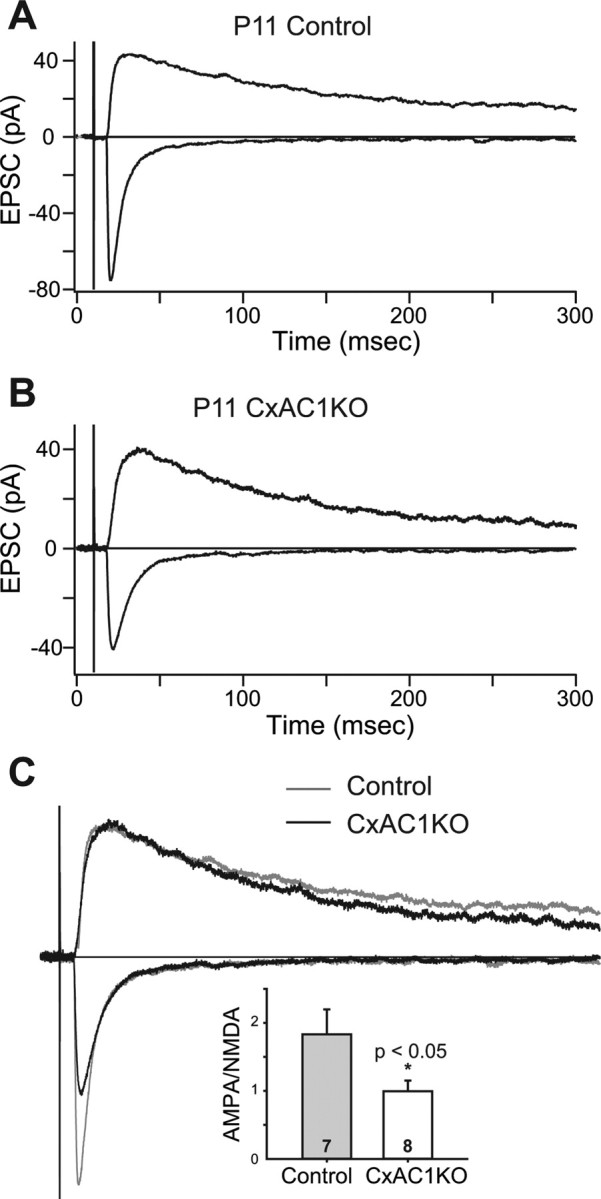
AMPA/NMDA current ratio is reduced in CxAC1KO mice. A, B, Sample whole-cell voltage-clamp measurements of AMPA receptor-mediated and NMDA receptor-mediated TC EPSCs in a P11 littermate control (A), and P11 CxAC1KO (B) mouse. C, Overlay of the responses in A and B scaled so that the NMDA receptor currents are the same amplitude. Note the AMPA receptor-mediated EPSC in CxAC1KO animal is small compared with the littermate control. Inset, Summary quantification of AMPA/NMDA current ratios of control (AC1flox/−) and CxAC1KO mice at P9–P11. The AMPA/NMDA current ratio of CxAC1KO mice is significantly smaller than that of control mice (*p < 0.05; t test). Error bars indicate SEM.
Figure 7.

AMPA receptor mini-EPSCs are smaller at CxAC1KO TC synapses. A, B, Sample traces of evoked AMPA receptor-mediated current responses in Ca2+-ACSF (gray) and Sr2+-ACSF (black) from recordings in P9 CxAC1KO (B) and littermate control (AC1flox/−) (A) neurons. In the presence of Sr2+-ACSF, responses to evoked synchronous release is lower and quantal events (evoked mini-EPSCs) in response to asynchronous release appear. The amplitudes of these quantal events are smaller on average in CxAC1KO mice. C, Average frequency histogram of evoked mini-EPSCs. The CxAC1KO histogram (white bars) peaks at a smaller amplitude than littermate controls (gray bars), and large amplitude events are almost absent in CxAC1KO mice. Inset, Mean evoked mini-EPSC amplitude is significantly lower (*p < 0.05, t test) for CxAC1KO mice (white) with respect to littermate controls (gray). Error bars indicate SEM. D, The cumulative probability distribution in CxAC1KO animals (white) also shows a shift toward smaller amplitudes compared with littermate controls (gray).
We found that the AMPA/NMDA ratio at P9–P11 for CxAC1KO mice (0.99 ± 0.16 pA; n = 8) was significantly lower than that of control mice (1.56 ± 0.17, n = 6 for AC1+/−; 1.61 ± 0.22, n = 6 for Emx-Cre; AC1+/−; 1.83 ± 0.36, n = 7 for AC1flox/−; and 1.67 ± 0.15, n = 19 for all controls; p < 0.05, t test) (Fig. 6C). AMPA receptor-mediated miniature events (AMPA receptor minis) at single TC synapses were examined by replacing Ca2+ with Sr2+ in the extracellular solution. In the presence of Sr2+, the neurotransmitter release at axon terminals is desynchronized and AMPA receptor minis are observed in response to this quantal release at single synapses (Fig. 7A,B) (Xu-Friedman and Regehr, 1999). Evoked mini-EPSC amplitudes of CxAC1KOs were on average smaller than littermate controls at P9–P11 (9.48 ± 0.76 pA for n = 7 AC1flox/− control vs 6.59 ± 0.33 pA for n = 10 CxAC1KO; p < 0.05, t test) (Fig. 7C, inset). Comparison of the evoked mini-EPSC amplitude histograms showed a shift in the peak of the distribution, with the histogram in CxAC1KO mice peaking at a smaller amplitude than littermate controls (Fig. 7C). In addition, CxAC1KO mice had very few responses with large amplitudes (Fig. 7C). The mean cumulative probability distributions also revealed a shift in evoked mini-EPSCs in CxAC1KO mice toward smaller amplitudes compared with littermate controls (Fig. 7D). These results indicate that, in the absence of AC1 signaling in cortical excitatory neurons, the number of functional AMPA receptors at TC synapses is reduced, leading to smaller AMPA receptor-mediated currents and smaller AMPA/NMDA ratios. These defects are similar to those observed in brl mice, which were therefore attributable at least in part to the absence of AC1 function in cortical neurons.
LTP is impaired in CxAC1KO mice during TC synapse development
The trafficking of AMPA receptors into synapses is thought to control synaptic strength during map refinement (Crair and Malenka, 1995; Feldman et al., 1998; Lu et al., 2003; Inan et al., 2006). LTP at the TC synapse, which can only be induced during a critical period that ends around P7 (Crair and Malenka, 1995), is thought to depend on AMPA receptor insertion into TC synapses. Our previous work showed that LTP cannot be induced at brl TC synapses, which is consistent with the fewer functional AMPA receptors observed at these synapses. Therefore, the lower AMPA receptor response observed in CxAC1KO mice might be associated with a defect in TC LTP. We examined this possibility using a standard LTP “pairing” protocol in TC slices from P4–P6 CxAC1KO and littermate control (AC1flox/−, Emx1-Cre;AC1+/−, AC1+/−, and AC1flox/+) mice (Fig. 8). All control groups showed a significant potentiation, whereas the CxAC1KO mice did not (p < 0.05 for control groups and p = 0.59 for CxAC1KOs, paired t test). We also found that the amount of potentiation (EPSC percentage change) was significantly lower in CxAC1KO mice (4.0 ± 2.4%; n = 8) compared with any of the control groups (AC1flox/+, 83.2 ± 8.5%, n = 3; AC1+/−, 49.2 ± 12.9%, n = 5; Emx1-Cre; AC1+/−, 65.4 ± 26.2%, n = 5; AC1flox/−, 53.9 ± 15.9%, n = 8; p < 0.05, Student's t test), which are pooled together in Figure 8 (60.3 ± 9.0%; p < 0.01, Student's t test). This suggests that AC1 function in layer IV neurons is responsible for the normal functional development of TC synapses, and that the impaired LTP observed in brl mice is attributable at least in part to the absence of AC1 function in cortical neurons.
Figure 8.

LTP is defective at CxAC1KO TC synapses. A, B, Example whole-cell voltage-clamp recordings from P6 littermate control (AC1flox/−) (A) and P6 CxAC1KO (B) neurons show that TC AMPA response in control but not CxAC1KO layer IV neurons can be potentiated using an LTP “pairing” protocol. Traces on the right (average of 20 sweeps) from before (1) and 35 min after (2) pairing show significant potentiation in control but not in CxAC1KO neurons. C, Summary graph of pairing experiments showing the EPSC percentage change for CxAC1KO mice (open circles) and controls (AC1flox/−, Emx1-Cre;AC1+/−, AC1+/−, and AC1flox/+) (filled circles). Control TC synapses reveal obvious potentiation, whereas CxAC1KOs cannot potentiate. Error bars indicate SEM. D, Summary histogram of EPSC percentage change from the average of 20 sweeps starting at 35 min after pairing for control and CxAC1KO TC synapses. CxAC1KO TC synapses show almost no potentiation and their EPSC percentage change is significantly lower (*p < 0.01, Student's t test) than that of control mice.
TC synaptic release efficacy is intact in CxAC1KO mice
We next examined whether CxAC1KO mice have normal presynaptic function at TC synapses. One estimate of presynaptic release is the short-term plasticity measured by comparing the responses to two closely spaced stimuli. This paired-pulse ratio (PPR), which is the amplitude of the second response relative to the first response, is indicative of the probability of release (Pr) at a synapse (Zucker, 1973, 1989). PPRs <1, called paired-pulse depression (PPD), indicate that a synapse has a relatively high Pr. TC synapses are high Pr synapses showing PPD. brl TC synapses have higher PPRs, or lower Pr, than control TC synapses (Lu et al., 2006). To investigate whether the low Pr at brl TC synapses is attributable to the lack of cortical AC1 function, we measured the NMDA receptor responses to paired stimuli at four different ISIs (Fig. 9). We found no difference in the PPRs of TC synapses of CxAC1KO mice compared with those of littermate control (AC1flox/−) mice at any ISI (Fig. 9A,B), indicating no defect in neurotransmitter release efficacy (Fig. 9C) [for littermate controls (n = 8) and for CxAC1KOs (n = 8), respectively: 0.53 ± 0.05 and 0.52 ± 0.06, p = 0.81, t test at ISI = 50 ms; 0.55 ± 0.04 and 0.57 ± 0.05, p = 0.75, t test at ISI = 75 ms; 0.60 ± 0.08 and 0.58 ± 0.04, p = 0.82, t test at ISI = 100 ms; 0.65 ± 0.05 and 0.63 ± 0.02, p = 0.70, t test at ISI = 500 ms].
Figure 9.
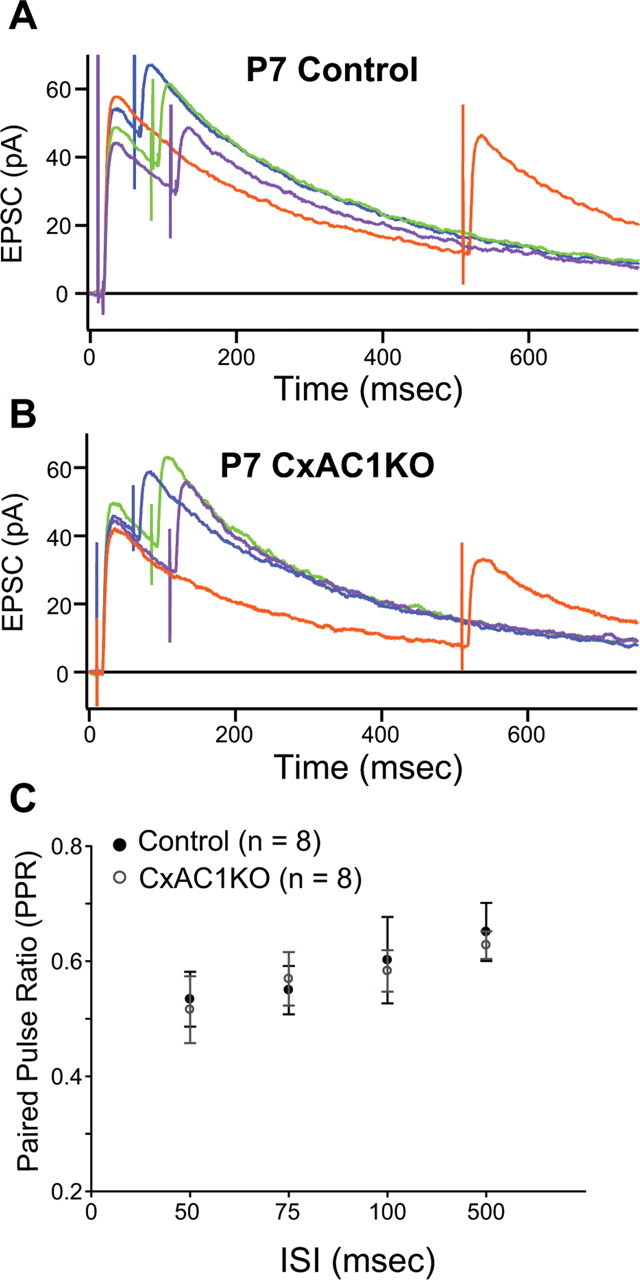
PPR at CxAC1KO TC synapses is similar to littermate controls. A, B, Average example EPSC responses of a P7 CxAC1KO (B) and a P7 littermate control (AC1flox/−) (A) cell to paired stimuli at ISIs 50 ms (blue), 75 ms (green), 100 ms (purple), and 500 ms (orange) reveal similar PPRs. C, Summary plot for average PPRs at ISIs at 50, 75, 100, and 500 ms are not different between CxAC1KO (open circles) and littermate controls (black circles) at P5–P7, suggesting that Pr at CxAC1KO TC synapses is not defective. Error bars indicate SEM.
Another estimate of presynaptic release is obtained by measuring the rate of NMDA receptor current decay in the presence of the irreversible open channel blocker MK801. The rate at which the amplitude of NMDA receptor-mediated EPSCs are attenuated by MK801 during repetitive stimulation is directly related to Pr (Rosenmund et al., 1993). Since MK801 is an irreversible open channel blocker, more NMDA receptors will be permanently blocked, and the blockade rate will be faster in high Pr synapses compared to synapses with lower Pr. NMDA receptor currents from TC synapses in brl mice take longer to block in the presence of MK801 relative to control synapses, which is consistent with a reduced Pr (Lu et al., 2006). We measured the rate of NMDA receptor block at CxAC1KO and littermate control (AC1flox/−) TC synapses using MK801 and observed a similar blockade rate (Fig. 10A,B). The summary plot of the percentage block calculated by normalizing each sweep to the amplitude of the first sweep after resuming stimulation also showed no difference (Fig. 10C). Comparison of the weighted tau, calculated by fitting a double exponential curve to each cell showed no significant difference between CxAC1KOs and their littermate controls (Fig. 10D) [9.68 ± 1.04 sweeps for littermate controls (n = 8) and 9.37 ± 1.41 sweeps for CxAC1KOs (n = 8); p = 0.86, t test]. These results all indicate that the lower neurotransmitter release efficacy observed at TC synapses in brl mice is not attributable to lack of AC1 function in excitatory cortical neurons, but is consistent with a functional role of AC1 in presynaptic TC afferent terminals.
Figure 10.
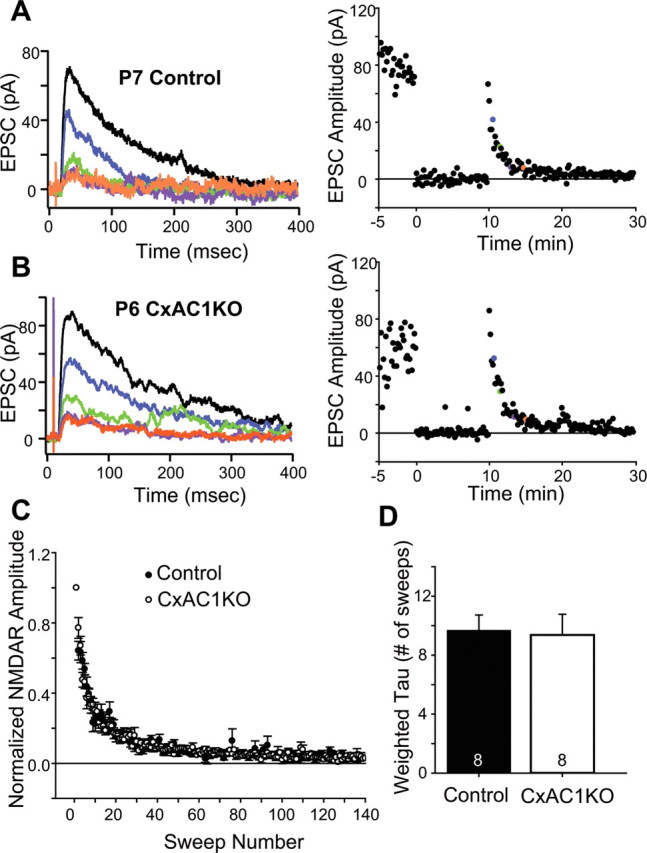
CxAC1KO TC synapses exhibit no difference in Pr. A, B, Sample traces of a P6 CxAC1KO (B) and a P7 littermate control (AC1flox/−) (A) taken at progressively later times after MK801 wash and the corresponding experiments with the NMDA receptor response amplitude versus time (right). A baseline NMDA receptor current is acquired before turning the stimulation off to wash on MK801 (10 μm). After resuming stimulation, the NMDA receptor current amplitude decreases steadily with a rate that is proportional to the probability of release. Color of the sample traces (A, B) corresponds to the points colored on the amplitude plot on the right panel. C, Comparison of littermate control and CxAC1KO mice at P5–P7 reveals no difference in the rate of NMDA receptor block by MK801, further indicating that the Pr is not different between CxAC1KO and control TC synapses. Error bars indicate SEM. D, Weighted time constants (tau) measured by fitting a double-exponential curve to the NMDA receptor amplitude decay over time are not different between genotypes.
Discussion
We generated CxAC1KO mice in which the AC1 gene is disrupted in the cortex but not in the thalamus or the brainstem. We found that these mice show a grossly normal barrel pattern, in contrast to global AC1 KO mice that lack any signature of cortical barrels. Whisker follicle lesion-induced critical period plasticity in the barrel cortex of CxAC1KO mice was also similar to that of control mice. These results suggest that AC1 in cortical excitatory neurons is not necessary for the development of an overtly normal barrel map or lesion-induced plasticity. However, we found that cortical AC1 plays an important role in development of the asymmetric dendritic morphology typical of layer IV barrel neurons and in the emergence of a fully normal layer IV barrel cytoarchitecture. Our physiological analyses of TC synapses in CxAC1KO mice revealed that cortical AC1 is required for induction of LTP and for strengthening of TC synapses through an increase in AMPA receptor-mediated currents, but not for regulation of presynaptic release efficacy. Thus, CxAC1KO mice revealed specific roles for cortical AC1 in the morphological and physiological maturation of layer IV barrel neurons.
How does postsynaptic AC1 regulate morphological and physiological maturation of barrel neurons?
Adenylyl cyclases are a family of broadly expressed proteins that catalyze the conversion of ATP to cAMP when stimulated. AC1 is a neuron-specific and membrane-bound form of adenylyl cyclase that is sensitive to changes in cytosolic Ca2+ concentration (Wang and Storm, 2003; Ferguson and Storm, 2004). Activation of AC1 by Ca2+ occurs in neurons through NMDA receptors and/or voltage-gated Ca2+ channels (Kovalchuk et al., 2000; Bloodgood and Sabatini, 2007), leading to an increase in cAMP production, a second messenger whose primary target is PKA. The cAMP/PKA signaling pathway has been implicated in a wide range of activity-dependent processes such as hippocampal and cerebellar synaptic plasticity in adulthood and developmental plasticity in the visual cortex (Storm et al., 1998; Villacres et al., 1998; Wong et al., 1999; Xia and Storm, 2005; Cui et al., 2007).
The location and position of the face map during the construction of barrels in the rodent somatosensory cortex is dictated by molecular gradients intrinsic to the neocortex (Grove and Fukuchi-Shimogori, 2003; Sur and Rubenstein, 2005; Inan and Crair, 2007). However, patterning of layer IV neurons to form barrels within the somatotopic map is to a large extent guided by activity-dependent mechanisms. Our current understanding is that TC axons are the first neural elements in the cortex to form a periphery-related pattern and provide a template of the whisker-specific pattern to the presumptive barrel field (Erzurumlu and Jhaveri, 1990; Senft and Woolsey, 1991; Agmon et al., 1993). Layer IV neurons then form clusters (walls) around the discrete patches of TC axon arbors and orient their dendrites toward them. Studies of the phenotypes of several lines of mutant mice revealed that glutamatergic synaptic transmission, its modulation by serotonin (5-HT), and signaling pathways regulated by neuronal activity play a major role in development of the barrel cortex (Erzurumlu and Kind, 2001; Erzurumlu and Iwasato, 2006; Inan and Crair, 2007). The barrel cortex phenotype of CxAC1KO mice described here suggests that cortical AC1 function plays a critical role in the preference for layer IV spiny stellate neurons to orient their dendrites toward TC axon arbors and form normal layer IV barrels.
Synaptic plasticity at developing somatosensory TC synapses is NMDA receptor and Ca2+-dependent, and the maturation of TC synapses involves an increase in AMPA receptor-mediated currents (Crair and Malenka, 1995; Feldman et al., 1998). Synaptic maturation is thought to occur through LTP-like mechanisms that regulate the trafficking of AMPA receptors into the synapse via PKA-dependent phosphorylation of AMPA receptors (Crair and Malenka, 1995; Lu et al., 2003; Inan et al., 2006). Mice with a global KO of PKARIIβ have deficits in synaptic plasticity and TC synapse maturation (Inan et al., 2006). Immunoelectron microscopic and biochemical evidence suggests that PKARIIβ is located and functions primarily on the postsynaptic side of developing TC synapses (Inan et al., 2006; Watson et al., 2006). In this study, we showed that CxAC1KO mice have physiological phenotypes similar to those observed in PKARIIβ KO mice. This suggests that postsynaptic AC1/cAMP/PKA signaling plays an important role in TC synapse maturation. Morphologically, layer IV neurons in the barrel cortex of both cortex-specific KO mice of NMDA receptor NR1 subunit (Datwani et al., 2002a) and PKARIIβ KO mice (Inan et al., 2006) show reduced dendritic asymmetry, similar to that of CxAC1KO mice. Therefore, it is likely that by linking NMDA receptor-mediated Ca2+ influx and PKA activation, cortical AC1 plays a role in postsynaptic physiological maturation of TC synapses, which underlies the emergence of asymmetric dendritic orientation in barrel neurons, a key feature of the morphological maturation of barrels.
Where and how does AC1 influence barrel map patterning?
We did not detect a gross impairment in cortical barrel patterning in CxAC1KO mice. Although quantitative analyses detected subtle differences in layer IV neuron barrel cytoarchitecture assayed with postsynaptic markers in CxAC1KO mice, it is clear that the absence of AC1 in excitatory neurons of the cortex is not sufficient to reproduce the severe barrel map phenotype observed in global AC1 KO and brl mice (Welker et al., 1996) (Figs. 1C–F, 3). Because the AC1 gene is highly expressed throughout the somatosensory system during early postnatal development (Matsuoka et al., 1997; Nicol et al., 2005), the lack of AC1 function in the thalamus and/or brainstem of brl mice, which is unaffected in CxAC1KO mice, may contribute to the complete disruption of the cortical barrel pattern in brl mice.
One route for the subcortical action of AC1 may be in TC afferents, because brl mice have impaired neurotransmitter release efficacy (Lu et al., 2006), which was not observed in CxAC1KO mice (Figs. 9, 10). This physiological impairment may be mediated by RIMs, active zone proteins that modulate neurotransmitter release and are phosphorylated by PKA downstream of AC1 (Lu et al., 2006). Consistent with this, Rim1α KO mice have a mild barrel phenotype and presynaptic functional impairments at TC synapses similar to those observed in brl mice (Lu et al., 2006). A presynaptic role for AC1 in sensory map refinement is also suggested by in vitro evidence in the visual system. In brl mice, the segregation of ipsilateral and contralateral retinotectal and retinogeniculate axons is impaired (Ravary et al., 2003; Plas et al., 2004; Nicol et al., 2006b). In vitro coculture assays using retinal and tectal explants from wild-type and brl mice show that wild-type retinal axons correctly target in the brl tectum but brl retinal axons fail to establish regional selectivity and refinement in the wild-type tectum, suggesting a presynaptic role for AC1 in sensory map refinement (Nicol et al., 2006a).
In addition to the complete disruption of cortical barrels, thalamic barreloid formation is also modestly impaired in AC1−/− (global AC1 KO and brl) mice. AC1 in thalamic or brainstem neurons may play a direct role in the formation of VB thalamic barreloids. Although brainstem barrelettes are intact in AC1−/− mice, it is possible that functional input from brainstem axons projecting to VB thalamic neurons is impaired. The progressive deterioration in the whisker pattern evident when ascending the trigeminal pathway from brainstem through thalamus to cortex in AC1−/− mice may indicate a cumulative role for AC1 in the transfer of information about the whisker pattern through the trigeminal system (for a summary of morphological and physiological phenotypes in AC1−/− and CxAC1KO mice, see Table 1).
Table 1.
Summary of barrel system phenotypes in AC1−/− (global knock-out or brl mice) and CxAC1KO mice
| Morphology |
TC physiology |
||||
|---|---|---|---|---|---|
| Barrelettes (brainstem) | Barreloids (thalamus) | Barrels (cortex) | Presynaptic | Postsynaptic | |
| AC1−/− | Normal | Partial | Absent | Impaired | Impaired |
| CxAC1KO | Normal | Normal | Grossly normal | Normal | Impaired |
In AC1−/− (global knock-out or brl mice), whisker-related patterning is completely disrupted in the S1 barrel cortex, partially disrupted in the VB thalamus, and normal in the brainstem. Both presynaptic and postsynaptic measures of TC synapse function are also disrupted in AC1−/− mice. In CxAC1KO mice, TC axon clustering and layer IV barrel morphology is grossly normal, although quantitative measures of layer IV barrel cytoarchitecture and barrel neuron morphology reveal small deficits. At the same time, thalamic barreloids and brainstem barrelettes are normal in CxAC1KO mice. Physiologically, the TC synapse has normal presynaptic but disrupted postsynaptic function in CxAC1KO mice.
It is intriguing that the extent of lesion-induced morphological plasticity and the duration of the critical period in CxAC1KO mice are indistinguishable from those of control mice. We previously demonstrated that CxNR1KO mice, in which NMDA receptor function is genetically impaired only in cortical excitatory neurons, showed normal lesion-induced TC axonal plasticity (Datwani et al., 2002b), and mutant mice for PKARIIβ, which is expressed in layer IV cortical neurons but not TC axon terminals, showed normal lesion-induced plasticity (Inan et al., 2006). Furthermore, parachlorophenylalanine-treated MAOA knock-out mice, in which the emergence of barrel patterning, but not barreloid and barrelette patterning, is delayed, show no delay in the closure of the lesion-induced plasticity period (Rebsam et al., 2005). Thus, it is likely that lesion-induced morphological plasticity is regulated at the subcortical level.
An alternative explanation for the relatively mild morphological phenotype and normal lesion-induced morphological plasticity in CxAC1KO mice could be the persistence of AC1 expression in cortical interneurons of CxAC1KO mice, because Cre-mediated gene disruption induced by Emx1-Cre mice is restricted to excitatory neurons in the cortex (Iwasato et al., 2000, 2004). Inhibitory synaptic transmission mediated by interneurons has emerged as an essential component for shaping aspects of the development and plasticity of cortical networks (Hensch and Stryker, 2004; Foeller et al., 2005; Gabernet et al., 2005). However, inhibitory neurons make up only 15% of the neuronal population in layer IV of cortex (Lin et al., 1985). Although inhibitory synaptic circuitry develops after the critical period for TC synapse maturation and barrel map formation (Daw et al., 2007), we cannot rule out a role for inhibitory interneurons in anatomical map development. To clarify these various possibilities, the role of AC1 in the thalamus, brainstem, and cortical inhibitory neurons needs to be explicitly determined.
Footnotes
This work was supported in part by a Grant-in-Aid for Scientific Research on Priority Areas (Elucidation of Neural Network Function in the Brain) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (T.I.); National Institutes of Health Grant R01 MH62639; and the Rett Syndrome Research Foundation (M.C.C.). We thank Reiko Ando, Mizuho Iwama, and Yoshikazu M. Saito for technical assistance, Brain Science Institute Research Resources Center for animal care and technical assistance, Akash Datwani for the Golgi protocol, Jun-ichi Miyazaki for CAG-CAT-Z mice, Yueyi Zhang for technical assistance, and members of the Crair Laboratory for helpful comments on this manuscript.
References
- Abdel-Majid RM, Leong WL, Schalkwyk LC, Smallman DS, Wong ST, Storm DR, Fine A, Dobson MJ, Guernsey DL, Neumann PE. Loss of adenylyl cyclase I activity disrupts patterning of mouse somatosensory cortex. Nat Genet. 1998;19:289–291. doi: 10.1038/980. [DOI] [PubMed] [Google Scholar]
- Agmon A, Yang LT, O'Dowd DK, Jones EG. Organized growth of thalamocortical axons from the deep tier of terminations into layer IV of developing mouse barrel cortex. J Neurosci. 1993;13:5365–5382. doi: 10.1523/JNEUROSCI.13-12-05365.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu C. Numerical data on neocortical neurons in adult rat, with special reference to the GABA population. Brain Res. 1993;609:284–292. doi: 10.1016/0006-8993(93)90884-p. [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL. Ca2+ signaling in dendritic spines. Curr Opin Neurobiol. 2007;17:345–351. doi: 10.1016/j.conb.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Chan CH, Godinho LN, Thomaidou D, Tan SS, Gulisano M, Parnavelas JG. Emx1 is a marker for pyramidal neurons of the cerebral cortex. Cereb Cortex. 2001;11:1191–1198. doi: 10.1093/cercor/11.12.1191. [DOI] [PubMed] [Google Scholar]
- Crair MC, Malenka RC. A critical period for long-term potentiation at thalamocortical synapses. Nature. 1995;375:325–328. doi: 10.1038/375325a0. [DOI] [PubMed] [Google Scholar]
- Cui W, Smith A, Darby-King A, Harley CW, McLean JH. A temporal-specific and transient cAMP increase characterizes odorant classical conditioning. Learn Mem. 2007;14:126–133. doi: 10.1101/lm.496007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datwani A, Iwasato T, Itohara S, Erzurumlu RS. NMDA receptor-dependent pattern transfer from afferents to postsynaptic cells and dendritic differentiation in the barrel cortex. Mol Cell Neurosci. 2002a;21:477–492. doi: 10.1006/mcne.2002.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datwani A, Iwasato T, Itohara S, Erzurumlu RS. Lesion-induced thalamocortical axonal plasticity in the S1 cortex is independent of NMDA receptor function in excitatory cortical neurons. J Neurosci. 2002b;22:9171–9175. doi: 10.1523/JNEUROSCI.22-21-09171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw MI, Ashby MC, Isaac JT. Coordinated developmental recruitment of latent fast spiking interneurons in layer IV barrel cortex. Nat Neurosci. 2007;10:453–461. doi: 10.1038/nn1866. [DOI] [PubMed] [Google Scholar]
- Durham D, Woolsey TA. Effects of neonatal whisker lesions on mouse central trigeminal pathways. J Comp Neurol. 1984;223:424–447. doi: 10.1002/cne.902230308. [DOI] [PubMed] [Google Scholar]
- Erzurumlu RS, Iwasato T. Patterning of the somatosensory maps with NMDA receptors. In: Erzurumlu RS, Guido W, Molnár Z, editors. Development and plasticity in sensory thalamus and cortex. New York: Springer; 2006. pp. 158–182. [Google Scholar]
- Erzurumlu RS, Jhaveri S. Thalamic axons confer a blueprint of the sensory periphery onto the developing rat somatosensory cortex. Brain Res Dev Brain Res. 1990;56:229–234. doi: 10.1016/0165-3806(90)90087-f. [DOI] [PubMed] [Google Scholar]
- Erzurumlu RS, Kind PC. Neural activity: sculptor of “barrels” in the neocortex. Trends Neurosci. 2001;24:589–595. doi: 10.1016/s0166-2236(00)01958-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE, Nicoll RA, Malenka RC, Isaac JT. Long-term depression at thalamocortical synapses in developing rat somatosensory cortex. Neuron. 1998;21:347–357. doi: 10.1016/s0896-6273(00)80544-9. [DOI] [PubMed] [Google Scholar]
- Ferguson GD, Storm DR. Why calcium-stimulated adenylyl cyclases? Physiology (Bethesda) 2004;19:271–276. doi: 10.1152/physiol.00010.2004. [DOI] [PubMed] [Google Scholar]
- Foeller E, Celikel T, Feldman DE. Inhibitory sharpening of receptive fields contributes to whisker map plasticity in rat somatosensory cortex. J Neurophysiol. 2005;94:4387–4400. doi: 10.1152/jn.00553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabernet L, Jadhav SP, Feldman DE, Carandini M, Scanziani M. Somatosensory integration controlled by dynamic thalamocortical feed-forward inhibition. Neuron. 2005;48:315–327. doi: 10.1016/j.neuron.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove EA, Fukuchi-Shimogori T. Generating the cerebral cortical area map. Annu Rev Neurosci. 2003;26:355–380. doi: 10.1146/annurev.neuro.26.041002.131137. [DOI] [PubMed] [Google Scholar]
- Hensch TK, Stryker MP. Columnar architecture sculpted by GABA circuits in developing cat visual cortex. Science. 2004;303:1678–1681. doi: 10.1126/science.1091031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inan M, Crair MC. Development of cortical maps: perspectives from the barrel cortex. Neuroscientist. 2007;13:49–61. doi: 10.1177/1073858406296257. [DOI] [PubMed] [Google Scholar]
- Inan M, Lu HC, Albright MJ, She WC, Crair MC. Barrel map development relies on protein kinase A regulatory subunit II beta-mediated cAMP signaling. J Neurosci. 2006;26:4338–4349. doi: 10.1523/JNEUROSCI.3745-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasato T, Erzurumlu RS, Huerta PT, Chen DF, Sasaoka T, Ulupinar E, Tonegawa S. NMDA receptor-dependent refinement of somatotopic maps. Neuron. 1997;19:1201–1210. doi: 10.1016/s0896-6273(00)80412-2. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knopfel T, Erzurumlu RS, Itohara S. Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature. 2000;406:726–731. doi: 10.1038/35021059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasato T, Nomura R, Ando R, Ikeda T, Tanaka M, Itohara S. Dorsal telencephalon-specific expression of Cre recombinase in PAC transgenic mice. Genesis. 2004;38:130–138. doi: 10.1002/gene.20009. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Katoh H, Nishimaru H, Ishikawa Y, Inoue H, Saito YM, Ando R, Iwama M, Takahashi R, Negishi M, Itohara S. Rac-GAP alpha-chimerin regulates motor-circuit formation as a key mediator of EphrinB3/EphA4 forward signaling. Cell. 2007;130:742–753. doi: 10.1016/j.cell.2007.07.022. [DOI] [PubMed] [Google Scholar]
- Kovalchuk Y, Eilers J, Lisman J, Konnerth A. NMDA receptor-mediated subthreshold Ca2+ signals in spines of hippocampal neurons. J Neurosci. 2000;20:1791–1799. doi: 10.1523/JNEUROSCI.20-05-01791.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrand C, Cases O, Wehrle R, Blakely RD, Edwards RH, Gaspar P. Transient developmental expression of monoamine transporters in the rodent forebrain. J Comp Neurol. 1998;401:506–524. [PubMed] [Google Scholar]
- Lin CS, Lu SM, Schmechel DE. Glutamic acid decarboxylase immunoreactivity in layer IV of barrel cortex of rat and mouse. J Neurosci. 1985;5:1934–1939. doi: 10.1523/JNEUROSCI.05-07-01934.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HC, Gonzalez E, Crair MC. Barrel cortex critical period plasticity is independent of changes in NMDA receptor subunit composition. Neuron. 2001;32:619–634. doi: 10.1016/s0896-6273(01)00501-3. [DOI] [PubMed] [Google Scholar]
- Lu HC, She WC, Plas DT, Neumann PE, Janz R, Crair MC. Adenylyl cyclase I regulates AMPA receptor trafficking during mouse cortical “barrel” map development. Nat Neurosci. 2003;6:939–947. doi: 10.1038/nn1106. [DOI] [PubMed] [Google Scholar]
- Lu HC, Butts DA, Kaeser PS, She WC, Janz R, Crair MC. Role of efficient neurotransmitter release in barrel map development. J Neurosci. 2006;26:2692–2703. doi: 10.1523/JNEUROSCI.3956-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Matsuoka I, Suzuki Y, Defer N, Nakanishi H, Hanoune J. Differential expression of type I, II, and V adenylyl cyclase gene in the postnatal developing rat brain. J Neurochem. 1997;68:498–506. doi: 10.1046/j.1471-4159.1997.68020498.x. [DOI] [PubMed] [Google Scholar]
- Nicol X, Muzerelle A, Bachy I, Ravary A, Gaspar P. Spatiotemporal localization of the calcium-stimulated adenylate cyclases, AC1 and AC8, during mouse brain development. J Comp Neurol. 2005;486:281–294. doi: 10.1002/cne.20528. [DOI] [PubMed] [Google Scholar]
- Nicol X, Muzerelle A, Rio JP, Metin C, Gaspar P. Requirement of adenylate cyclase 1 for the ephrin-A5-dependent retraction of exuberant retinal axons. J Neurosci. 2006a;26:862–872. doi: 10.1523/JNEUROSCI.3385-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol X, Bennis M, Ishikawa Y, Chan GC, Reperant J, Storm DR, Gaspar P. Role of the calcium modulated cyclases in the development of the retinal projections. Eur J Neurosci. 2006b;24:3401–3414. doi: 10.1111/j.1460-9568.2006.05227.x. [DOI] [PubMed] [Google Scholar]
- Plas DT, Visel A, Gonzalez E, She WC, Crair MC. Adenylate cyclase 1 dependent refinement of retinotopic maps in the mouse. Vision Res. 2004;44:3357–3364. doi: 10.1016/j.visres.2004.09.036. [DOI] [PubMed] [Google Scholar]
- Ravary A, Muzerelle A, Herve D, Pascoli V, Ba-Charvet KN, Girault JA, Welker E, Gaspar P. Adenylate cyclase 1 as a key actor in the refinement of retinal projection maps. J Neurosci. 2003;23:2228–2238. doi: 10.1523/JNEUROSCI.23-06-02228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsam A, Seif I, Gaspar P. Refinement of thalamocortical arbors and emergence of barrel domains in the primary somatosensory cortex: a study of normal and monoamine oxidase a knock-out mice. J Neurosci. 2002;22:8541–8552. doi: 10.1523/JNEUROSCI.22-19-08541.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsam A, Seif I, Gaspar P. Dissociating barrel development and lesion-induced plasticity in the mouse somatosensory cortex. J Neurosci. 2005;25:706–710. doi: 10.1523/JNEUROSCI.4191-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- Schlaggar BL, Fox K, O'Leary DD. Postsynaptic control of plasticity in developing somatosensory cortex. Nature. 1993;364:623–626. doi: 10.1038/364623a0. [DOI] [PubMed] [Google Scholar]
- Senft SL, Woolsey TA. Growth of thalamic afferents into mouse barrel cortex. Cereb Cortex. 1991;1:308–335. doi: 10.1093/cercor/1.4.308. [DOI] [PubMed] [Google Scholar]
- Storm DR, Hansel C, Hacker B, Parent A, Linden DJ. Impaired cerebellar long-term potentiation in type I adenylyl cyclase mutant mice. Neuron. 1998;20:1199–1210. doi: 10.1016/s0896-6273(00)80500-0. [DOI] [PubMed] [Google Scholar]
- Strominger RN, Woolsey TA. Templates for locating the whisker area in fresh flattened mouse and rat cortex. J Neurosci Methods. 1987;22:113–118. doi: 10.1016/0165-0270(87)90004-5. [DOI] [PubMed] [Google Scholar]
- Sur M, Rubenstein JL. Patterning and plasticity of the cerebral cortex. Science. 2005;310:805–810. doi: 10.1126/science.1112070. [DOI] [PubMed] [Google Scholar]
- Van der Loos H, Woolsey TA. Somatosensory cortex: structural alterations following early injury to sense organs. Science. 1973;179:395–398. doi: 10.1126/science.179.4071.395. [DOI] [PubMed] [Google Scholar]
- Villacres EC, Wong ST, Chavkin C, Storm DR. Type I adenylyl cyclase mutant mice have impaired mossy fiber long-term potentiation. J Neurosci. 1998;18:3186–3194. doi: 10.1523/JNEUROSCI.18-09-03186.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Storm DR. Calmodulin-regulated adenylyl cyclases: cross-talk and plasticity in the central nervous system. Mol Pharmacol. 2003;63:463–468. doi: 10.1124/mol.63.3.463. [DOI] [PubMed] [Google Scholar]
- Watson RF, Abdel-Majid RM, Barnett MW, Willis BS, Katsnelson A, Gillingwater TH, McKnight GS, Kind PC, Neumann PE. Involvement of protein kinase A in patterning of the mouse somatosensory cortex. J Neurosci. 2006;26:5393–5401. doi: 10.1523/JNEUROSCI.0750-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker E, Armstrong-James M, Bronchti G, Ourednik W, Gheorghita-Baechler F, Dubois R, Guernsey DL, Van der Loos H, Neumann PE. Altered sensory processing in the somatosensory cortex of the mouse mutant barrelless. Science. 1996;271:1864–1867. doi: 10.1126/science.271.5257.1864. [DOI] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron. 1999;23:787–798. doi: 10.1016/s0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MT, Merzenich MM, Leake PA. Changes in endogenous enzymatic reactivity to DAB induced by neuronal inactivity. Brain Res. 1978;141:185–192. doi: 10.1016/0006-8993(78)90629-7. [DOI] [PubMed] [Google Scholar]
- Woolsey TA, Van der Loos H. The structural organization of layer IV in the somatosensory region (SI) of mouse cerebral cortex. The description of a cortical field composed of discrete cytoarchitectonic units. Brain Res. 1970;17:205–242. doi: 10.1016/0006-8993(70)90079-x. [DOI] [PubMed] [Google Scholar]
- Woolsey TA, Wann JR. Areal changes in mouse cortical barrels following vibrissal damage at different postnatal ages. J Comp Neurol. 1976;170:53–66. doi: 10.1002/cne.901700105. [DOI] [PubMed] [Google Scholar]
- Xia Z, Storm DR. The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci. 2005;6:267–276. doi: 10.1038/nrn1647. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG. Presynaptic strontium dynamics and synaptic transmission. Biophys J. 1999;76:2029–2042. doi: 10.1016/S0006-3495(99)77360-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Changes in the statistics of transmitter release during facilitation. J Physiol (Lond) 1973;229:787–810. doi: 10.1113/jphysiol.1973.sp010167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]