

Abstract
Diagnosis of the type of glomerular disease that causes the nephrotic syndrome is necessary for appropriate treatment and typically requires a renal biopsy. The goal of this study was to identify candidate protein biomarkers to diagnose glomerular diseases. Proteomic methods and informatic analysis were used to identify patterns of urine proteins that are characteristic of the diseases. Urine proteins were separated by two-dimensional electrophoresis in 32 patients with FSGS, lupus nephritis, membranous nephropathy, or diabetic nephropathy. Protein abundances from 16 patients were used to train an artificial neural network to create a prediction algorithm. The remaining 16 patients were used as an external validation set to test the accuracy of the prediction algorithm. In the validation set, the model predicted the presence of the diseases with sensitivities between 75 and 86% and specificities from 92 to 67%. The probability of obtaining these results in the novel set by chance is 5 × 10−8. Twenty-one gel spots were most important for the differentiation of the diseases. The spots were cut from the gel, and 20 were identified by mass spectrometry as charge forms of 11 plasma proteins: Orosomucoid, transferrin, α-1 microglobulin, zinc α-2 glycoprotein, α-1 antitrypsin, complement factor B, haptoglobin, transthyretin, plasma retinol binding protein, albumin, and hemopexin. These data show that diseases that cause nephrotic syndrome change glomerular protein permeability in characteristic patterns. The fingerprint of urine protein charge forms identifies the glomerular disease. The identified proteins are candidate biomarkers that can be tested in assays that are more amenable to clinical testing.
Glomerular diseases such as diabetic nephropathy and FSGS are associated with proteinuria that is caused by increased glomerular permeability. Of the 100,000 patients who developed ESRD in the United States in 2003, more than half had some form of glomerular disease (1). The glomerulus consists of a tuft of capillaries and associated cells that are responsible for filtration of small molecules while preventing the loss of larger molecules. The glomerular wall contains three layers: Endothelial cells, basement membrane, and epithelial cells. Much of the selectivity of filtration occurs in the basement membrane, where the barrier excludes proteins on the basis of both their size and their charge. Uncharged molecules pass through the basement membrane more readily than negatively charged proteins of a similar size (2,3). Selective permeability related to the size of the molecule and its charge has been shown with charged dextrans (4). The permeability barrier is damaged in glomerular diseases that lead to proteinuria. Because treatment is disease specific, it requires knowledge of the underlying process. A renal biopsy is needed to make a definitive diagnosis of the cause of the disease. The utility of renal biopsy is limited by several factors. Because of comorbid conditions such as bleeding disorders and obesity, some patients are not suitable or are at higher risk for a biopsy (5,6). Because the biopsy obtains only a small portion of the kidney, in some cases it may not accurately portray the disease if the affected portion of the kidney is not sampled. In other cases, the disease may be so far advanced that diagnostic features are obscured. Urine testing for biomarkers could replace renal biopsy as a simple, safe, and accurate test that could be repeated to follow progression of the disease and monitor response to therapy.
Potential urine markers for diagnosis of glomerular disease have been proposed, but none has been confirmed to differentiate between causes of glomerular diseases. Many of the proposed markers are urinary cytokines. Levels of monocyte chemoattractant protein-1 (MCP-1) (7), IL-6 (8), vascular cellular adhesion molecule-1 (9), the complement degradation product C3d (10), and urinary free light chains (11) have been proposed as markers of renal activity of lupus. Urinary concentrations of vascular endothelial growth factor (12-15), IL receptor 1 antagonist (16), IL-17 (17), TNF-α (18), and CD46 (19) have been proposed as markers of specific glomerular diseases. Urinary macrophages (20,21), podocytes, and associated proteins are also potential markers for glomerular diseases (22-24). An expression ratio of two genes that are expressed in podocytes, podocin and synaptopodin differentiated patients with FSGS from those with minimal-change disease (25). Although many studies have compared levels of candidate markers between two diseases, none has reliably used urine protein markers to differentiate between a group of glomerular diseases. The presence of plasma proteins in urine of patients with nephrotic syndrome provides an opportunity for discovery of new biomarkers. Differences in the charge of proteins such as ferritin and horseradish peroxidase are known to affect their permeability at the glomerulus (26,27). In nephrotic syndrome, the glomerular permeability barrier is altered, resulting in proteinuria. If changes in size and charge permeability occur independently and are specific to the disease, then these changes could be used to predict the cause of glomerular diseases.
The pathway from biomarker discovery to clinically useful assay has three phases. The first phase is the discovery of candidate biomarkers. An unbiased approach that is not limited to known candidates is best for this stage. The false discovery rate in this phase may be high because of noise in the system, a necessarily small sample size, and introduction of systematic bias during analysis. Unlike the first stage, in which many proteins are measured simultaneously, in the second phase, only the candidate biomarkers are measured. Because the number of analytes measured is smaller, a more reproducible, rapid and accurate assay can be used. In this phase, some candidate markers may be discarded because they are not useful. Finally, in the third phase, the successful biomarkers (and the algorithm to interpret them) can be tested in a large, novel set of patients under true clinical conditions.
Proteomic techniques such as two-dimensional gel electrophoresis (2DE) are well suited to the discovery phase of biomarker identification. 2DE is a high-resolution separation technique that can be coupled with protein identification by mass spectrometry. A particular strength of 2DE for urine biomarker discovery is the ability to visualize differences in posttranslational modifications when these changes alter the isoelectric point of the proteins. Posttranslational modifications affect the charge on the protein and the ability of a given protein to pass through the glomerular permeability barrier. Because many plasma proteins exist as glycosylation forms with different charges, they may be filtered differentially. We used 2DE to identify patterns of candidate biomarkers that can differentiate from among four glomerular disease. The candidate markers can predict the disease with a relatively high degree of sensitivity and specificity in a population of patients who were not used to derive the algorithm.
Materials and Methods
Sample Collection and Protein Separation
We examined urine from 32 patients with proteinuria of >3 g/d. Twenty-seven of the patients, including three with diabetic nephropathy, had a renal biopsy to confirm the diagnosis. Because patients with typical diabetic nephropathy are not routinely biopsied at our center, we obtained urine from five additional patients with a typical presentation of diabetic nephropathy and without suggestion of any other disease process. These patients all had diabetes for at least 15 yr, diabetic retinopathy with a history of laser photocoagulation therapy, an absence of microscopic hematuria, and negative serologies for hepatitis and HIV. Samples were collected at the Medical University of South Carolina and Ralph H. Johnson VA hospitals under a protocol that was approved by the appropriate institutional review boards. Urine samples were collected immediately before biopsy. Since the urine was collected at the time of biopsy, samples were retained in the bladder for variable periods of time. The samples were centrifuged at 1000 × g for 10 min to remove cellular and particulate matter and immediately frozen at −80°C until processing. No protease inhibitors were added. These are conditions that we have refined in our laboratory to optimize reproducibility (unpublished data). Two milliliters of urine was injected into a Biologic Duo Flow HPLC system (Bio-Rad, Hercules, CA), and buffer exchange with 100 mM ammonium acetate at a flow rate of 5 ml/min was done. The sample was passed through a HiTrap Desalting column (Amersham, Uppsala, Sweden), and a fraction of 1.75 ml was collected with desalting verified by conductivity tracings. The sample was then frozen at −80°C and lyophilized. Urine protein concentration was adjusted to 100 μg in 185 μl with a buffer that contained 9 M urea, 4% NP-40, 0.2% 3 to 10 ampholytes, and 50 mM dithiothreitol. The samples were centrifuged at 100,000 × g for 30 min. The supernatant was applied to Bio-Rad IPG strips (11 cm, pH 4 to 7). Strips were incubated at room temperature for 1 h. After 1 h, the strips were covered with mineral oil and rehydrated for an additional 12 h at room temperature. After focusing, strips were equilibrated sequentially in buffers that contained dithiothreitol and iodoacetamide and separated by SDS-PAGE on an 8 to 16% gradient gel using a Criterion Doceca cell (Bio-Rad). Proteins were stained with Sypro Ruby, the gels were imaged, and individual spots were aligned across the gel using PDQuest (Bio-Rad). Urine concentrations of IL-6, IL-8, and MCP-1 were measured with a Bio-Plex System (Bio-Rad) using Luminex xMAP technology according to the manufacturer’s instructions.
Exploratory Multivariate Statistical Analysis
To determine whether unsupervised groupings of patients on the basis of the urine protein spots present would correlate with the disease process that caused the nephrotic syndrome, we performed an exploratory analysis using clustering by the bottom-up approach of unsupervised simultaneous clustering of gels and spots by unweighted pair group average. The clustering was performed using code that was written in Matlab. Clustering of patient samples was compared with the order of sample collection, disease, race, age, and serum creatinine to determine potential sources of variability within the samples.
Artificial Neural Network Analysis
Protein spot intensities were ranked by intensity and expressed as quantiles as described previously (28). Four patients with each disease were randomly selected for the training set using the random-number-generator function in Microsoft Excel. The artificial neural network (ANN) algorithm was trained on urine concentrations of IL-6, IL-8, MCP-1, and the set of ranked protein spot abundance data from 16 patients. The data from the remaining 16 patients (external validation set) were not seen by the ANN during the training phase. An input was assigned for each of the four diseases, where 0 was disease absent and 1 was disease present. The identification of ANN models was performed by Matlab code that was written along the guidelines previously proposed (29), which includes bootstrapped cross-validation as an early stop criterion and screening for optimal topology. The predictive value of each spot was evaluated by sensitivity analysis by determining, for each ith spot in each jth gel/patient, Si,j = (dOj/dIi,j) × (Ii,j/Oj). A cross-validation scheme with leave one ninth out was used in which every ninth sample was used for internal validation. The median performing ANN was selected from each run.
The external data set was kept completely independent from the training procedures with the purpose of having an unbiased assessment of model predictability. Data from the external validation set were analyzed using the network that was obtained with the training set, and an output for each disease category for each patient was obtained. The output was a number between 0 and 1. A threshold value for each disease was chosen such that values that were greater than the threshold were disease positive and values that were less than the threshold were disease negative. Because each prediction of disease was independent, each patient could be predicted to be disease positive in zero to four diseases. The prediction was compared with the known disease for each patient, and each prediction was determined to be true positive, false positive, true negative, or false negative.
To determine the importance of the number of spots included in the analysis, we sequentially removed spots from the analysis. Spots were divided into two groups: The 21 spots that provided the most sensitivity to the analysis and the remaining 103 spots. Spots in the 103 group were ordered using a random-number generator and removed from the analysis in groups of five spots. For each removal of five spots, a new ANN was trained using the training set. Each network was trained only once with the data for each number of spots. The network was used to analyze the external test set. Threshold values for each test were set at the value that would best optimize sensitivity. After the initial 103 spots were removed, the same process was done with the final 21 spots. These spots were removed one at a time in the order of increasing sensitivity as determined by the initial analysis. Total accuracy of the test was calculated as the number of correct predictions divided by the number of tests (n = 64). Sensitivity was calculated as the number of correct predictions from the tests of the disease that was present (true positives).
The initial evaluation of the output gave a true or false value for each of the four diseases but did not give a single disease output. To determine the accuracy of the test to be able to predict one disease, we designed a simple voting scheme so that a single disease output could be obtained. In patients for whom only one of the four test results was positive, the positive test was chosen to predict the disease. In cases in which more than one test was positive, the ANN output value was compared with the threshold value. The test for which the difference between the threshold value and the output value was greatest was chosen as the predicted disease.
Protein Identification
Protein spots were picked from the gels and digested with trypsin as described previously (30). Digests were concentrated using C18 Zip Tips (Millipore, Billerica, MA). Proteins were identified using a matrix-assisted laser desorption ionization–time of flight (MALDI-TOF-MS) mass spectrometer (MS), an ABI 4700 MALDI-TOF/TOF MS, or a Finnigan LTQ linear ion trap MS as described previously (31). Initial protein identification was done using the Mascot search engine, whereby the Mascot score is a descriptor of the quality of the match of the spectra to a protein. Identification by peptide mass fingerprinting required a Mascot score >62. Protein coverage was calculated as the percentage of the total number of amino acids in the protein that were accounted for by the predicted matches. Proteins that were identified with Mascot scores <90 were confirmed by tandem MS with a total ion score of at least 80. Protein identification from the ion trap MS was done using the Turbo-SEQUEST algorithm. Criteria for identification of peptides was XCorr >1.5 for singly charged ions, >2.0 for doubly charged, and >2.5 for triply charged ions. All proteins that were identified by the linear ion trap had at least three peptides.
Results
Urine samples were collected from 32 patients. Demographic and clinical information of the patients in the study is shown in Table 1. Patients with lupus nephritis were younger and more likely to be female. Patients with diabetic nephropathy had higher serum creatinine values. In the group with membranous nephropathy, there was a trend toward increased proteinuria that did not reach statistical significance. All of the patients except six were on either an angiotensin-converting enzyme inhibitor or an angiotensin receptor blocker at the time urine was collected (diabetes six of eight; lupus nephritis eight of 11; membranous nephropathy four of five; FSGS eight of eight). Levels of the three urinary cytokines (IL-6, IL-8, and MCP-1) were not different between groups.
Table 1.
Characteristics of patients in the disease groupsa
| Characteristic | FSGS |
Lupus Nephritis |
Membranous Nephropathy |
Diabetic Nephropathy |
P | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All | Train | Test | All | Train | Test | All | Train | Test | All | Train | Test | ||
| Gender (M:F) | 4:4 | 1:3 | 3:1 | 1:10 | 1:3 | 0:7 | 4:1 | 3:1 | 1:0 | 4:4 | 1:3 | 3:1 | |
| Race (white:black) | 4:4 | 2:2 | 2:2 | 6:5 | 2:2 | 4:3 | 2:3 | 2:2 | 0:1 | 3:5 | 1:3 | 2:2 | |
| Age | 38 ± 5 | 41 ± 9 | 34 ± 5 | 27 ± 3 | 26 ± 4 | 27 ± 3 | 63 ± 8 | 65 ± 10 | 54 | 57 ± 7 | 63 ± 4 | 52 ± 13 | 0.001 |
| Creatinine | 2.2 ± 1.0 | 1.7 ± 0.5 | 2.7 ± 2.0 | 1.8 ± 0.3 | 1.4 ± 0.4 | 2.1 ± 0.5 | 2.2 ± 0.8 | 2.4 ± 1.0 | 1.6 | 4.7 ± 0.9 | 4.0 ± 0.5 | 5.3 ± 1.9 | 0.039 |
| UA Pr/Cr | 5.1 ± 1.8 | 6.4 ± 2.3 | 3.8 ± 2.8 | 6.9 ± 2.5 | 3.9 ± 2.0 | 8.6 ± 3.7 | 12.1 ± 3.9 | 13.0 ± 4.8 | 8.5 | 5.6 ± 2.3 | 3.2 ± 2.3 | 8.0 ± 4.0 | 0.358 |
| IL-6 | 369 ± 48 | 401 ± 80 | 337 ± 58 | 543 ± 64 | 559 ± 100 | 533 ± 88 | 535 ± 128 | 591 ± 148 | 312 | 527 ± 115 | 568 ± 204 | 486 ± 139 | 0.427 |
| IL-8 | 187 ± 56 | 187 ± 80 | 189 ± 91 | 275 ± 59 | 257 ± 92 | 285 ±82 | 122 ± 17 | 116 ± 21 | 145 | 381 ± 193 | 645 ± 357 | 137 ± 49 | 0.419 |
| MCP-1 | 427 ± 23 | 437 ± 68 | 418 ± 62 | 752 ± 186 | 1044 ± 499 | 585 ± 82 | 655 ± 204 | 707 ± 254 | 448 | 56 ± 97 | 576 ± 134 | 547 ± 160 | 0.446 |
MCP-1, monocyte chemoattractant protein-1; UA Pr/Cr, urine protein:creatinine.
Urine proteins were separated by 2D gel electrophoresis (Figure 1), and the abundance of the protein spots was determined. Protein abundance was compared between groups to identify differences in protein expression. No differences in protein expression of single spots that could differentiate all four diseases were found. To identify aggregate variation in the samples and to determine whether clinical diagnosis or specific clinical and demographic characteristics contributed to the overall variability, we performed a double-cluster analysis of the gels and spots. To identify associations with the variability, we compared sample collection order, diagnosis, race, age, and serum creatinine with the position in the cluster analysis (Figure 2). No correlations were observed, demonstrating that none of these factors was the major influence on variability within the samples. Because analysis of individual spot abundances was unable to identify biomarkers that could differentiate the diseases, we used ANN to find differences in patterns of spots and to identify markers. Particular attention was given to data analysis safeguards against overfitting in the test set. The external validation set (16 patients) was selected once and was analyzed once, with no inclusion in any internal cross-validation procedure. Data about four patients from each of the four groups were used to train the self-configurable ANN to predict the disease. The external validation set was used to test the accuracy of its predictions. The threshold was set as the value that minimized false discovery rate. The output value from the ANN for the external validation set was compared with the threshold value, and the prediction was determined. The output values and predictions for the validation set are shown in Table 2. Each row shows a patient’s disease and the output value for the test for each of the four diseases. The true disease is in the column on the left side of the table, and the output value for diagnosis of each of the diseases is shown in the remaining columns. The output value for each of the four diseases was compared with the threshold value at the bottom of each column to obtain the prediction of disease positive or disease negative. False predictions are shown in bold. The International Society of Nephrology/Renal Pathology Society disease classification is shown for patients with lupus nephritis. The area under the receiver operating characteristics curve for the test data was found to be 0.69 for FSGS, 0.84 for lupus nephritis, and 0.73 for diabetic nephropathy. The area under the receiver operating characteristics curve for membranous nephropathy is not reported because there was only one case in the test set. Sixty-four predictions were made (16 patients for each of four diseases), 11 of which were incorrect (accuracy 83%). Two incorrect predictions each were made for FSGS, systemic lupus erythematosus, and diabetic nephropathy and five for membranous nephropathy. Sensitivity of the assay in patients in the external test set ranged from 75 to 86%, and specificity ranged from 92 to 67% (Figure 3).
Figure 1.

Proteins that are necessary for diagnosis of the cause of the nephrotic syndrome. Representative gel from a patient with the nephrotic syndrome. Proteins were separated in two dimensions by isoelectric point and molecular weight. The 21 numbered spots provided the most sensitivity to the analysis of the cause of the glomerular disease by artificial neural network (ANN). Numbers correspond to protein identifications in Table 3.
Figure 2.
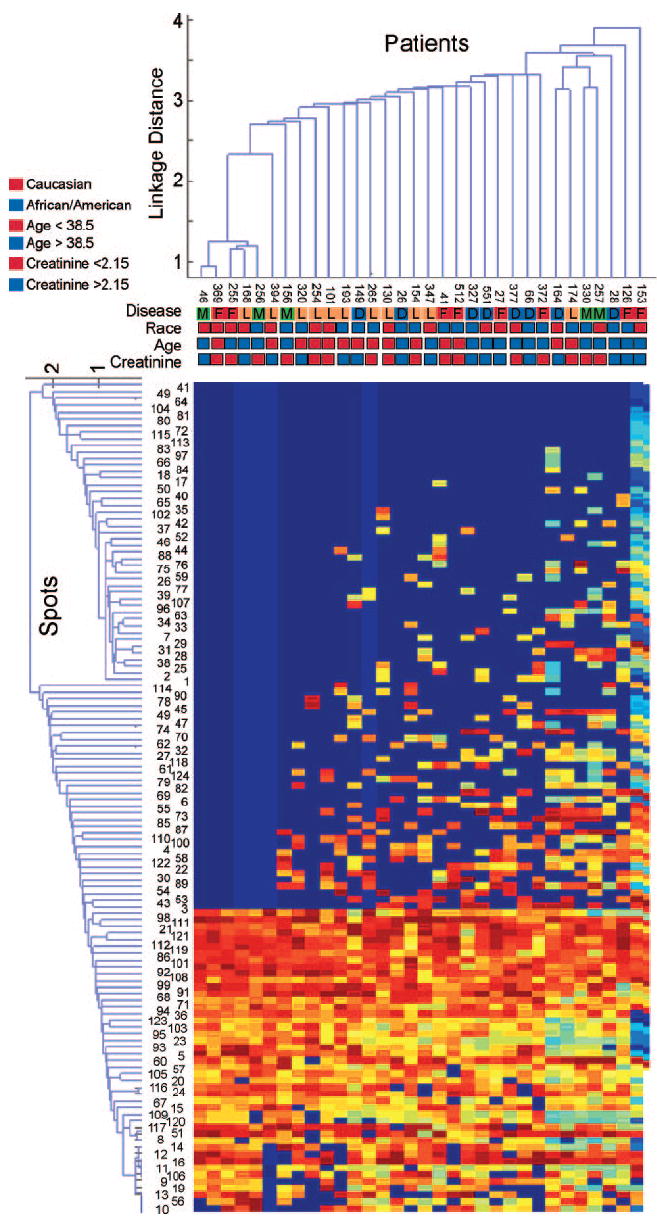
Unsupervised cluster analysis of protein expression in patients with glomerular diseases. Patterns of clustering did not occur on the basis of collection order, disease, race, age, or serum creatinine of patients. The colored boxes represent disease, race, age, and serum creatinine values. The creatinine values are color coded for those above or below the median value. Numbers in the line above the disease represent the sequential order in which the samples were collected.
Table 2.
Output values for prediction of glomerular diseasesa
| Disease | Test |
Disease | |||||||
|---|---|---|---|---|---|---|---|---|---|
| FSGS |
SLE |
Membranous |
Diabetic Nephropathy |
||||||
| Output | Interpretation | Output | Interpretation | Output | Interpretation | Output | Interpretation | ||
| FSGS | 5.8E-03 | Positive | 0.17 | Negative | 0.25 | Negative | 9.7E-08 | Negative | FSGS |
| FSGS | 5.4E-08 | Positive | 0.36 | Negative | 0.26 | Negative | 5.9E-08 | Negative | FSGS |
| FSGS | 4.7E-10 | Negative | 0.28 | Negative | 0.33 | Negative | 0.75 | Positive | Diabetes |
| FSGS | 0.12 | Positive | 0.59 | Positive | 0.28 | Negative | 4.4E-10 | Negative | FSGS |
| SLE III | 3.1E-10 | Negative | 0.50 | Positive | 0.39 | Positive | 4.6E-10 | Negative | Membranous |
| SLE IV | 1.2E-09 | Negative | 0.67 | Positive | 0.23 | Negative | 2.8E-10 | Negative | SLE |
| SLE IV | 5.7E-10 | Negative | 0.67 | Positive | 0.34 | Negative | 3.1E-10 | Negative | SLE |
| SLE IV | 2.2E-10 | Negative | 0.65 | Positive | 0.53 | Positive | 2.4E-10 | Negative | Membranous |
| SLE V | 4.0E-09 | Negative | 0.52 | Positive | 0.32 | Negative | 1.5E-08 | Negative | SLE |
| SLE V | 1.8E-10 | Negative | 0.77 | Positive | 0.49 | Positive | 2.1E-10 | Negative | SLE |
| SLE V | 1.0 | Positive | 0.24 | Negative | 0.19 | Negative | 2.1E-07 | Negative | FSGS |
| Membranous | 1.9E-10 | Negative | 0.48 | Negative | 0.37 | Positive | 1.2E-09 | Negative | Membranous |
| Diabetes | 1.4E-09 | Negative | 0.34 | Negative | 0.22 | Negative | 1 | Positive | Diabetes |
| Diabetes | 2.1E-09 | Negative | 0.42 | Negative | 0.43 | Positive | 3.2E-05 | Positive | Membranous |
| Diabetes | 2.2E-10 | Negative | 0.32 | Negative | 0.23 | Negative | 1 | Positive | Diabetes |
| Diabetes | 3.6E-10 | Negative | 0.40 | Negative | 0.45 | Positive | 1.6E-07 | Negative | Membranous |
| Threshold | 5.3E-08 | 0.5 | 0.36 | 3.0E-05 | |||||
SLE, systemic lupus erythematosus. Bold text denotes incorrect prediction.
Figure 3.
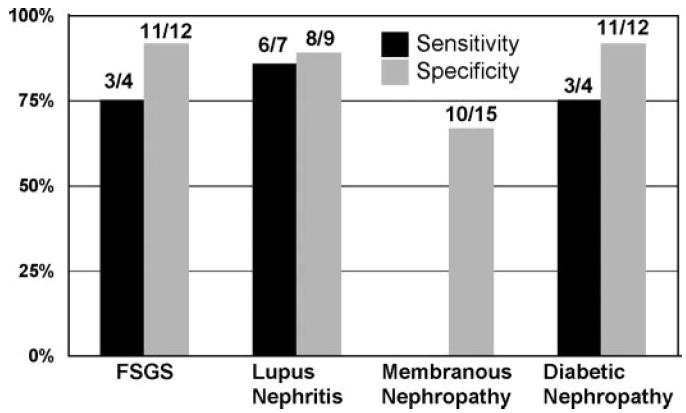
Sensitivity and specificity of biomarkers to predict four glomerular diseases. Calculations were made for patients in the set of patients who were not used to train the ANN. Sensitivity for membranous nephropathy is not reported because only one patient was tested. The legend shows the number of true positives/patients with the disease for the sensitivity bars and the number of true negatives/number of patients without the disease for specificity bars.
To determine the importance of individual spots to the analysis, we sequentially removed spot values from the data set and retrained the network. As in the initial analysis, the network was always trained with the training set and the results were observed from the test set that was not used to train the network. The number of correct diagnoses was determined, and accuracy and sensitivity of the assay were determined for each number of spots. The results are shown in Figure 4. Total accuracy of the test began to decline when fewer than 50 spots were included and decreased more rapidly toward 50% (chance) when fewer than 20 spots were included. Sensitivity of the assay was maintained at >70% until fewer than five spots were included.
Figure 4.
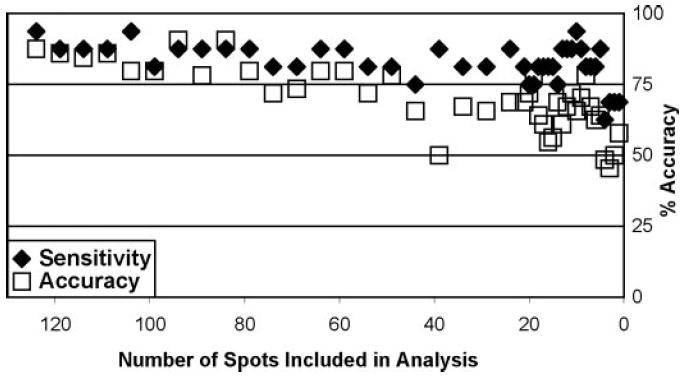
Relationship of number of spots included in the analysis to the sensitivity and total accuracy for the assay. An ANN was trained for each sequential removal of spots from the data set. Sensitivity was calculated as the percentage of true-positive diagnoses from 16. Total accuracy was calculated as the percentage of correct test from all 64 possible.
Because the tests provided only a true/false prediction for the presence of each disease and did not give a final prediction of which single disease was present, we used the difference between threshold values and the ANN output to predict which of the four diseases was present. From the 16 patients in the test set, eight had only the expected test greater than threshold and three had only tests for diseases that were not present greater than threshold. The remaining five had tests for two diseases that were greater than threshold (positive), and the voting scheme was used. Two of the five votes predicted the correct disease, and three predicted the incorrect disease. The final predicted disease is shown in the last column of Table 1. Of the 16 patients, the test correctly predicted the single disease present in 10. It is interesting that the test was always correct when it predicted lupus nephritis (four of four) and usually correct for FSGS (three of four) and diabetic nephropathy (two of three) but poor for prediction of membranous nephropathy (one of five).
We determined from the ANN the amount of sensitivity that each spot provided to the analysis. The top 11 inputs for each disease were chosen for identification. These spots included all inputs that provided >2.5% of the sensitivity for any diagnosis. None of the cytokine inputs was among the top 11 sensitivities. Twenty-one protein spots accounted for all of the inputs in the top 11 because several spots were among the top sensitivities for more than one disease. These spots are shown on the gel in Figure 1. For determination of the identity of the proteins, spots were cut from the gel, digested with trypsin, and analyzed by mass spectrometry. We identified 20 of the 21 proteins. Table 3 shows the protein identification and the molecular weight and isoelectric point of the protein spot on the gel. Mascot scores are shown for proteins that were identified by peptide mass fingerprinting and total ions scores for proteins that were identified by MALDI-TOF MS/MS. Three proteins were identified by electrospray MS/MS on a linear ion trap instrument (LTQ). It is interesting that all of the identified proteins are plasma proteins and many are present as multiple charge forms on the gel. Many of these proteins are known to contain glycosylation posttranslational modifications that produce variable charges depending on the specific glycosylation present, including zinc α-2 glycoprotein, α-1 antitrypsin, haptoglobin, transferrin, albumin, and α-1 microglobulin. Two of the spots are protein fragments because they are present at a lower molecular weight on the gel than the predicted size of the protein (6210 [albumin] and 7209 [orosomucoid]).
Table 3.
Protein identifications
| SSP | MW | pI | Protein Identification | Total Ion Score | Mascot Score | % Protein Coverage |
|---|---|---|---|---|---|---|
| 4812 | 79 | 5.3 | Albumin | 128 | 47 | |
| 6210 | 24 | 5.8 | Albumin | LTQ | ||
| 2514 | 51 | 4.9 | α-1 antitrypsin | 153 | 50 | |
| 2606 | 52 | 4.9 | α-1 antitrypsin | 176 | 54 | |
| 3609 | 52 | 5.0 | α-1 antitrypsin | 112 | 47 | |
| 2308 | 31 | 5.0 | α-I microglobulin | 168 | 68 | 56 |
| 3306 | 34 | 5.1 | Complement factor B Ba fragment | LTQ | ||
| 3512 | 42 | 5.0 | Haptoglobin | 103 | 50 | |
| 5811 | 82 | 5.4 | Hemopexin | 88 | 97 | 45 |
| 6004 | 14 | 5.8 | Not identified | |||
| 0506 | 44 | 4.1 | Orosomucoid | 204 | 71 | 43 |
| 7209 | 27 | 6.2 | Orosomucoid | LTQ | ||
| 4107 | 21 | 5.3 | Plasma retinol binding protein | 115 | 99 | 87 |
| 0814 | 75 | 4.1 | Transferrin | 92 | 37 | |
| 8908 | 94 | 6.3 | Transferrin | 137 | 46 | |
| 8910 | 92 | 6.4 | Transferrin | 170 | 51 | |
| 4009 | 15 | 5.3 | Transthyretin | 103 | 87 | |
| 4609 | 55 | 5.3 | Vitamin D binding protein | 99 | 56 | |
| 2420 | 40 | 4.9 | Zinc α2 glycoprotein | 109 | 66 | |
| 2421 | 41 | 5.0 | Zinc α2 glycoprotein | 101 | 54 | |
| 2512 | 41 | 4.8 | Zinc α2 glycoprotein | 97 | 59 |
LTQ, identification made with linear ion trap mass spectrometer; MW, molecular weight of spot in gel; pI, isoelectric point of spot in gel; SSP, spot number.
Finally, we examined the differences in mean quantile values between the diseases to determine whether differences in individual proteins could be diagnostic themselves. As expected from the data shown in Figure 4, large differences in individual spot abundance between diseases were not seen. Quantile values are the ranked spot abundances expressed as a number between 0 and 1. The smallest mean differences in abundance was seen for spot 0506 (orosomucoid), for which the mean value for all four diseases was within 0.05 of each other, to spot 8908 (transferrin), for which the maximum and minimum differed by 0.35.
Discussion
We identified a set of protein spots that differentiate between patients with one of four common renal glomerular diseases with a relatively high degree of sensitivity and specificity. These proteins are candidate biomarkers because they have not yet been tested in a more reproducible assay on a larger set of patients. Nevertheless, the markers were confirmed in an independent set of patients in whom they showed an impressive level of accuracy despite the variability present in 2DE. The analytic ability of the candidate biomarkers in this relatively small set of patients with heterogeneity within diseases demonstrates that there is a strong diagnostic signal in these markers.
Several studies have used proteomics to characterize candidate urine markers for glomerular diseases. Capillary electrophoresis coupled to mass spectrometry has been used to identify candidate polypeptides in diabetic nephropathy (32-34) and other glomerular diseases (35-38). These studies have defined patterns of polypeptides that are associated with glomerular diseases but have not yet confirmed the validity of the patterns in an independent set. Two-dimensional gel electrophoresis has been used to define a reference map of proteins that are different between healthy subjects and those with IgA nephropathy but did not characterize differences between diseases (39). Our previous study in lupus nephritis and a study from Thongboonkerd et al. (40) are the most similar to this study. We identified a set of urine that can differentiate between classes of lupus nephritis (31). We did not confirm the results in a new set of patients however. Thongboonkerd et al. (40) used 2DE to identify a growth hormone and a protein that they were unable to identify as differentially expressed in FSGS compared with healthy subjects and those with lupus nephritis or diabetic nephropathy. They did not characterize the diagnostic ability of these proteins or confirm the finding in an independent set. This study is the first to find markers for glomerular diseases, identify the proteins, and confirm that the pattern of candidate markers is valid in an independent set.
In this study, a single marker was not sufficient to distinguish from among the diseases. In fact, unsupervised clustering, as shown in Figure 2, did not segregate the diseases, demonstrating that the relationship between the diagnostic markers is complex. Only when we used the ANN analysis were we able to differentiate the diseases from each other. An important concern when using ANN for analysis is the concern of overfitting (29). Overfitting occurs when the network is trained to identify not just the signal but also the noise within the signal. We have taken several approaches to avoid this. The ANN that we used has been written and implemented by one of us (J.S.A.) and includes bootstrapped cross-validation as an early stop criterion and screening for optimal topology to minimize overfitting. More important, we have tested the algorithm using a set of patients who were not used to train the network, and patients were placed into the training and test sets randomly. Despite these safeguards, it is possible that a systematic factor is introduced by collection or some other phenomenon that is associated with the samples from patients with a disease but not with the disease itself. To test whether we could find such a factor, we performed the unsupervised clustering analysis that is shown in Figure 2. There do not seem to be any factors that are associated with a given disease. The final support for the relevance of these proteins is that there is a physiologic rationale for why they may be biomarkers for glomerular diseases.
The proteins that we identified as diagnostic markers are plasma proteins that are filtered at the glomerulus. It is interesting that many proteins were identified from multiple spots at different isoelectric points on the gel. It is possible that these charge forms represent modifications of the proteins as they transit the tubule because this is known to occur. However, these proteins also exist in plasma in multiple charge forms, whereby the differences in isoelectric point are related to variable glycosylation of the proteins (41). This suggests that the reason that urine proteins can predict specific glomerular diseases is that there are differences in the relationship between the glomerular size and charge permeability barriers that are specific to a given disease. Independent changes for size and charge permeability in glomerulonephritis have previously been reported (42,43). We recently showed that charge forms of plasma proteins in the urine can differentiate between classes of lupus nephritis (31). Changes in permeability have previously been reported in a single disease but have not been shown to have diagnostic specificity among a group of diseases. This study demonstrates two important advantages of 2D gel electrophoresis as a biomarker discovery tool. First, not only can it discover spots that are candidate biomarkers, but also the proteins that make up the spots can be identified, so the proteins can be used for the development of diagnostic assays. Second, it can differentiate between posttranslationally modified proteins when they result in differences in the isoelectric point.
The assay that we used performs four tests for each patient: A true or false test for each of the four diseases. When a larger number of patients are tested in a higher throughput test, a decision tree analysis could be used to predict the single disease present. However, the approach that we used allows diagnosis of more than one disease simultaneously. We identified among patients with nephrotic syndrome diagnostic markers that correctly identified the presence or absence of the disease in 53 of 64 tests because each of the 16 patients had an independent determination of the presence of each of the four diseases. Although the accuracy is not yet as good as it needs to be for a diagnostic assay, the probability of obtaining a result this good or better in a novel set of patients by chance is 5 × 10−8. This is the probability of any combination of 53 correct results of 64: {[64!/(53! × 11!)]/2 [64] = 4.03 × 10−8}. By adding to that the probability of higher numbers of correct results that diminish and become smaller (8.2 × 10−9, 1.5 × 10−9, 2.3 × 10−10, etc), one obtains the cumulative probability of a result this good or better. These results demonstrate the power of this combination of techniques and analysis to uncover a diagnostic signal behind the noise that is produced by biologic and technical variability. They suggest a high probability of success when the candidate markers are assayed with a more reproducible assay. Even more exciting is that the assay was done in a group of patients with a variety of coexisting diseases and heterogeneity within the diseases; for instance, among the patients with lupus were patients with three International Society of Nephrology/ Renal Pathology Society classes of lupus nephritis (Table 1). This demonstrates that the assay was able to identify patterns that are common to the disease despite the heterogeneity. The accuracy of the test is likely to increase when the candidate markers are tested using a more reproducible assay. In addition to the 64 true or false answers for the individual diseases, we used a voting scheme to predict a single disease. Although this approach is premature, until a better test is developed using the candidate markers, the correct identification of 10 of 16 diseases demonstrates the promise of the approach. A potential shortcoming of the study was the inclusion of five patients among the eight patients with diabetic nephropathy who did not have renal biopsies. We included these patients because they have a high probability of having diabetic nephropathy because they had a typical presentation, long history of diabetes, and the presence of diabetic retinopathy. The presence of diabetic retinopathy strongly suggests the presence of diabetic nephropathy among patients with proteinuria. In a study of nephrotic syndrome among patients with diabetes and diabetic retinopathy, all of the patients with retinopathy had diabetic nephropathy (44). We cannot rule out the possibility, however, that the patients had a second renal disease in addition to diabetic nephropathy. Despite this question, this study has identified candidate markers that can identify the patients who have diabetic nephropathy (with or without an additional disease). In the next phase of biomarker discovery, the biomarkers can be tested in a more facile format for their ability to differentiate diabetic nephropathy from diabetic nephropathy plus another disease.
These findings not only have potential significance for understanding the pathophysiology of glomerular changes in specific diseases but also are a promising way to diagnose specific glomerular diseases without a renal biopsy. The candidate biomarkers that we identified can be used to develop a multiplexed assay that can identify the glomerular disease in patients with the nephrotic syndrome. Addition of markers that can differentiate minimal-change disease from the other diseases included here will be important for a clinically meaningful assay. Confirmation in larger sets of patients and development of a useful clinical assay will require development of tests that can differentiate between charge forms of a protein. The test will use protein-binding molecules such as antibodies. An assay that is based on these proteins should have greater accuracy because of the increased sensitivity, specificity, and dynamic range of antibodies and the larger number of patients who could be used to train an algorithm. The development of this assay presents a challenge because many of the proteins are identical but the charge form is different. Potential approaches to development of this assay will include use of antibodies that recognize the specific modification of the protein, the combination of an antibody that recognizes all forms of the protein with a substance such as a lectin that recognizes a specific glycosylation, or the combination of an antibody with a chip that contains features with affinities for many different modifications of proteins.
Acknowledgments
Support for this project came from the Medical University of South Carolina (MUSC) General Clinical Research Center (RR01070); Department of Veterans Affairs; and grants from Dialysis Clinics, Inc., and the National Institutes of Health (R21 AR051719). The informatics pipeline for data analysis was developed with federal funds as part of the National Heart, Lung, and Blood Institute Proteomics Initiative, National Institutes of Health, under contract N01-HV-28181.
Tandem mass spectrometry was done in the MUSC mass spectrometry facility with the assistance of Dr. Kevin Schey and Jennifer Bethard. We are grateful to Tim Taylor and the MUSC nephrology fellows for help with collection of samples and identification of patients. The Bio-Plex was purchased with funds from the Inflammatory Mediators of Glomerular Diseases Research Enhancement Award Program from the Department of Veterans Affairs.
Footnotes
Disclosures None.
References
- 1.US Renal Data System. USRDS 2004 Annual Data Report: Atlas of End-Stage Renal Disease in the United States. Bethesda: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2004. [Google Scholar]
- 2.Brenner BM, Hostetter TH, Humes HD. Glomerular perm-selectivity: Barrier function based on discrimination of molecular size and charge. Am J Physiol. 1978;234:F455–F460. doi: 10.1152/ajprenal.1978.234.6.F455. [DOI] [PubMed] [Google Scholar]
- 3.Brenner BM, Hostetter TH, Humes HD. Molecular basis of proteinuria of glomerular origin. N Engl J Med. 1978;298:826–833. doi: 10.1056/NEJM197804132981507. [DOI] [PubMed] [Google Scholar]
- 4.Caulfield JP, Farquhar MG. The permeability of glomerular capillaries to graded dextrans. Identification of the basement membrane as the primary filtration barrier. J Cell Biol. 1974;63:883–903. doi: 10.1083/jcb.63.3.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eiro M, Katoh T, Watanabe T. Risk factors for bleeding complications in percutaneous renal biopsy. Clin Exp Nephrol. 2005;9:40–45. doi: 10.1007/s10157-004-0326-7. [DOI] [PubMed] [Google Scholar]
- 6.Stiles KP, Yuan CM, Chung EM, Lyon RD, Lane JD, Abbott KC. Renal biopsy in high-risk patients with medical diseases of the kidney. Am J Kidney Dis. 2000;36:419–433. doi: 10.1053/ajkd.2000.8998. [DOI] [PubMed] [Google Scholar]
- 7.Rovin BH, Song H, Birmingham DJ, Hebert LA, Yu CY, Nagaraja HN. Urine chemokines as biomarkers of human systemic lupus erythematosus activity. J Am Soc Nephrol. 2005;16:467–473. doi: 10.1681/ASN.2004080658. [DOI] [PubMed] [Google Scholar]
- 8.Peterson E, Robertson AD, Emlen W. Serum and urinary interleukin-6 in systemic lupus erythematosus. Lupus. 1996;5:571–575. doi: 10.1177/096120339600500603. [DOI] [PubMed] [Google Scholar]
- 9.Molad Y, Miroshnik E, Sulkes J, Pitlik S, Weinberger A, Monselise Y. Urinary soluble VCAM-1 in systemic lupus erythematosus: A clinical marker for monitoring disease activity and damage. Clin Exp Rheumatol. 2002;20:403–406. [PubMed] [Google Scholar]
- 10.Negi VS, Aggarwal A, Dayal R, Naik S, Misra R. Complement degradation product C3d in urine: Marker of lupus nephritis. J Rheumatol. 2000;27:380–383. [PubMed] [Google Scholar]
- 11.Hopper JE, Sequeira W, Martellotto J, Papagiannes E, Perna L, Skosey JL. Clinical relapse in systemic lupus erythematosus: Correlation with antecedent elevation of urinary free light-chain immunoglobulin. J Clin Immunol. 1989;9:338–350. doi: 10.1007/BF00918666. [DOI] [PubMed] [Google Scholar]
- 12.Honkanen E, von Willebrand E, Koskinen P, Teppo AM, Tornroth T, Ruutu M, Gronhagen-Riska C. Decreased expression of vascular endothelial growth factor in idiopathic membranous glomerulonephritis: Relationships to clinical course. Am J Kidney Dis. 2003;42:1139–1148. doi: 10.1053/j.ajkd.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 13.Honkanen EO, Teppo AM, Gronhagen-Riska C. Decreased urinary excretion of vascular endothelial growth factor in idiopathic membranous glomerulonephritis. Kidney Int. 2000;57:2343–2349. doi: 10.1046/j.1523-1755.2000.00094.x. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto K, Kanmatsuse K. Elevated vascular endothelial growth factor levels in the urine of patients with minimal-change nephrotic syndrome. Clin Nephrol. 2001;55:269–274. [PubMed] [Google Scholar]
- 15.Cha DR, Kang YS, Han SY, Jee YH, Han KH, Han JY, Kim YS, Kim NH. Vascular endothelial growth factor is increased during early stage of diabetic nephropathy in type II diabetic rats. J Endocrinol. 2004;183:183–194. doi: 10.1677/joe.1.05647. [DOI] [PubMed] [Google Scholar]
- 16.Rauta V, Teppo AM, Tornroth T, Honkanen E, Gronhagen-Riska C. Lower urinary-interleukin-1 receptor-antagonist excretion in IgA nephropathy than in Henoch-Schonlein nephritis. Nephrol Dial Transplant. 2003;18:1785–1791. doi: 10.1093/ndt/gfg234. [DOI] [PubMed] [Google Scholar]
- 17.Matsumoto K, Kanmatsuse K. Increased urinary excretion of interleukin-17 in nephrotic patients. Nephron. 2002;91:243–249. doi: 10.1159/000058399. [DOI] [PubMed] [Google Scholar]
- 18.Kalantarinia K, Awad AS, Siragy HM. Urinary and renal interstitial concentrations of TNF-alpha increase prior to the rise in albuminuria in diabetic rats. Kidney Int. 2003;64:1208–1213. doi: 10.1046/j.1523-1755.2003.00237.x. [DOI] [PubMed] [Google Scholar]
- 19.Shoji T, Nakanishi I, Kunitou K, Tsubakihara Y, Hirooka Y, Kishi Y, Hatanaka M, Matsumoto M, Toyoshima K, Seya T. Urine levels of CD46 (membrane cofactor protein) are increased in patients with glomerular diseases. Clin Immunol. 2000;95:163–169. doi: 10.1006/clim.2000.4847. [DOI] [PubMed] [Google Scholar]
- 20.Hotta O, Yusa N, Kitamura H, Taguma Y. Urinary macrophages as activity markers of renal injury. Clin Chim Acta. 2000;297:123–133. doi: 10.1016/s0009-8981(00)00239-4. [DOI] [PubMed] [Google Scholar]
- 21.Hotta O, Yusa N, Ooyama M, Unno K, Furuta T, Taguma Y. Detection of urinary macrophages expressing the CD16 (Fc gamma RIII) molecule: A novel marker of acute inflammatory glomerular injury. Kidney Int. 1999;55:1927–1934. doi: 10.1046/j.1523-1755.1999.00431.x. [DOI] [PubMed] [Google Scholar]
- 22.Hara M, Yanagihara T, Takada T, Itoh M, Matsuno M, Yamamoto T, Kihara I. Urinary excretion of podocytes reflects disease activity in children with glomerulonephritis. Am J Nephrol. 1998;18:35–41. doi: 10.1159/000013302. [DOI] [PubMed] [Google Scholar]
- 23.Vogelmann SU, Nelson WJ, Myers BD, Lemley KV. Urinary excretion of viable podocytes in health and renal disease. Am J Physiol Renal Physiol. 2003;285:F40–F48. doi: 10.1152/ajprenal.00404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hara M, Yanagihara T, Itoh M, Matsuno M, Kihara I. Immunohistochemical and urinary markers of podocyte injury. Pediatr Nephrol. 1998;12:43–48. doi: 10.1007/s004670050401. [DOI] [PubMed] [Google Scholar]
- 25.Schmid H, Henger A, Cohen CD, Frach K, Grone HJ, Schlondorff D, Kretzler M. Gene expression profiles of podocyte-associated molecules as diagnostic markers in acquired proteinuric diseases. J Am Soc Nephrol. 2003;14:2958–2966. doi: 10.1097/01.asn.0000090745.85482.06. [DOI] [PubMed] [Google Scholar]
- 26.Rennke HG, Patel Y, Venkatachalam MA. Glomerular filtration of proteins: Clearance of anionic, neutral, and cationic horseradish peroxidase in the rat. Kidney Int. 1978;13:278–288. doi: 10.1038/ki.1978.41. [DOI] [PubMed] [Google Scholar]
- 27.Rennke HG, Venkatachalam MA. Glomerular permeability: In vivo tracer studies with polyanionic and polycationic ferritins. Kidney Int. 1977;11:44–53. doi: 10.1038/ki.1977.6. [DOI] [PubMed] [Google Scholar]
- 28.Almeida JS, Stanislaus R, Krug EL, Arthur JM. Normalization and analysis of residual variation in 2D gel electrophoresis for quantitative differential proteomics. Proteomics. 2005;5:1242–1249. doi: 10.1002/pmic.200401003. [DOI] [PubMed] [Google Scholar]
- 29.Almeida JS. Predictive non-linear modeling of complex data by artificial neural networks. Curr Opin Biotechnol. 2002;13:72–76. doi: 10.1016/s0958-1669(02)00288-4. [DOI] [PubMed] [Google Scholar]
- 30.Lefler DM, Pafford RG, Black NA, Raymond JR, Arthur JM. Identification of proteins in slow continuous ultrafiltrate by reversed-phase chromatography and proteomics. J Proteome Res. 2004;3:1254–1260. doi: 10.1021/pr0498640. [DOI] [PubMed] [Google Scholar]
- 31.Oates JC, Varghese SA, Bland AM, Taylor TP, Self SE, Stanislaus R, Almeida JS, Arthur JM. Prediction of urinary protein markers in lupus nephritis. Kidney Int. 2005;65:2588–2592. doi: 10.1111/j.1523-1755.2005.00730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rossing K, Mischak H, Parving HH, Christensen PK, Walden M, Hillmann M, Kaiser T. Impact of diabetic nephropathy and angiotensin II receptor blockade on urinary polypeptide patterns. Kidney Int. 2005;68:193–205. doi: 10.1111/j.1523-1755.2005.00394.x. [DOI] [PubMed] [Google Scholar]
- 33.Mischak H, Kaiser T, Walden M, Hillmann M, Wittke S, Herrmann A, Knueppel S, Haller H, Fliser D. Proteomic analysis for the assessment of diabetic renal damage in humans. Clin Sci (Lond) 2004;107:485–495. doi: 10.1042/CS20040103. [DOI] [PubMed] [Google Scholar]
- 34.Meier M, Kaiser T, Herrmann A, Knueppel S, Hillmann M, Koester P, Danne T, Haller H, Fliser D, Mischak H. Identification of urinary protein pattern in type 1 diabetic adolescents with early diabetic nephropathy by a novel combined proteome analysis. J Diabetes Complications. 2005;19:223–232. doi: 10.1016/j.jdiacomp.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 35.Haubitz M, Wittke S, Weissinger EM, Walden M, Rupprecht HD, Floege J, Haller H, Mischak H. Urine protein patterns can serve as diagnostic tools in patients with IgA nephropathy. Kidney Int. 2005;67:2313–2320. doi: 10.1111/j.1523-1755.2005.00335.x. [DOI] [PubMed] [Google Scholar]
- 36.Weissinger EM, Wittke S, Kaiser T, Haller H, Bartel S, Krebs R, Golovko I, Rupprecht HD, Haubitz M, Hecker H, Mischak H, Fliser D. Proteomic patterns established with capillary electrophoresis and mass spectrometry for diagnostic purposes. Kidney Int. 2004;65:2426–2434. doi: 10.1111/j.1523-1755.2004.00659.x. [DOI] [PubMed] [Google Scholar]
- 37.Wittke S, Mischak H, Walden M, Kolch W, Radler T, Wiedemann K. Discovery of biomarkers in human urine and cerebrospinal fluid by capillary electrophoresis coupled to mass spectrometry: Towards new diagnostic and therapeutic approaches. Electrophoresis. 2005;26:1476–1487. doi: 10.1002/elps.200410140. [DOI] [PubMed] [Google Scholar]
- 38.Wittke S, Fliser D, Haubitz M, Bartel S, Krebs R, Hausadel F, Hillmann M, Golovko I, Koester P, Haller H, Kaiser T, Mischak H, Weissinger EM. Determination of peptides and proteins in human urine with capillary electrophoresis-mass spectrometry, a suitable tool for the establishment of new diagnostic markers. J Chromatogr A. 2003;1013:173–181. doi: 10.1016/s0021-9673(03)00713-1. [DOI] [PubMed] [Google Scholar]
- 39.Park MR, Wang EH, Jin DC, Cha JH, Lee KH, Yang CW, Kang CS, Choi YJ. Establishment of a 2-D human urinary proteomic map in IgA nephropathy. Proteomics. 2006;6:1066–1076. doi: 10.1002/pmic.200500023. [DOI] [PubMed] [Google Scholar]
- 40.Thongboonkerd V, Klein JB, Jevans AW, McLeish KR. Urinary proteomics and biomarker discovery for glomerular diseases. Contrib Nephrol. 2004;141:292–307. doi: 10.1159/000074606. [DOI] [PubMed] [Google Scholar]
- 41.Wilson NL, Schulz BL, Karlsson NG, Packer NH. Sequential analysis of N- and O-linked glycosylation of 2D-PAGE separated glycoproteins. J Proteome Res. 2002;1:521–529. doi: 10.1021/pr025538d. [DOI] [PubMed] [Google Scholar]
- 42.Machii R, Sakatume M, Kubota R, Kobayashi S, Gejyo F, Shiba K. Examination of the molecular diversity of alpha1 antitrypsin in urine: Deficit of an alpha1 globulin fraction on cellulose acetate membrane electrophoresis. J Clin Lab Anal. 2005;19:16–21. doi: 10.1002/jcla.20049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Myers BD, Okarma TB, Friedman S, Bridges C, Ross J, Asseff S, Deen WM. Mechanisms of proteinuria in human glomerulonephritis. J Clin Invest. 1982;70:732–746. doi: 10.1172/JCI110669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parving HH, Gall MA, Skott P, Jorgensen HE, Lokkegaard H, Jorgensen F, Nielsen B, Larsen S. Prevalence and causes of albuminuria in non-insulin-dependent diabetic patients. Kidney Int. 1992;41:758–762. doi: 10.1038/ki.1992.118. [DOI] [PubMed] [Google Scholar]