

Abstract
Brain-derived neurotrophic factor (BDNF) and its receptor TrkB play an important function in neuronal development and synaptic plasticity. Recently we have established that cyclothiazide (CTZ) is a novel convulsant drug inducing robust epileptiform activity in hippocampal neurons both in vitro and in vivo. However, the molecular mechanisms underlying such convulsant action of CTZ is unknown. Here, we investigated potential roles of BDNF-TrkB signaling pathway in the CTZ-induction of epileptiform activity. In anaesthetized rats, CTZ dose-dependently induced epileptiform activity characterized by progressing of multiple peaks of population spikes, spontaneous spiking events, and synchronized epileptiform bursts. Pre-injection of a receptor tyrosine kinase inhibitor K252a or a specific antibody for TrkB receptors before intracerebroventricular injection of CTZ significantly suppressed the epileptiform activity induced by CTZ. Similarly, in cultured hippocampal pyramidal neurons, pretreatment with CTZ together with K252a or TrkB receptor antibody also inhibited the CTZ-induction of epileptiform activity. Furthermore, we demonstrated that acute application of K252a in hippocampal cultures inhibited epileptiform bursts and action potential firing. We conclude that activation of BDNF-TrkB signaling pathway is fundamentally important during the CTZ-induction of epileptiform activity both in vitro and in vivo.
Keywords: Cyclothiazide, BDNF, TrkB receptor, epileptiform activity, hippocampal neuron
Introduction
Brain-derived neurotrophic factor (BDNF) and its specific receptor TrkB are critically involved in neural development and synaptic plasticity (Huang and Reichardt, 2001; Poo, 2001; Lu, 2003). BDNF-TrkB signaling pathway has also been found to play an important role in epileptogenesis (Binder et al., 2001). BDNF release depends on neuronal activity (Goodman et al., 1996; Balkowiec and Katz, 2000). Epileptiform activity increases the expression level of BDNF mRNA as well as proteins (Gall et al., 1991; Nawa et al., 1995; Elmer et al., 1998; He et al., 2002). In BDNF heterozygous (+/-) mice, the kindling rate of epileptiform activity is decreased two-fold compared to wild type controls (Kokaia et al., 1995). In addition, in vivo studies have demonstrated that infusion of BDNF into the adult rat hippocampus results in seizures (Scharfman et al., 2002). The effect of BDNF is generally believed to be mediated by the activation of TrkB receptors. Transgenic overexpression of a truncated dominant negative form of Trk-B receptors interferes with epileptiform activity in the kainic acid (KA) model of epilepsy in mice (Lahteinen et al., 2002). Recent work reported that conditional knockout of TrkB receptor, but not BDNF, significantly suppresses the formation of epileptiform activity in the kindling model of epilepsy (He et al., 2004). While substantial evidences suggest that BDNF-TrkB signaling is proepileptic, some studies also suggest possible antiepileptic effects of neurotrophins including BDNF (Simonato et al., 2006). The precise mechanisms of BDNF-TrkB signaling during epileptogenesis are not fully understood yet. Here, we employ a novel epilepsy model recently established in our lab to further investigate the functional role of BDNF-TrkB in epileptogenesis.
The convulsant drug we identified is cyclothiazide (CTZ). CTZ has long been known as an AMPA receptor desensitization blocker and thus prolongs glutamate excitatory responses (Partin et al., 1993; Trussell et al., 1993; Yamada and Tang, 1993; Zorumski et al., 1993). CTZ also increases presynaptic glutamate release (Diamond and Jahr, 1995; Bellingham and Walmsley, 1999; Ishikawa and Takahashi, 2001). In addition, we have demonstrated that CTZ can directly inhibit GABAA receptor function, acting as a GABAA receptor blocker (Deng and Chen, 2003). Furthermore, we demonstrated that CTZ induces epileptiform bursts in hippocampal neurons both in vitro and in vivo (Qi et al., 2006a), partly due to downregulation of tonic GABAA receptor function (Qi et al., 2006b). Thus, the opposite actions of CTZ on glutamatergic and GABAergic neurotransmission provide a unique model for studying mechanisms of epileptogenesis. Here, we report that BDNF-TrkB signaling pathway is critically involved in the CTZ-induction of epileptiform bursts. Blocking TrkB receptors significantly reduced epileptiform bursts induced by CTZ in hippocampal neurons both in vivo and in vitro. Patch clamp recordings also revealed a significant block of epileptiform bursts and action potential firing after acutely inhibiting receptor tyrosine kinases with K252a. Our studies suggest that BDNF-TrkB signaling is likely a common pathway during epileptogenesis that mediates the transition from individual action potentials to epileptiform bursts.
Materials and Methods
Animal preparation
In vivo experiments were performed on urethane anaesthetized (1.2 g kg-1, i.p.) male Sprague Dawley rats (280-350 g). The level of anaesthesia was assessed by the absence of a withdrawal reflex, and additional anaesthetic (urethane, 0.2–0.6 mg kg-1, i.p.) was administered as necessary. Body temperature was maintained at 37 ± 0.5 °C with a Harvard Homoeothermic Blanket (Harvard Apparatus Limited, Kent, UK). Animals were housed in a regulated environment (21 ± 1 °C) with a 12 hour light-dark cycle, and food and water available ad libitum. All experiments were approved by the local committees of Laboratory Animals, Fudan University and Chongqing Technology and Business University and carried out in accordance with Chinese National Science Foundation animal research regulation. At the end of experiments, animals were euthanized with overdose of urethane.
Animals were prepared as previously described (Qi et al., 2006a, Wang et al., 2006). Animals had their lateral tail vein cannulated for drug administration and were mounted in a stereotaxic frame. An incision was made in the midline of the head to expose the top part of the skull. For implantation of the i.c.v. cannula, a drill hole was made on the skull above the left side of the lateral-ventricle (0.3 mm posterior to bregma, 1.3 mm lateral to the midline). A guide cannula (Bilaney Consultants Ltd, TN, USA) was then placed 4 mm below the skull surface for drug delivery, and secured by the dental cement.
Recording and Data Acquisition
For recording and stimulating, a large burr hole was made in the left side of the incised skull above the hippocampal area, and the dura was pierced and removed. The stereotaxic coordinates were determined from the stereotaxic atlas of the rat brain (Paxinos & Watson, 1986). A concentric bipolar metal electrode (Harvard Apparatus Ltd, Kent, UK) was placed close to CA3 region (3.8-4.5 mm posterior to bregma, 3.5-4.0 mm lateral to the midline, and 3.0-3.8 mm below the brain surface) in order to stimulate the CA3 pryamidal cell layer and/or Shaffer collateral pathway. For recording in the CA1 pyramidal cell layer, a tungsten electrode (0.5 MΩ, WPI, Stevenage, UK) was placed 3.5-4.2 mm posterior to bregma, 2.0-3.0 mm lateral to the midline. The depth of the recording electrode was approximately 2.0-2.5 mm below the brain surface as determined by the sudden change of the electrical noise and the shape of the evoked field excitatory postsynaptic potential (fEPSP) and population spike (PS). For CA3-CA1 Shaffer collateral stimulation, a constant current generator passed a square-wave pulse (0.2 ms in duration) through the stimulating electrode (test pulse). Test pulses evoked a positive excitatory postsynaptic field potential in the CA1 with a population spike superimposed as a negative deflection on the rising phase. After initial recording and stimulating tests, both electrodes were adjusted to obtain the maximal evoked EPSP and population spike amplitude. The PS amplitude was used as a measure of postsynaptic responses, with a stimulation intensity set at the current required to produce the maximal response, pre-determined by input-output curves. During experiments, CA1 pyramidal cell excitability was sampled every 60 seconds by a test pulse. In-between stimulations, the baseline activity was recorded for evidence of spontaneous activity. The electrophysiological signals were amplified (200 times) and filtered (0.3-3 kHz) using a NeuroLog System (Digitimer Ltd., Hearts, UK) and visualised and stored in a PC computer through an A-D converter, CED 1401 micro (Cambridge Electronic Design, Cambridge, UK). Once both electrodes were in the right place, the fEPSPs and PS were monitored for at least 30 minutes (20 min of baseline recording without stimulation and 10 min evoked EPSP/PS recording) until a stable recording was achieved. Following a 30 minute recorded baseline of all responses, drugs or vehicles were administered i.c.v. (intracerebral ventricle) at volume of 5 μl via the pre-implanted guide cannula into the lateral ventricle. Pharmacologically induced epileptiform activity was monitored for 3 hours after either CTZ or vehicle DMSO injection by observing changes of evoked potentials transform from single PS into multi-peaked display and spontaneous epileptiform burst activity in CA1 pyramidal neurons (Wheal et al., 1998). The anaesthetic level was monitored and maintained throughout the course of experiments, in particular, after convulsant drug administration. On some occasions, the brain was taken for histological validation of the injection and recording/stimulating sites.
Animals were divided into following 10 groups for drug test:
Group 1: DMSO control;
Groups 2-3: CTZ 1 μmol and 5 μmol;
Group 4: K252a alone;
Group 5-7: K252a 30, 60 and 180 min pre-treatment plus CTZ 5 μmol;
Group 8: TrkB antibody alone;
Group 9-10: TrkB antibody 30 and 60 min pre-treatment plus CTZ 5 μmol.
Analysis of data
Epileptiform activity within CA1 pyramidal cells was analyzed offline using Spike2 (an analysing program for CED 1401, Cambridge, UK) and specific scripts designed for this study with Spike2. Multiple population spike peaks typically represent epileptiform evoked responses (Traub & Jefferys, 1994). Therefore, the evoked multiple population spike peaks, defined as the negative deflection peaks superimposed on the rising phase of the evoked EPSPs, were counted and the latency for evoking these multiple population spike peaks was analyzed. The spontaneous high amplitude spiking events (>0.5 mV) were defined as those containing 1 or 2 spikes occurring at low frequency (<1 Hz) (Fig 1). The latency for spontaneous high amplitude spikes was recorded from the usual ‘silent’ baseline. The highly synchronized bursting activity was defined, in distinguishing from spontaneous spiking events, as having high frequency multiple high amplitude spikes (>0.5 mV) with an initial interspike interval of less than 0.2 s, a minimum of 5 spikes, and burst duration over 1s.
Figure 1.
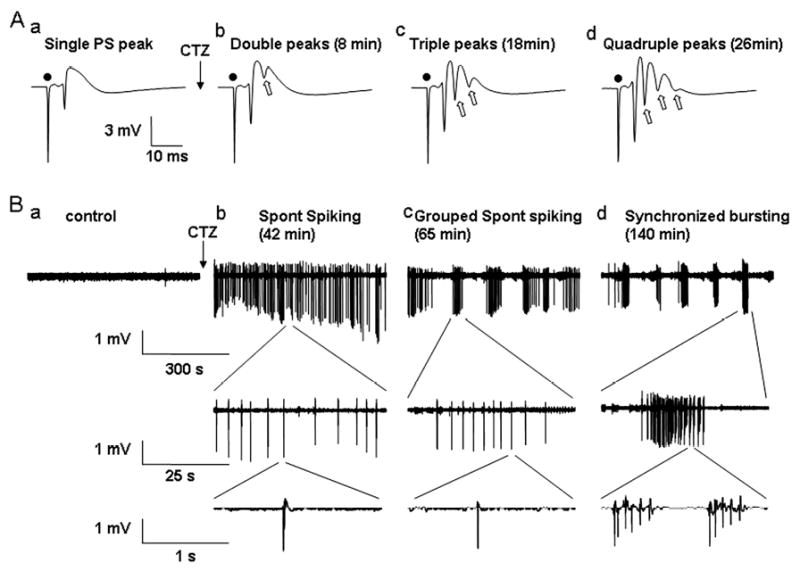
CTZ induced progressive epileptiform bursts including evoked multiple population spikes and spontaneous seizure bursts in hippocampal CA1 pyramidal region in in vivo recordings. (A) Typical recordings showing that the evoked population spikes recorded from CA1 pyramidal layer in urethane-anesthetised rats transformed from single peak at control condition to double, triple, and quadruple multiple peaks (the extra peaks are indicated by hollow arrows) after CTZ injection (5 μmol, 5 μl, i.c.v.) (● indicates the stimulus artefact). The time in parenthesis indicates the latency of the multiple PS peaks after CTZ injection. (B) Spontaneous discharges recorded in the same rat as in (A). Before CTZ injection, the base line activity was usually silent in CA1 pyramidal cells (a). After CTZ injection, some high amplitude spontaneous spiking activity appeared, first in continuous but individual mode (b), and then became partially grouped (c), and finally formed highly synchronized epileptiform bursts (d). Each large burst was consisted of many smaller bursts of discharges.
Group data were expressed as the mean ± SEM. Across groups of data, statistical significance between means was determined using one-way ANOVA with Tukey HSD post hoc analysis (GraphPad Prism, GraphPad Software Inc.). Comparisons within a group used a paired two-tail t-test. Significance level was set at P <0.05.
Histology
At the end of experiments Pontamine Sky Blue (1 μl) was injected though the pre-implanted cannula to verify the cannula position before the brain was removed and fixed in 10% formal saline. Frozen sections (80 μm) were cut and stained with Neutral Red. The Pontamine Sky Blue marked sites and the stimulating and recording electrode tracks were visualised and mapped onto standard sections of the brain (Paxinos & Watson, 1998). For those successfully recovered recording/stimulating tracks, they were all localised in the right place in CA1 pyramidal cell layer and CA3 area, respectively, and the marked cannula sites were in the lateral ventricle.
In vitro electrophysiology
The detailed in vitro electrophysiology protocol has been described previously (Qi et al., 2006 a, b). Briefly, whole-cell recordings were performed on cultured hippocampal neurons from CA1-CA3 region in current clamp mode using a MultiClamp 700A amplifier (Axon Instrument). Patch pipettes were pulled using Sutter P-97 pipette puller and the final resistance of pipettes was 3-6 MΩ. The recording chamber was continuously perfused with a bath solution containing 128 mM NaCl, 30 mM Glucose, 25 mM HEPES, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, pH 7.3 adjusted with NaOH. The pipette solution contained 125 mM KGluconate, 10 mM KCl, 2 mM EGTA, 10 mM HEPES, 10 mM Tris-phosphocreatine, 4 mM MgATP, 0.5 mM Na2GTP, pH 7.3 adjusted with KOH. The series resistance was typically 10-20 MΩ and partially compensated by 30-50%. The membrane potential was held around -70 mV in current clamp recordings. Data were acquired using pClamp 9 software, sampled at 2-10 kHz, and filtered at 1 kHz. Off-line analysis was done with Clampfit 9 software (Axon Instrument). An epileptiform burst is defined by at least 5 consecutive action potentials overlaying on top of a large depolarization shift (>10 mV). When quantifying percentage of neurons showing epileptiform activity, the criteria is at least 4 repeated epileptiform bursts occurring during 20 min of recording. Pyramidal shape neurons were selected for recordings.
Drugs and solutions
The following drugs were freshly made before each experiment: Cyclothiazide (1 M or 0.2 M in DMSO for in vivo injection) and K252a (0.25 M in DMSO for in vivo injection) were purchased from Tocris (Northpoint, Bristol); anti-TrkB mouse antibody (TrkB antibody) was from BD Biosciences (San Jose, California); Pontamine sky blue dye (20 mg ml-1; BDH, Poole) was dissolved in 0.5 M sodium acetate; Urethane (25%; Sigma Aldrich Chemical Co., Poole, Dorset) was dissolved in distilled water.
Results
CTZ-evoked epileptiform activity in hippocampal CA1 neurons in vivo
In anesthetized rats, recordings were made from CA1 pyramidal cell layer with stimulating rate at 1/60 seconds, if applied, in the area of CA3. In control condition, only a single PS was embedded on top of the evoked EPSP and there was no spontaneous activity in the baseline (Fig. 1 Aa and Ba). Microinjection of CTZ (5 μmol in 5 μl DMSO) into left-side lateral ventricle induced multiple evoked PS peaks in a time-dependent manner (Fig. 1 Ab, Ac, Ad). Furthermore, after ∼40 min of CTZ injection, spontaneous high amplitude spikes appeared in the baseline, which gradually formed grouped spikes and finally became highly synchronized epileptiform bursts (Fig 1Bb, Bc, Bd). Interestingly, the evoked multiple peaks always appeared first after CTZ injection, before the occurrence of high amplitude spontaneous spikes, although sometimes the evoked 4 peaks appeared concurrently with the spontaneous spikes. We also noted that after CTZ injection, it usually took 1-2 hours for the initial spontaneous spikes to be gradually transformed into highly synchronized epileptiform bursts (Fig 1B). After the bursting activity occurred, it was usually impossible to evoke EPSPs and PSs.
We investigated the potent epileptogenic effect of CTZ through i.c.v. administration at two different doses, 1 μmol and 5 μmol, respectively. Injection of higher dose of CTZ (5 μmol) showed an increased probability and shortened latency in inducing epileptiform bursts (Fig. 2). CTZ injection resulted in double PS peaks in all rats tested (n=10 at 1 μmol and n=12 at 5 μmol), and triple PS peaks in 8 out of 10 (80%) rats at 1 μmol and 12 out of 12 (100%) rats at 5 μmol dose (Fig. 2 Aa). CTZ injection at both 1 and 5 μmol doses induced spontaneous high amplitude spikes in all rats tested (Fig. 2 Ab). In contrast, 1 μmol CTZ injection only induced synchronized epileptiform bursts in 5 out of 10 (50%) rats, whereas 5 μmol CTZ injection induced epileptiform bursts in 10 out 12 (83%) rats (Fig 2 Ac). The latency for evoking triple PS peaks was 51.9 ± 7.7 min (n=8) after 1 μmol CTZ injection, and greatly shortened to 22.2 ± 2.3 min (n=12) after 5 μmol CTZ injection (Fig. 2 Ba, p<0.001, Student's t test). The latency for inducing spontaneous high amplitude spikes was 51.2 ± 1.6 min (n=10) after 1 μmol CTZ injection, and 39.9 ± 2.8 min (n=12) after 5 μmol CTZ injection (Fig. 2 Bb, p<0.01). Furthermore, the latency for inducing synchronized epileptiform bursts was 102.9 ± 8.1 min (n=5) after 1 μmol CTZ injection, and 85.5 ± 8.2 min (n=10) after 5 μmol CTZ injection (Fig 2 Bc, p>0.2). Overall, the latency for evoking epileptiform activity was shortened at 5 μmol group in comparison with the 1 μmol group, indicating that the epileptogenic effect of CTZ is dose-dependent. For control experiments, DMSO (5 μl, i.c.v.), the vehicle for dissolving CTZ, was found not to induce any multiple PS peaks nor spontaneous spikes or synchronized bursts in 3 hours recording period in all rats tested (n=6) (Fig. 2A).
Figure 2.
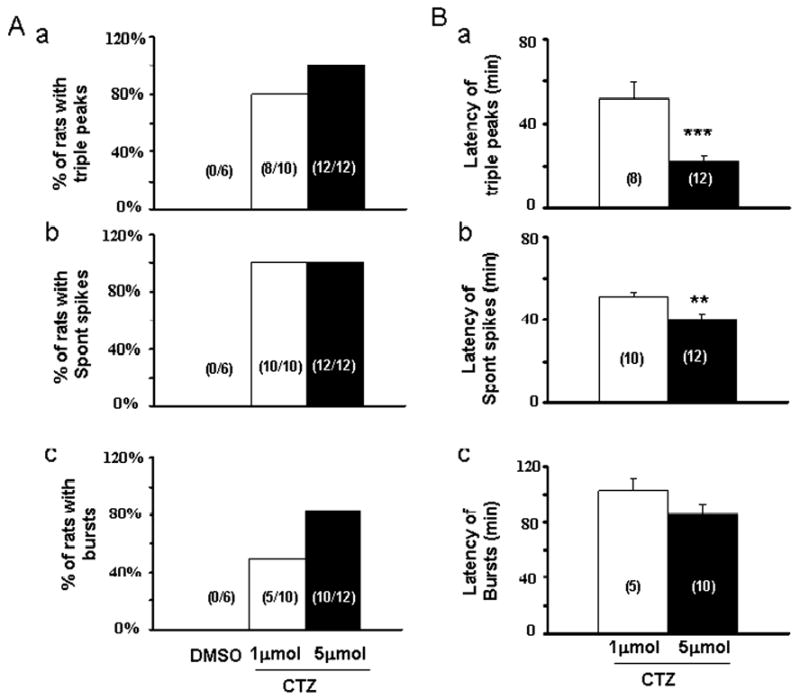
Bar histograms showing the pooled data of CTZ-induced epileptiform activity. (A) The percentage of rats showing triple PS peaks (a), spontaneous spiking (b), and synchronized bursts (c) after injecting DMSO vehicle or CTZ (1 or 5 μmol, i.c.v.). (B) The latency of CTZ-evoked triple PS peaks (a), spontaneous spiking (b), and synchronized bursts (c). **, P<0.01 and ***, P<0.001.
Effects of K252a on CTZ induced epileptiform bursts in hippocampal CA1 neurons in vivo
To test the hypothesis that BDNF-TrkB receptor signaling pathway mediates the CTZ induction of epileptiform bursts, we first employed K252a, a non-competitive inhibitor for receptor tyrosine kinases including Trk B receptors, to investigate its effect in hippocampal CA1 neurons (Kang et al., 1997; Zhu and Roper, 2001; Cheng and Yeh, 2003). K252a was administered (250 μM, 5 μl, i.c.v.) either 30 min (n=6), 1 hour (n=9), or 3 hours (n=5) prior to CTZ injection (5 μmol). In 30 min pre-injection group, K252a almost completely blocked CTZ-induced epileptiform bursts, but the 60 min pre-injection group showed persistent spontaneous spikes after longer delay following CTZ injection (Fig. 3A-C). The percentage of animals showing triple PS peaks, spontaneous spikes, and synchronized bursts after K252a and CTZ injection was summarized in the middle columns of Fig. 4. For control experiments, K252a injection alone did not induce any epileptiform activity as expected. Injection of K252a 30 min before CTZ injection suppressed triple PS peaks and epileptiform bursts in all 6 rats tested, while only 1 out of 6 rats showed spontaneous spikes after a prolonged latency of >100 min following CTZ-injection (Fig. 4, middle columns). If K252a was injected 60 min or 180 min before CTZ injection, most animals showed triple PS peaks (60 min, 78% ; 180 min, 60%) and spontaneous high amplitude spikes (60 min, 89% ; 180 min, 80%) (Fig. 4A-B, middle columns). However, synchronized epileptiform bursts were greatly reduced in 60 min pre-injection group (44%) and slightly reduced in 180 min group (60%) (Fig. 4C). For those rats still showing some forms of CTZ-induced epileptiform bursts after K252a pre-injection, we further analyzed whether K252a altered the latency of the epileptiform activity after CTZ injection (Fig. 5, middle columns). Pre-injection of K252a significantly increased the latency for subsequent CTZ-evoked triple PS peaks (CTZ alone, 22.2 ± 2.3 min ; 60 min, 56.1 ± 4.0 min, n=7 ; 180 min, 54.7 ± 5.8 min, n=3; P<0.001 for both 60 and 180 min groups), and for spontaneous high amplitude spikes (CTZ alone, 39.9 ± 2.8 min ; 60 min, 69.7 ± 5.4 min, n=8 ; 180 min, 66.5 ± 2.2 min, n=4; P<0.001 for both 60 and 180 min groups) (Fig. 5A-B, middle columns). Interestingly, the synchronized epileptiform bursts had a long latency after CTZ injection alone (85.5 ± 8.2 min, n=10), and K252a pre-injection tended to increase the latency for epileptiform bursts (60 min, 110.4 ± 6.4 min, n=4 ; 180 min, 96.5 ± 7.0 min, n=3) but without reaching a statistic significance (60 min, P=0.097; 180 min, P=0.50) (Fig 5C, Table 1). These results suggest that CTZ-induction of epileptiform activity is mediated by the activation of receptor tyrosine kinases, including TrkB receptors. Since K252a is a broad spectrum receptor tyrosine kinase inhibitor, in the next set of experiments we used more specific TrkB receptor antibodies to investigate the role of TrkB receptors during CTZ-induction of epileptiform activity.
Figure 3.
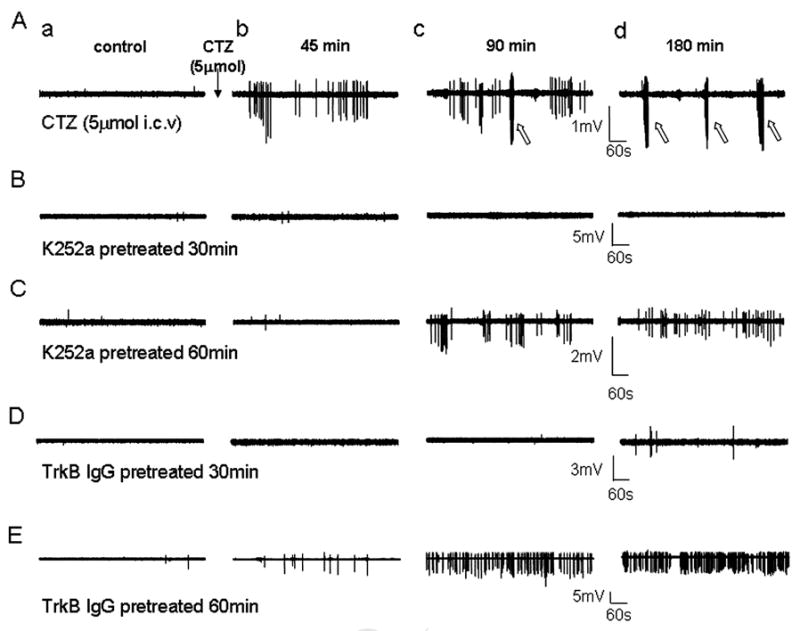
Effects of K252a and TrkB antibody on CTZ-induced spontaneous epileptiform bursts in hippocampal CA1 pyramidal cell layer in anaesthetised rats sampled before, 45, 90 and 180 min after CTZ injection (5 μmol, i.c.v.). (A) Original traces from a control rat. Note the spontaneous spiking events occurring first, followed by the highly synchronized epileptiform bursts (open arrows). (B) Original traces from a rat injected first with K252a (1 μmol, i.c.v) 30 min before the injection of CTZ. No spontaneous spikes or synchronized bursts were observed during 180 min recordings after CTZ injection. (C) When K252a was injected 60 min before CTZ injection, some spontaneous spiking events, but not synchronized bursts, were detected with a significant latency. (D) Original traces showing that injection with TrkB antibody (0.5 μg in 5 μl, i.c.v.) 30 min before the injection of CTZ significantly suppressed epileptiform bursts. (E) Injection of TrkB antibody 60 min before CTZ injection did not prevent the induction of spontaneous spikes, but still inhibited synchronized bursts.
Figure 4.
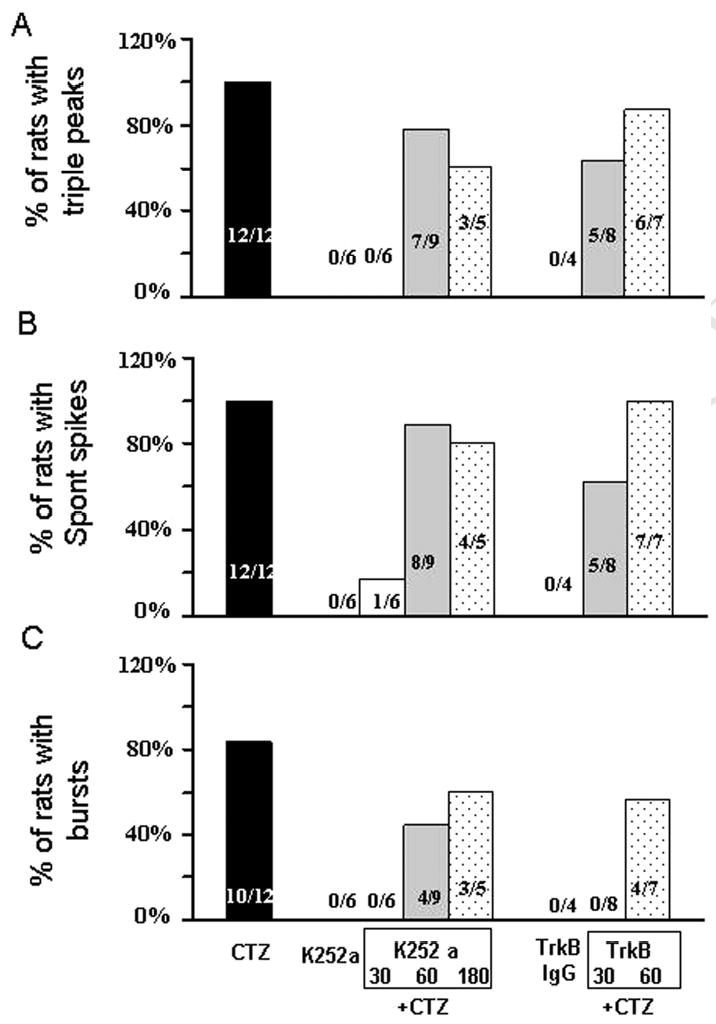
Bar histograms showing the group data of the effects of K252a and TrkB antibody on the CTZ-induction of epileptiform activity in anaesthetised rats. All CTZ injection was at the dose of 5 μmol. Injection of K252a 30 min prior to the injection of CTZ essentially suppressed triple PS peaks (A), spontaneous spikes (B), and synchronized bursts (C) in all rats tested. If K252a was injected 60 or 180 min prior to the injection of CTZ, the anti-convulsant effect was less prominent, possibly due to the metabolism or clearance of the drug during the longer interval. Similarly, injection of TrkB antibody 30 min prior to the CTZ injection also showed stronger effect than the 60 min interval in inhibiting the percentage of rats with epileptiform activity.
Figure 5.
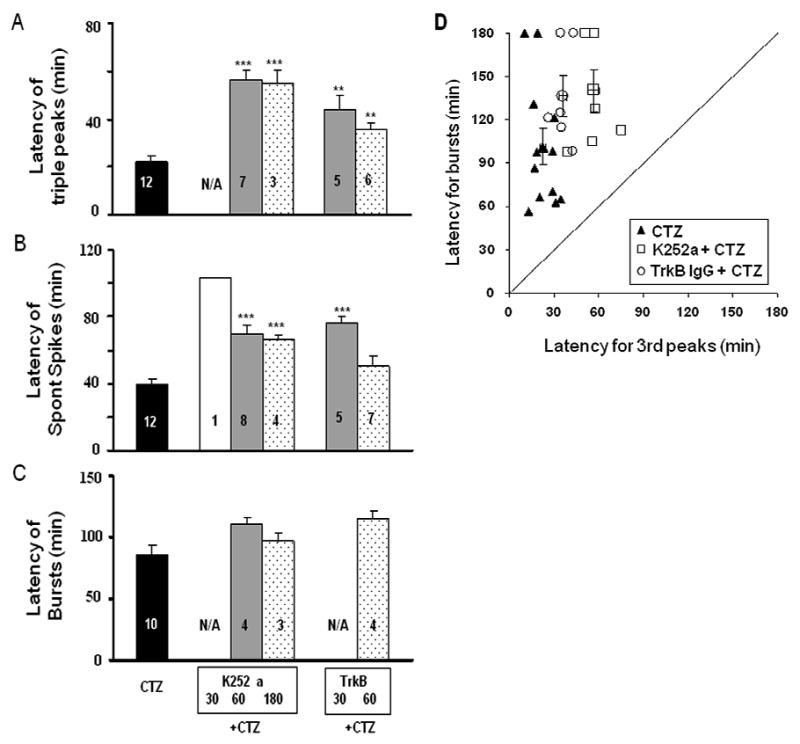
Bar histograms showing the group data of the effects of K252a and TrkB antibody on the latency of epileptiform activity induced by CTZ in anaesthetised rats. Comparing to the injection of CTZ (5 μmol, i.c.v.) alone, rats pre-injected with K252a or TrkB antibody significantly prolonged the latency in evoking triple PS peaks (A), spontaneous spikes (B), but not synchronized bursts (C). ** P<0.01 and *** P<0.001, in comparison with those of injecting CTZ alone. (D). Correlation between the latency for CTZ-evoked triple PS peaks and the latency for CTZ-induced spontaneous epileptiform bursts with or without K252a and TrkB IgG pre-treatment.
Effects of TrkB antibody on CTZ-induced epileptiform activity in hippocampal CA1 neurons in vivo
TrkB receptor specific antibody (TrkB antibody, Transduction Laboratories, BD Biosciences) was injected (1.25 μg in 5 μl, i.c.v.) in 2 groups with 30 and 60 min prior to CTZ (5 μmol) administration. In 30 min pre-injection group, TrkB antibody essentially inhibited all synchronized epileptiform bursts induced by CTZ, similar to that of K252a (Fig. 3D, in comparison with Fig. 3B). However, when CTZ was injected after 60 min interval following TrkB antibody injection, the inhibitory effect of TrkB antibody was partially diminished, suggesting that TrkB antibody might have been significantly cleared from lateral ventricle or inactivated after one hour (Fig. 3E). Pooled data showed that the 30 min TrkB antibody pre-injection group had a modest decrease in the percentage of animals showing triple PS peaks and spontaneous spikes (both 63%), but 60 min pre-injection did not show significant effect (Fig. 4A-B, right columns). In contrast, synchronized bursts were suppressed in all animals tested (n=8) in the 30 min pre-injection of TrkB antibody group, and about half (57%) of the animals in the 60 min pre-injection group (Fig. 4C, right columns). Despite the presence of triple PS peaks and spontaneous spikes in most animals pre-injected with TrkB antibody, the latency was nevertheless significantly prolonged for these activities (Fig. 5, right columns). In 30 min pre-injection group, TrkB antibody significantly increased the latency for triple PS peaks to 43.6 ± 6.2 min (n=5, P<0.01) and spontaneous spikes to 76.5 ± 3.6 (n=5, P<0.001). After 60 min pre-injection, the latency for triple PS peaks was still significantly increased (35.7 ± 2.5 min, n=6, P<0.01), but not the latency for spontaneous spikes (50.8 ± 5.4 min, n=7, P=0.063) (Fig. 5A-B, right columns). The latency for the synchronized bursts, in the rats where bursts persisted, tended to be prolonged after 60 min pre-injection of TrkB antibody but without reaching statistic significance (114.8 ± 6.0 min, n=4, P=0.054) (Fig. 5C, right column). To understand the relationship between the latency for epileptiform bursts and triple peaks, we plotted the latency for epileptiform bursts against the latency for triple peaks (Fig. 5D). It is clearly shown that the latency for epileptiform bursts is much longer than the latency for triple peaks, and both are prolonged by TrkB IgG and K252a. Injection of TrkB antibody (1.25 μg in 5μl, i.c.v.) alone did not induce any spontaneous activity in all 4 rats tested.
Because some animals showed CTZ-induced synchronized epileptiform bursts in the 60 min pre-injection groups of K252a or TrkB antibody, we have further quantified the frequency and duration of bursts in control and testing groups (Fig. 6). The injection of CTZ alone at two different doses, 1 and 5 μmol, induced synchronized bursts at a similar frequency (1 μmol, 1.98 ± 0.42 / 10 min, n=5 ; 5 μmol, 2.76 ± 0.36 / 10 min, n=10) and duration (1 μmol, 16.7 ± 3.2 s ; 5 μmol, 19.0 ± 1.3 s) (Fig. 6A-B, left columns). Pre-injection of K252a (60 min) significantly reduced CTZ (5 μmol) evoked burst frequency (1.26 ± 0.18 / 10 min, n=4, P<0.05) and burst duration (8.8 ± 0.6 s, n=4, P<0.01) (Fig. 6A-B, middle column). The inhibitory effects of TrkB antibody were modest comparing to K252a, with a modest decrease in the burst frequency (1.68 ± 0.30 Hz, n=4, P=0.11) but a significant decrease in the burst duration (11.6 ± 1.1 s, n=4, P<0.01) (Fig. 6A-B, right column).
Figure 6.
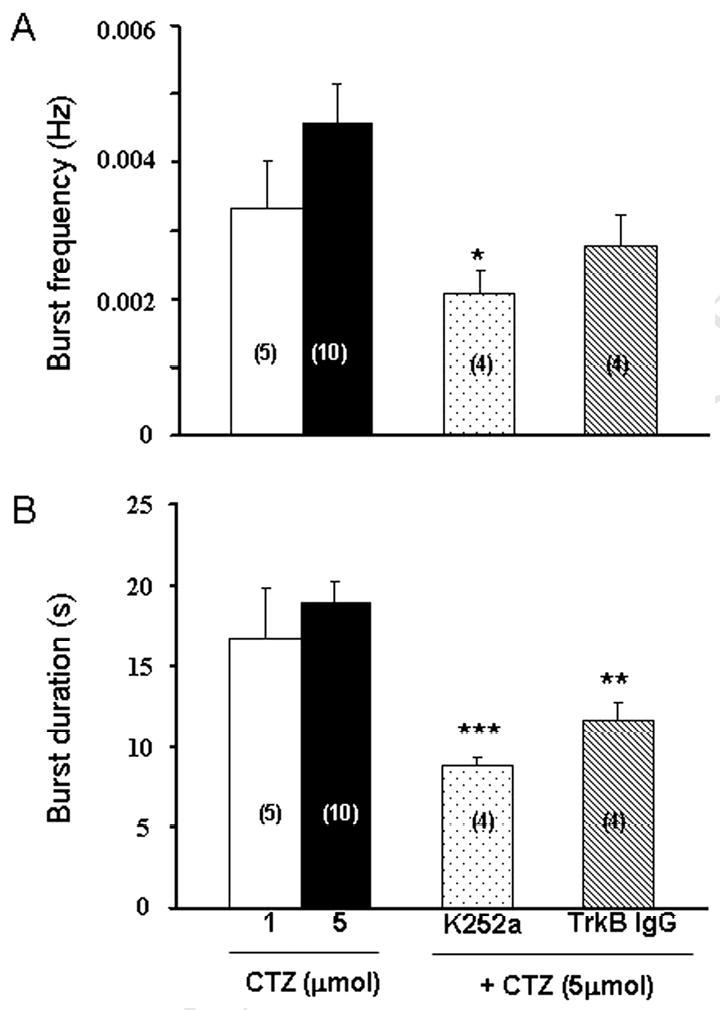
Bar histograms showing the effect of K252a and TrkB antibody on CTZ-induced synchronized bursts. (A) Pre-injection of K252a 60 min prior to CTZ (5 μmol) injection significantly reduced the frequency of synchronized bursts. (B) Pre-injection of K252a or TrkB antibody 60 min prior to CTZ injection both shortened the duration of synchronized bursts. * P<0.05, ** P<0.01 and *** P<0.001 in comparison with those of injecting CTZ alone. Note that pretreatment with K252a or TrkB antibody 30 min prior to CTZ-injection totally prevented the synchronized bursts (see Fig. 5).
Together, our results demonstrate that BDNF-TrkB signaling pathway is critically involved in the CTZ-induction of epileptiform activity in in vivo conditions.
Effects of K252a and TrkB antibody on CTZ-induced epileptiform activity in cultured hippocampal neurons
We further employed in vitro hippocampal cultures to investigate the effects of K252a and TrkB antibody on epileptiform bursts at the single cell level. Previous work from our lab have reported that CTZ-treatment of hippocampal cultures induced robust epileptiform bursts (Qi et al., 2006a). In the first set of experiments, K252a (500 nM) or TrkB-receptor specific antibody (5 μg/ml) was added together with CTZ (5 μM) during chronic treatment (48 hrs). After 48 hr-treatment, all drugs were washed away and neuronal activity was examined with whole-cell patch clamp recordings in normal bath solution. CTZ treatment alone induced robust epileptiform activity as reported before (Fig. 7A) (Qi et al., 2006a). However, K252a+CTZ or TrkB antibody+CTZ both resulted in a significant attenuation of epileptiform activity (Fig. 7B-C). The percentage of neurons displaying epileptiform activity was reduced from 85.7% with CTZ alone to about half with K252a+CTZ (40.9%) and one third of the control with TrkB antibody+CTZ (26.7%) (Fig. 7D). Among those neurons which did show epileptiform activity with K252a or TrkB antibody chronic treatment, the frequency of epileptiform bursts was also significantly reduced (CTZ alone, 0.13 ± 0.02 Hz; K252a+CTZ, 0.026 ± 0.006 Hz, p<0.001; TrkB antibody+CTZ, 0.058 ± 0.009 Hz, p<0.01) (Fig. 7E). In addition to chronic treatment, we also tested whether acute treatment of TrkB antibody has any effect on epileptiform activity. Hippocampal cultures were pretreated with TrkB IgG (5 μg/ml) for 1 hr before treating with CTZ (20 μM) for 2 hrs. After the treatment, coverslips were transferred to recording chamber for experiments in normal bath solution. We found that acute treatment of TrkB IgG significantly inhibited the epileptiform burst frequency induced by CTZ (CTZ alone, 0.038±0.01 Hz, n=8; CTZ+TrkB IgG, 0.017±0.006 Hz, n=9; p<0.002). These in vitro data in hippocampal cultures are consistent with our in vivo experimental results, suggesting an important role of BDNF-TrkB signaling in CTZ-induction of epileptiform activity.
Fig. 7.
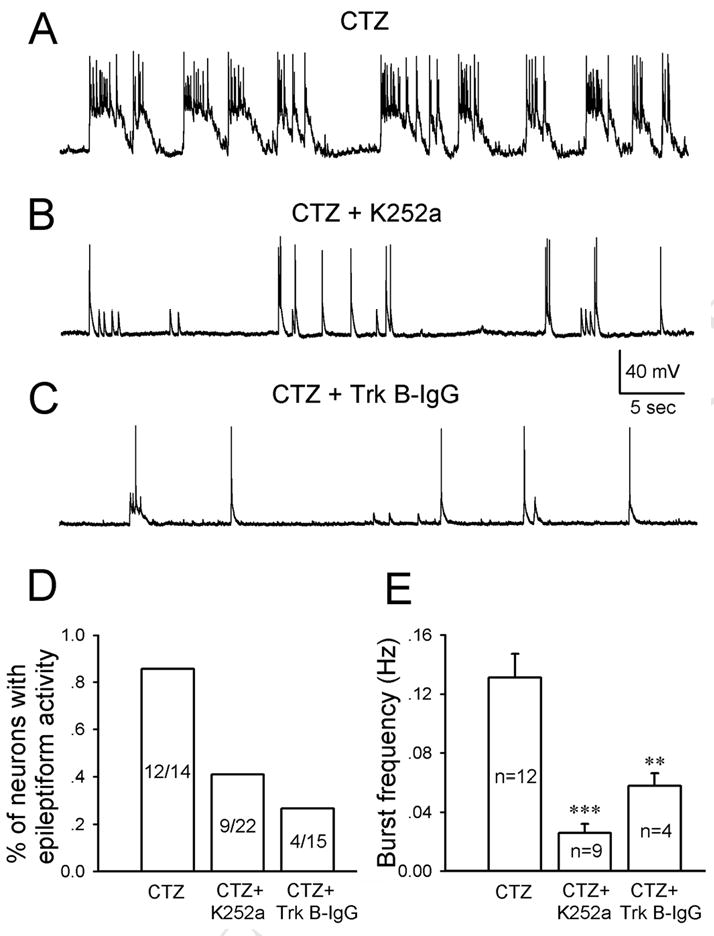
Chronic blocking TrkB receptors suppressed CTZ-induced epileptiform activity in cultured hippocampal neurons. (A-C) Typical neuronal activity after chronic treatment with CTZ alone (5 μM, 48 hrs) (A), CTZ+K252a (500 nM) (B), or CTZ+TrkB antibody (5 μg/ml) (C). The robust epileptiform activity induced by CTZ was significantly inhibited by both K252a and TrkB antibody. (D) Bar graph showing the grouped data on the percentage of neurons having epileptiform activity under CTZ alone or CTZ plus K252a or TrkB antibody. (E) Pooled data showing a significant decrease of the frequency of epileptiform bursts by K252a and TrkB antibody. ** P<0.01, and *** P<0.001 in comparison with the data of CTZ alone.
We next tested the acute effect of K252a on chronic or acute CTZ treatment-induced epileptiform activity. Acute application of K252a (500 nM) showed a direct inhibitory effect on the epileptiform activity already established by chronic or acute CTZ treatment (Fig. 8). We demonstrate here that long-term treatment (7 days) of hippocampal cultures with CTZ (20 μM) resulted in robust epileptiform bursts similar to that with 2-day treatment reported before (Qi et al., 2006a) (Fig. 8A, n=4). Acute bath application of K252a abolished most of the epileptiform bursts (Fig. 8B). Similarly, acute application of K252a (500 nM) also inhibited the epileptiform bursts induced by acute CTZ treatment (20 μM, 2 hrs) (Fig. 8C).
Fig. 8.
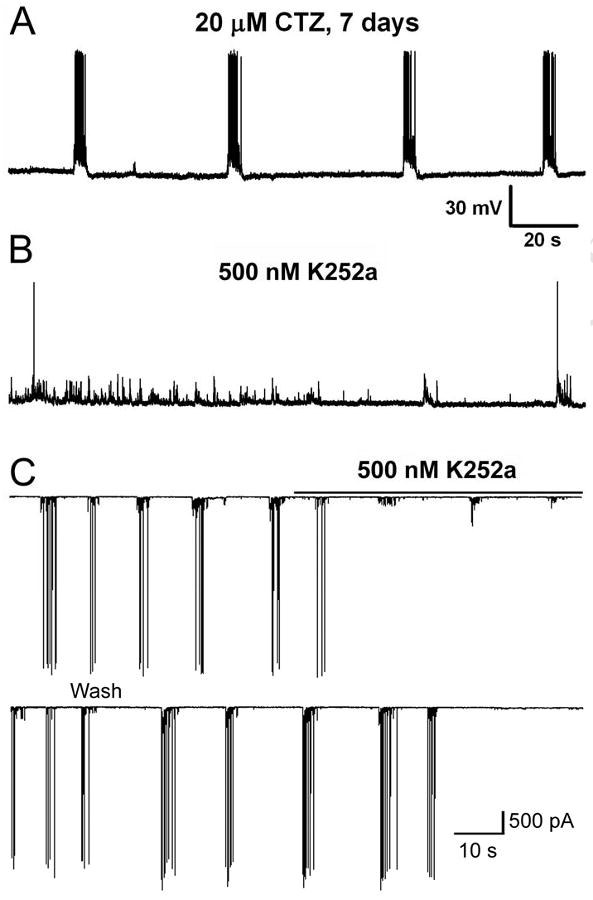
Representative traces illustrating that acute application of K252a suppressed the epileptiform activity induced by chronic or acute treatment of CTZ in cultured hippocampal neurons. (A) Epileptiform bursts persisted after long-term CTZ treatment (7 days). (B) Acute application of K252a (500 nM) inhibited the highly synchronized epileptiform bursts. Note that there were spontaneous synaptic potentials remaining after the inhibition of epileptiform bursts. (A) and (B) were in current clamp mode. (C) Acute application of K252a (500 nM) also abolished the epileptiform bursts induced by acute treatment of CTZ (20 μM, 2 hrs). Voltage clamp mode with holding potential at -70 mV.
One interesting observation arising from our K252a experiments is that action potentials associated with epileptiform bursts appeared to be inhibited in the presence of K252a but spontaneous synaptic potentials or currents persisted (Fig. 8 B & C). Therefore, we decided to investigate whether K252a has any direct effect on the basal neuronal activity in hippocampal cultures without CTZ treatment. Interestingly, bath application of 250 nM K252a significantly decreased the action potential firing frequency in all of the neurons recorded (Fig. 9A, n=7; control, 0.98 ± 0.28 Hz; during K252a, 0.31 ± 0.17; p<0.001, paired t test). The application of higher concentration of 500 nM K252a further suppressed the action potential firing and in some neurons even completely blocked action potentials (Fig. 9B, n=4). This inhibitory effect is readily reversible after washing off K252a. The precise mechanism underlying the K252a inhibition of action potential firing is currently under further study. Preliminary experiments did not find such inhibition of action potentials in the presence of TrkB antibodies, suggesting that K252a has unique effect on action potential firing that is likely beyond its effect on TrkB receptors.
Fig. 9.
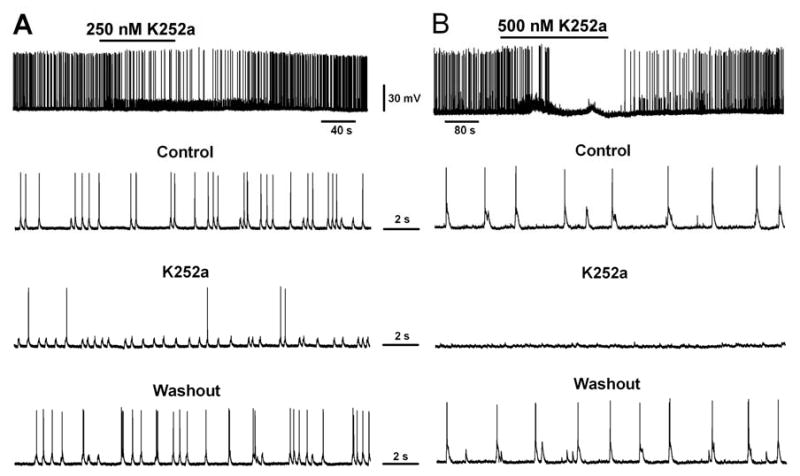
Acute application of K252a blocked action potential firing in cultured hippocampal neurons. (A) One example showing that acute application of K252a (250 nM) significantly reduced the action potential firing rate when bath applied to cultured hippocampal neurons. (B) Another example showing a more significant block of action potentials after acute application of higher concentration of K252a (500 nM).
Discussion
In this work we have demonstrated that BDNF-TrkB signaling pathway plays a critical role in mediating the induction of epileptiform activity by CTZ. Intracerebroventricular injection of CTZ induces progression of epileptiform bursts, starting from an early onset of multiple evoked PS peaks, to highly synchronized epileptiform bursts within 180 min. Infusion of receptor tyrosine kinase inhibitor K252a or specific TrkB receptor antibody essentially abolished the epileptiform bursts induced by CTZ. Similarly, in cultured hippocampal neurons, chronic or acute application of K252a or TrkB IgG together with CTZ significantly inhibited the epileptiform bursts induced by CTZ. Our results support that BDNF-TrkB signaling is a common pathway during epileptogenesis.
CTZ is a potent convulsant inducing epileptiform bursts in hippocampal neurons
The i.c.v. administration of CTZ induces epileptiform bursts in a dose-dependent manner in urethane anaesthetized rats. Multiple peaks of PSs, a traditional determinant of epileptogenesis, have been observed in 100% of animals after the injection of CTZ. In addition, spontaneous seizure-like activity occurred in 50% of animals administered with 1 μmol and ∼80% of animals dosed with 5 μmol CTZ. Such prompt seizure activity was comprised of high amplitude and high frequency synchronized burst firing, exhibiting much similarity to both pentylenetetrazole and KA models as well as human temporal lobe epilepsy (Avoli et al., 2002; Avoli et al., 2005). Before the high frequency synchronized bursting activity, there were low frequency (∼1 Hz), high amplitude individual spikes spontaneously occurring. Thus, in vivo recordings from anaesthetized rats showed that, after i.c.v. injection of CTZ, there was a time-dependent progression of epileptiform bursts, from the multiple PS peaks to spontaneous individual spiking, to final highly synchronized burst firing. The long delay of the synchronized bursts (80-100 min) suggests that neuronal activity has to reach certain threshold before being abnormally synchronized. On the other hand, the delay for the synchronization appears to be less dose-dependent. Although CTZ injection at higher dose (5 μmol) induced triple peaks PSs with a much shorter latency (∼20 min) than the low dose (1 μmol, latency >50 min), the latency for epileptiform bursts at both doses was about 80-100 min, with no statistical difference (Fig. 2B). Furthermore, the burst frequency and the burst duration were also similar between the 1 and 5 μmol doses (Fig. 6). These results suggest that the formation of epileptiform bursts depends on certain activity threshold. Once surpassing the threshold, the network activity will be synchronized according to its intrinsic properties regardless of the initial dose of convulsant drugs.
It is noteworthy to point out that although CTZ is a potent convulsant drug as we have reported before (Qi et al., 2006a), our initial experimentation with subcutaneous (50 mg kg-1, n=2) or intravenous (2.5 mg kg-1, 25 mg kg-1, and 200 mg kg-1; n=5) injection of CTZ produced neither evoked multiple PS peaks nor spontaneous burst firing in all of the animals tested. In contrast, our later experiments with i.c.v. administration of CTZ, as presented in the current study, showed that CTZ can induce epileptiform bursts. These results suggest that CTZ, once prescribed as a clinical anti-hypertension drug, is not capable of passing through blood-brain barrier. This is perhaps why such epileptic side effect of CTZ was not reported in previous clinical trials.
Blocking receptor tyrosine kinases and BDNF-TrkB pathway on CTZ-induced epileptiform bursts
Our findings that blocking TrkB receptors abolished the induction of epileptiform activity by CTZ, a convulsant identified in our lab, suggest that BDNF-TrkB signaling is likely a common pathway involved in many different epilepsy models (Binder et al., 2001). We found that the time interval between the pre-injection of K252a or TrkB antibody and the injection of CTZ is critical for the suppression of epileptiform bursts. If CTZ was injected within 30 min after blocking TrkB receptors, no epileptiform bursts can be elicited by CTZ. However, if CTZ was injected 60 min after pre-injection of K252a or TrkB antibody, about half of animals still displayed epileptiform bursts, suggesting that the blocking effect on TrkB receptors have been partially relieved within 60 min. Therefore, the metabolic rate of K252a or TrkB antibody in the cerebroventricules is somewhere between 30 and 60 min. We also noticed that the inhibitory effect of K252a is more potent than TrkB antibody in terms of suppressing multiple peaks of PSs and spontaneous spiking events. Although drug doses may be a contributing factor, the fact that K252a inhibits not only TrkB receptors but also other receptor tyrosine kinases suggests that signaling pathways other than BDNF-TrkB may also play a role in regulating neuronal activity and epileptiform bursts.
Common properties of BDNF and CTZ in regulating neural activity
It is interesting to note that BDNF potentiates glutamatergic neurotransmission but in the mean time inhibits GABAergic neurotransmission (Kang and Schuman, 1995; Tanaka et al., 1997; Binder et al., 2001; Zhu and Roper, 2001). Similarly, we have demonstrated that CTZ also has such dual effects in enhancing glutamatergic neurotransmission and depressing GABAergic neurotransmission (Deng and Chen, 2003). It is thus not a coincidence that both BDNF and CTZ have been found to be able to induce epileptiform bursts (Scharfman et al., 2002; Qi et al., 2006a). However, the acting mechanisms are distinctly different between BDNF and CTZ. BDNF acts by binding with TrkB receptors and activate down-stream protein kinases to phosphorylate substrates, leading to increased presynaptic release probability or changes in postsynaptic receptor function (Binder and Scharfman, 2004; Binder, 2007). Rapid application of BDNF to neurons can cause direct membrane depolarization or even firing of action potentials (Kafitz et al., 1999; Rose et al., 2004). In contrast, application of CTZ alone does not activate ion channels or receptors directly (Deng and Chen, 2003; Qi et al., 2006a). Instead, CTZ acts as a modulator by prolonging AMPA receptor responses or inhibiting GABAA receptor responses (Qi et al., 2006a). Despite the fundamental differences in their acting mechanisms, both BDNF and CTZ can substantially increase neural activity by positive regulation of glutamatergic and negative regulation of GABAergic neurotransmission. Our present study further suggests that the BDNF receptor TrkB is essential for CTZ-induction of epileptiform activity. Together, we propose that CTZ-induced initial increase of neuronal activity may trigger BDNF release, which activates TrkB receptors and further increases neuronal activity. Such positive feedback effects on neuronal activity may eventually lead to the formation of abnormally synchronized epileptiform bursts.
Acknowledgments
This work was supported by grants from National Institute of Health (NS054858) and National Science Foundation of the United States of America to G.C. (0236429); and grants from National Natural Science Foundation of China (30770672 to Y.W.; 30828014 to Y.W. and G.C.), Educational Ministry China (KG0707021) and Chongqing Science and Technology Commission (CHTC 2007BA5025) to Y.W. It is also partly supported by a grant from Shanghai Pujiang Program (07PJ14015) to Y.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Avoli M, Louvel J, Pumain R, Kohling R. Cellular and molecular mechanisms of epilepsy in the human brain. Prog Neurobiol. 2005;77:166–200. doi: 10.1016/j.pneurobio.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Avoli M, D'Antuono M, Louvel J, Kohling R, Biagini G, Pumain R, D'Arcangelo G, Tancredi V. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog Neurobiol. 2002;68:167–207. doi: 10.1016/s0301-0082(02)00077-1. [DOI] [PubMed] [Google Scholar]
- Balkowiec A, Katz DM. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J Neurosci. 2000;20:7417–7423. doi: 10.1523/JNEUROSCI.20-19-07417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham MC, Walmsley B. A novel presynaptic inhibitory mechanism underlies paired pulse depression at a fast central synapse. Neuron. 1999;23:159–170. doi: 10.1016/s0896-6273(00)80762-x. [DOI] [PubMed] [Google Scholar]
- Binder DK. Neurotrophins in the dentate gyrus. Prog Brain Res. 2007;163:371–397. doi: 10.1016/S0079-6123(07)63022-2. [DOI] [PubMed] [Google Scholar]
- Binder DK, Scharfman HE. Brain-derived neurotrophic factor. Growth Factors. 2004;22:123–131. doi: 10.1080/08977190410001723308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Croll SD, Gall CM, Scharfman HE. BDNF and epilepsy: too much of a good thing? Trends Neurosci. 2001;24:47–53. doi: 10.1016/s0166-2236(00)01682-9. [DOI] [PubMed] [Google Scholar]
- Cheng Q, Yeh HH. Brain-derived neurotrophic factor attenuates mouse cerebellar granule cell GABA(A) receptor-mediated responses via postsynaptic mechanisms. J Physiol. 2003;548:711–721. doi: 10.1113/jphysiol.2002.037846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng LB, Chen G. Cyclothiazide potently inhibits gamma-aminobutyric acid type A receptors in addition to enhancing glutamate responses. PNAS. 2003;100:13025–13029. doi: 10.1073/pnas.2133370100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron. 1995;15:1097–1107. doi: 10.1016/0896-6273(95)90098-5. [DOI] [PubMed] [Google Scholar]
- Elmer E, Kokaia Z, Kokaia M, Carnahan J, Nawa H, Lindvall O. Dynamic changes of brain-derived neurotrophic factor protein levels in the rat forebrain after single and recurring kindling-induced seizures. Neuroscience. 1998;83:351–362. doi: 10.1016/s0306-4522(97)00387-4. [DOI] [PubMed] [Google Scholar]
- Gall C, Lauterborn J, Bundman M, Murray K, Isackson P. Seizures and the regulation of neurotrophic factor and neuropeptide gene expression in brain. Epilepsy Res Suppl. 1991;4:225–245. [PubMed] [Google Scholar]
- Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996;7:222–238. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- He XP, Minichiello L, Klein R, McNamara JO. Immunohistochemical evidence of seizure-induced activation of trkB receptors in the mossy fiber pathway of adult mouse hippocampus. J Neurosci. 2002;22:7502–7508. doi: 10.1523/JNEUROSCI.22-17-07502.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XP, Kotloski R, Nef S, Luikart BW, Parada LF, McNamara JO. Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron. 2004;43:31–42. doi: 10.1016/j.neuron.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Takahashi T. Mechanisms underlying presynaptic facilitatory effect of cyclothiazide at the calyx of Held of juvenile rats. J Physiol. 2001;533:423–431. doi: 10.1111/j.1469-7793.2001.0423a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafitz KW, Rose CR, Thoenen H, Konnerth A. Neurotrophin-evoked rapid excitation through TrkB receptors. Nature. 1999;401:918–921. doi: 10.1038/44847. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- Kokaia M, Ernfors P, Kokaia Z, Elmer E, Jaenisch R, Lindvall O. Suppressed epileptogenesis in BDNF mutant mice. Exp Neurol. 1995;133:215–224. doi: 10.1006/exnr.1995.1024. [DOI] [PubMed] [Google Scholar]
- Lahteinen S, Pitkanen A, Saarelainen T, Nissinen J, Koponen E, Castren E. Decreased BDNF signalling in transgenic mice reduces epileptogenesis. Eur J Neurosci. 2002;15:721–734. doi: 10.1046/j.1460-9568.2002.01897.x. [DOI] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawa H, Carnahan J, Gall C. BDNF protein measured by a novel enzyme immunoassay in normal brain and after seizure: partial disagreement with mRNA levels. Eur J Neurosci. 1995;7:1527–1535. doi: 10.1111/j.1460-9568.1995.tb01148.x. [DOI] [PubMed] [Google Scholar]
- Partin KM, Patneau DK, Winters CA, Mayer ML, Buonanno A. Selective modulation of desensitization at AMPA versus kainate receptors by cyclothiazide and concanavalin. A Neuron. 1993;11:1069–1082. doi: 10.1016/0896-6273(93)90220-l. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain: in Stereotaxic Coordinates. 2nd. New York: Academic Press; 1986. [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Qi J, Wang Y, Jiang M, Warren P, Chen G. Cyclothiazide induces robust epileptiform activity in rat hippocampal neurons both in vitro and in vivo. J Physiol. 2006a;571:605–618. doi: 10.1113/jphysiol.2005.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi JS, Yao J, Fang C, Luscher B, Chen G. Downregulation of tonic GABA currents following epileptogenic stimulation of rat hippocampal cultures. J Physiol. 2006b;577:579–590. doi: 10.1113/jphysiol.2006.113134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Blum R, Kafitz KW, Kovalchuk Y, Konnerth A. From modulator to mediator: rapid effects of BDNF on ion channels. Bioessays. 2004;26:1185–1194. doi: 10.1002/bies.20118. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Sollas AL, Croll SD. Spontaneous limbic seizures after intrahippocampal infusion of brain-derived neurotrophic factor. Exp Neurol. 2002;174:201–214. doi: 10.1006/exnr.2002.7869. [DOI] [PubMed] [Google Scholar]
- Simonato M, Tongiorgi E, Kokaia M. Angels and demons: neurotrophic factors and epilepsy. Trends Pharmacol Sci. 2006;27:631–638. doi: 10.1016/j.tips.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trussell LO, Zhang S, Raman IM. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron. 1993;10:1185–1196. doi: 10.1016/0896-6273(93)90066-z. [DOI] [PubMed] [Google Scholar]
- Wheal HV, Bernard C, Chad JE, Cannon RC. Pro-epileptic changes in synaptic function can be accompanied by pro-epileptic changes in neuronal excitability. Trends Neurosci. 1998;21:167–174. doi: 10.1016/s0166-2236(97)01182-x. [DOI] [PubMed] [Google Scholar]
- Yamada KA, Tang CM. Benzothiadiazides inhibit rapid glutamate receptor desensitization and enhance glutamatergic synaptic currents. J Neurosci. 1993;13:3904–3915. doi: 10.1523/JNEUROSCI.13-09-03904.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WJ, Roper SN. Brain-derived neurotrophic factor enhances fast excitatory synaptic transmission in human epileptic dentate gyrus. Ann Neurol. 2001;50:188–194. doi: 10.1002/ana.1074. [DOI] [PubMed] [Google Scholar]
- Zorumski CF, Yamada KA, Price MT, Olney JW. A benzodiazepine recognition site associated with the non-NMDA glutamate receptor. Neuron. 1993;10:61–67. doi: 10.1016/0896-6273(93)90242-j. [DOI] [PubMed] [Google Scholar]