

Abstract
Contributions of lipases to CTL function have been debated, including if T cell lipases damage target cells. Expression of the lipase pancreatic lipase-related protein 2 (PLRP2) was previously found in IL-4 cultured lymphocyte cell lines but absent from IL-2 cultured lymphocytes. Here, we evaluated IL-2 and IL-4 induced CTLs for hydrolysis of target cell lipids and killing. Using anti-CD3 redirected lysis of [3H]-oleic acid-labeled P815 tumor cells, we detected the release of the radioactive fatty acid (FA). When PLRP2+/+ and PLRP2−/− CTLs were compared, there was more killing by the PLRP2+/+ CTLs. However, [3H]-oleic acid release was similar per dead P815, suggesting that lipid hydrolysis was produced by the dead P815s rather than by PLRP2. The FA release and death were completely dependent on perforin and also occurred when P815s were killed by perforin-containing T cell granule extracts that lacked lipase activity. Death by the cytotoxic granules extracts was unaffected by the addition of lipases. A lipase inhibitor, tetrahydrolipstatin, blocked FA release without affecting CTL-mediated cytotoxicity. Also, CTL-mediated death caused as much FA release as death by disruption of cells by freeze–thawing. The released oleic acid may be sufficient to promote secondary apoptotic responses after CTL-induced trauma.
Keywords: fatty acid release, IL-4, lipases, pancreatic lipase-related protein 2, Tc2 lymphocytes
Introduction
Function in the immune system to clear tumors or virally infected cells. In their arsenal, CTLs possess biochemical weaponry for several methods of killing, including release of the potent proteins such as perforin and granzymes for the exocytosis-induced pathway (1) and engagement of the plasma membrane Fas ligand for the Fas death receptor pathway (2). Contributions of lipases to CTL function have been debated since 1971 when Koren et al. (3) observed release of oleic acid from dying P815 target cells. Lipase activities that are activated only by perforin-dependent CTL killing remain to be characterized, while lipase activities activated by the Fas pathway have been characterized as phospholipases A2 and C as well as diacylglycerol lipase (4). The possibility that lipases may be secreted as killing enzymes is supported by the expression of a lipase, pancreatic lipase-related protein 2 (PLRP2), that is induced in CTLs by IL-4 (5), a cytokine that supports type 2 cytotoxic T lymphocyte (Tc2) differentiation (6). Additionally, knock out of the PLRP2 gene results in reduced killing capability (7). As of yet, the exact function of PLRP2 in activated T cells remains unknown; however, the lipase is a promising candidate as a membrane-damaging enzyme because it can hydrolyze multiple lipids, including triglycerides (8), phospholipids (8–10) and galactolipids (11, 12). Furthermore, cell death produced by IL-2 induced type 1 (Tc1) CTLs, which lack PLRP2 expression (5), results in substantial hydrolysis of lipids of the tumor cells ‘targeted’ for killing (13). Our paper extends the key observations by Kleinfeld et al. (13) concerning Tc1 CTL-initiated release of target cell fatty acids (FAs) to the examination of Tc2 PLRP2-associated hydrolysis of the tumor cell lipids.
Despite the cited observations, the connection between general lipase-mediated membrane hydrolysis and T cell-mediated cytotoxicity remains unclear. Several tantalizing, but unexplored, ideas in T cell immunology could potentially be explained by lipases. For example, it is still unknown exactly how perforin and granzymes are transferred from a CTL into the target cell cytoplasm. Traditional models involve perforin-dependent plasma membrane pores through which granzymes can diffuse. However, at physiological conditions, perforin alone is unable to produce pores with sufficient diameter to allow granzyme entry (14). Lipases from the CTLs may be able to alter these pores and facilitate granzyme entry into cells. Other models involving perforin-induced cell death are based on evidence that granzymes can enter cells through receptor-mediated endocytosis, but the internalized granzymes cannot be released from the endosomes without a permeabilizing agent such as perforin (14). Again, lipases from the CTLs could have a cytotoxic function, potentially co-localizing in and disrupting the granzyme-laden endosomes. Lipases of the target cells may also be directly involved in the induction of apoptosis. For example, tumor necrosis factor-α may induce apoptosis in part by activation of target cell phospholipase A2 enzymes, although there is conflicting evidence on the connection between lipid hydrolysis and apoptosis (15, 16). Thus, lipases contributing to cell death can be from either the CTL or the target (or from both). In this study, we tested this possibility using the lipase inhibitor tetrahydrolipstatin (THL).
We have selected immunological and biochemical approaches to address the potential for lipases of different CTLs to damage tumor cell membranes. We activated mouse T cells with con A to induce CTLs from all T cells regardless of their antigen-restricted specificity and cultured the cells with either IL-2 or IL-4, to polarize the CTLs toward cytotoxic type 1 or type 2 T cells, respectively. Also, we detected lysis by CTLs regardless of their antigen specificity, by using an anti-CD3 antibody redirected method to initiate lysis (17). Thus, by using con A activation and redirected lysis, we were able to characterize the rapidly induced lipases of CTLs and avoid the variables associated with long-term differentiation that occurs during the time needed in vitro to produce large numbers of T cells specific for a single antigen. Wild type (WT), PLRP2−/− or perforin (Pfn−/−) CTLs were used to evaluate T cell-derived lipase. The PLRP2−/− CTLs were used to determine the role of this lipase in CTL killing and the perforin−/− CTLs were used to determine if perforin lesions were required for target cell lipid hydrolysis.
The release of CTL lipases was detected two ways. The first was to monitor the release of any triglyceride lipases (including PLRP2) by T cells after stimulation to secrete proteins and to release granules (18). The second approach was to assay both triglyceride and phospholipase activities by monitoring hydrolysis of lipids of P815 cells, using radiolabeled oleic acid that is incorporated into many different triglycerides and phospholipids. For the second approach, it became necessary to extract the cell-free supernatants to distinguish between soluble FAs and intact lipids associated with small apoptotic vesicles that are also found in the supernatants (3, 13, 19). Here, we report that both Tc1 and Tc2 CTLs release triglyceride lipases. Cells dying after Tc1 and Tc2 attack release substantial amounts of FA. However, it appears that during CTL-mediated killing, both CTL lipases and lipases of the target cells fail to contribute to tumor cell death.
Methods
CTLs and tumor cells
CTLs were generated from mouse splenocytes; BALB/c mice (Jackson Laboratories, Bar Harbor, ME, USA), PLRP2−/− BALB/c (7) and their PLRP2+/+ littermate controls (from Dr Mark Lowe at the University of Pittsburgh, USA), C57BL6 WT or Pfn−/− mice (order #003505, Prf1tm1Sdz, Jackson Laboratories). T cells were stimulated and grown in complete RPMI 1640 media (Sigma, St. Louis, MO, USA) containing 10 mM HEPES buffer (Sigma), 24 mM sodium bicarbonate (Fisher Laboratories, Waltham, MA, USA), 1% penicillin/streptomycin (Sigma), 10% FCS from Hyclone (Logan, UT, USA), 25 μM 2-mercaptoethanol (Sigma), 2.5 μg ml−1 con A (Sigma), and 500 U ml−1 of either mouse recombinant IL-2 (2.9 × 10−9 M) or IL-4 (1.4 × 10−9 M) from eBiosciences (San Diego, CA, USA) or BD Biosciences (San Jose, CA, USA). The specific activity of r-IL-2 was 1 × 107 U mg−1 (1.7 × 1011 U mmole−1); the specific activity of r-IL-4 was 2.5 × 107 U mg−1 (3.5× 1011 U mmole−1). Following culture for 3 days at a concentration of 0.5 × 106 cells ml−1, CTLs were passaged into mitogen-free complete media with the respective cytokines. Cells were incubated at 37°C and 5% CO2. eGFP-P815 mastocytoma tumor cells (20) were maintained in continuous passage in DMEM (Gibco, North Andover, MA, USA) with 10% FCS.
Lipase assays
Triglyceride lipase assays.
Lipase activity was determined by release of FA from [3H]-labeled triolein. An assay solution containing 30 mM Tris–HCl (Fisher), 1 mM CaCl2 (Sigma) at pH 8.5 and 0.32 mM total triolein with 0.312 mM unlabeled (from 1.016 M stock, Sigma) mixed with [3H]-triolein (from 0.1 mM stock, specific activity 52.6 Ci mmol−1, Perkin Elmer (Waltham, MA, USA) at a 50:1 ratio. The micelles were incubated with the cell-free supernatants or granule extracts of CTLs. Assays were incubated at 37oC and stopped at 1 h. FAs were extracted using chloroform/methanol/heptane (14.5 parts/12 parts/10 parts; Fisher) and made basic with a pH 10 carbonate solution. The aqueous phase was formed by centrifugation at 2600 r.p.m. (1040 × g) for 5 min at room temperature. From the top phase, 200 μl were collected, mixed with 500 μl of scintillation cocktail (MicroScint-20 Perkin Elmer) and counted in a Beckman scintillation counter for 3 min per sample. To calibrate and standardize the activity of different assays, a titration of r-PLRP2 from 50 ng to 1.6 ng was carried out and stopped after 10 min to create a standard curve. Release of CTL lipases was triggered by either a combination of phorbol myristic acid (100 ng ml−1, Sigma) and ionomycin (1 μg ml−1, Sigma) or by bead-bound anti-CD3 antibody (clone 2C11, eBiosciences).
Inhibition of r-PLRP2 by THL.
Recombinant (r-) human PLRP2 (10) (a generous gift from F. Carriere, PhD) was inhibited with the irreversible lipase inhibitor THL (21) (Sigma). The inhibitor was dissolved in 100% dimethyl sulfoxide (DMSO, Sigma) and then added to r-PLRP2 in HEPES buffered saline with 1 μg ml−1 BSA at varying concentrations up to 8 μg ml−1 and incubated for 10 min at 37oC. Following this incubation, the lipase was assayed with Di-FMU octanoate (22) (Invitrogen, Carlsbad, CA, USA).
[3H]-oleic acid release from labeled P815 tumor cell lipids.
A feature of P815 cells, as mast cell-derived tumors, facilitates T cell-mediated killing regardless of the CTLs’ antigen specificity. P815s have immunoglobulin Fc receptors that bind antibodies, in this case specific to CD3 epsilon, a signaling component of the T cell receptor for antigen. Anti-CD3 epsilon bound simultaneously to both the T cells’ CD3 and to the P815 Fc-Ig receptors triggers CTL lysis, cytotoxic granule release and secretion of other T cell mediators. P815 cells were labeled overnight with 25 μCi [3H]-oleic acid (Moravek Biochemicals, Brea, CA, USA) per million cells in DMEM media containing 1 mg ml−1 delipidated BSA (Sigma). Labeled P815 cells were washed and mixed with 2 μg ml−1 anti-CD3 clone 2C11 monoclonal antibody (eBiosciences) to support CTL-redirected lysis. The assay media used were complete RPMI 1640 media with 1 mg ml−1 delipidated BSA. In quadruplicate assays, 104 P815 cells were added in a 100 μl volume to wells of 96-well microtiter plates (Becton Dickinson, Fullerton, CA, USA). CTLs were added to each well in a 100 μl volume at varying effector:target ratios. Following 4 h of incubation, the cell-free supernatant was separated by centrifugation at 1200 r.p.m. and the released [3H]-oleic acid (and any labeled lipids in small apoptotic bodies) was measured by liquid scintillation. The total counts per minute (CPM) labeled was measured by adding 1% SDS detergent to labeled tumor cells. Spontaneous release was measured with an equal volume of media in place of CTLs. Percent specific release of oleic acid was calculated as follows:
% specific release = [(experimental CPM − spontaneous CPM)/(total CPM − spontaneous CPM)] × 100.
For inhibition of lipase-generated radiolabeled oleic acid in the CTL assays, THL (Sigma) was first dissolved in DMSO and then added to the assays at 10 μg ml−1 final concentration, thus potentially affecting both effector CTL and target cell lipases. To determine maximal endogenous lipase activity in the tumor cells, 105 radiolabeled P815 cells in 1 ml of RPMI 1640 + 1 mg ml−1 BSA were 2× frozen in liquid N2, thawed and then incubated for 4 h as below for the cytotoxicity assays.
To monitor labeled FA release following death caused by extracts of isolated cytotoxic granules, we used extracts of cytotoxic lymphocyte granules from rat RNK-16 cells (23) at a concentration of 0.057 mg ml−1 (final 280 hemolytic U ml−1) and oleic acid-labeled P815 cells. The cells were incubated with the perforin-containing granule extracts for 4 h in the presence of the metabolic inhibitors 5 mM 2-deoxyglucose (Sigma), 1.5 mM NaN3 (Sigma) and 1 mM KCN (Sigma) to increase the cellular susceptibility to isolated perforin (24).
Purified bovine pancreatic triglyceride lipase (PTL) was obtained from Sigma, and bovine lipase co-lipase was obtained from AbD Serotec (Raleigh, NC, USA). These lipases were used in combination with cytotoxic granule extras. Without granule extracts, these proteins lack cytotoxicity and the ability to hydrolyze lipids of intact targets cells.
51Cr cytotoxicity assays
Lysis of target cells was determined by redirected lysis in a similar manner to the [3H]-oleic acid release assays. The tumor target cells were P815s, which express low levels of the Fas receptor on their surface (25) and express Fc receptors. The P815 cells were labeled with Na251CrO4 (Perkin Elmer) (17, 26) and used in anti-CD3 redirected lysis assays. Following 4 h of redirected lysis, 100 μl of supernatant was collected for detection in a Cobra-II auto gamma counter. In order to determine if the oleic acid release assay was proportional to the dead cells (as opposed to the lymphocyte cell density), in some experiments we normalized FA release to the number of dead cells, using the ratio of WT and PLRP2−/− CTL lytic activities [in lytic units (LU)] as a correction factor to make the number of dead cells equivalent. Lytic units were calculated as number of lytic units per 10 × 106 lymphocytes, with 1 lytic unit defined as the number of lymphocytes required to lyse half (5 × 104) the targets cells (27).
Determination of FAs versus apoptotic membrane fragments released after CTL-mediated killing
The [3H] radiolabel released into the cell-free supernatants could be either FAs or lipid vesicles from apoptotic cells (28). Lipids and FAs were separated by extraction with organic solvents (29, 30). In 24-well plates (Corning), [3H]-oleic acid labeled, 105 tumor cells and 8 × 105 CTL effectors were incubated in 1 ml volumes with or without anti-CD3 antibodies. Following a 4-h incubation, the cells were collected, centrifuged at 28 000 × g in a microcentrifuge, and the supernatant and cell pellet were separated by decanting. Cell pellets were re-suspended in 1 ml of chloroform. The supernatant or the cell pellet (200 μl) was added to a fresh mixture of chloroform:methanol:heptane (12.5/10/14) in 4 ml glass vials and vortexed. Carbonate pH 10 (250 μl) was added to the mixture and vortexed. The glass vials were centrifuged using a Thermo IEC-7000M (International equipment company, Needham, MA, USA) at 1040 × g for 5 min to separate the phases. The aqueous (upper) and organic (lower) phases (100 μl) were removed for scintillation counts using a Beckman scintillation counter instrument.
Correction factors were made for the FAs remaining in the chloroform phase. For determination of FA and triglyceride partitioning, 5 μCi [3H]-oleic acid (Movarek Biochemicals) or 5 μCi [3H]-triolein (Perkin Elmer) were brought up in 1 ml RPMI 1640 containing 1 mg ml−1 BSA and extracted as above. Corrections were made to the experimental aqueous phase scintillation data by adding the percentage of oleic acid predicted to remain in the organic layer. The equations are listed below:
1. Total CPM as FAs = (Experimentally determined CPM in the aqueous layer)/(the fraction of total oleic acid normally partitioning into the aqueous layer).
2. Total CPM as lipids = (Experimentally determined CPM in the organic layer) − (the oleic acid partitioned into the organic layer).
In addition to the detection of hydrolyzed lipids by organic extraction, we used thin layer chromatography (TLC) to verify that the released radiolabeled oleic acid was both a free FA and found in different lipids (data not illustrated).
Statistical analyses
Results are expressed as mean ± SD of the collected data. Statistical evaluations of the data were performed with Excel and Sigma plot software. The differences in mean values were evaluated by Student's t-test. Differences were considered significant if the P was <0.05.
Results
Release of lipase activity from CTLs
Tc1 have higher cytotoxicity than Tc2 CTLs and might also differ in their release of lipases. Evidence supporting a difference is found with the presence of the pancreatic lipase, PLRP2, which is induced with the type 2 cytokine, IL-4. We have determined that IL-4 induced Tc2 CTLs rapidly and selectively express PLRP2 within 4 days of activation, while IL-2 induced Tc1 CTLs lack PLPR2 expression (manuscript in preparation). When freshly isolated T cells are activated with the con A, all T cells respond, bypassing their need for specific antigens. The T cells will become polarized to become Tc1 CTLs by the presence of endogenous gamma interferon and require addition of IL-2 to grow (in the absence of added IL-4) or become polarized to become Tc2 CTLs if there are high levels of IL-4 (which can support T cell growth for up to 14 days).
To determine if CTLs release lipase activity, day 6 Tc1 and Tc2 CTLs were induced to undergo exocytosis via stimulation with anti-CD3 antibodies covalently coupled to beads. At 0 and 2.5 h, the T cell-free supernatants were collected and assayed for triglyceride lipase activity. Both Tc1 and Tc2 CTLs released lipase activity (Fig. 1). There was measurable lipase activity in the tissue culture media alone and in the media removed from the CTLs at the initiation of culture time zero (Tzero)]. The Tc1 cells secreted lipase activity without anti-CD3 stimulation, which was decreased after anti-CD3 induced stimulation. In contrast, the Tc2 cells secreted more lipase activity after anti-CD3 stimulation. In 3 of 6 experiments with Tc2 CTLs from WT and PLRP2−/− littermates, the released lipase activity was greater for the WT; however, the differences had a P values >0.05. Thus, we were unable to detect lipase activity attributable to PLRP2 and the identities of the lipases are unknown. The anti-CD3 stimulation was accompanied by release of granzyme proteases (not indicated).
Fig. 1.

CTL release of triglyceride lipases. Splenocytes were activated and cultured with 500 U ml−1 of either murine r-IL-2 (to generate Tc1 CTLs) or r-IL-4 (to generate Tc2 CTLs). CTLs were incubated for 2.5 h with immobilized anti-CD3 to stimulate the T cell receptors for antigen and trigger exocytosis of cytotoxic granules and protein secretion. Each lipase assay contained 3.2 million cell equivalents. As a control for endogenous lipase activity present in the media and contributed by the washed CTLs, cell-free supernatants were taken at the Tzero points. The cell-free supernatants from the Tzero, Tc1 and Tc2 cell after stimulation, were concentrated and immediately assayed for lipase activity using radioactive triolein for 60 min at 37°C. Tc1 CTLs secreted a constitutive lipase activity that was decreased after anti-CD3 stimulation. In contrast, Tc2 cells secreted detectable lipase activity after anti-CD3 induced exocytosis. All P-values were determined using Student’s t-tests.
Tc1 and Tc2 CTLs release radiolabeled FAs and/or lipids from tumor cells during killing
The next issue was whether death mediated by CTLs would be accompanied by major hydrolysis of tumor target cell lipids, particularly since perforin damage occurs at the plasma membrane and apoptosis might leave organelles like lipase-containing lysosomes intact. Tumor cell lipids were labeled with [3H]-oleic acid and used to monitor release of oleic acid during killing. Parallel assays were performed with CTLs and P815 tumor targets labeled with 51Cr to monitor cell death. Tc1 and Tc2 CTLs, cultured with either IL-2 or IL-4, effected substantial release of [3H]-oleic acid from tumor cell membranes that paralleled cytotoxicity (Fig. 2A and B). As might be expected, the Tc1 cells were more active killers than the Tc2 cells (31). Both oleic acid release and cytotoxicity required the presence of anti-CD3 antibody that was needed to support the redirected lysis. The release of oleic acid suggests that both types of CTLs can release lipid products as either FAs generated by lipases and/or as lipids associated with apoptotic bodies or membrane vesicles. The Tc1 CTLs had more lytic activity per T cell (Fig. 2A). Tc1 CTLs also released more oleic acid radiolabel than the Tc2 cells (Fig. 2B) when compared at the same effector to target cell (E:T) ratios. It should be noted that the oleic acid radiolabel released is a slight underestimate of the actual release because of potential reincorporation of the released FAs into the live P815 cells and into the varying numbers of CTLs.
Fig. 2.

Release of [3H]-oleic acid from tumor cells after attack by cytotoxic T cells. P815 cells were incubated overnight with [3H]-oleic acid to label their lipids or labeled with 51Cr for 4 h to monitor cell death. The [3H]-radiolabel released into the cell-free supernatant was monitored as an initial indication of lipase activity (that would release oleic acid). Subsequently, we found that apoptotic bodies and membrane vesicles contributed to ∼50% of the [3H]-radiolabel in these supernatant (see Fig. 3). Release was proportional to the ratio of effector CTLs to P815 target cells (E:T). Tc1 or Tc2 CTLs, derived from BALB/c spleens and stimulated with either 500 U ml−1 IL-2 (A) or 500 U ml−1 IL-4 (B), respectively, were used to redirect lysis of [3H]-oleic acid-labeled (closed triangles) or 51Cr-labeled P815 tumors (closed squares).
The [3H]-oleic acid radiolabel released into the supernatants represents both FA products and membrane-associated lipids
Zhang et al. (28) showed that released radiolabel in the supernatant of apoptotic cells (killed by etopside stimulus), that was purported to represent detection of FAs, was actually reflective of floating apoptotic bodies with intact lipids as well as reflective of soluble FAs that were produced by cellular phospholipase A2. We wanted to distinguish oleic acid released by lipase activities from the membrane fragments and vesicles of dead cells. To determine the fraction of the cell-free supernatant that was truly FA, we performed chloroform:methanol:heptane extractions to isolate the aqueous phase FAs. In preliminary studies, we found that radiolabeled triolein and phospholipids were completely retained in the organic phase and that oleic acid partitioned 84–90% in the aqueous layer and 10–16% in the organic phase. Extractions of the cell-free supernatant indicate that roughly half of the released radiolabel was soluble FAs (Fig. 3A). We speculate that the other half of the released label, the radiolabel retained in the organic phase, might be apoptotic bodies. When we used centrifugation to precipitate apoptotic bodies and membrane vesicles from the cell-free supernatant, ∼50% of the radiolabel released by Tc1 CTLs and tumor cells at an E:T of 8:1 with anti-CD3 was precipitated (data not illustrated), consistent with half of the released radiolabel as membrane-bound vesicles. The release of hydrolyzed membrane FAs was determined independently by TLC analysis of the resulting centrifuged cell-free supernatant (data not illustrated). An increase in effector to target ratio to 32:1 resulted in an enhanced FA to lipid ratio present in the supernatant (data not illustrated). Extractions of the cell pellet demonstrated that the majority of biologically incorporated FA radiolabel (95% or greater in all conditions) partitioned into the organic layer, reflecting label incorporated into phospholipids or triglycerides (Fig. 3B). CTL-mediated cytotoxicity resulted in loss of radiolabel from cell pellet concurrent with the release of soluble radiolabel from dead cells into the supernatant. The dead cells and cell fragments of the targets incubated with CTLs and anti-CD3 in Fig. 3(B) retained less radiolabel than the targets without CTLs or without anti-CD3. Thus, the cell-free supernatants contained substantial FAs, as originally reported by A. Kleinfeld, as well as substantial membrane fragments associated with cell death that contained un-degraded lipids. Similar results were observed with cells killed by Tc2 CTLs. Based on the specific activity of the oleic acid, the soluble FA concentrations reached 1.7 × 10−9 M concentrations after 2.5 h of killing at lymphocyte to P815 ratios of 8:1. The volume of the soluble FAs was 1 ml for 5 × 105 target cells, while physiologically these cells would probably be surrounded by 2 μl or less tissue fluid creating the potential for local oleic acid production to exceed 200 μM in 2.5 h. (This concentration becomes significant for FA-induced apoptosis, see Discussion.)
Fig. 3.

The cell-free supernatant contained soluble FAs after CTL attack. (A) Extractions of the cell-free supernatant indicated that roughly half of the released label was water-soluble oleic acid and that oleic acid-containing lipids were also elevated in the supernatants after cell death. (B) Extractions of cell pellets yielded nearly all oleic acid into the organic phase, where lipids partitioned. A loss in the cell-associated lipid counts was observed after redirected lysis.
[3H]-oleic acid release is proportional to the number of dead P815 cells and is PLRP2 independent
An important issue is whether the release of the FA-derived radiolabel was mediated in part by the CTL lipase, PLRP2. To address this issue, we questioned whether [3H]-oleic acid radiolabel released would differ between PLRP2+/+ and PLRP2−/− CTLs when normalized to a constant number of dead P815 cells. More activity by the WT CTLs would be consistent with PLRP2 activity. PLRP2+/+ CTLs generally displayed more [3H]-oleic acid release when compared with the oleic acid released by the PLRP2−/− CTLs at similar E:T ratios, regardless of whether the CTLs in question were Tc1 or Tc2 CTLs (Fig. 4A and B). Reduction in cytotoxicity by the PLRP2−/− CTLs, as previously reported, was quantified for this experiment and other representative experiments, using short-term 51Cr release assays and lytic units (Table 1). Differences in cell death mediated by Tc2 PLRP2+/+ CTLs versus PLRP2−/− CTLs were greater than for WT compared with KO Tc1 cells and were 3.5-fold better for WT Tc2 cells than PLRP2−/− cells (Table 1). To address the issue of whether the lipases came from the Tc2 CTLs or from P815s (or from both cells), we normalized the [3H]-oleic acid release to the number of dead P815 cells with the idea that PLRP2 activity would be reflected by more lipid hydrolysis by PLRP2+/+ Tc2 than PLRP2−/− Tc2 CTLs. When the correction factor (of 3.5) was applied to the [3H]-oleic acid release mediated in Tc2 killing in Fig. 4(D), there was similar release of label per dead P815 cell, killed by WT or by PLRP2−/− CTLs. Thus, overall, the data of Fig. 4 are consistent with the majority of lipase being independent of PLRP2 and potentially being from P815 cells rather than T cells. Later, we illustrate that dead P815s have substantial auto-degrading lipase activity.
Fig. 4.

Release of the oleic acid-derived radiolabel was independent of PLRP2. CTLs derived from either PLRP2+/+ (solid line) or PLRP2−/− (dashed line) mice were cultured with 500 U ml−1 IL-2 (Tc1, A and B) or 500 U ml−1 IL-4 (Tc2, C and D) and used for redirected lysis of [3H]-oleic acid-labeled P815 cells (A–D). In the right column (B and D), the oleic acid release has been normalized to the death of the tumor cells, such that the graphs represent the amount of FA released relative to the number of cells killed. The arrows between the left and right columns indicate the correction factor used to normalize the oleic acid release to the number of dead cells. The correction factor was determined from the fold reduction in death exhibited by PLRP2−/− CTL when compared with PLRP2+/+ CTLs (Table 1). The WT CTLs with the PLRP2 gene were more lytic than the PLRP2−/− CTLs, particularly after culture with IL-4 to induce PLRP2 (3.5× more lytic). However, when the oleic acid release was normalized to cell death (see B and D), PLRP2 had insignificant effects on the amount of lipid radiolabel released during killing.
Table 1.
| LU/10 M PLRP2+/+ CTLs |
LU/10 M PLRP2−/− CTLs |
Ratios PLRP2+/+ CTLs versus PLRP2−/− CTLsb |
|||
| PLRP2+/+ Tc1 | PLRP2+/+ Tc2 | PLRP2−/− Tc1 | PLRP2−/− Tc2 | Tc1 | Tc2 |
| 2049 | 386 | 1276 | 110 | 1.6 | 3.5 |
The lytic activities of Tc1 and Tc2 CTL generated from PLRP2+/+ and PLRP2−/− mice were measured by 4-h 51Cr release.
Ratios of the lytic activity were used to determine the fold difference in lytic activity exhibited by PLRP2+/+ and PLRP2−/− for both the Tc1 and Tc2 CTLs. This experiment is representative of five experiments.
The FAs released from P815 cells may be caused by endogenous lipase activities
Both apoptosis and necrosis can activate endogenous lipases. To assess further whether the lipase activity was derived from CTLs or from dead tumor cells, we evaluated an extreme model for endogenous lipases released using cells that were 100% disrupted at the start of incubation and then incubated for the same time as the CTL assays. We compared the [3H]-oleic acids released by live P815 cells alone, by 2× frozen and thawed P815 tumor cells, by cytotoxic granules with P815 cells and by tumor cells after CTL-mediated killing (Table 2). The FA released by live P815 cells alone were ∼2% of all the [3H]-oleic acid incorporated, while in the extreme case of 100% cell death caused by freezing and thawing the P815s, another 2% of free FAs were released by endogenous lipases. When CTLs mediated the killing (at 8:1), a total of ∼5% free FAs were released, with similar release mediated by either Tc1 or Tc2 CTLs. Using granules isolated from cytotoxic lymphocytes, we tested if FAs release could be carried out by the action of extracts of cytotoxic lymphocyte granules containing perforin that alone will disrupt plasma membranes and induce tumor death. To increase P815s tumor cells susceptibility to the cytotoxic granule extracts, the P815 cells were treated with 5 mM 2-Deoxy-D-glucose, 1.5 mM NaN3 and 1 mM KCN to hinder cellular repairs of cytotoxic granule-mediated damage (24). Granules at a concentration of .0574 mg ml−1 resulted in 74% cell death in the pre-treated P815s and mediated a release of 9% of the total FA. Treated P815s alone released 6% of the total FA, indicating that cytotoxic granule-mediated cell death yielded a 3% specific release of FAs. The granule extracts lacked lipase activities as evaluated by two assays. Furthermore, addition of 1 μg ml−1 of r-PLRP2 or 2 μg ml−1 PTL in combination with excess (10 μg ml−1) co-lipase failed to increase the cytotoxicity of the cytotoxic granules although these lipases did increase the oleic acid release (data not illustrated). Thus, the death by CTL attack activates endogenous P815 lipases and there would have been sufficient un-hydrolyzed substrate to observe additional lipases.
Table 2.
FA released from P815s under varying conditionsa
| Effector cells or mediator used in experiment |
||||
| Tc1 CTLs | Tc2 CTLs | Cytotoxic granule extract | ||
| Control live P815sb | 1.80% | 2.00% | 6.00% | |
| 2× freeze–thawc | 4.16% | 4.02% | N/D | |
| 8:1 CTL:P815d | 4.97% | 5.18% | N/D | |
| 1/20 granule additione | N/D | N/D | 9% | |
The FAs were extracted from the cell-free supernatants at the end of 4 h incubations of the cells.
P815 were labeled with [3H]-oleic acid and allowed to incubate alone for 4 h.
Release of FAs by P815 cells after 2× freeze–thaw cycles.
Redirected lysis was at an effector to target ratio of 8 to 1. For Tc1 cells, 83% of the P815 cells were killed and for Tc2 cells, 67% of the P815 cells were killed. The data are representative of two similar experiments.
Cytotoxic granule extract (0.0574 μg ml−1) was added to P815 targets that were treated with 5 mM 2-deoxyglucose, 1.5 mM NaN3 and 1 mM KCN to increase tumor cell sensitivity to perforin. The lysis by granule extracts was 74%, respectively.
CTL-associated P815 lipase activity was perforin dependent
To determine if the observed lipase release is a result of CTL-mediated cellular death of the tumor cell, we used Pfn−/− CTLs to prevent target cell lysis (and release of endogenous P815 lipases). In the absence of perforin, triggering of CTL cell receptors for antigen causes cytotoxic granule proteins, including granzymes, to be exocytosed but the tumor cells remain intact (as measured by 51Cr release, data not illustrated). To eliminate the apoptotic-induced cell death by the predominant death ligand, fasL, on the surface of the activated T cell, we used P815 target cells. P815s have low expression of the Fas receptor and would be less inclined to undergo apoptotic death in the presence of T cell death ligands (25). This would reduce the possibility of [3H]-apoptotic bodies interfering with our results. Lipase activities are released by CTLs upon anti-CD3 stimulation (data not illustrated). We compared the release of FAs using both Tc1 and Tc2 Pfn−/− CTLs. There was no detectable release of radiolabel (over that secreted by the live P815 cells) by either Tc1 or Tc2 Pfn−/− CTLs. These data indicate that the Tc1 and Tc2 derived lipases were unable to mediate substantial hydrolysis of lipids within intact tumor target cells (Fig. 5).
Fig. 5.

CTL release of FA was perforin dependent. CTLs were derived from WT and perforin knockout mice of the same C57BL/6 background, using conditions described for Fig. 2 and assayed for redirected lysis of [3H]-oleic acid-labeled or 51Cr-labeled P815 cells. There was substantial 51Cr lytic activity for the WT Tc1 and Tc2 CTLs that was totally absent from the Pfn−/− CTLs (data not illustrated). (A and B) In the absence of perforin, release of oleic acid was also undetectable for both Tc1 and Tc2 CTLs even at the highest ratio E:T ratio of 1:1.
THL, an inhibitor of the pancreatic α/β hydrolase fold-containing subclass of lipases, affects the lipases of P815 cells and the lipase activity associated with CTL-mediated cytotoxicity
We next asked whether the release of P815 FAs was reflective of a lipase activity and if THL (THL) inhibition of lipase activity would decrease the cytotoxic activities of either Tc1 or Tc2 CTLs. THL, (also known by the pharmaceutical name OrlistatR) is an irreversible lipase inhibitor that binds to Ser152 in the catalytic triad of lipases of the α/β hydrolase fold-containing lipases (32–34). THL has been also shown to inhibit PLRP2 activity (9) and under the pH of our tissue culture media at 8 μg ml−1 (3.2 × 10-7 M), THL inactivated 88% of r-PLRP2 in 10 min (data not illustrated). THL is actually a slow-acting inhibitor, particularly under the tissue culture conditions. With 1 μm THL, 25% of r-PLRP2 was still active after 10 min. Both endogenous P815 lipase activities (from dead cells alone) and CTL-initiated lipase activities (from P815 cells killed by CTLs) were inhibited by THL. When [3H]-oleic acid-radiolabeled P815 cells were disrupted by freeze–thawing, and then incubated for 4 h with or without 10 μg ml−1 THL, the lipase activity, which released 4% of total radiolabel, was inhibited 43% (data not illustrated). At 10 μg ml−1, THL dramatically inhibited the release of radiolabel mediated by either Tc1 or Tc2 CTLs of either WT or PLRP2−/− origin (Fig. 6A–D). Thus, there were active lipases of P815 cells or CTLs, other than PLRP2, that were sensitive to THL. The cytotoxic activity of Tc1 and Tc2 CTL was unaffected by treatment with THL (Fig. 6E and F). It should be noted that the induction of cytotoxicity occurs in only minutes (35) and that by comparison, THL was a slow inhibitor. Thus, THL was able to inactivate the lipases that substantially degraded P815 lipids but might have been insufficient to reduce any rapidly induced biological functions that might be dependent upon the T cell lipases. Furthermore, THL was unsuitable as a probe to distinguish PLRP2 from other cellular lipases.
Fig. 6.
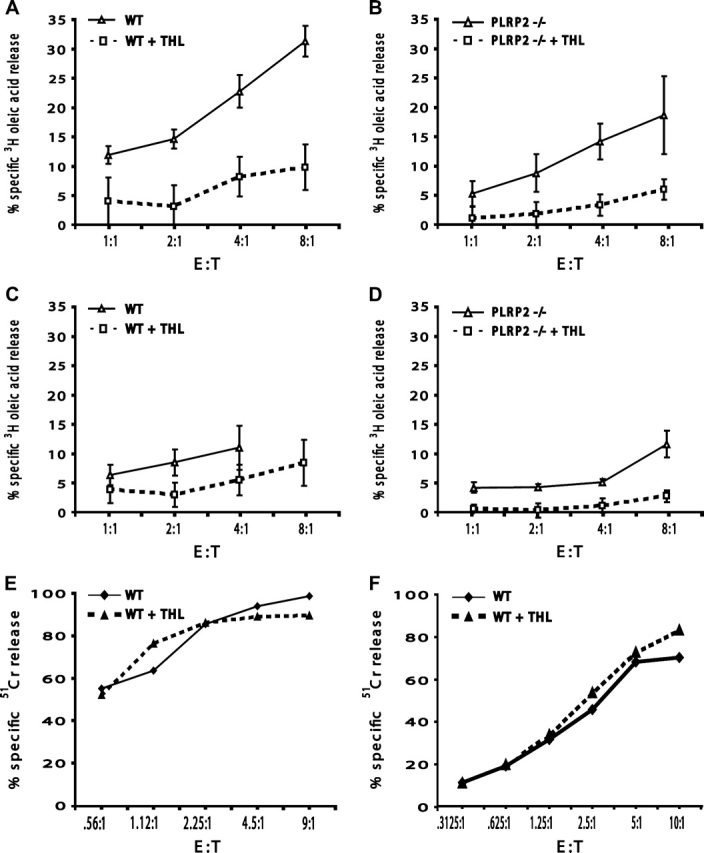
Oleic acid release was sensitive to the lipase inhibitor THL, regardless of the WT or PLRP2−/− status of the CTLs. However, THL lacked effects on CTL-mediated cytotoxicity. CTLs were derived and tested as in Fig. 2. Inhibited assays received 10 μg ml−1 THL (dashed line), while control assays received DMSO as a control for the solvent added with THL (solid line). Both WT (A and C) and PLRP2−/− CTL release of oleic acid (B and D) were affected by THL. In (E and F), the P815s were labeled with 51Cr to monitor CTL-mediated lysis in the presence or absence of 10 μg ml−1 THL.
Discussion
In this study, we centered our attention on lipase damage to tumor cell membranes after CTL-mediated cytotoxicity and on the function of the Tc2 CTL-associated lipase, PLRP2. We found evidence showing the release of lipase activity by both Tc1 and Tc2 CTLs. Using [3H]-oleic acid-labeled P815 targets, we observed the release of FAs after CTL-mediated killing by both Tc1 and Tc2 CTLs. The contribution of PLRP2 from Tc2 CTLs to the hydrolysis of the lipids of the tumor cells was undetectable. We determined that a significant proportion of the lipid hydrolysis could be produced by endogenous tumor lipases when cells were disrupted in the absence of CTLs and that, when cells remained intact after anti-CD3 conjugation with Pfn−/− CTLs, the tumor cell lipids were refractory to the CTL lipases. These two observations suggest that CTL lipases have modest (if any) general lipase activity toward the P815 cell lipids. It should be noted that we found that r-PLRP2 is stable in the presence of granule extracts containing active granzyme proteases (data not illustrated), so it is likely that released PLRP2 lipase would remain intact. PLRP2 activity would also be expected to be low as the tumor lipids contain only low amounts of the triglycerides and phosphatidylethanolamine (compared with phosphatidylcholine), which are the lipids that PLRP2 favors as substrates (8, 36). Thus, the issue remains open whether PLRP2 mediates any lipid hydrolysis when Tc2 CTLs deliver their cytotoxic mediators.
Our data suggest the presence of another CTL lipase or multiple lipases that are secreted by Tc1 and by Tc2 CTLs; however, the observations with the Pfn−/− CTLs also suggest that these lipases are also unable to hydrolyze the lipids within the membranes of intact P815 cells. We observed oleic acid release from disrupted tumor targets, when burst by freezing and thawing or when attacked by CTLs. However, the major questions that persist are (i) whether all the lipase actually originates from the dead tumor cells and (ii) whether CTL lipases play a role in cytotoxicity or have another yet-to-be-discovered function. Answers may be attainable when the tumors are derived from animals that are genetically ablated for the endogenous lipases. These animals remain to be derived. Although our THL experiments indicate that tumor death is unaffected even by an inhibitor concentration sufficient to block the released lipase activity by 8-fold, which would suggest that the lipase is irrelevant to death, caution is needed before concluding that the CTL lipases are totally irrelevant to cell death. CTL killing can occur within minutes following TCR engagement (35), and inactivation of CTL lipase may be too slow to affect cytotoxicity. THL is a slow-acting inhibitor (37) and thus may be an inadequate probe. When new lipase-deficient tumors and better lipase inhibitors become available, the issue of the roles of lipases in CTL cytotoxicity should be re-evaluated.
The fact that perforin was required for detectable lipase activity suggests that the lipases may be acting synergistically or in conjunction with cytotoxic CTL molecules. Perforin may act to initiate cell membrane damage by forming foci as start sites for lipid hydrolysis. We have found that PLRP2 is located separately from the dense cytotoxic granules containing perforin (our unpublished observations) and so synchronized release of PLRP2 and cytotoxic granules would be necessary if PLRP2 were one of the lipases. If release were synchronized, the sulfated proteoglycans of the cytotoxic granules (38) would be available to bind PLRP2, which binds well to heparin (39).
In this paper, we detected lipases that are released by Tc1 and Tc2 CTLs and noted differences, in that only the Tc2 cells seemed to release lipases after T cell antigen receptor stimulation. Little of this lipase appears to be PLRP2, which means that our experiments with the PLRP2-deficient CTLs provide no insight into functions of this lipase. Of note is the observation that perforin−/− Tc2 CTLs were effective against tumors in vivo (40). Tc2 cells secrete a different cytokine profile from Tc1 cells, secreting IL-4, IL-5, IL-6 and IL-10 (6); however, cytotoxicity is independent of these cytokines. Furthermore, adoptively transferred immune anti-tumor Tc2 CTLs were able to increase the primary host Tc1 CTL response to tumors (40), indicating that Tc2 CTLs may orchestrate immune events as well as kill tumor cells. The Tc2 lipases may have a role in these events. Previous studies have been focused on the discovery of lipases in Tc1 CTLs, because IL-2 supports higher expression of perforin and granzymes, the proteins that support short-term lytic assay in vitro, compared to IL-4 induced Tc2 CTLs (41).
The biological significance of PLRP2 found in Tc2 CTLs remains a mystery. It may be relevant that PLRP2 of goat semen mediates toxicity, but only when provided exogenous lipid substrates are available to support generation of toxic byproducts such as oleic and linoleic acid. The goat spermatids remained viable in the presence of PLRP2 alone, and triglycerides alone, but their acrosomal membranes were disrupted when PLRP2 was co-incubated with triglycerides (42, 43). The FA products of physiologically available triglycerides, oleic and linoleic acids, can mediate apoptosis of tumor cells but these FAs require >24 h for death to occur (44–48). Thus, it is possible that PLRP2 released by CTLs could produce lipid byproducts to affect tumor cell survival indirectly, provided that the appropriate triglyceride substrates are available in situ. Our future experiments will explore this possibility.
The most striking finding of our studies is the amount of FA release from cells killed by lymphocytes. The findings withTc1 CTLs corroborate the findings of Drs Anel A (49) and Kleinfeld et al. (13, 19). Our data with the Tc2 CTLs are novel and indicate similar release per dead cell, regardless of whether it was killed by a Tc1 or a Tc2 CTL. Our interpretation, shared by Dr Kleinfeld (13) who also used oleic acid-radiolabeled targets cells, is that the lipases are of target cell origin. These lipases appear to be activated by cell death, even when the original injury started with perforin damage at the target cell plasma membrane. However, initiation of the lipases may be promoted by biochemical pathways within the dying cells that are linked to CTL activity (49, 50) namely, (i) CTL-derived granzyme B-mediated activation of intracellular target cell procaspases to initiate apoptosis, followed by (ii) apoptosis-associated activation of lipases, including sphingomyelinase, as part of apoptosis to release FAs. Thus, regardless of the mechanisms, CTL killing releases FAs that will become immediate mediators of inflammation. Elevated concentrations of oleic and linoleic FAs >100 μM (with albumin from 10% serum) cause cells to undergo apoptosis (46–48, 51). Thus, FAs released by cells dying from specific CTL attack, provided that the FAs exceed the buffering capacity of localized albumin (52), have potential to promote additional death of adjacent cells.
Funding
NIH (R01CA38942-19 to D.H., R01HD33060 and DK053100 to M.L. and T32 09563 to B.N.A. and D.T.); University of Nevada Undergraduate Fellowship (to J.L.); NIH Nevada INBRE (P20RR016463) for flow cytometry.
Acknowledgments
The authors thank William H. Welch, PhD and Stephanie A. Fraser, PhD for their critical comments on the experimental design and manuscript, Rita Miller for husbandry of the PLRP2−/− mice and Dr Frederick Carriere for the generous gift of r-PLRP2.
Glossary
Abbreviations
- CPM
counts per minute
- DMSO
dimethyl sulfoxide
- FA
fatty acid
- LU
lytic units
- PLRP2
pancreatic lipase-related protein 2
- PTL
pancreatic triglyceride lipase
- THL
tetrahydrolipstatin
- TLC
thin layer chromatography
- Tc1
T cytotoxic type 1 T lymphocyte
- Tc2
T cytotoxic type 2 T lymphocyte
- Tzero
time zero
- WT
wild type
References
- 1.Raja SM, Metkar SS, Froelich CJ. Cytotoxic granule-mediated apoptosis: unraveling the complex mechanism. Curr. Opin. Immunol. 2003;15:528. doi: 10.1016/s0952-7915(03)00111-0. [DOI] [PubMed] [Google Scholar]
- 2.Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 3.Koren HS, Ferber E, Fischer H. Changes in phospholipid metabolism of a tumor target cell during a cell-mediated cytotoxic reaction. Biochim. Biophys. Acta. 1971;231:520. doi: 10.1016/0005-2760(71)90120-2. [DOI] [PubMed] [Google Scholar]
- 4.Iturralde M, Pardo J, Lacasa E, et al. Characterization of the lipolytic pathways that mediate free fatty acid release during Fas/CD95-induced apoptosis. Apoptosis. 2005;10:1369. doi: 10.1007/s10495-005-1511-1. [DOI] [PubMed] [Google Scholar]
- 5.Grusby MJ, Nabavi N, Wong H, et al. Cloning of an interleukin-4 inducible gene from cytotoxic T lymphocytes and its identification as a lipase. Cell. 1990;60:451. doi: 10.1016/0092-8674(90)90596-7. [DOI] [PubMed] [Google Scholar]
- 6.Sad S, Marcotte R, Mosmann TR. Cytokine-induced differentiation of precursor mouse CD8+ T cells into cytotoxic CD8+ T cells secreting Th1 or Th2 cytokines. Immunity. 1995;2:271. doi: 10.1016/1074-7613(95)90051-9. [DOI] [PubMed] [Google Scholar]
- 7.Lowe ME, Kaplan MH, Jackson-Grusby L, D'Agostino D, Grusby MJ. Decreased neonatal dietary fat absorption and T cell cytotoxicity in pancreatic lipase-related protein 2-deficient mice. J. Biol. Chem. 1998;273:31215. doi: 10.1074/jbc.273.47.31215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jennens ML, Lowe ME. Rat GP-3 is a pancreatic lipase with kinetic properties that differ from colipase-dependent pancreatic lipase. J. Lipid Res. 1995;36:2374. [PubMed] [Google Scholar]
- 9.Giller T, Buchwald P, Blum-Kaelin D, Hunziker W. Two novel human pancreatic lipase related proteins, hPLRP1 and hPLRP2. Differences in colipase dependence and in lipase activity. J. Biol. Chem. 1992;267:16509. [PubMed] [Google Scholar]
- 10.Eydoux C, De Caro J, Ferrato F, et al. Further biochemical characterization of human pancreatic lipase-related protein 2 expressed in yeast cells. J. Lipid Res. 2007;48:1539. doi: 10.1194/jlr.M600486-JLR200. [DOI] [PubMed] [Google Scholar]
- 11.Andersson L, Carriere F, Lowe ME, Nilsson A, Verger R. Pancreatic lipase-related protein 2 but not classical pancreatic lipase hydrolyzes galactolipids. Biochim. Biophys. Acta. 1996;1302:236. doi: 10.1016/0005-2760(96)00068-9. [DOI] [PubMed] [Google Scholar]
- 12.Sias B, Ferrato F, Grandval P, et al. Human pancreatic lipase-related protein 2 is a galactolipase. Biochemistry. 2004;43:10138. doi: 10.1021/bi049818d. [DOI] [PubMed] [Google Scholar]
- 13.Richieri GV, Kleinfeld AM. Free fatty acids are produced in and secreted from target cells very early in cytotoxic T lymphocyte-mediated killing. J. Immunol. 1991;147:2809. [PubMed] [Google Scholar]
- 14.Froelich CJ, Orth K, Turbov J, et al. New paradigm for lymphocyte granule-mediated cytotoxicity. Target cells bind and internalize granzyme B, but an endosomolytic agent is necessary for cytosolic delivery and subsequent apoptosis. J. Biol. Chem. 1996;271:29073. doi: 10.1074/jbc.271.46.29073. [DOI] [PubMed] [Google Scholar]
- 15.Brekke OL, Sagen E, Bjerve KS. Specificity of endogenous fatty acid release during tumor necrosis factor-induced apoptosis in WEHI 164 fibrosarcoma cells. J. Lipid Res. 1999;40:2223. [PubMed] [Google Scholar]
- 16.Hayakawa M, Ishida N, Takeuchi K, et al. Arachidonic acid-selective cytosolic phospholipase A2 is crucial in the cytotoxic action of tumor necrosis factor. J. Biol. Chem. 1993;268:11290. [PubMed] [Google Scholar]
- 17.Segal DM. SPDP crosslinking of antibodies to form heteroconjugates mediating redirected cytotoxicity. In: Sitkovsky MV, Henkart PA, editors. Cytotoxic cells: Recognition, Effector Function, Generation, and Methods. Boston: Birkhauser; 1993. p. 485. [Google Scholar]
- 18.Evans DL, Harris DT, Staton DL, Jaso-Friedmann L. Detection of function-associated molecules on rat leukemic NK cells: activation by monoclonal antibody or phorbol ester. Nat. Immun. Cell Growth Regul. 1990;9:353. [PubMed] [Google Scholar]
- 19.Kleinfeld AM, Okada C. Free fatty acid release from human breast cancer tissue inhibits cytotoxic T-lymphocyte-mediated killing. J. Lipid Res. 2005;46:1983. doi: 10.1194/jlr.M500151-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Kienzle N, Olver S, Buttigieg K, Kelso A. The fluorolysis assay, a highly sensitive method for measuring the cytolytic activity of T cells at very low numbers. J. Immunol. Methods. 2002;267:99. doi: 10.1016/s0022-1759(02)00150-3. [DOI] [PubMed] [Google Scholar]
- 21.Borgstrom B. Mode of action of tetrahydrolipstatin: a derivative of the naturally occurring lipase inhibitor lipstatin. Biochim. Biophys. Acta. 1988;962:308. doi: 10.1016/0005-2760(88)90260-3. [DOI] [PubMed] [Google Scholar]
- 22.Shah DS, Russell RR. A novel glucan-binding protein with lipase activity from the oral pathogen Streptococcus mutans. Microbiology. 2004;150:1947. doi: 10.1099/mic.0.26955-0. [DOI] [PubMed] [Google Scholar]
- 23.Hudig D, Callewaert DM, Redelman D, Allison NJ, Krump M, Tardieu B. Lysis by RNK-16 cytotoxic lymphocyte granules. Rate assays and conditions to study control of cytolysis. J. Immunol. Methods. 1988;115:169. doi: 10.1016/0022-1759(88)90285-2. [DOI] [PubMed] [Google Scholar]
- 24.Verret CR, Firmenich AA, Kranz DM, Eisen HN. Resistance of cytotoxic T lymphocytes to the lytic effects of their toxic granules. J. Exp. Med. 1987;166:1536. doi: 10.1084/jem.166.5.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saxena RK, Adler WH. Cytolytic activity of mitogen activated old and young mouse spleen cells against tumor target cells expressing high or low levels of Fas antigen. Exp. Mol. Med. 1999;31:137. doi: 10.1038/emm.1999.23. [DOI] [PubMed] [Google Scholar]
- 26.Segal DM, Snider DP. Targeting and activation of cytotoxic lymphocytes. Chem. Immunol. 1989;47:179. [PubMed] [Google Scholar]
- 27.Pross HF, Maroun JA. The standardization of NK cell assays for use in studies of biological response modifiers. J. Immunol. Methods. 1984;68:235. doi: 10.1016/0022-1759(84)90154-6. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Driscoll TA, Hannun YA, Obeid LM. Regulation of membrane release in apoptosis. Biochem. J. 1998;334:479. doi: 10.1042/bj3340479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lowe ME. The catalytic site residues and interfacial binding of human pancreatic lipase. J. Biol. Chem. 1992;267:17069. [PubMed] [Google Scholar]
- 30.Lowe ME. Assays for pancreatic triglyceride lipase and colipase. Methods Mol. Biol. 1999;109:59. doi: 10.1385/1-59259-581-2:59. [DOI] [PubMed] [Google Scholar]
- 31.Kienzle N, Buttigieg K, Groves P, Kawula T, Kelso A. A clonal culture system demonstrates that IL-4 induces a subpopulation of noncytolytic T cells with low CD8, perforin, and granzyme expression. J. Immunol. 2002;168:1672. doi: 10.4049/jimmunol.168.4.1672. [DOI] [PubMed] [Google Scholar]
- 32.Hadvary P, Lengsfeld H, Wolfer H. Inhibition of pancreatic lipase in vitro by the covalent inhibitor tetrahydrolipstatin. Biochem. J. 1988;256:357. doi: 10.1042/bj2560357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hadvary P, Sidler W, Meister W, Vetter W, Wolfer H. The lipase inhibitor tetrahydrolipstatin binds covalently to the putative active site serine of pancreatic lipase. J. Biol. Chem. 1991;266:2021. [PubMed] [Google Scholar]
- 34.Ransac S, Gargouri Y, Moreau H, Verger R. Inactivation of pancreatic and gastric lipases by tetrahydrolipstatin and alkyl-dithio-5-(2-nitrobenzoic acid). A kinetic study with 1,2-didecanoyl-sn-glycerol monolayers. Eur. J. Biochem. 1991;202:395. doi: 10.1111/j.1432-1033.1991.tb16387.x. [DOI] [PubMed] [Google Scholar]
- 35.Martz E. Early steps in specific tumor cell lysis by sensitized mouse T lymphocytes. II. Electrolyte permeability increase in the target cell membrane concomitant with programming for lysis. J. Immunol. 1976;117:1023. [PubMed] [Google Scholar]
- 36.Roussel A, Yang Y, Ferrato F, Verger R, Cambillau C, Lowe M. Structure and activity of rat pancreatic lipase-related protein 2. J. Biol. Chem. 1998;273:32121. doi: 10.1074/jbc.273.48.32121. [DOI] [PubMed] [Google Scholar]
- 37.Lookene A, Skottova N, Olivecrona G. Interactions of lipoprotein lipase with the active-site inhibitor tetrahydrolipstatin (Orlistat) Eur. J. Biochem. 1994;222:395. doi: 10.1111/j.1432-1033.1994.tb18878.x. [DOI] [PubMed] [Google Scholar]
- 38.MacDermott RP, Schmidt RE, Caulfield JP, et al. Proteoglycans in cell-mediated cytotoxicity. Identification, localization, and exocytosis of a chondroitin sulfate proteoglycan from human cloned natural killer cells during target cell lysis. J. Exp. Med. 1985;162:1771. doi: 10.1084/jem.162.6.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sias B, Ferrato F, Pellicer-Rubio MT, et al. Cloning and seasonal secretion of the pancreatic lipase-related protein 2 present in goat seminal plasma. Biochim. Biophys. Acta. 2005;1686:169. doi: 10.1016/j.bbalip.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 40.Dobrzanski MJ, Reome JB, Dutton RW. Role of effector cell-derived IL-4, IL-5, and perforin in early and late stages of type 2 CD8 effector cell-mediated tumor rejection. J. Immunol. 2001;167:424. doi: 10.4049/jimmunol.167.1.424. [DOI] [PubMed] [Google Scholar]
- 41.Kienzle N, Olver S, Buttigieg K, et al. Progressive differentiation and commitment of CD8+ T cells to a poorly cytolytic CD8low phenotype in the presence of IL-4. J. Immunol. 2005;174:2021. doi: 10.4049/jimmunol.174.4.2021. [DOI] [PubMed] [Google Scholar]
- 42.Pellicer-Rubio MT, Magallon T, Combarnous Y. Deterioration of goat sperm viability in milk extenders is due to a bulbourethral 60-kilodalton glycoprotein with triglyceride lipase activity. Biol. Reprod. 1997;57:1023. doi: 10.1095/biolreprod57.5.1023. [DOI] [PubMed] [Google Scholar]
- 43.Pellicer-Rubio MT, Combarnous Y. Deterioration of goat spermatozoa in skimmed milk-based extenders as a result of oleic acid released by the bulbourethral lipase BUSgp60. J. Reprod. Fertil. 1998;112:95. doi: 10.1530/jrf.0.1120095. [DOI] [PubMed] [Google Scholar]
- 44.Fernanda Cury-Boaventura M, Cristine KC, Gorjao R, Martinsd L, Curi R. Mechanisms involved in Jurkat cell death induced by oleic and linoleic acids. Clin. Nutr. 2006;25:1004. doi: 10.1016/j.clnu.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 45.Lima TM, Kanunfre CC, Pompeia C, Verlengia R, Curi R. Ranking the toxicity of fatty acids on Jurkat and Raji cells by flow cytometric analysis. Toxicol. In Vitro. 2002;16:741. doi: 10.1016/s0887-2333(02)00095-4. [DOI] [PubMed] [Google Scholar]
- 46.Andrade LN, de Lima TM, Curi R, Castrucci AM. Toxicity of fatty acids on murine and human melanoma cell lines. Toxicol. In Vitro. 2005;19:553. doi: 10.1016/j.tiv.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 47.Cury-Boaventura MF, Pompeia C, Curi R. Comparative toxicity of oleic acid and linoleic acid on Jurkat cells. Clin. Nutr. 2004;23:721. doi: 10.1016/j.clnu.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 48.Cury-Boaventura MF, Pompeia C, Curi R. Comparative toxicity of oleic acid and linoleic acid on Raji cells. Nutrition. 2005;21:395. doi: 10.1016/j.nut.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 49.Iturralde M, Gamen S, Pardo J, et al. Saturated free fatty acid release and intracellular ceramide generation during apoptosis induction are closely related processes. Biochim. Biophys. Acta. 2003;20:40. doi: 10.1016/j.bbalip.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 50.Gamen S, Marzo I, Anel A, Pineiro A, Naval J. CPP32 inhibition prevents Fas-induced ceramide generation and apoptosis in human cells. FEBS Lett. 1996;390:232. doi: 10.1016/0014-5793(96)00666-7. [DOI] [PubMed] [Google Scholar]
- 51.Anel A, Naval J, Desportes P, Gonzalez B, Uriel J, Pineiro A. Increased cytotoxicity of polyunsaturated fatty acids on human tumoral B and T-cell lines compared with normal lymphocytes. Leukemia. 1992;6:680. [PubMed] [Google Scholar]
- 52.Huber AH, Kampf JP, Kwan T, Zhu B, Kleinfeld AM. Fatty acid-specific fluorescent probes and their use in resolving mixtures of unbound free fatty acids in equilibrium with albumin. Biochemistry. 2006;45:14263. doi: 10.1021/bi060703e. [DOI] [PMC free article] [PubMed] [Google Scholar]