

Abstract
Phylloquinone (vitamin K1) is a lipophilic compound present in plasma at low concentrations, which presents technical challenges for determining its bioavailability or metabolic fate using stable isotopes. We developed a method to simultaneously measure unlabeled and deuterium-labeled phylloquinone concentrations in plasma specimens using high-performance liquid chromatography/mass spectrometry with atmospheric pressure chemical ionization (LC–APCI/MS). Phylloquinone was extracted from plasma using hexane, further purified by solid-phase extraction, and then quantified using high-performance liquid chromatography with an APCI/MS as a detector. Plotting the expected versus the measured amount of serial dilutions of either unlabeled or labeled phylloquinone gave correlation coefficients (R) of 0.999 for both compounds. The minimum detectable concentrations of unlabeled and labeled phylloquinone were 0.05 and 0.08 pmol/injection, respectively. Pooled plasma samples spiked with between 0.5 and 32 nmol phylloquinone/L gave average recoveries of 96.7% with 5.4% relative standard deviation (RSD) for unlabeled phylloquinone and 96.2% with 6.6% RSD for labeled phylloquinone. Plasma phylloquinone concentrations determined by LC-fluorescence and LC–APCI/MS methods from healthy subjects (n = 17) were not statistically different (P = 0.13). The LC–PCI/MS method is a sensitive technique for simultaneous determination of both unlabeled and labeled phylloquinone and can be applied to bioavailability studies.
Vitamin K is a cofactor necessary for the enzymatic modification of glutamic acid residues to γ-carboxyglutamic acid (Gla) residues in proteins that have been implicated in hemostasis, bone metabolism, tissue calcification, and cell cycle regulation.1–3 Phylloquinone is the primary dietary source of vitamin K and is present at highest concentrations in green leafy vegetables and certain plant oils.4 The ability to establish dietary recommendations for vitamin K is limited by our inadequate understanding of the metabolic fate of phylloquinone from the diet.5,6 Direct quantification of phylloquinone in plasma is the best indicator of recent dietary intake.7,8 Efforts to collect these data to establish recommendations have been hampered by the difficulty in accurate detection of very low endogenous concentrations in plasma. The ability to administer stable isotope labeled phylloquinone and accurately monitor plasma concentrations would facilitate these determinations. Thus, a sensitive and specific method is required to measure phylloquinone labeled with a stable isotope, such as deuterium, for studies of vitamin K bioavailability.
Several techniques are used for the measurement of vitamin K in human plasma. High-performance liquid chromatography (HPLC) with fluorescence detection after postcolumn reduction of vitamin K is a common method used to quantify plasma phylloquinone.9 The use of stable isotope labeled phylloquinone requires both chromatographic and mass spectrometry techniques to measure the concentration of unlabeled and labeled molecules. Recently, some new techniques, such as gas chromatography/mass spectrometry (GC/MS),5,6,10–12 liquid chromatography/mass spectrometry (LC/MS),13 and HPLC/tandem mass spectrometry with atmospheric pressure chemical ionization (LC–APCI/MS/MS) have been used to detect vitamin K.14,15 An LC/MS system for the quantitative analysis of 13C-phylloquinone in plasma has also been described. However, it has limited applicability because uniform endogenous labeling of a dietary source of 13C-labeled phylloquinone is very costly.13 Among the methods using GC/MS, technical challenges were reported in plasma samples containing high concentrations of lipophilic compounds (e.g., in nonfasting samples), which precluded the determination of phylloquinone concentrations in these samples.5,6 Enzyme hydrolysis and subsequent derivatization of phylloquinone before analysis were used to resolve this problem.11,12 However, the disadvantages of this approach included the complicated and labor-intensive nature of sample preparation. We also have noted contamination of the cholesterol esterase with phylloquinone (150 pmol of phylloquinone per gram of enzyme powder; unpublished data).
In order to study phylloquinone absorption and clearance rates at physiological doses, we have extended the method reported by our group5,6 and developed a specific and sensitive technique to quantify unlabeled and deuterium-labeled phylloquinone in plasma and in lipid plasma fractions using LC–APCI/MS.
EXPERIMENTAL SECTION
Reagents and Standards
Solvents used for extraction and chromatography were HPLC grade (Fisher Scientific, Springfield, NJ). Stock standards, phylloquinone (Sigma, St. Louis, MO) and internal standard, vitamin K1(25) (GL Synthesis, Worcester, MA), were prepared in methanol and were characterized spectrophotometrically and chromatographically before use.9 All operations were performed under yellow light.
Deuterium-Labeled Phylloquinone
Deuterium-labeled phylloquinone was extracted from deuterium-labeled collard greens, which were grown hydroponically in an environmental growth chamber at USDA/ARS Children’s Nutrition Research Center in Houston Texas, as previously described.5 Deuterium-labeled phylloquinone was extracted from collard greens by liquid–liquid extraction and purified by solid-phase extraction.5 Then it was stored in methanol and used as stock solution. The concentration of deuterium-labeled phylloquinone from collard greens was determined by HPLC with fluorescence detection.9
Plasma Preparation
The internal standard, K1(25), was added in amounts of 2.3 ng to 0.5 mL of sample. An amount of 1 mL of ethanol was added followed by 5–10 s of vigorous mixing to denature the protein, and then 1 mL water and 3 mL of hexane were added, followed by vortex mixing for 5 min. The resulting mixture was centrifuged at 3000 rpm for 5 min, and the upper (hexane) layer was removed and evaporated under reduced pressure. The samples were reconstituted in 0.5 mL of hexane and applied to Bond Elut 500 mg silica SPE columns (part no. 12113036 Varian, U.S.A.), conditioned with 4.0 mL of ethyl ether/hexane (3.5/96.5 v/v) and 4.0 mL of hexane. The columns were washed with 4.0 mL of hexane, and the phylloquinone was eluted with 8.0 mL of 3.5% ethyl ether in hexane. The column conditioning, absorption, washing, and elution steps were performed using a Vac Elut SPS 24 manifold (Varian, U.S.A.). The eluate was evaporated using a Savant Speed Vac plus instrument (Savant Inc., U.S.A.). The residue was dissolved in 30 μL of methylene chloride followed by 100 μL of aqueous methanol solution. Aliquots (100 μL) were automatically injected into the LC/MS system.
LC–APCI/MS Conditions
The LC/MS consisted of an Agilent HP series 1100 G1946D MSD with an APCI source connected to an Agilent series 1200 HPLC instrument. The temperature-controlled HPLC column compartment was set to 20 °C. We used a ProntoSil C30 column (5 μm, 250 mm × mm) (MAC-MOD Analytical Inc., Chadds Ford, PA). The mobile phase was solvent A (5.5 mM acetic acid in methanol) and solvent B (100% methylene chloride). A linear gradient was run as follows: 0% solvent B at 0 min to 10% solvent B at 25 min with flow rate 1.0 mL/min. The mobile phase composition was changed to 70:30 (A/B) at 26 min, and flow rate was increased to 1.2 mL/min to remove lipophilic compounds from the column. At 35 min solvent B was decreased to 0%, and the flow rate was decreased to 1.0 mL/min to re-equilibrate the column. The cycle was finished at 40 min.
The MSD conditions were as follows: the MS ion source was positive APCI with the spray chamber gas temperature set at 350 °C, vaporizer temperature set at 400 °C, the drying nitrogen 7.0 L/min, capillary voltage was 3800 V, nitrogen nebulizer pressure was set to 45 psi, and corona current set at 5 μA. Selected ion monitoring (SIM) was used to detect isotopomers of phylloquinone (m/z 451) and deuterium-labeled phylloquinone (m/z 450–470). The most abundant labeled isotopomers in collard greens were at m/z 459–463 (64% of total in collard greens). These isotopomers were chosen for SIM because they produced the highest signal-to-noise ratio for deuterium-labeled phylloquinone. Data were collected using Agilent Chemstation software (version B.03.01).
Validation Procedure and Quantification
Solutions for the determination of linearity, dilution recovery, and precision were made by spiking pooled plasma with either unlabeled phylloquinone or deuterium-labeled phylloquinone, followed by serial dilution (between 0.5 and 32.0 nmol/L phylloquinone) with unspiked pooled plasma.
The response factor (RF) of unlabeled phylloquinone calibration standard was defined as the phylloquinone peak area (total area of trans- plus cis-phylloquinone) divided by the peak area of the internal standard (area of trans-K1(25) only). The RF of the sample phylloquinone was defined as the phylloquinone peak area (which was trans-phylloquinone only) divided by the peak area of the internal standard (area of trans-K1(25) only). Quantification was achieved by dividing the sample RF by the RF of the standard and multiplying by the total amount of phylloquinone standard injected (trans plus cis) and the dilution factor.
Clinical Applications
To compare the conventional fluorescence HPLC method9 with LC–APCI/MS detection for determination of unlabeled phylloquinone concentrations, 17 archived fasting plasma samples from a previously published study16 were analyzed using both methods.
To determine feasibility of using the LC–APCI/MS detection for deuterium-labeled fasting and nonfasting samples, five healthy subjects were each fed 120 g of deuterium-labeled collard greens containing 180 μg of labeled phylloquinone in the Metabolic Research Unit at the Jean Mayer USDA Human Nutrition Research Center at Tufts University. Fasting blood samples were drawn at 0 h before ingestion of the deuterium-labeled collard greens, 24 and 48 h postingestion, following a 12 h fast. Nonfasting samples were drawn at 4, 5, 6, 7, 9, 12, and 16 h. Plasma was separated by centrifugation for 20 min at 1300g and 4 °C. Lipoprotein fractions representing very low density lipoprotein (VLDL), low-density lipoprotein (LDL), and high-density lipoprotein (HDL) were isolated from plasma by sequential ultracentrifugation at 4 °C, as previously described.17
Statistical Analysis
Linear correlation coefficients were used to compare (1) the expected and the measured amount of serial dilutions of either unlabeled or labeled phylloquinone and (2) phylloquinone concentrations, as determined by the LC–APCI/MS method and conventional HPLC method with fluorescence detection. Student’s paired t test was used for comparing differences in phylloquinone concentration as determined by the LC–APCI/MS and HPLC method. All statistical analyses were performed using Microsoft Excel 2003. Results were considered statistically significant if the observed significance value was less than 0.05 (P < 0.05).
RESULTS AND DISCUSSION
LC-APCI/MS
Trans- and cis-phylloquinone are separated by C30 columns, but not C18 columns.18 In this LC–APCI/MS system, we use peak area (abundance) to quantify phylloquinone concentration. Therefore, the total abundance of trans-plus cis-phylloquinone in the calibration standards was used to quantify unlabeled or labeled phylloquinone. Synthetic phylloquinone and K1(25) contained about 15% and 25% cis isomers, respectively (Figure 1A). However, deuterium-labeled phylloquinone from collard greens and unlabeled phylloquinone from natural sources only contained trans-phylloquinone (Figure 1B).
Figure 1.

LC-APCI/MS chromatographic profiles of (A) the calibration standards containing phylloquinone (VK) and K1(25) and (B) plasma sample collected from a healthy subject 7 h after the consumption of a meal containing deuterium-labeled collard greens.
Cooling the C30 column to 20 °C improved separation of vitamin K compound peaks from other lipophilic compound peaks in the mass chromatogram. A gradient elution protocol was used to remove lipophilic compounds, achieving a consistently more stable baseline which optimized the signal-to-noise ratio. Representative LC–APCI/MS chromatograms of the calibration standards, the unlabeled trans form of phylloquinone, and the deuterium-labeled trans form of phylloquinone and K1(25) from archived human plasma are shown in Figure 1. Phylloquinone and internal standard peaks were clearly resolved from other unidentified compounds that were present in plasma. Under the conditions as described, average retention time for unlabeled trans-phylloquinone, deuterium-labeled trans-phylloquinone, and the internal standard trans-K1(25) were 15.4, 15.3, and 25.9 min, respectively.
Linearity, Precision, and Accuracy
The linearity of unlabeled or deuterium-labeled phylloquinone measurements was assessed by testing pooled plasma spiked with phylloquinone in concentrations that ranged from 0.5 to 32.0 nmol phylloquinone/L (Figure 2). The linear regression comparison of measured to expected concentrations of phylloquinone gave correlation coefficients (R) of 0.999 for both unlabeled phylloquinone and deuterium-labeled phylloquinone.
Figure 2.

Linearity of unlabeled and deuterium-labeled phylloquinone in series of spiked plasma by LC–APCI/MS: (A) unlabeled phylloquinone; (B) deuterium-labeled phylloquinone.
The limits of detection for unlabeled and labeled phylloquinone were determined by the incremental dilution of working standard until a concentration was injected that could no longer be detected by this system. The minimum detectable concentration of unlabeled and labeled phylloquinone was 0.05 and 0.08 pmol/injection (equal to plasma concentration of about 0.2 and 0.5 nmol/L), respectively. Population reference intervals ranging from 0.22 to 8.88 nmol/L have been reported for plasma phylloquinone.8,19,20
As shown in Table 1, for pooled plasma samples spiked with 0.5–32.0 nmol phylloquinone/L, the mean relative recoveries ranged from 91% to 103% for unlabeled phylloquinone and 88% to 102% for labeled phylloquinone. The precision was measured as the percent relative standard deviation (RSD). The RSD for unlabeled and deuterium-labeled phylloquinone at concentrations from 1.0 to 32 nmol/L was less than 10%.
Table 1.
Accuracy and Precision of the LC–APCI/MS Method, Based on Recovery of Known Amounts of Phylloquinone Added to Plasma Samples (n = 3)
| unlabeled phylloquinone |
deuterium-labeled phylloquinone |
|||
|---|---|---|---|---|
| spiked concentration (nmol/L) | mean recovery (%)a | RSD (%)b | mean recovery (%) | RSD (%) |
| 0.5 | 97.8 ± 15.1 | 15.4 | 92.2 ± 7.2 | 7.8 |
| 1.0 | 92.4 ± 3.2 | 3.5 | 87.7 ± 7.7 | 8.8 |
| 2.0 | 98.8 ±4.3 | 4.4 | 96.0 ± 7.7 | 7.9 |
| 4.0 | 97.7 ± 3.1 | 3.1 | 93.5 ± 9.0 | 9.6 |
| 8.0 | 91.2 ± 1.2 | 1.4 | 99.6 ± 8.8 | 8.7 |
| 16.0 | 95.9 ± 7.1 | 7.3 | 97.6 ± 2.6 | 2.7 |
| 32.0 | 103.2 ± 2.8 | 2.7 | 102.0 ± 3.4 | 3.3 |
Mean percent relative recovery (mean % ± SD) represents the accuracy of the method.
Percent relative standard deviation (RSD) represents the precision of the method.
Archived plasma samples (n = 17) were used to compare HPLC with fluorescence detection to the LC–APCI/MS method. The phylloquinone concentrations determined by the two methods were compared using the paired t test and were not statistically different (P = 0.13, Figure 3). Similarly, phylloquinone concentrations were highly correlated between the two methods (R = 0.935).
Figure 3.
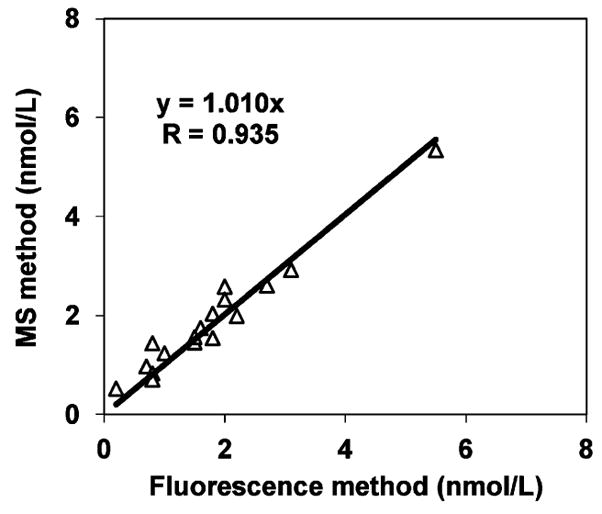
Comparison of plasma phylloquinone concentrations (nmol/L) in human plasma between the LC–APCI/MS method (MS method) and HPLC with fluorescence detection method (fluorescence method) (n = 17).
Some published assays for phylloquinone determination do not use internal standards.6,11 However, in our experience, use of an internal standard is essential for accurate determination of plasma phylloquinone concentration. Extraction efficiency from plasma can be corrected for using K1(25) as an internal standard. This compound and phylloquinone have similar chemical properties.
Clinical Applications
We measured phylloquinone in plasma samples obtained from healthy subjects (n = 5) in response to intake of deuterium-labeled collard greens. The scans of all labeled isotopomers in plasma samples were also analyzed for isotopomer profiles. The most abundant isotopomer in plasma (m/z 461) corresponded to that in the collard greens (Figure 4, parts A and B). Labeled phylloquinone was also measured in the VLDL, LDL, and HDL fractions, as illustrated in Figure 4C. Deuterium-labeled phylloquinone concentrations were measurable in all VLDL, LDL, and HDL fractions between 5 and 16 h following intake of the deuterium-labeled collard greens. Both LDL and HDL fractions carried smaller amounts of phylloquinone compared with the VLDL fraction, which was the major carrier of phylloquinone in plasma. This technique resolved the analysis problem previously reported using GC/MS, in which the different lipoprotein fractions clogged the GC column, so the phylloquinone levels could not be measured.6 This new method allows us to measure both unlabeled and deuterium-labeled phylloquinone in fasting and nonfasting plasma and lipoprotein fractions, regardless of the lipoprotein profile of the samples. This highlights the main advantage of the LC–APCI/MS system for determination of plasma phylloquinone in bioavailability studies.
Figure 4.

Phylloquinone absorption in healthy subjects using LC–APCI/MS. Isotope distributions in (A) labeled collard greens and (B) plasma at 7 h following intake of deuterium-labeled collard greens. The isotopomers that were measured (m/z 459–463) in selected ion monitoring (SIM) are presented in gray bars. (C) Deuterium-labeled phylloquinone concentrations in lipoprotein fractions (VLDL, LDL, and HDL) at different time points following intake of deuterium-labeled collard greens (n = 5).
CONCLUSIONS
We developed a method to simultaneously measure unlabeled and deuterium-labeled phylloquinone concentrations using LC–APCI/MS. Our current method can be used directly for determination of phylloquinone in nonfasting samples containing high concentrations of lipophilic compounds and in lipoprotein fractions without enzyme hydrolysis or HPLC semipurification. The use of an internal standard improved the reliability of the determination. Thus, the LC–APCI/MS method is a sensitive, specific, and accurate technique for simultaneous measurement of both unlabeled and labeled phylloquinone and can be applied to bioavailability studies.
Acknowledgments
This work was supported by the U.S. Department of Agriculture, Agricultural Research Service under Cooperative Agreement No. 58-1950-7-707 and the National Institutes of Health (DK069341). Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the authors and do not necessarily reflect the view of the U.S. Department of Agriculture. No authors report a conflict of interest.
References
- 1.Ferland G. Nutr Rev. 1998;56:223–230. doi: 10.1111/j.1753-4887.1998.tb01753.x. [DOI] [PubMed] [Google Scholar]
- 2.Furie B, Bouchard BA, Furie BC. Blood. 1999;93:1798–1808. [PubMed] [Google Scholar]
- 3.Vermeer C, Gijsbers BL, Craciun AM, Groenenvan Dooren MM, Knapen MH. J Nutr. 1996;126:1187S–1191S. doi: 10.1093/jn/126.suppl_4.1187S. [DOI] [PubMed] [Google Scholar]
- 4.Booth SL, Suttie JW. J Nutr. 1998;128:785–788. doi: 10.1093/jn/128.5.785. [DOI] [PubMed] [Google Scholar]
- 5.Dolnikowski GG, Sun Z, Grusak MA, Peterson JW, Booth SLJ. Nutr Biochem. 2002;13:168–174. doi: 10.1016/s0955-2863(01)00210-8. [DOI] [PubMed] [Google Scholar]
- 6.Erkkila AT, Lichtenstein AH, Dolnikowski GG, Grusak MA, Jalbert SM, Aquino KA, Peterson JW, Booth SL. Metabolism. 2004;53:215–221. doi: 10.1016/j.metabol.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 7.Booth SL, Tucker KL, McKeown NM, Davidson KW, Dallal GE, Sadowski JA. J Nutr. 1997;127:587–592. doi: 10.1093/jn/127.4.587. [DOI] [PubMed] [Google Scholar]
- 8.McKeown NM, Jacques PF, Gundberg CM, Peterson JW, Tucker KL, Kiel DP, Wilson PW, Booth SL. J Nutr. 2002;132:1329–1334. doi: 10.1093/jn/132.6.1329. [DOI] [PubMed] [Google Scholar]
- 9.Davidson KW, Sadowski JA. Methods Enzymol. 1997;282:408–421. doi: 10.1016/s0076-6879(97)82124-6. [DOI] [PubMed] [Google Scholar]
- 10.Fauler G, Leis HJ, Schalamon J, Muntean W, Gleispach H. J Mass Spectrom. 1996;31:655–660. doi: 10.1002/(SICI)1096-9888(199606)31:6<655::AID-JMS339>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 11.Jones KS, Bluck LJ, Coward WA. Rapid Commun Mass Spectrom. 2006;20:1894–1898. doi: 10.1002/rcm.2538. [DOI] [PubMed] [Google Scholar]
- 12.Jones KS, Bluck LJC, Wang LY, Coward WA. Eur J Clin Nutr. 2008;62:1273–1281. doi: 10.1038/sj.ejcn.1602859. [DOI] [PubMed] [Google Scholar]
- 13.Kurilich AC, Britz SJ, Clevidence BA, Novotny JA. J Agric Food Chem. 2003;51:4877–4883. doi: 10.1021/jf021245t. [DOI] [PubMed] [Google Scholar]
- 14.Kang W, Jeong JH, Ma E, Kwon KI. J Pharm Biomed Anal. 2007;44:1178–1182. doi: 10.1016/j.jpba.2007.04.028. [DOI] [PubMed] [Google Scholar]
- 15.Suhara Y, Kamao M, Tsugawa N, Okano T. Anal Chem. 2005;77:757–763. doi: 10.1021/ac0489667. [DOI] [PubMed] [Google Scholar]
- 16.Shea MK, Dallal GE, Dawson-Hughes B, Ordovas JM, O’Donnell CJ, Gundberg CM, Peterson JW, Booth SL. Am J Clin Nutr. 2008;88:356–363. doi: 10.1093/ajcn/88.2.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Havel RJ, Eder HA, Bragdon JH. J Clin Invest. 1955;34:1345–1353. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woollard DC, Indyk HE, Fong BY, Cook KK. J AOAC Int. 2002;85:682–691. [PubMed] [Google Scholar]
- 19.Azharuddin MK, O’Reilly DS, Gray A, Talwar D. Clin Chem. 2007;53:1706–1713. doi: 10.1373/clinchem.2007.086280. [DOI] [PubMed] [Google Scholar]
- 20.Sadowski JA, Hood SJ, Dallal GE, Garry PJ. Am J Clin Nutr. 1989;50:100–108. doi: 10.1093/ajcn/50.1.100. [DOI] [PubMed] [Google Scholar]