

Abstract

N-Heterocyclic carbenes have been demonstrated to react through divergent pathways under the same conditions. Experimental and computational evidence demonstrates that the ability to favor generation of homoenolate equivalents from α,β-unsaturated aldehydes versus the oxidation of aldehydes to esters is highly dependent upon the choice of solvent. The solvation environment plays an important role due to the mechanistic differences in these processes, with polar protic solvent favoring the oxidation process due to solvation of intermediates with greater charge separation.
The discovery and understanding of new catalytic processes is an essential pursuit in science. Nucleophilic carbene catalysis has emerged as a powerful method to access a broad class of transformations with unique modes of reactivity.1 The ability to predict and control the myriad of potential reaction outcomes is a vital objective in maximizing the impact of these Lewis base processes (Figure 1).2 Two important, yet divergent, N-heterocyclic carbene (NHC) catalysis redox pathways have recently garnered significant interest. The addition of carbenes to α,β-unsaturated aldehydes can promote oxidation of the aldehyde and/or internal redox through functionalization at the β-carbon (homoenolate reactivity) with concomitant oxidation of the carbonyl. Our interests include developing new reactions based on both pathways. Even with the recent surge of new carbene catalyzed processes, the identification of optimized conditions that signficantly favor the β-protonation product (saturated ester) has remained elusive. This limitation has prevented the development of high yielding and enantioselective homoenolate protonations to afford β,β-disubstituted esters without oxidation products. Herein, we disclose an experimental investigation along with the first computational studies of these specific pathways to understand the factors that control the observed reactivity preferences between homoenolate and oxidation reactivities.
Figure 1.

Product Distribution in NHC-Catalyzed Reactions
The divergent pathways operative in carbene catalysis represent both an opportunity and a challenge. In the first option (path A, Figure 2), the generation of homoenolate equivalents is possible by the addition of an NHC to an α,β-unsaturated aldehyde,3 which involves a formal 1,2-proton shift to generate an extended enediamine intermediate (2 to 3). Alternatively, the oxidation of the carbonyl of aldehydes has also been explored4 (path B, Figure 2). The tetrahedral addition intermediate (2) collapses, consequently generating acyl azolium intermediate (7) with concomitant production of a formal reducing equivalent which can add to organic oxidants (e.g., aldehyde 1) in solution5 or molecular oxygen.6 A significant challenge to the further development of carbene catalysis is that the homoenolate and redox pathways are operative under the same reaction conditions,7 indicating that the factors controlling these courses are complex and interdependent.
Figure 2.

Divergent Pathways in NHC-Catalyzed Reactions
Our own investigations necessitated the minimization of the oxidation product within an NHC-catalyzed homoenolate protonation. We envisioned accomplishing this through careful consideration of the reaction conditions and proposed mechanisms. However, these proposed mechanisms involve multiple proton transfer steps and association and dissociation steps that significantly challenge mechanistic investigations. Thus, computational models including solvation effects, a powerful, and heretofore unexplored, avenue8 for understanding this intrinsic dichotomy of NHC reactivity at the microscopic level, were applied toward the understanding of the influence of solvent polarity and hydrogen bonding on the two divergent pathways.
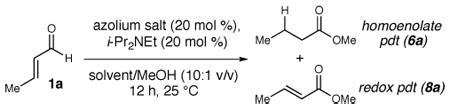
Archetypal imidazolium, triazolium, and benzimidazolium salts were surveyed in the reaction of crotonaldehyde in the presence of methanol to promote catalyst turnover (Table 1).9 Crotonaldehyde, with an alkyl substituent at the β-position, was selected for the experimental studies since it is the simplest unsaturated aldehyde for relevant calculations (vide infra). A limited range of polar protic to nonpolar solvents was investigated. Azolium salts A and D showed the greatest propensity to promote the oxidation pathway, with methyl crotonate (8a) being observed as the predominant product in nearly all of the solvents surveyed. In contrast, azolium salts B and C showed an interesting reversal in reactivity dependent on the solvent employed in the reaction.10
Table 1.
Reaction of NHC Catalysts With Crotonaldehyde
 | ||||
|---|---|---|---|---|
| entry | azolium salt | solvent | 6a:8a | yield (GC)a |
| 1 | 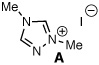 |
MeOH | 1:7.4 | 33 |
| 2 | THF | 1:1.4 | 18 | |
| 3 | CH2Cl2 | 1:1.9 | 29 | |
| 4 | PhMe | 2.1:1 | 20 | |
| 5 | 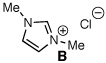 |
MeOH | 1:11.8 | 61 |
| 6 | THF | 2.2:1 | 12 | |
| 7 | CH2Cl2 | 2.1:1 | 32 | |
| 8 | PhMe | 3.0:1 | 22 | |
| 9 | 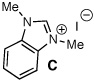 |
MeOH | 1:9.9 | 41 |
| 10 | THF | 2.2:1 | 9 | |
| 11 | CH2Cl2 | 2.2:1 | 35 | |
| 12 | PhMe | 2.9:1 | 15 | |
| 13 | 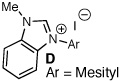 |
MeOH | 1:11.6 | 54 |
| 14 | THF | 1:2.1 | 17 | |
| 15 | CH2Cl2 | 1:2.2 | 22 | |
| 16 | PhMe | 1:1.7 | 18 | |
All yields represent amounts of 6a+8a at 12 h reaction time and are calculated using GC with dodecane as an internal standard.
In methanol, all catalysts (A–D) greatly favor the oxidation pathway (producing 8a) as observed by GC analysis (Figure 3). Reactions with non-protic solvents (THF, CH2Cl2, PhMe) resulted in mixtures of both methyl butyrate from the homoenolate pathway and methyl crotonate from the redox pathway. As the polarity of the solvent decreases, homoenolate products are increasingly favored, although the variation is not substantial (e.g., entries 6–8 and 10–12 and Figure 3). Degassing of the solvent to preclude oxidation via an interaction of the intermediates with molecular oxygen showed no significant effect on the ratios of 6a to 8a.
Figure 3.

Dependence of Oxidation Process on Solvent Polarity
While the observed GC yields of methyl butyrate (6a) were poor in all cases (<25%), the ratio of products (6a:8a) is the critical data and indicates a) which azolium structures favor each product and b) how solvent hydrogen bonding capability impacts the outcome.11 The difference in the observed ratio of products originates from partial suppression of the oxidation pathway in non-polar solvents.
For a systematic elucidation of the experimental results, the competitive formation of 3 (ΔH2) or 7 (ΔH5) from 2 was probed for the carbenes of A and B using density functional calculations.12 In the formation of 7, crotonaldehyde (1a) was used as the oxidant, giving 7 plus the alkoxide of crotyl alcohol. A single explicit methanol molecule was complexed with each reactant or product (placed at the most favorable position) to mimic the likely solution environment. Enthalpies at 298 K for 1, 2, 3, 7 and the alkoxide of crotyl alcohol were determined in the gas phase using the M06L/MIDI!(6D)/Auto level of theory, with M06-2X/6-311+G(2df,p) single-point energy corrections.13 These species were reoptimized in both MeOH and CH2Cl2 using the SM8 solvation model14 at the M06L/MIDI!(6D)/Auto level to provide free energies of solvation. Composite energies were determined as the gas-phase enthalpy plus the solvation correction (Figure 4). All calculations were performed with a locally modified version of Gaussian 03 (Univ. of Minnesota).15
Figure 4.

Calculated Enthalpies For NHC-Catalyzed Pathways
Collectively, the experimental and computational data indicate a weak solvent effect at play in the homoenolate pathway, while the oxidation pathway is strongly disfavored by non-polar solvent. The importance of solvent polarity in carbene-catalyzed Umpolung and oxidation reactions can be described in relation to the mechanisms proposed in Figure 2. From the initial addition intermediate (2) proton transfer results in a charge neutral homoenolate equivalent (3), while generation of a reducing equivalent in the oxidation pathway results in a cationic acyl azolium intermediate (7). The greater charge separation in the oxidation pathway makes this process more sensitive to solvation; more polar solvents, like methanol, better solvate the charged intermediates. The homoenolate pathway is less dependent upon the choice of solvent and is more affected by the choice of catalyst.
These results were put to immediate use and applied to the asymmetric protonation of NHC-generated homoenolate equivalents (eq 1, Figure 5). While the desired product (10) was observed, the ratio of the saturated and unsaturated esters was poor. A change in the solvent based on our investigations improved the reaction to a 13:1 ratio of 10 to 11 and improved the overall yield of the desired saturated ester. This result demonstrates a) the application of the results of the crotonaldehyde model system to a substrate much more suited to the generation of homoenolate equivalents and b) provides an essential foundation for future asymmetric catalysis with N-heterocyclic carbenes.
Figure 5.

Protonation of Homoenolate Equivalents
The solvent dependence of the oxidation pathway presents a clear opportunity to suppress or promote this process in known reactions through modifying reaction conditions. While the trends reported herein for carbene-catalyzed processes may not be universal, the evidence that reaction parameters can be rationally modulated to favor the oxidation or homoenolate pathway can now be applied to concentrate and optimize the evaluation of possible solvents and catalyst structures for related reactions. While optimization of reaction conditions is inherently empirical, these studies point the way to a more systematic analysis and provide a more predictive approach. In a broader sense, the integration of these results with known NHC-catalyzed processes should lead to the discovery of new and unique transformations. Further studies that expand and leverage these observations in the area of carbene catalysis are ongoing and will be reported in due course.
Supplementary Material
Acknowledgment
Research support was generously provided by NIH/NIGMS (K.A.S., GM73072), the National Science Foundation (C.JC., CHE-0610183) GlaxoSmithKline, AstraZeneca, and the Sloan Foundation. B.E.M. was supported by a GAANN Fellowship.
Footnotes
Supporting Information Available Experimental procedures and spectral data for all new compounds, Cartesian coordinates and computed energies for relevant species. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Nair V, Vellalath S, Babu BP. Chem. Soc. Rev. 2008;37:2691–2698. doi: 10.1039/b719083m. [DOI] [PubMed] [Google Scholar]; (b) Enders D, Niemeier O, Henseler A. Chem. Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]; (c) Marion N, Diez-Gonzalez S, Nolan SP. Angew. Chem., Int. Ed. 2007;46:2988–3000. doi: 10.1002/anie.200603380. [DOI] [PubMed] [Google Scholar]; (d) Zeitler K. Angew. Chem., Int. Ed. 2005;44:7506–7510. doi: 10.1002/anie.200502617. [DOI] [PubMed] [Google Scholar]
- 2.Denmark SE, Beutner GL. Angew. Chem., Int. Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 3.(a) Chan A, Scheidt KA. Org. Lett. 2005;7:905–908. doi: 10.1021/ol050100f. [DOI] [PubMed] [Google Scholar]; (b) Chan A, Scheidt KA. J. Am. Chem. Soc. 2007;129:5334–5335. doi: 10.1021/ja0709167. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Phillips EM, Reynolds TE, Scheidt KA. J. Am. Chem. Soc. 2008;130:2416–2417. doi: 10.1021/ja710521m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Castells J, Llitjos H, Morenomanas M. Tetrahedron Lett. 1977:205–206. [Google Scholar]; (b) Inoue H, Higashiura K. Chem. Commun. 1980:549–550. [Google Scholar]; (c) Maki BE, Chan A, Phillips EM, Scheidt KA. Org. Lett. 2007;9:371–374. doi: 10.1021/ol062940f. [DOI] [PubMed] [Google Scholar]; (d) Maki BE, Scheidt KA. Org. Lett. 2008;10:4331–4334. doi: 10.1021/ol8018488. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Maki BE, Chan A, Phillips EM, Scheidt KA. Tetrahedron. 2009;65:3102–3109. doi: 10.1016/j.tet.2008.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Guin J, De Sarkar S, Grimme S, Studer A. Angew. Chem., Int. Ed. 2008;47:8727–8730. doi: 10.1002/anie.200802735. [DOI] [PubMed] [Google Scholar]
- 5.(a) Miyashita A, Suzuki Y, Nagasaki I, Ishiguro C, Iwamoto K, Higashino T. Chem. Pharm. Bull. 1997;45:1254–1258. [Google Scholar]; (b) Chan A, Scheidt KA. J. Am. Chem. Soc. 2006;128:4558–4559. doi: 10.1021/ja060833a. [DOI] [PubMed] [Google Scholar]; (c) Noonan C, Baragwanath L, Connon SJ. Tetrahedron Lett. 2008;49:4003–4006. [Google Scholar]
- 6.Liu YK, Li R, Yue L, Li BJ, Chen YC, Wu Y, Ding LS. Org. Lett. 2006;8:1521–1524. doi: 10.1021/ol0529905. [DOI] [PubMed] [Google Scholar]
- 7.(a) Maki BE, Chan A, Scheidt KA. Synthesis. 2008:1306–1315. doi: 10.1055/s-2008-1072516. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chan A, Scheidt KA. J. Am. Chem. Soc. 2008;130:2740–2741. doi: 10.1021/ja711130p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zeitler K, Rose CA. J. Org. Chem. 2009;74:1759–1762. doi: 10.1021/jo802285r. [DOI] [PubMed] [Google Scholar]
- 8.For computational investigations of NHC-catalyzed esterifications, see: Lai CL, Lee HM, Hu CH. Tetrahedron Lett. 2005;46:6265–6270..
- 9.For a discussion of reaction pathways for these processes, see refs. 3c and 4c. Other catalysts, notably 1,3-dimesitylimidazolium chloride and 3,4-dimethyl-1-mesityl-1,2,4-triazolium tetrafluoroborate, were investigated and showed similar reactivity patterns to azolium salts B and C.
- 10.Over the range of solvents, the variation in the 6a:8a ratio expressed as a 298 K equilibrium free energy change is 1.7, 2.2, 2.0, and 1.2 kcal/mol for A, B, C, and D, respectively.
- 11.For isolated yields, crotonaldehyde is a poor substrate for the protonation of the homoenolate intermediate, see reference 3c.
- 12.The neutral spiro intermediate of 2 was not utilized since both homoenolate and oxidation pathways require loss of the former aldehyde CH. For details on related proposed thiazolium spiro structures see: Gronert S. Org. Lett. 2007;9:3065–3068. doi: 10.1021/ol0711467..
- 13.Zhao Y, Truhlar DG. Theor. Chem. Acc. 2008;120:215–241. [Google Scholar]
- 14.Marenich AV, Olson RM, Kelly CP, Cramer CJ, Truhlar DG. J. Chem. Theory Comput. 2007;3:2011–2033. doi: 10.1021/ct7001418. [DOI] [PubMed] [Google Scholar]
- 15.Frisch MJ, et al. Gaussian 03, Revision E.01. Wallingford, CT: Gaussian, Inc.; 2004. . For full citation, see Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.