

Abstract
Chromosome 9 is highly structurally polymorphic. It contains the largest autosomal block of heterochromatin, which is heteromorphic in 6–8% of humans, whereas pericentric inversions occur in more than 1% of the population. The finished euchromatic sequence of chromosome 9 comprises 109,044,351 base pairs and represents >99.6% of the region. Analysis of the sequence reveals many intra- and interchromosomal duplications, including segmental duplications adjacent to both the centromere and the large heterochromatic block. We have annotated 1,149 genes, including genes implicated in male-to-female sex reversal, cancer and neurodegenerative disease, and 426 pseudogenes. The chromosome contains the largest interferon gene cluster in the human genome. There is also a region of exceptionally high gene and G + C content including genes paralogous to those in the major histocompatibility complex. We have also detected recently duplicated genes that exhibit different rates of sequence divergence, presumably reflecting natural selection.
With the completion of the reference sequence for the human genome, the focus now is on detailed analysis to produce a precise and comprehensive depiction of the genome and to identify biologically important features. We present here the highly accurate finished sequence (>99.99%1) and analysis of chromosome 9, adding to the completed individual chromosome sequences2-8. In accordance with the goals of the Human Genome Project, our primary aim was to sequence and analyse the euchromatic or gene-containing region of this sub-metacentric chromosome. However, we have also mapped and sequenced substantial portions of the pericentromeric segmentally duplicated regions, which constitute approximately 7% of the chromosome.
Genomic sequence and landscape
A physical map of six contigs spanning the euchromatic part of chromosome 9 (Table 1) was assembled using restriction enzyme fingerprinting and marker content analysis of clones identified by screening up to 90 genomic equivalents of bacterial, P1-derived and yeast artificial chromosome (BAC, PAC and YAC) cosmid and fosmid clone libraries9 (Supplementary Table S1). A total of 925 minimally overlapping clones were selected from the map and sequenced (Supplementary Table S2). The latest sequence assembly and the versions analysed here are available at http://www.sanger.ac.uk/HGP/Chr9. The features identified in our analysis are shown in Fig. 1 (rollfold) (for a more detailed view see Supplementary Fig. S1). The sequence of the short arm is contiguous. It contains the 9pter (TTAGGG)n telomeric repeat, obtained using YACs containing the captured telomere (H. Riethman, personal communication), and copies of the pericentromeric duplicated sequences. On the long arm there are four small gaps within an 8-megabase (Mb) subtelomeric region (9q34.1–34.3, 128.6–136.5 Mb). The total extent of these gaps (determined by fluorescent in situ hydridization of flanking clones to DNA fibres) is <300 kilobases (kb). The absence of clones representing these gaps is probably a consequence of the exceptionally high G+C content in this region (see below). Similar subtelomeric unclonable gaps in (G+C)-rich regions have been seen previously2-4. The most telomeric sequence obtained at 9qter is contiguous with the shortest allelic variant of the subtelomeric repeat. The proximal end of the sequence of the long arm extends into representative blocks of the segmentally duplicated pericentromeric repeats.
Table 1.
Sequence contigs on chromosome 9
| Contig* | Size (Mb) | Gap size (kb) |
|---|---|---|
| AL928970–BX005214 | 39.4 | – |
| Pericentromere/centromere | – | ND |
| AL353608–AL360004 | 62.0 | – |
| Gap 1 | – | 50 |
| AL354898–AL591386 | 3.8 | – |
| Gap 2 | – | 200 |
| AL354796–AL683798 | 0.2 | – |
| Gap 3 | – | 13 |
| AL669970–AL138781 | 1.8 | – |
| Gap 4 | – | 30 |
| AL603784–AL954642 | 1.9 | – |
| Total | 109.1 | 293 |
| Total euchromatic region | 109.4 | – |
Contigs are indicated by the first and last sequence accession.
ND, not determined.
Figure 1.
Chromosome 9 sequence features (see rollfold). Tracks from top to bottom are: (1) sequence scale (Mb); (2) coverage of chromosome 9 sequence (black) and gaps (grey); (3) synteny to mouse (top track) and rat (bottom track) chromosomes, with chromosomes colour-coded and coordinate range (Mb) indicated (Un/random indicate that there is no current chromosome location for the homologous mouse or rat sequences); (4) position of predicted CpG islands (brown); (5) location of ECRs showing sequence homology to Fugu (blue), zebrafish (dark blue) and Tetraodon (dark pink); (6) placement of ‘known’ (dark blue) and ‘novel coding sequence’ (black) annotated gene structures (official gene symbols used when available). Owing to space restrictions this figure represents an abbreviated set of features and we therefore recommend downloading Supplementary Fig. S1 to follow the text accurately.
A total of 46.15% of chromosome 9 is repeat, similar to previous reports on other chromosomes (for a detailed breakdown of the repeat content see Supplementary Table S3). The G+C content along the chromosome is 41.4% but fluctuates widely, with (G+C)-rich regions correlating with a higher density of both exons and short interspersed nucleotide elements (SINEs). An 8-Mb subtelomeric region of the long arm (in 9q34) has a very high G+C content (54.2%). Part of this region (near the telomere, 134.7–135.6 Mb) is exceptional, with a G+C content of 59%. In contrast the sequence from 9.0 to 11.0 Mb has a G+C content of 35.1% and a SINE content of just 6.6%.
The completeness and quality of the final sequence assembly was verified by alignment to fosmid and BAC end sequences (http://genome.cse.ucsc.edu/) and by comparison with independent genetic and gene map information. We accounted for all of the 603 genes assigned to chromosome 9 in the RefSeq database. All known chromosome 9 markers from three genetic maps10-12 were identified in the finished sequence. Comparison of marker order in the sequence with the 193 markers placed on the deCODE genetic map (Fig. 2) identified just one discordant marker placement, D9S1786 relative to D9S1851. On re-examining the deCODE map, these two markers were barely resolved by the genetic data. We conclude that the order of these two markers is as determined in the finished sequence.
Figure 2.
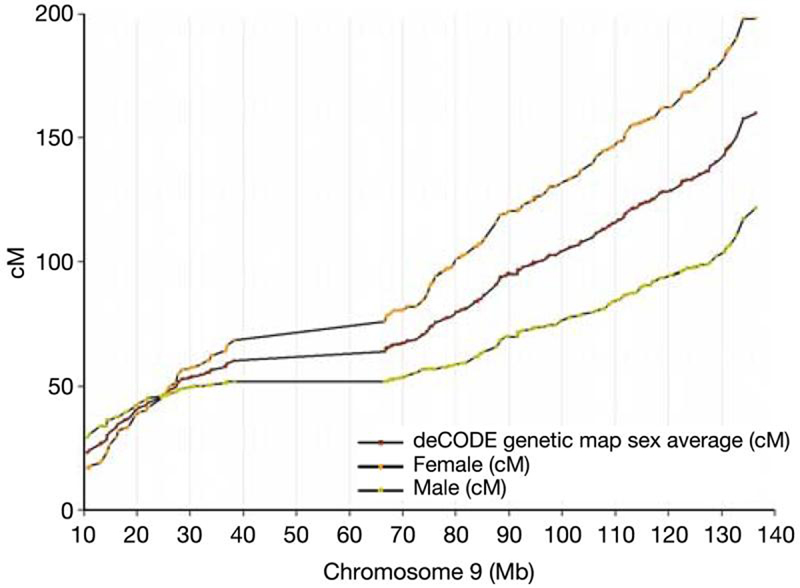
The relationship between the physical and deCODE10 genetic distance. The physical location of each genetic marker is shown on the female, male and sex-averaged genetic maps
Comparison of the genetic map with the finished sequence also enabled us to assess recombination rate along the chromosome (Fig. 2). On the basis of the deCODE map, the mean sex-averaged recombination rate of chromosome 9 is 1.33 cM Mb−1. With the benefit of the finished sequence, we were able to identify a previously unrecognized region of high recombination (as previously defined13) between D9S164 and D9S1826 in the subtelomeric region 9q34 (recombination rate 4.5 cM Mb−1).
Gene index
Gene structures were manually annotated in the finished sequence on the basis of computational analysis using predictive algorithms and supporting evidence from expressed sequence tags (ESTs), complementary DNAs and proteins. A total of 1,575 structures, including 1,149 genes and 426 pseudogenes, were categorized as described previously4 and are listed in Table 2. The average gene density of the chromosome is 10.5 genes Mb−1, close to the genome average (Supplementary Table S4), but ranges from 3 genes Mb−1 in 9p23 to 22 genes Mb−1 in the subtelomeric region 9q34. Part of the subtelomeric region (134.7–135.6 Mb) is exceptional, with a gene density of 66 genes Mb−1, which is associated with a very high G+C content of 59%. Annotated genes (excluding pseudogenes) cover 47% of the sequence, whereas exons occupy 2.5% of the total and have a mean length of 326 base pairs (bp). The longest exon (10,135 bp) lies in a gene of unknown function (KIAA1958), whereas the longest intron spans 582 kb in TRPM3. The longest gene is protein tyrosine phosphatase receptor type D (PTPRD), a gene implicated in synaptic plasticity and new memory acquisition14 that spans 2.3 Mb and contains 46 exons. This gene, which occurs in the gene-poor 9p23 region, is one of the largest genes found in the human genome so far, being comparable in size to the 2.3-Mb dystrophin gene on Xp12. We identified 432 CpG islands (see Supplementary Methods) within a window of 5 kb upstream and 1 kb downstream of the 613 ‘known’ genes (that is, genes that match a known messenger RNA or protein sequence); thus 70% of these genes have an associated CpG island.
Table 2.
Annotation summary
| Class* | Gene count |
Total length in bp (% coverage) |
Mean gene length (bp) |
Mean exon length |
Number of exons |
|---|---|---|---|---|---|
| Known genes | 613 | 41,424,864 (37.9) | 68,440 | 322 | 6,628 |
| Novel genes | 147 | 4,707,061 (4.3) | 32,024 | 474 | 605 |
| Novel transcripts | 176 | 4,655,574 (4.3) | 26,974 | 296 | 683 |
| Putative genes | 213 | 1,912,134 (1.7) | 8,980 | 244 | 488 |
| Total genes | 1,149 | 51,435,292 (47.0) | 46,407 | 326 | 8,404 |
| Processed pseudogenes | 397 | 459,658 (0.4) | 1,157 | 709 | 475 |
| Unprocessed pseudogenes | 29 | 405,851 (0.4) | 13,994 | 165 | 216 |
| Total structures | 1,575 | 52,022,232 (48.0) | 34,405 | 342 | 9,095 |
Classes follow those laid down in ref. 4 and are based on the longest transcript of each gene; the total number of exons from all alternative transcripts is 14,640. Known genes, identical to known human cDNA or protein and have an entry in Locuslink; novel genes, have an ORF and are identical to spliced human ESTs or have some similarity to other cDNA/proteins (all species); novel transcripts, similar to novel genes but have ambiguous ORFs or multiple evidence for non-coding RNA; putative genes, with identity to 1–3 spliced ESTs but do not contain evidence for an ORF; pseudogenes, similar to known proteins but contain a frameshift and/or stop codon that disrupts the ORF.
Alternative splicing of genes generates multiple transcripts, leading to diversity of protein structures. On chromosome 9 the mean number of transcripts annotated per gene is 3.09. In some cases alternative splicing was extensive; for example, we annotated 26 different transcripts in the CIZ1 gene, which encodes a nuclear protein potentially involved in brain tumorigenesis15. Eight of these transcripts have open reading frames (ORFs) encoding different protein isoforms, two of which are partial and do not contain the zinc finger domain.
MicroRNA (miRNA) genes encode RNA products of around 22 nucleotides (http://www.sanger.ac.uk/Software/Rfam/mirna/index.shtml) and have been implicated in gene regulation. We have detected 14 miRNA genes on chromosome 9 including two clusters of three genes in 9q22. All 14 miRNAs are conserved in mouse with respect to gene order and orientation, and the two human clusters have counterparts on mouse chromosome 13. We also identified eight transfer RNA genes using tRNAscan-SE distributed along the chromosome.
Comparative analysis was used as an independent measure of the completeness of gene annotation of the protein-coding genes. We identified 4,190 evolutionarily conserved regions (ECRs; see Supplementary Methods) that are conserved in the sequence of human chromosome 9 and the genomic sequence of five other species (mouse, rat, Fugu, Tetraodon and zebrafish; see Supplementary Table S5). A total of 4,091 of the ECRs overlapped 4,516 exons that had previously been annotated, whereas no evidence was found for the existence of coding transcripts on either DNA strand of the human sequence for 99 of the ECRs. On the basis of this observation we conclude that the annotation of the protein-coding exons is at least 97.9% (=4,516/(4,516 + 99)) complete. There was a notable cluster of 22 ECRs in the introns of two flanking genes, MAPKAP1 and PBX3 (123.7–124.3 Mb, see Supplementary Fig. S2a, b), and these ECRs are therefore good candidates to test for possible regulatory function.
Sequence duplication
Gene duplication events give rise to multigene families. In some cases, subsequent evolutionary divergence of duplicated genes gives rise to new or altered protein function16. We searched the sequence for gene clusters likely to have arisen by duplication using BLASTP (as previously defined8) and on the basis of known protein domains using InterProScan (http://www.ebi.ac.uk/interpro/scan.html) (Supplementary Table S6). The BLASTP analysis identified 31 clusters containing 98 genes (Supplementary Fig. S1 and Table S7). There are 16 interferon type 1 genes in the human genome, all of which are on chromosome 9. Fifteen are clustered in a single 364-kb region (21.0–21.4 Mb) within a genomic duplication (Fig. 3a; see also Supplementary Fig. S1). This gene cluster is also present in the orthologous mouse sequence, indicating that some expansion of the type 1 interferons occurred before divergence of human and mouse from a common mammalian ancestor. The top-ranking InterProScan hits (listed in Supplementary Table S6) show good agreement with the clustering by BLASTP.
Figure 3.
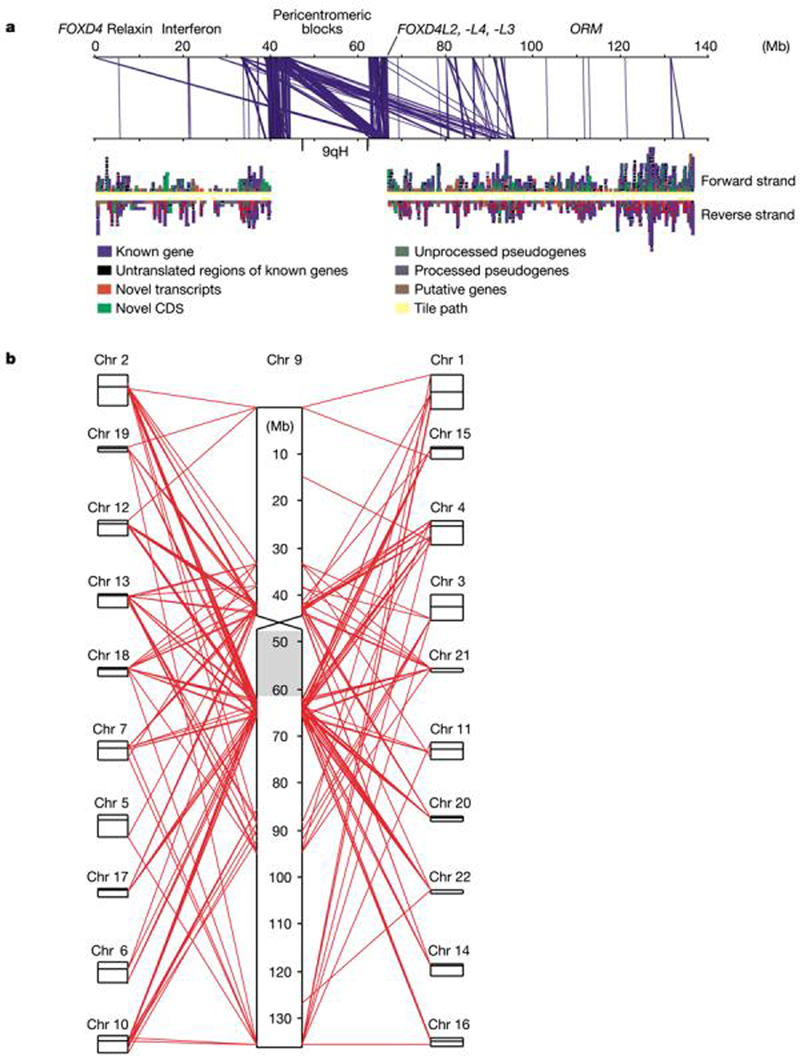
Segmental duplications on chromosome 9. a, Intrachromosomal duplications. The top panel shows duplications plotted against the sequence coordinates (matches of >1 kb in length), whereas the bottom panel shows annotated genes on the forward and reverse strand. Some of the gene duplications we identified are also noted. The view is produced using Apollo (http://www.fruitfly.org/annot/apollo). b, Interchromosomal duplications. Large duplications (>10 kb) are shown, with chromosome 9 magnified. Other chromosomes are arranged in the order in which they match to chromosome 9. Chromosomes 8, 14, X and Y are not detected at this level. The grey rectangle is heterochromatin (47–62 Mb). Supplementary Fig. S3 shows interchromosomal duplication matches at >1 kb.
Some gene duplications are observed in human but not mouse, indicating that they occurred since the divergence of these two species. On the basis of this criterion we identified 18 sets of recently duplicated genes on chromosome 9 (both intra- and interchromosomal duplications; see Methods and Supplementary Table S8). These included two relaxin genes, five forkhead genes, two distinct sets of olfactory receptor genes, a pair of orosomucoid (ORM) genes, and the 13 interferon A genes within the type 1 cluster. Note that in the case of the interferon genes, there is evidence of expansion of these genes since the human–mouse divergence as well as duplication earlier in mammalian evolution (described above). The accumulation of synonymous changes between duplicated genes gives an indication of the time that has elapsed since the duplication event, on the assumption that there is no selection acting on such changes. The rate of synonymous base substitutions between duplicated sequences is given by the parameter Ks (ref. 17). For example, comparison of the olfactory receptor genes OR13C3 and OR13C4 gives a high Ks value (0.372), suggesting that this duplication is less recent than the duplication that gave rise to the forkhead genes FOXD4L2 and FOXD4L4, which have a much lower Ks value (0.0134) (Supplementary Table S8).
Evolutionary divergence of genes after duplication may arise as a result of natural selection acting on new mutations at either locus. The accumulation of non-synonymous changes (given by the parameter Ka (ref. 17)) relative to the rate of synonymous change (Ks) gives an indication of selection expressed as the ratio Ka/Ks. Comparison of the two relaxin genes (RLN1 and RLN2) gives a Ks of 0.119 and a Ka of 0.0999, and thus one of the highest Ka/Ks ratios of 0.84 among the recent duplications. By contrast the olfactory receptors OR13C3 and OR13C4, despite being the result of a much less recent duplication, appear to be more conserved (Ks = 0.372, Ka = 0.0843, Ka/Ks = 0.23) compared with the relaxin genes. Note, however, that these results are based on small numbers of nucleotide substitutions (Supplementary Table S8).
Phylogenetic reconstruction using the relaxin genes along with additional sequences from chimpanzee (Z27225, Z27245), rhesus macaque18, lemur (AF317625) and pig (J02792) demonstrated that the relaxin duplication occurred after the divergence of lemurs from other primates but at least before divergence of higher primates from Old World monkeys. A likelihood ratio test indicated that positive selection has acted on the relaxin sequences since duplication (P < 0.001) (see Methods and ref. 19). There is evidence for RLN2 being selectively expressed in the ovary during pregnancy where it acts to facilitate parturition20. There is no evidence that RLN1 is expressed in the ovaries but it may be expressed in other tissues21. Hence, any evolved differences in function of the duplicated relaxin genes may represent an example of tissue-specific sub-functionalization. The differing levels of sequence similarity seen across the genes (Fig. 4) may reflect these alternative functions. Further analysis of these genes may uncover functional explanations for this positive selection.
Figure 4.

Alignments between duplicated chromosome 9 sequences using MultiPipMaker50, indicating percentage identity for each alignment. A genomic duplication event seems to have taken place with a 7,144-bp sequence (5,289,576–5,296,720 bp) and a 7,125-bp region (5,324,724–5,331,849 bp), duplicating the two-exon 5-kb relaxin (RLN) genes. The sequence homology across the duplication shows variation, with the region around the first exon and into the intron matching at 97%, whereas there is a marked decrease in similarity towards exon 2. In comparison, another recently duplicated gene pair, orosomucoid (ORM), duplicated within a 6,212-bp sequence (112,627,806–112,634,018 bp) and 6,220 bp (112,634,577–112,640,797 bp) show more homogeneous sequence alignment.
Extensive genomic segmental duplications were reported previously for the draft sequence22,23, with intra- and interchromosomal repeats estimated to involve from 5.5% to 7.1% and 3.6% to 4.7% of chromosome 9, respectively, the fourth highest level among the autosomes. The centromere of chromosome 9 is flanked by heterochromatic C bands rich in satellite sequences. Heteromorphism of 9qh is common and pericentric inversions occur in 1% of the population24. We generated over 7 Mb of finished sequence from the pericentromeric regions, which currently lie in 33 contigs of mean size 262 kb. This sequence has been included in an analysis of intra- (Fig. 3a) and interchromosomal (Fig. 3b) duplications based on the method of ref. 23.
The pericentromeric contigs contain no unique DNA, with all sequences sharing high (>95%) sequence identity to sequences within chromosome 9 or elsewhere. Many pericentromeric sequences are duplicated in the region and we identified large blocks of duplications within and between the two pericentromeric regions at 9p11–12 and 9q11–12/13 with an average sequence identity of >97% (Fig. 3a). There are also matches to pericentromeric and non-pericentromeric regions of other chromosomes; for example, duplication of a set of genes from the ancestral chromosome fusion site at 2q13 (ref. 25) including an active FOXD4 gene at 9p24 and copies in the pericentromeric sequences (Fig. 3a, b). These characteristics are consistent with formation of intrachromosomal and interchromosomal duplications26,27.
Despite the duplicated nature of the chromosome 9 pericentromeric sequence, 138 gene features were annotated within it (summarized in Supplementary Table S9). There are only two known genes, one of which contains an SST1 satellite sequence within its predicted ORF and is only expressed in neoplastic tissue. A total of 120 annotated structures are pseudogenes or transcripts with no associated ORF. The remaining 16 structures include many transcripts specific to either testis or neoplastic tissue. This restricted transcription pattern is consistent with previous analyses of similar regions on chromosomes 10 (ref. 28) and 21 (ref. 29).
This analysis revealed much recent segmental duplication; however, it has also been proposed that 9q may have been involved in a more ancient event, leading to 9q34 containing genes paralogous to the major histocompatibility complex on chromosome 6. ‘Block duplication’ has recently been proposed as an alternative to ‘chromosome/genome duplication’ or ‘selection-based clustering’ for the underlying duplication mechanism(s)30. We have placed 20 novel or previously unlocalized putative major histocompatibility complex gene paralogues within a block of 24 Mb on 9q34 (112.9–136.5 Mb), bringing the total to 31 putative paralogues between this and the corresponding region of chromosome 6 (Supplementary Table S10). Considering an estimated duplication time of around 500 million years ago, it is not surprising that the overall gene order is poorly conserved. Nevertheless, the clustering of these paralogues is highly nonrandom (P < 0.0001).
Sequence variation
Inherited sequence variations have an important role in determining variable disease and drug response, and provide the basis for studies of disease association and population genetics31. The most abundant form of sequence variation is single nucleotide polymorphisms (SNPs). We have mapped 117,405 SNPs on the sequence (1,073 SNPs Mb−1). A total of 51,122 SNPs (43.5%) were generated during the sequencing by comparing sequence in clone overlaps, whereas the remaining 91,217 were extracted from dbSNP (http://www.ncbi.nlm.nih.gov/SNP). The density of mapped random SNPs is clearly higher within the duplicated sequences (Supplementary Fig. S1) at 9p24, those close to the AQP7 locus (33.5–33.6 Mb) at 9p13, and those flanking the pericentromeric sequences on 9p (38.8–39.4 Mb) and 9q (66.5–66.7 Mb). The elevated SNP density in these regions probably reflects a high rate of misplaced SNPs or paralogous sequence variants23. After adjusting for bias by removing SNPs generated by directed sequencing, we established MCART1 (mitochondrial carrier triple repeat 1) as the most polymorphic gene on chromosome 9 with 16 coding SNPs kb−1.
We have also mapped 617,071 sites of sequence divergence between chimpanzee and human from a set of 14 million chimpanzee sequences, released by the Broad Institute and Washington University Genome Sequencing Centre. These sequence differences are evenly distributed except in three regions: two close to 9pter and another one in the 9q pericentromeric region at 66.6 Mb (Supplementary Fig. S1). Two of these regions also have a higher human SNP density and are probably caused by mismapping due to duplicated sequences. A total of 0.6% of the unmatched variations (where there is no corresponding human SNP) are in coding regions (Supplementary Table S11). We assessed selection levels for the chromosome 9 genes by calculating Ka/Ks values compared to chimpanzee. Examples of genes outside duplicated regions that have elevated Ka/Ks values, and may therefore have undergone marked divergence since their common ancestor, include C9orf37 (Ka/Ks of 2.37) and IL11RA (Ka/Ks of 2.12) (Supplementary Table S12).
Medical implications
There are 95 genes on chromosome 9 that are known to be associated with a disease (OMIM and GeneCard). Sixty-eight of these associations have been detected in the sequence (Supplementary Table S13). The remaining 27 disease loci are currently defined by linkage analysis (Supplementary Table S14). The complete sequence and annotation of the chromosome provides a valuable tool for identifying the genes involved in disease as well as a starting point for genetic analysis of multifactorial diseases. The finished sequence has already enabled the identification of several new genes involved in disease. Chorea-acanthocytosis is an autosomal recessive neurodegenerative disorder with neurological symptoms resembling Huntington's chorea. A novel causative gene (CHAC; also known as VPS13A) involved in protein sorting was identified from the genomic sequence32. CHAC is also notable for having 72 exons, the highest number for a chromosome 9 gene. Sequences generated from the autosomal recessive Hereditary Inclusion Body Myopathy (HIBM) critical region were used to identify a 13-exon gene, GNE, which has been established as the gene involved in the disorder33.
There are three genes on 9p24, DMRT1, -2 and -3, which code for proteins with a DNA-binding motif (DM domain) and are expressed in testis. DM domain genes control sex determination in many species, including Drosophila and Caenorhabditis elegans34. High DMRT1 expression is necessary for testicular differentiation35, and hemizygous deletion of 9p24 results in haploinsufficiency and human XY sex reversal36. There is also evidence for another gene in the region involved in XY sex reversal, NR5A1 (ref. 37). The presence of two such loci may reflect extensive conserved synteny between parts of chromosome 9 and the chicken sex-determining chromosome Z38, because 17 of the 24 known chicken Z genes have chromosome 9 orthologues39.
Chromosome 9p21 harbours the tumour suppressor gene CDKN2A and is often involved in translocations or deletions in tumour cell lines. Mutation or loss of CDKN2A is linked to familial melanoma and other tumours40. Reciprocal translocation between chromosomes 9 and 22 leads to generation of a derivative chromosome, the Philadelphia chromosome, in >90% of chronic myelogenous leukaemia and 5–15% of acute B cell lymphoblastic leukaemias41. The translocation results in fusion of ABL1 and the chromosome 22 gene BCR42 to give a chimaeric oncogene with deregulated tyrosine kinase activity43. Inhibition of the BCR-ABL tyrosine kinase is the basis of the anti-cancer activity of imatinib mesylate (Gleevec)44. The ability to use genomic information to develop effective new therapies such as this will benefit enormously from finished human chromosome sequences.
Methods
Establishing sequence-ready bacterial contigs covering chromosome 9 was a prerequisite for clone-by-clone sequencing, and the methods have been described previously9. Briefly, publicly available markers used included 916 ESTs, 763 of which mapped onto the Genemap '98 radiation hybrid map, 229 polymorphic microsatellites from genetic maps, and 226 from RHmaps (Stanford and WI), which were used to screen initially the RPCI-11 BAC library (http://bacpac.chori.org/hmale11.htm). These were later augmented with 633 sequence-tagged sites (STSs) derived from single pass sequences of plasmid subclones made from flow-sorted chromosomes. Owing to the similarity in base composition and size of chromosomes 9–12, a pool of samples containing the four chromosomes was used. Fingerprints for positive clones were extracted from humanmap (http://genome.wustl.edu/projects/human/index.php?fpc = 1) or generated in-house45, then assembled into contigs in FPC46 on the basis of shared HindIII fragments and individual clone marker content. Novel STSs were designed from sequences at the ends of each contig and used in further iterative walking experiments onto an increasing number of different libraries to generate new coverage across gaps. In cases where no bacterial clone coverage could be identified YAC and chromosome-specific cosmid libraries were screened. Extensive checks were performed on each clone before sequencing.
Methods used for shotgun sequencing, finishing strategies, comparative analysis and identification of ECRs, sequence annotation predications and SNP identification are as previously described8, and are available as Supplementary Methods.
Gene duplications
A database of manually annotated chromosome 9 genes and Ensembl genes from human (build 15.33, excluding genes from chromosome 9), mouse (build 15.2) and rat (build 15.2) was generated. Homologues of each chromosome 9 gene, excluding pseudogenes, were found using BLASTN47, and the genetic distance between the full extent of these sequences was determined17.
Matches between a chromosome 9 gene and another human sequence were classed as probable recent duplications in cases where the synonymous genetic distance between the human sequences was less than the synonymous genetic distance between the human sequence and the closest mouse or rat sequence. In cases where there was no match to a rodent an arbitrary, synonymous genetic distance cut-off of 0.6 was applied. Likely duplications were disregarded where a substantial disagreement in the number of exons between genes existed or where a match sequence was annotated as a possible pseudogene.
Phylogenetic reconstruction of the relaxin sequences was performed using fastDNAml48. The likelihood ratio test of positive selection was performed following the method of ref. 19 using nested models M1 and M2.
SsahaSNP49 was used to map chimpanzee reads (approximately 14 million) and call single base sequence differences against the human NCBI31 assembly. Those that mapped to chromosome 9 were remapped to the latest Sanger assembly.
Segmental duplication
Various methods have been proposed for the detection of segmental duplications in the human genome22,23. Our method is based on that described by ref. 23. WU-BLASTN47 (http://blast.wustl.edu) was used to perform all searches.
To detect intrachromosomal duplications, the sequence of chromosome 9 (SANGER_10 assembly) was searched against itself, and ‘self’ matches (that is, occurring at the same position in the query and target) were discarded. Interchromosomal duplications were detected by searching the chromosome sequence against the non-chromsome-9 parts of the NCBI34 build of the human genome (http://www.ensembl.org/Homo_sapiens/).
Acknowledgements
We thank the Fugu and Tetraodon sequencing groups, the Mouse Sequencing Consortium, the Zebrafish sequencing groups at the Wellcome Trust Sanger Institute, the Rat Genome project at Baylor College of Medicine, and the Broad Institute and Washington University Genome Sequencing Centre for their early release of the chimpanzee sequences. We also thank H. Riethman for the provision of clones, V. K. Khodiyar, H. M. Wain, E. A. Bruford, M. W. Wright, R. C. Lovering, C. C. Talbot and M. J. Lush from the HUGO Gene Nomenclature Committee for the official gene nomenclature, the German Federal Ministry for Education and Research (BMBF) through DLR for financial support of the GBF group, and the EMBL and Ensembl database teams at the European Bioinformatics Institute. Work at the Institute of Human Genetics was funded by the Wellcome Trust, and work at HUGO was funded by the NIH and the MRC. Work at the Sanger Institute was funded by the Wellcome Trust.
Footnotes
Accession numbers for the sequences analysed for this paper can be found in Supplementary Fig. S1. All reported DNA sequences have been deposited in EMBL, GenBank or DDBJ.
Supplementary Information accompanies the paper on www.nature.com/nature.
Competing interests statement The authors declare that they have no competing financial interests.
References
- 1.Felsenfeld A, Peterson J, Schloss J, Guyer M. Assessing the quality of the DNA sequence from the Human Genome Project. Genome Res. 1999;9:1–4. [PubMed] [Google Scholar]
- 2.Dunham I, et al. The DNA sequence of human chromosome 22. Nature. 1999;402:489–495. doi: 10.1038/990031. [DOI] [PubMed] [Google Scholar]
- 3.Chromosome 21 Mapping and Sequencing Consortium. The DNA sequence of human chromosome 21. Nature. 2000;405:311–319. doi: 10.1038/35012518. [DOI] [PubMed] [Google Scholar]
- 4.Deloukas P, et al. The DNA sequence and comparative analysis of human chromosome 20. Nature. 2001;414:865–871. doi: 10.1038/414865a. [DOI] [PubMed] [Google Scholar]
- 5.Heilig R, et al. The DNA sequence and analysis of human chromosome 14. Nature. 2003;421:601–607. doi: 10.1038/nature01348. [DOI] [PubMed] [Google Scholar]
- 6.Skaletsky H, et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423:825–837. doi: 10.1038/nature01722. [DOI] [PubMed] [Google Scholar]
- 7.Hillier LW, et al. The DNA sequence of human chromosome 7. Nature. 2003;424:157–164. doi: 10.1038/nature01782. [DOI] [PubMed] [Google Scholar]
- 8.Mungall AJ, et al. The DNA sequence and analysis of human chromosome 6. Nature. 2003;425:805–811. doi: 10.1038/nature02055. [DOI] [PubMed] [Google Scholar]
- 9.Bentley DR, et al. The physical maps for sequencing human chromosomes 1, 6, 9, 10, 13, 20 and X. Nature. 2001;409:942–943. doi: 10.1038/35057165. [DOI] [PubMed] [Google Scholar]
- 10.Kong A, et al. A high-resolution recombination map of the human genome. Nature Genet. 2002;31:241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- 11.Dib C, et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature. 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- 12.Broman KW, Murray JC, Sheffield VC, White RL, Weber JL. Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am. J. Hum. Genet. 1998;63:861–869. doi: 10.1086/302011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu A, et al. Comparison of human genetic and sequence-based physical maps. Nature. 2001;409:951–953. doi: 10.1038/35057185. [DOI] [PubMed] [Google Scholar]
- 14.Uetani N, et al. Impaired learning with enhanced hippocampal long-term potentiation in PTPδ-deficient mice. EMBO J. 2000;19:2775–2785. doi: 10.1093/emboj/19.12.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warder DE, Keherly MJ. Ciz1, Cip1 interacting zinc finger protein 1 binds the consensus DNA sequence ARYSR(0-2)YYAC. J. Biomed. Sci. 2003;10:406–417. doi: 10.1007/BF02256432. [DOI] [PubMed] [Google Scholar]
- 16.Ohno S, Wolf U, Atkin NB. Evolution from fish to mammals by gene duplication. Hereditas. 1968;59:169–187. doi: 10.1111/j.1601-5223.1968.tb02169.x. [DOI] [PubMed] [Google Scholar]
- 17.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 18.Crawford RJ, Hammond VE, Roche PJ, Johnston PD, Tregear GW. Structure of rhesus monkey relaxin predicted by analysis of the single-copy rhesus monkey relaxin gene. J. Mol. Endocrinol. 1989;3:169–174. doi: 10.1677/jme.0.0030169. [DOI] [PubMed] [Google Scholar]
- 19.Yang Z, Nielsen R, Goldman N, Pedersen AM. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hudson P, et al. Structure of a genomic clone encoding biologically active human relaxin. Nature. 1983;301:628–631. doi: 10.1038/301628a0. [DOI] [PubMed] [Google Scholar]
- 21.Bathgate RA, Samuel CS, Burazin TC, Gundlach AL, Tregear GW. Relaxin: new peptides, receptors and novel actions. Trends Endocrinol. Metab. 2003;14:207–213. doi: 10.1016/s1043-2760(03)00081-x. [DOI] [PubMed] [Google Scholar]
- 22.Bailey JA, et al. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 23.Cheung J, et al. Genome-wide detection of segmental duplications and potential assembly errors in the human genome sequence. Genome Biol. 2003;4:R25. doi: 10.1186/gb-2003-4-4-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Madan K, Bobrow M. Structural variation in chromosome No 9. Ann. Genet. 1974;17:81–86. [PubMed] [Google Scholar]
- 25.Fan Y, Newman T, Linardopoulou E, Trask BJ. Gene content and function of the ancestral chromosome fusion site in human chromosome 2q13-2q14.1 and paralogous regions. Genome Res. 2002;12:1663–1672. doi: 10.1101/gr.338402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horvath JE, Bailey JA, Locke DP, Eichler EE. Lessons from the human genome: transitions between euchromatin and heterochromatin. Hum. Mol. Genet. 2001;10:2215–2223. doi: 10.1093/hmg/10.20.2215. [DOI] [PubMed] [Google Scholar]
- 27.Park JP, Wojiski SA, Spellman RA, Rhodes CH, Mohandas TK. Human chromosome 9 pericentric homologies: implications for chromosome 9 heteromorphisms. Cytogenet. Cell Genet. 1998;182:192–194. doi: 10.1159/000015097. [DOI] [PubMed] [Google Scholar]
- 28.Guy J, et al. Genomic sequence and transcriptional profile of the boundary between pericentromeric satellites and genes on human chromosome arm 10p. Genome Res. 2003;13:159–172. doi: 10.1101/gr.644503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brun ME, Ruault M, Ventura M, Roizes G, De Sario A. Juxtacentromeric region of human chromosome 21: a boundary between centromeric heterochromatin and euchromatic chromosome arms. Gene. 2003;312:41–50. doi: 10.1016/s0378-1119(03)00530-4. [DOI] [PubMed] [Google Scholar]
- 30.Flajnik MF, Kasahara M. Comparative genomics of the MHC: glimpses into the evolution of the adaptive immune system. Immunity. 2001;15:351–362. doi: 10.1016/s1074-7613(01)00198-4. [DOI] [PubMed] [Google Scholar]
- 31.Sachidanandam R, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- 32.Rampoldi L, et al. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nature Genet. 2001;28:119–120. doi: 10.1038/88821. [DOI] [PubMed] [Google Scholar]
- 33.Eisenberg I, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nature Genet. 2001;29:83–87. doi: 10.1038/ng718. [DOI] [PubMed] [Google Scholar]
- 34.Raymond CS, et al. Evidence for evolutionary conservation of sex-determining genes. Nature. 1998;391:691–695. doi: 10.1038/35618. [DOI] [PubMed] [Google Scholar]
- 35.Smith CA, McClive PJ, Western PS, Reed KJ, Sinclair AH. Conservation of a sex-determining gene. Nature. 1999;402:601–602. doi: 10.1038/45130. [DOI] [PubMed] [Google Scholar]
- 36.Shan Z, et al. FISH mapping of the sex-reversal region on human chromosome 9p in two XY females and in primates. Eur. J. Hum. Genet. 2000;8:167–173. doi: 10.1038/sj.ejhg.5200431. [DOI] [PubMed] [Google Scholar]
- 37.Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77:481–490. doi: 10.1016/0092-8674(94)90211-9. [DOI] [PubMed] [Google Scholar]
- 38.Nanda I, et al. Conserved synteny between the chicken Z sex chromosome and human chromosome 9 includes the male regulatory gene DMRT1: a comparative (re)view on avian sex determination. Cytogenet. Cell Genet. 2000;89:67–78. doi: 10.1159/000015567. [DOI] [PubMed] [Google Scholar]
- 39.Schmid M, et al. First report on chicken genes and chromosomes 2000. Cytogenet. Cell Genet. 2000;90:169–218. doi: 10.1159/000056772. [DOI] [PubMed] [Google Scholar]
- 40.Della Torre G, et al. CDKN2A and CDK4 mutation analysis in Italian melanoma-prone families: functional characterization of a novel CDKN2A germ line mutation. Br. J. Cancer. 2001;85:836–844. doi: 10.1054/bjoc.2001.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong S, et al. IL-3 receptor signaling is dispensable for BCR-ABL-induced myeloproliferative disease. Proc. Natl Acad. Sci. USA. 2003;100:11630–11635. doi: 10.1073/pnas.2035020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chissoe SL, et al. Sequence and analysis of the human ABL gene, the BCR gene, and regions involved in the Philadelphia chromosomal translocation. Genomics. 1995;27:67–82. doi: 10.1006/geno.1995.1008. [DOI] [PubMed] [Google Scholar]
- 43.Pendergast AM, Gishizky ML, Havlik MH, Witte ON. SH1 domain autophosphorylation of P210 BCR/ABL is required for transformation but not growth factor independence. Mol. Cell. Biol. 1993;13:1728–1736. doi: 10.1128/mcb.13.3.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Druker BJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 45.Humphray SJ, Knaggs SJ, Ragoussis I. Contiguation of bacterial clones. Methods Mol. Biol. 2001;175:69–108. doi: 10.1385/1-59259-235-X:069. [DOI] [PubMed] [Google Scholar]
- 46.Soderlund C, Humphray S, Dunham A, French L. Contigs built with fingerprints, markers, and FPC V4.7. Genome Res. 2000;10:1772–1787. doi: 10.1101/gr.gr-1375r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 48.Olsen GJ, Matsuda H, Hagstrom R, Overbeek R. fastDNAmL: a tool for construction of phylogenetic trees of DNA sequences using maximum likelihood. Comput. Appl. Biosci. 1994;10:41–48. doi: 10.1093/bioinformatics/10.1.41. [DOI] [PubMed] [Google Scholar]
- 49.Ning Z, Cox AJ, Mullikin JC. SSAHA: a fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwartz S, et al. PipMaker—a web server for aligning two genomic DNA sequences. Genome Res. 2000;10:577–586. doi: 10.1101/gr.10.4.577. [DOI] [PMC free article] [PubMed] [Google Scholar]