

Abstract
Background
Secondary analyses of two randomized, controlled phase III trials demonstrated that selenium and vitamin E could reduce prostate cancer incidence. To characterize pharmacodynamic and gene expression effects associated with use of selenium and vitamin E, we undertook a randomized, placebo-controlled phase IIA study of prostate cancer patients before prostatectomy and created a preoperative model for prostatectomy tissue interrogation.
Methods
Thirty-nine men with prostate cancer were randomly assigned to treatment with 200 μg of selenium, 400 IU of vitamin E, both, or placebo. Laser capture microdissection of prostatectomy biopsy specimens was used to isolate normal, stromal, and tumor cells. Gene expression in each cell type was studied with microarray analysis and validated with a real-time polymerase chain reaction (PCR) and immunohistochemistry. An analysis of variance model was fit to identify genes differentially expressed between treatments and cell types. A beta-uniform mixture model was used to analyze differential expression of genes and to assess the false discovery rate. All statistical tests were two-sided.
Results
The highest numbers of differentially expressed genes by treatment were 1329 (63%) of 2109 genes in normal epithelial cells after selenium treatment, 1354 (66%) of 2051 genes in stromal cells after vitamin E treatment, and 329 (56%) of 587 genes in tumor cells after combination treatment (false discovery rate = 2%). Validation of 21 representative genes across all treatments and all cell types yielded Spearman correlation coefficients between the microarray analysis and the PCR validation ranging from 0.64 (95% confidence interval [CI] = 0.31 to 0.79) for the vitamin E group to 0.87 (95% CI = 0.53 to 0.99) for the selenium group. The increase in the mean percentage of p53-positive tumor cells in the selenium-treated group (26.3%), compared with that in the placebo-treated group (5%), showed borderline statistical significance (difference = 21.3%; 95% CI = 0.7 to 41.8; P = .051).
Conclusions
We have demonstrated the feasibility and efficiency of the preoperative model and its power as a hypothesis-generating engine. We have also identified cell type– and zone-specific tissue effects of interventions with selenium and vitamin E that may have clinical implications.
CONTEXT AND CAVEATS
Prior knowledge
Selenium and vitamin E have been associated with reduced incidence of prostate cancer in secondary analyses of two randomized, controlled phase III trials.
Study design
In this randomized, double-blind phase IIA chemoprevention trial, 39 men with prostate cancer were treated with 200 μg of selenium, 400 IU of vitamin E, both, or placebo for 3 to 6 weeks before surgery. The gene expression profile of each cell type and of each treatment group was determined.
Contribution
This study provided a proof-of-principle that prostate biopsy specimens can serve as a source of tissue for molecular interrogation. Differential gene expression related to selenium and/or vitamin E treatments was identified that was cell type specific and tissue zone specific and that may have clinical implications.
Implications
This preoperative model can be used to investigate differential gene expression associated with other treatments for prostate cancer.
Limitations
The study period was only 3 to 6 weeks long and contained only 39 patients. The expression of biomarkers that are modulated after a short intervention may not correlate with a clinical endpoint in large clinical trials. Sampling errors caused by the presence of multifocal tumors and by the diverse nature of the tumors can lead to errors in identification of biomarkers and false-negative findings. A large chemoprevention trial of selenium and vitamin E showed no evidence of a preventive effect, and so any supplement-induced changes noted in gene expression are not likely to predict for prevention efficacy.
From the Editors
Prostate cancer is the most commonly diagnosed nondermatologic cancer among men in the United States and was expected to represent 25% of all newly diagnosed cancers in men in 2008 (1). This percentage may be an underestimate because occult prostate cancers are frequently detected at autopsy (2). The prevalence and long latency period of prostate cancer make it an ideal candidate for chemoprevention, but its characteristic multiple tumor foci and pathological zones (3) create challenges to meaningful comparison of interventions and chemoprevention efforts.
The Selenium and Vitamin E Cancer Prevention Trial (SELECT) was based on epidemiological and clinical evidence that selenium and vitamin E may have protective effects (4–10) and on secondary findings from the Nutritional Prevention of Cancer study (11,12) and the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study Group trial (13–15) that have linked reduced prostate cancer incidence with selenium and vitamin E use, respectively. To identify potential antioxidant therapeutic targets and biomarkers for prostate cancer, we undertook a randomized, placebo-controlled phase IIA preprostatectomy study that complements SELECT. Its aim was to create a model for prostatectomy tissue interrogation that could be used to characterize pharmacodynamic and gene expression effects associated with selenium and vitamin E use. We also studied cell type– and zone-specific tissue effects of selenium, vitamin E, and combination treatment with both. Cell types included normal, tumor, and stromal cells. The prostate zones included the transition, the central, and the peripheral zones.
Patients and Methods
Clinical Trial Design, Patient Selection, and Evaluation
We conducted a 2 × 2 factorial, randomized, placebo-controlled, double-blind phase IIA trial of selenium and vitamin E in patients with clinically organ-confined prostate cancer. From February 1, 2001, through April 30, 2002, 48 patients were enrolled consecutively to ensure a final sample size of 40 patients because of the likelihood that some patients would not start the treatment regimen or undergo prostatectomy after enrollment. Entry criteria required histologically confirmed adenocarcinoma of the prostate, clinical stage T1c or T2, low- or intermediate-grade disease (Gleason score of 7 or less) on initial biopsy examination, a prostate-specific antigen level of less than 10 ng/mL, a life expectancy of at least 10 years, a performance status of less than 2 (Zubrod scale), no history of thyroid disease, and scheduled radical prostatectomy. Taking more than 50 μg of selenium and/or 300 IU of vitamin E daily for more than 3 days consecutively within 1 month before registration prompted exclusion. Patients were randomly assigned to treatment by use of a computerized random number generator. Treatments were administered during the 3- to 6-week period from the time of enrollment until prostatectomy and included one of the following four daily oral regimens: 200 μg of L-selenomethionine (selenium), 400 IU of all-rac-alpha-tocopheryl acetate (vitamin E), a combination of 200 μg of L-selenomethionine and 400 IU of vitamin E, or placebo. These agents and their doses were identical to those used in the SELECT. All patients also received a multiple vitamin and 250 mg of vitamin C each day.
Patients were removed from the study if they had any toxic effect that was greater than grade 1 according to the National Cancer Institute Common Toxicity Criteria scale. No dose modification was allowed.
This trial was approved by the institutional review board of The University of Texas M. D. Anderson Cancer Center in Houston, Texas. All patients gave written informed consent, in keeping with the policies of the institution, acknowledging the study's investigational nature, and were enrolled by physicians.
Blood and Tissue Collection
For selenium analysis, pretreatment and posttreatment blood samples from all four treatment groups (selenium, vitamin E, both, or placebo) were collected into 7-mL Vacutainers containing disodium EDTA (product 369736; Becton Dickinson, Franklin Lakes, NJ). Plasma was isolated by centrifugation (2000g at 4°C for 15 minutes), placed into 7-mL trace element–free Vacutainers (product 369735; Becton Dickinson), and stored at 4ºC until they were shipped to Texas A&M University (College Station, TX) for determination of selenium levels. For vitamin E analysis, blood samples from all four treatment groups were collected in 10-mL Vacutainers containing sodium heparin (product 366480; Becton Dickinson). After centrifugation (2000g at 4°C for 15 minutes), aliquots of supernatant were stored at −80°C until analyzed.
After prostatectomy, core biopsy tissue samples were obtained from the surgical specimens by use of a Multiple Biopsy Device (Engineered Medical Systems, Indianapolis, IN), which has an 18-gauge 20-cm biopsy needle and so simulates the clinical biopsy technique. Core biopsy samples were either snap frozen or fixed in formalin and embedded in paraffin. For histological examination, the prostatectomy specimens were embedded entirely in paraffin as previously described (16). Briefly, after the apex of the prostate was separated and sectioned radially, the remaining gland was sectioned with a commercial meat slicer at 4.0-mm intervals in a transverse plane perpendicular to the posterior surface. Each cross section was subdivided into portions to fit four to six standard cassettes, and a schematic map of the cross sections was prepared as described previously (17). Each tumor focus was outlined. The total number of tumor foci, the zone of origin, the Gleason score, and the pathological stage of each tumor focus were noted.
Apoptotic Index
Prostatectomy specimens were fixed in neutral-buffered formalin and then embedded in paraffin blocks from which 4-μm sections were cut and stained with hematoxylin–eosin. Both mitosis and apoptosis were scored by microscopic examination at 400× magnification. The morphological features used for histological identification of apoptotic bodies have been described and illustrated elsewhere (18). Briefly, in apoptosis, cells shrink, chromatin condenses and marginates against the nuclear membrane, and nuclear budding results in the formation of discrete membrane-bound apoptotic bodies. Cells displaying any of these morphological features were scored as being apoptotic. Five fields of non-necrotic areas were randomly selected in each histological section, and the numbers of apoptotic bodies and mitotic cells per 100 nuclei in each field were counted and then expressed as the percentage of apoptotic or mitotic cells.
Proliferation Index
Ki67 expression was used to determine the proliferation index. Briefly, unstained sections of formalin-fixed, paraffin-embedded prostate tissue were baked in an oven overnight at 60°C and then deparaffinized in xylene and an alcohol series. Endogenous peroxidase activity in tissue sections was blocked by incubation in a solution of 3% H2O2 in methanol for 5 minutes at room temperature, followed by washing in water. For antigen retrieval, slides were heated for three 45-minute periods in a 500 W microwave in 10 mM sodium citrate (pH 6.0), with a 20-minute cooling period at room temperature in between each treatment and after the last treatment.
Slides were then incubated with mouse anti-human Ki67 polyclonal antibody (MIB1; Dako, Glostrup, Denmark; 1:100 dilution) in Chemmate antibody dilution buffer (Ventana Medical Systems, Tuscon, AZ) for 60 minutes at room temperature. Biotinylated anti-mouse antibody (LSAB kit; Dako) was applied for 30 minutes at room temperature, followed by incubation with streptavidin peroxidase (LSAB2 system; Dako) for 16 minutes at room temperature. The slides were then rinsed in phosphate-buffered saline and stained with diaminobenzidine chromogen solution (ResGen; Invitrogen, Carlsbad, CA), and the color reaction was observed under a light microscope. Sections were then counterstained with hematoxylin. Negative-staining controls were slides not incubated with primary antibody. Positive controls included tonsil tissue specimens with proliferating cells that exhibited brown staining. Five fields of prostate cancer and five fields of normal prostate were randomly selected in each case. In each field, the number of Ki67-positive nuclei was recorded as number per 100 nuclei. A total number of 500 nuclei were scored, and the number of Ki67-positive nuclei was recorded and expressed as a percentage.
Determination of Selenium Levels
Selenium levels were determined in blood samples that were collected from all 39 prostate cancer patients before and after treatments. Posttreatment blood was collected within 24 hours of surgery. Archived blood samples were stored at 4°C until they were shipped to Texas A&M University (College Station, TX) for analysis. For selenium analysis, plasma samples were digested with ultrapure nitric acid in a microwave oven (OI Model MDS; OI Analytical, College Station, TX) under high pressure (160 pounds per square inch) at 180°C. After samples cooled to room temperature, ultrapure hydrogen peroxide was added, and the samples were reheated to 90°C under atmospheric pressure to digest lipids. Samples were then cooled to room temperature, diluted to 20 mL with deionized water, and transferred to polyethylene bottles for storage until subsequent analysis.
Reducing selenium (VI) to selenium (IV) required that 5-mL digest samples be moved to polypropylene vessels. After the addition of 5 mL concentrated HCl (Baker Instra-Analyzed grade; J. T. Baker, Phillipsburg, NJ) to these samples, they were heated in a graphite heating block (CPI International, Santa Rosa, CA) for 60 minutes at 95°C. Deionized water was then added to samples after they were cooled to room temperature to a final concentration of 3 M HCl. For quality control, each batch of 20 or fewer samples included one blank and one sample to which known amounts of Se were added, one procedural blank (ie, a blank treated as a sample through preparation and analysis to evaluate contamination), one duplicate sample, and one sample of certified reference material, which were processed through the digestion and selenium reduction steps with the samples. Blank and procedural blank controls differ in that a known amount of Se is added to the blank, whereas a procedural control contains no Se. Calibration standards were prepared from commercial standards obtained from CPI International.
An A P S Analytical Millennium Excalibur atomic fluorescence spectrometer (Deerfield Beach, FL) was used to measure selenium levels in the samples and standards. Briefly, samples were mixed 1:1 with a solution of 0.7% (mass/volume) sodium borohydride in 0.1 M sodium hydroxide through flow injection analysis. A gas–liquid separator was used to transfer selenium hydride to an argon gas stream, and an air–hydrogen flame decomposed it to free Se atoms (Se0). A boosted hollow cathode lamp and a solar blind detector were used to identify selenium by its atomic fluorescence. Concentrations of selenium, reported as parts per million (wet weight), were determined from the peak area of the fluorescence signal by use of unweighted linear regression analysis.
Determination of Vitamin E Levels
Vitamin E levels were determined in pretreatment and posttreatment plasma samples from all 39 patients in all treatment groups (selenium, vitamin E, both, or placebo) by simultaneously measuring both alpha-tocopherol and gamma-tocopherol by use of high-performance liquid chromatography (HPLC), as described previously by Sowell et al. (19). Posttreatment blood was collected within 24 hours of surgery. In brief, 200 μL of serum was added to 10 μL of 10% l-ascorbic acid, followed by the addition of 200 μL of an internal standard (retinyl butyrate at 7.5 μg/mL), and the mixture was vortex-mixed for 10–15 seconds. Next, 1 mL of hexane was added, and the mixture was vortex-mixed for another 5 minutes and then centrifuged for 10 minutes at 1800g at room temperature. The upper hexane layer was collected, and the extraction was repeated two more times; the three upper layers were pooled and dried in a SpeedVac (Savant Instruments, Inc., Farmingdale, NY) without heating. The dried sample was dissolved in 150 μL of mobile-phase solution (acetonitrile and ethanol, 50% and 50% by volume, in 0.01% triethylamine; Fisher Scientific, Fairlawn, NJ) and vortex-mixed for 10 seconds. The dissolved extract was filtered and then subjected to HPLC analysis with a Waters HPLC system equipped with 717 antosamples, a 996-photodiode array detector (Waters, Milford, MA), and a Customsil octadecylsyline C18 diameter, 150 × 4.6–mm column (Phenomenex, Torrance, CA) that with 5-μm particles has a mobile-phase flow rate of 1.0 mL/min under isocratic conditions. For both alpha- and gamma-tocopherols, chromatograms were obtained for any HPLC-purified samples with an absorbance at 300 nm. The concentration of alpha- or gamma-tocopherol was quantified by comparing the peak height of the analyte in the experimental sample to that of the corresponding alpha- or gamma-tocopherol standard. The concentration of vitamin E was presented as individual tocopherol isoforms.
RNA Preparation and Microarray Hybridization
Cancer, stromal, and non-neoplastic epithelial cells were dissected from 5-μm serial sections of fresh-frozen prostate tissue by use of laser-pulse capture microdissection (30 μm diameter) and a PixCell laser capture microscope (Arcturus Engineering, Mountain View, CA, or P.A.L.M. Microlaser Technologies, Bernried, Germany). RNA was prepared from captured cells after lysis in a buffer of 100 mM Tris–HCl, 500 mM lithium chloride, 10 mM EDTA, 1% lithium dodecyl sulfate, and 5 mM dithithreitol, and vortex-mixing for 4 minutes at room temperature. The lysate was centrifuged (12 000g for 15 minutes) at 4°C, and the supernatant was mixed with oligo-d(T) Dynabeads (Invitrogen) to bind mRNA. Incubation, washing, and elution steps were performed as described by the manufacturer's protocol. The extracted RNA was amplified according to the kit manufacturer's protocol (RiboAmp RNA Amplification kit; Molecular Devices, Sunnyvale, CA). Double-stranded cDNA was prepared from total RNA by use of the SuperScript Choice System for cDNA synthesis (Gibco–BRL Life Technologies, Carlsbad, CA), according to the manufacturer's instructions. Biotin-labeled complementary RNA was transcribed from the cDNA by use of the Enzo BioArray HighYield RNA transcript labeling kit (Enzo Life Sciences, Farmingdale, NY) and purified by use of an RNeasy spin column (Qiagen, Valencia, CA) and ethanol precipitation. The purified complementary RNA was dissolved in nuclease-free water and fragmented with fragmentation buffer (Enzo BioArray HighYield RNA transcript labeling kit), and the fragments were hybridized to the GeneChip Hu133A microarray (Affymetrix, Santa Clara, CA), both procedures according to the manufacturer's protocol. Microarrays were subsequently washed and stained in an Affymetrix GeneChip Fluidics station as described in the manufacturer's protocol and then scanned with an Affymetrix GeneChip Scanner 3000, which provides an image of the microarray and stores high-resolution fluorescence intensity data. The original CEL files that were generated after scanning were submitted to ArrayExpress (accession number E-MEXP-1327, at http://www.ebi.ac.uk/microarray-as/ae/).
Oligonucleotide Microarray Data Analysis
Genes were represented by 22 283 probe sets spotted on the Hu133A array (Affymetrix). The expression levels of genes were determined from hybridized probe set intensities by use of a positional-dependent nearest-neighbor model (20) that uses probe sequence information to estimate probe binding affinities. Genes that were absent or ubiquitously expressed at low levels and genes with little variation across samples were excluded from further analysis because variation in gene expression at low levels is not reproducible. The excluded low-intensity genes had a median logarithmic expression level of more than 5.35 in at most three array experiments. We also excluded genes with SD in logarithmic expression levels across tested samples of less than 0.07 (ie, 40% of the SD of log expression levels across experiments from all genes). Our analysis proceeded with the remaining 13 158 genes, after 9125 genes were excluded.
Pathway Analysis
To prioritize genes for further study, we subjected to pathway analysis the data for the differentially expressed genes from the microarray hybridization experiments with a false discovery rate of 2% to pathway analysis. Tables containing the names of genes, array probe set identifiers, and corresponding t scores of differential expression were uploaded in Ingenuity Pathways Analysis software (Ingenuity Systems, Redwood City, CA) for an Ingenuity analysis that generates networks of gene interactions and assigns gene ontology descriptions of biological function on the basis of information retrieved from the software's literature database. Right-tailed Fisher exact test was used to determine the probability that each biological function assigned to each network was matched by chance alone, a determination achieved by comparing the number of genes from the gene expression profile that participates in a given biological function with the total number of occurrences of those genes in all functional annotations stored in the software's database. To narrow the potential candidates for validation of gene expression even further, we uploaded in a Microsoft Access database annotations (from the Affymetrix Hu133A array annotation file, available at http://www.affymetrix.com/support/technical/annotationfilesmain.affx) and expression levels of the differentially expressed genes (from the microarray analysis), which had a false discovery rate of 2% between treatment groups (ie, selenium, vitamin E, or a combination of both selenium and vitamin E vs placebo). We then initiated queries via structured query language (according to Microsoft Access software manual; Microsoft Corp., Redmond, WA) to select the genes that, compared with placebo treatment, were expressed at a higher or lower level in both a single-agent treatment group and the combination treatment group in a specific cell type (ie, tumor, stromal, or normal epithelial cells). Unsupervised hierarchical clustering was performed with open-source software, Cluster version 2.51 and TreeView, and Ingenuity analysis was subsequently performed as described above. These analyses enabled the organization of genes in groups by their expression patterns across samples and according to their network interactions.
Real-Time Quantitative Polymerase Chain Reaction
Real-time quantitative polymerase chain reaction (RT-PCR) was performed by use of the One-Step Reverse Transcription PCR Taqman Master Mix Reagents Kit (Applied Biosystems, Foster City, CA), with primers and probe sets specific for genes that were identified by the microarray data analyses (ACADS, ANGPT1, ARPC4, B2M, CFLAR, CREB1, CUL4A, DNAJA3, EDNRA, ERCC3, GRN, GSK3B, HIPK2, HRAS, MAP3K8, NFKBIB, TP53, PDE8A, PRKCI, ST14, and TOP1) and delineated by assays identified by using Applied Biosystems’ FileBuilder software. The RNA templates used in the RT-PCRs were obtained from pooled RNAs isolated from normal, tumor, or stromal prostate cells.
We determined mRNA levels by use of the ABI PRISM 7700 Sequence Detection System (Applied Biosystems, Foster City, CA) with β-actin mRNA as the reference. The PCR incubation conditions were as follows: for stage I (reverse transcription), 48°C for 30 minutes; for stage II (denaturation), 95°C for 10 minutes; for stage III, 40 cycles of 95°C for 15 seconds (denaturation), followed by 60°C for 1 minute (primer annealing and extension). We used the comparative method for determining the threshold cycle (Ct) in the software Sequence Detector, version 1.7a (Applied Biosystems), to calculate the normalized RNA expression level (ΔCt) as the Ct of the gene of interest in a specific drug treatment and cell type group minus the Ct of β-actin for the corresponding sample group. The fold change in RNA expression between treatment and placebo groups (ΔΔCt) was the difference between the ΔCt of the gene in the placebo group and the ΔCt of the gene in the treatment group. Duplicate RT-PCR experiments were performed for 13 (ACADS, ANGPT1, CFLAR, CREB1, CUL4A, GSK3B, HIPK2, HRAS, NFKBIB, TP53, PDE8A, PRKCI, and ST14) of 21 genes tested, with an average SD of 28.5% over the mean value. Limited available RNA prevented running duplicate tests for all genes.
Immunohistochemistry for p53
Thirty-six prostatectomy specimens were available from all treatment groups. Unstained, 4-μm, formalin-fixed paraffin-embedded tissue sections were air-dried and then heated at 56°C overnight in an oven. Briefly, sections were brought to room temperature, endogenous peroxidases were blocked with 3% hydrogen peroxide in methanol for 10 minutes, and then sections were rinsed in distilled water. Tissue sections were treated with target retrieval solution (Dako, Glostrup, Denmark) and heated at 100°C for 30 minutes in a Pascal pressure cooker to unmask epitopes. Slides were cooled to room temperature, rinsed three times in deionized water, and then rinsed in Tris-buffered saline containing 0.05% Tween-20 (pH 8.0) (Sigma, St Louis, MO). Next, slides were incubated first with mouse anti-human p53 monoclonal antibody (clone DO-7; Dako, Glostrup, Denmark; dilution 1:1000) at room temperature for 1 hour, then with peroxidase-labeled dextran polymer conjugated to goat anti-mouse and anti-rabbit immunoglobulins (DAKO EnVision + Dual link System Peroxidase; Dako, Glostrup, Denmark) for 30 minutes, and finally with 3,3′-diaminobenzidine for 10 minutes. The slides were rinsed in Tris-buffered saline containing 0.05% Tween-20 between each step. The slides were counterstained for 1 minute with Mayer hematoxylin (Poly Scientific, Bayshore, NY; diluted 1:10 with deionized water) and rinsed twice with distilled water. Nuclei in the tissue sections were stained with Richard–Allan bluing reagent (Thermo Scientific, Waltham, MA) for 1 minute and then sections were dehydrated through an increasing ethanol series in xylene. A cover slip was placed over the sections and mounted with xylene-based mounting medium. The individual scoring (E.E.) immunostaining was blinded with respect to treatment groups.
Statistical Analysis
A 2 × 2 factorial trial design was used to evaluate the four treatments to which patients were randomly assigned, including selenium, vitamin E, both, or placebo. Patients were randomly assigned by use of a double-blind randomization procedure, with 10 patients per treatment group. Financial limitations confined the sample size to 40 patients. This sample size ensured that, for example, the estimated Ki67 value (SD = 2.5%; historical baseline mean = 12.4%) would have a 95% confidence interval with a width of 1.55% (= [2 × 1.96 × s]/401/2), where s is the SD. The 95% confidence interval estimates for other quantities were computed similarly. With 10 patients per group, a two-sided t test, with a statistical significance level at .05 to compare two of the four treatment groups, would detect a drop of 2%, 4%, or 6% in the Ki67 value compared with the historical mean of 12.4%, with a power of 43%, 95%, or 99%, respectively.
Patient characteristics were summarized as the median and range for continuous variables and the frequency and percentage for categorical variables. The Wilcoxon signed rank test was used to assess the changes in levels of selenium and vitamin E (as alpha- and gamma-tocopherols) between before- and after-treatment samples. A multiple linear regression model was fit to assess associations by treatment between baseline covariates and posttreatment selenium, posttreatment alpha-tocopherol, or posttreatment gamma-tocopherol levels. Step-down variable selection with a P value cutoff of .05 was performed to determine whether a variable should be retained in the model. Pairwise association between continuous variables was examined with scatter plots and smoothed Lowess curves (21); the statistical significance of associations was assessed by use of the Spearman correlation test, with 95% confidence intervals for the estimated nonparametric Spearman correlation coefficients computed by the bootstrap method. Differences in continuous variables among groups of patients were evaluated by use of the Kruskal–Wallis test (for more than two groups) or the Wilcoxon rank sum test (for two groups), with P values of less than .05 considered to be statistically significant. Exploratory and descriptive statistical methods were initially used for gene expression data, including hierarchical clustering and estimation of proportions, means, and SDs. To identify differentially expressed genes between treatments and tumor types, we fit the following analysis of variance (ANOVA) model with an interaction term for each gene:
where Y is gene expression; X1 and X2 are treatments and tumor types, respectively, for each gene; and ϵ is a measurement error term under the assumptions that errors are independent and normally distributed with a mean of 0 and that variance is homogeneous among treatment groups. Specific comparisons of gene expression between treatments and tumor types were evaluated by use of the ANOVA model and contrast analysis (22). To adjust for multiple comparisons, we used a beta-uniform mixture model to analyze the resulting subscribes P values for the comparisons from the ANOVA model with appropriate control for a false discovery rate. All statistical analyses were carried out in SPLUS (23,24).
Specific comparisons of gene expression between treatments and tissue types were evaluated by use of an ANOVA model with contrast analysis. We used a beta-uniform mixture model (24) to adjust for multiple comparisons by use of a false discovery rate and to obtain P values from the ANOVA model. This ANOVA model could also be used to calculate t scores for each comparison, which reflect fold changes in the expression levels of genes between the compared sample groups.
An ANOVA model was fit to assess p53 expression among treatment groups. Because gene expression arrays are not a reliable source from which to determine mutation status of p53, we obtained the percentage of tumor cells exhibiting detectable nuclear staining for p53 protein by counting cells in tissue sections. We treated this value as a continuous variable. All statistical tests were two-sided.
Results
Patient Characteristics
Forty-eight patients with prostate cancer were enrolled and randomly assigned to take selenium, vitamin E, both selenium and vitamin E, or placebo (Figure 1). Because of financial limitations, we planned to enroll a total of 40 patients, with 10 patients per treatment group. Nine patients were lost to assessment: three withdrew, two chose to have radiation therapy, two failed to supply tissue or blood samples, one never took the assigned agent, and one was lost to follow-up. Overall, 39 completed treatment and were evaluated in the apoptotic and proliferation indices studies, and of these, 36 supplied adequate specimens for microarray analysis.
Figure 1.

Study flow chart. Patient enrollment and disposition in the presurgical study of selenium and vitamin E in clinically organ-confined prostate cancer. Patients are treated with placebo, selenium only, vitamin E only, or the combination of selenium and vitamin E.
Detailed patient characteristics are shown in Table 1, which include age, race, body mass index, previous use of vitamin supplementation, clinical tumor stage, Gleason score, and smoking history. Patient characteristics were well balanced across the four treatment arms.
Table 1.
Patient characteristics
| Variable | All (n = 39) | Selenium arm (n = 9) | Vitamin E arm (n = 11) | Selenium and vitamin E arm (n = 10) | Placebo (n = 9) | P value* |
| Age, median (range), y | 59 (44–70) | 58 (47–67) | 58 (50–68) | 59 (47–65) | 66 (44–70) | .45 |
| Race or ethnicity, No. (%) | .78 | |||||
| White | 33 (84.6) | 7 (77.8) | 10 (90.9) | 9 (90) | 7 (77.8) | |
| Black | 1 (2.6) | 0 (0) | 0 (0) | 0 (0) | 1 (11.1) | |
| Hispanic | 2 (5.1) | 1 (11.1) | 0 (0) | 0 (0) | 1 (11.1) | |
| Asian | 3 (7.7) | 1 (11.1) | 1 (9.1) | 1 (10) | 0 (0) | |
| Body mass index, median (range), kg/m2 | 29.2 (21.8–44.9) | 28.8 (24.1–34.5) | 30.4 (22.5–44.9) | 29.4 (25.4–32.8) | 28.9 (21.8–40.0) | .93 |
| Previous use of vitamin supplementation, No. (%) | .55 | |||||
| No | 20 (51.3) | 6 (66.7) | 4 (36.4) | 6 (60) | 4 (44.4) | |
| Yes | 19 (48.7) | 3 (33.3) | 7 (63.6) | 4 (40) | 5 (55.6) | |
| Clinical tumor stage, No. (%) | .78 | |||||
| T1c | 25 (88.9) | 6 (66.7) | 8 (72.7) | 5 (50) | 6 (66.7) | |
| T2 | 14 (8.3) | 3 (33.3) | 3 (27.3) | 5 (50) | 3 (33.3) | |
| Gleason score, No. (%) | .38 | |||||
| 6 | 18 (46.2) | 5 (55.6) | 7 (63.6) | 3 (30) | 3 (33.3) | |
| 7 | 21 (53.8) | 4 (44.4) | 4 (36.4) | 7 (70) | 6 (66.7) | |
| Smoking history, No. (%) | .07 | |||||
| Current | 5 (12.8) | 4 (44.4) | 0 (0) | 0 (0) | 1 (11.1) | |
| Ever | 20 (51.3) | 4 (44.4) | 7 (63.6) | 6 (60) | 3 (33.3) | |
| Never | 14 (35.9) | 1 (11.1) | 4 (36.4) | 4 (40) | 5 (55.6) | |
| Treatment group, No. (%) | ||||||
| Selenium | 9 (23.1) | |||||
| Vitamin E | 11 (28.2) | |||||
| Selenium and vitamin E | 10 (25.6) | |||||
| Placebo | 9 (23.1) | |||||
Kruskal–Wallis test for continuous variables and generalized Fisher exact test for categorical variables. All statistical tests were two-sided.
Selenium and Vitamin E Levels
There was little variation in pretreatment selenium levels among patients in each of the four treatment groups. However, in the combination treatment group, the selenium level was statistically significantly higher after treatment (median = 0.19 ppm) than before treatment (median = 0.14 ppm) (difference = 0.05 ppm; 95% confidence interval [CI] = 0.01 to 0.07; P = .001) (Table 2). In the selenium treatment group, the selenium level was higher after treatment (median = 0.21 ppm) than before treatment (median = 0.15 ppm), but with borderline statistical significance (P = .06). Both alpha- and gamma-tocopherols were assessed to determine vitamin E levels. There was a statistically significant increase in the alpha-tocopherol levels between posttreatment (median = 11.59 μM) and pretreatment (median = 7.47 μM) in the vitamin E group (difference = 4.12 μM, 95% CI = 0.50 to 9.94 μM; P < .001) and in the selenium plus vitamin E group (posttreatment median = 15.53 μM and pretreatment median = 8.98 μM; difference = 6.55 μM, 95% CI = 0.58 to 11.78 μM; P < .001). Posttreatment gamma-tocopherol values were statistically significantly reduced, compared with pretreatment levels, in both the vitamin E group (posttreatment median = 0.36 μM and pretreatment median = 1.84 μM; difference = −1.06 μM, 95% CI = −2.87 to −0.57 μM; P < .001) and the combination treatment group (posttreatment median = 0.35 μM and pretreatment median = 1.74 μM; difference = −1.39 μM, 95% CI = −3.40 to −0.29 μM; P = .002).
Table 2.
Comparison of pre- and posttreatment selenium and vitamin E levels in plasma*
| Measurement and treatment group | Pretreatment median value (95% CI) | Posttreatment median value (95% CI) | Median change value (95% CI) | P value† |
| Selenium, ppm | ||||
| Selenium (n = 9) | 0.15 (0.11 to 0.22) | 0.21 (0.15 to 0.24) | +0.06 (−0.07 to 0.08) | .06 |
| Vitamin E (n = 11) | 0.14 (0.11 to 0.21) | 0.13 (0.11 to 0.17) | −0.01 (−0.07 to 0.02) | .07 |
| Selenium plus vitamin E (n =10) | 0.14 (0.11 to 0.19) | 0.19 (0.14 to 0.23) | +0.05 (0.01 to 0.07) | .001 |
| Placebo (n = 9) | 0.14 (0.12 to 0.17) | 0.15 (0.11 to 0.17) | +0.01 (−0.01 to 0.03) | |
| Alpha-tocopherol, μM | ||||
| Selenium (n = 9) | 8.15 (6.81 to 18.34) | 9.35 (6.21 to 19.84) | +1.37 (−1.90 to 2.60) | .09 |
| Vitamin E (n = 11) | 7.47 (4.16 to 12.04) | 11.59 (7.54 to 18.86) | +4.12 (0.50 to 9.94) | <.001 |
| Selenium plus vitamin E (n = 10) | 8.98 (7.19 to 15.48) | 15.53 (12.14 to 23.10) | +6.55 (0.58 to 11.78) | <.001 |
| Placebo (n = 8) | 9.98 (5.37 to 14.61) | 9.11 (6.11 to 11.22) | −0.87 (−3.84 to 1.65) | |
| Gamma-tocopherol, μM | ||||
| Selenium (n = 9) | 1.80 (0.46 to 2.82) | 1.58 (0.32 to 2.60) | −0.34 (−0.94 to 1.20) | .67 |
| Vitamin E (n =11) | 1.84 (0.82 to 3.54) | 0.36 (0.19 to 2.25) | −1.06 (−2.87 to −0.57) | <.001 |
| Selenium plus vitamin E (n = 10) | 1.74 (0.63 to 3.85) | 0.35 (0.15 to 0.78) | −1.39 (−3.40 to −0.29) | .002 |
| Placebo (n = 8) | 1.77 (0.57 to 2.50) | 2.22 (0.66 to 6.92) | +0.45 (0.13 to 4.67) | |
CI = confidence interval.
Wilcoxon signed rank test comparing the posttreatment levels with the pretreatment levels in each treatment group. All statistical tests were two-sided.
The association between the posttreatment selenium level (or alpha- or gamma-tocopherol level) and the pretreatment selenium level (or alpha- or gamma-tocopherol level) and age or body mass index was assessed by constructing smoothed Lowess curves and by use of a Spearman correlation test. No association was found between pre- and posttreatment selenium levels (data not shown), whereas a stronger positive association was found between pre- and posttreatment levels of alpha-tocopherol (Spearman correlation coefficient = 0.40; P = .02) and a weaker positive association was found between pre- and posttreatment levels for gamma-tocopherol (Spearman correlation coefficient = 0.26; P = .08). No association was found between age or body mass index and the posttreatment level of alpha- and gamma-tocopherol or selenium. We next simultaneously assessed the association of baseline covariates and treatment assignment with the posttreatment selenium (or alpha- or gamma-tocopherol) level by fitting a multiple linear regression model for the posttreatment selenium level (or posttreatment alpha- or gamma-tocopherol level). We found a borderline statistically significant association between the pre- and posttreatment selenium levels (ie, a higher pretreatment selenium level was associated with a higher posttreatment selenium level; P = .08) and a statistically significant association between selenium treatment and posttreatment selenium levels (ie, patients treated with selenium or selenium and vitamin E had a higher posttreatment selenium level than those not treated with selenium) (regression coefficient = 0.05, 95% CI = 0.02 to 0.07; P = .001). There was no statistically significant association between vitamin E level and posttreatment selenium level (P = .26), and no statistically significant interaction was detected between selenium and vitamin E (P = .53).
A statistically significant association between the pretreatment alpha-tocopherol level and the posttreatment alpha-tocopherol level was observed (P < .001). After adjusting for the pretreatment alpha-tocopherol level, statistically significant associations were found between the selenium level and the posttreatment alpha-tocopherol level (P < .04) and between the vitamin E level and the posttreatment alpha-tocopherol level (P < .001). In addition, a statistically significant inverse association was found between smoking (current and ever) and posttreatment alpha-tocopherol levels (regression coefficient = −2.23, 95% CI = −3.96 to −0.50; P = .02) (data not shown). No statistically significant association between pretreatment gamma-tocopherol level and posttreatment gamma-tocopherol level was detected (P = .16). After adjusting for the pretreatment gamma-tocopherol level, we found a statistically significant association between vitamin E treatment and posttreatment gamma-tocopherol level (regression coefficient = −1.67, 95% CI = −2.76 to −0.58; P = .005) but not between selenium treatment and posttreatment gamma-tocopherol level (P = .25). In the gamma-tocopherol analysis, we removed one patient from the analysis who was in the placebo group because his gamma-tocopherol level was 7.93 μM, a value that was an extreme outlier.
The association between pretreatment alpha-tocopherol levels and pretreatment gamma-tocopherol levels was assessed with the Spearman rank correlation test. A similar test was performed for the posttreatment association between alpha- and gamma-tocopherol levels. We found a statistically significant correlation between posttreatment alpha- and gamma-tocopherol levels (Spearman correlation coefficient = −0.36; P = .03) but not between pretreatment levels of alpha- and gamma-tocopherols (Spearman correlation coefficient = 0.15; P = .35).
Toxicity
Except for one patient who had grade 4 central nervous system toxic effects (ie, cerebrovascular ischemia) that were thought to be unrelated to the study drugs, no toxic effects were recorded.
Histological Features
Thirty of the 39 patients had two to four tumor foci in their prostatectomy specimens, and these foci were most likely located in the peripheral zone. Six patients had a single tumor in the peripheral zone of the prostate, and information about tumor foci was not available for three patients. No statistically significant difference in total tumor volume was found among treatment groups (P = .66, by the Kruskal–Wallis test). When apoptotic and proliferation indices were calculated for the largest tumor in the peripheral zone of the prostatectomy specimens across all treatment groups, overall values for the apoptotic index were 1.25–2 times higher in cancer cells than in normal cells, and overall values for the proliferation index were 3.3–5.5 times higher in cancer cells (data not shown). Values for both indices in both cancer and normal cells were highest in the selenium group. However, using different variables in a fitted regression failed to yield a statistically significant correlation between pretreatment selenium and vitamin E levels and the apoptotic index in posttreatment tissue, probably because of the short duration of therapy.
In contrast, statistically significant differences in the apoptotic indices were noted in other histological comparisons (Table 3). The apoptotic index was statistically significantly higher in cancer cells in the peripheral zone than in the central zone (regression coefficient = 0.36, 95% CI = 0.11 to 0.61; P = .009) and higher in normal epithelial cells in the transition zone than in the central zone (regression coefficient = 0.27, 95% CI = 0.09 to 0.45; P = .006), a difference that was independent of treatment effects (P = .01). The proliferation index in cancer cells of prostatectomy specimens was statistically significantly higher after vitamin E treatment than after placebo treatment (regression coefficient = −1.05, 95% CI = −0.09 to −2.01; P = .04) (Table 3) and statistically significantly higher in the peripheral zone than in the transition zone (regression coefficient = 1.92, 95% CI = 1.10 to 2.74; P < .001). A statistically significant association was found between body mass index and proliferation index (P = .01) (ie, a higher body mass index was correlated with a lower proliferation index) (regression coefficient = −4.49, 95% CI = −7.88 to −1.10; P = .01). In addition to the statistically significantly higher proliferation indices in cancer cells between smokers (current and former) and nonsmokers (regression coefficient = 1.18, 95% CI = 0.16 to 2.20; P = .03) (Table 3), we also found that smoking was associated with a higher proliferation index in normal cells, but with borderline statistical significance (P = .06) (data not shown).
Table 3.
Fitted linear mixed model for AI and PI in cancer and normal tissue samples from radical prostatectomy specimens*
| Index and cell type | Comparison(s) | Regression coefficient (95% CI) | P value† |
| AI and cancer cells | Peripheral zone vs central zone | 0.36 (0.11 to 0.61) | .009 |
| AI and normal cells | Transition zone vs central zone | 0.27 (0.09 to 0.45) | .006 |
| log(body mass index)‡ | 0.70 (0.03 to 1.37) | .05 | |
| PI and cancer cells | Vitamin E vs placebo | −1.05 (−2.01 to −0.09) | .04 |
| Peripheral zone vs transition zone | 1.92 (1.10 to 2.74) | <.001 | |
| Smoker vs nonsmoker‡ | 1.18 (0.16 to 2.20) | .03 | |
| log(body mass index)§ | −4.49 (−7.88 to −1.10) | .01 |
For each fitted model, only those covariates that were statistically significant are shown. The covariates under consideration include treatment group, tissue zone, smoking status, and body mass index. AI = apoptotic index; PI = proliferation index; CI = confidence interval.
P values were based on Wald tests of the regression coefficients estimates. All statistical tests were two-sided.
Smokers = current or former smokers; nonsmokers = never smokers.
Higher log(body mass index) corresponds to lower proliferation index.
Differential Gene Expression
Harvested prostatic normal epithelium, stromal, and tumor cells from patients were subjected to microarray-based gene expression analysis (Table 4) to investigate genes that were differentially expressed according to treatment and cell type. Overall, global gene expression patterns were not statistically significantly different by treatment group, as demonstrated by unsupervised hierarchical clustering. However, segregation patterns by cell type were detected (eg, gene expression in stromal cells was similar across treatment groups, and gene expression in normal tissue cells from the placebo group was distinct from that of other treatment groups and from cancer cells) (Supplementary Figure 1, available online). Of the 13 158 genes studied, 2109 genes in normal epithelial cells, 2051 genes in stromal cells, and 587 genes in tumor cells were differentially expressed overall and between treatment and placebo groups, with an estimated false discovery rate of 2% (P < .005) and a change in gene expression of at least 2.8-fold, which was estimated from ANOVA model with justification by SD (for detailed gene data, see ArrayExpress at http://www.ebi.ac.uk/microarray-as/ae/, and Supplementary Tables 1–9, available online). Gene expression in the normal epithelium was affected most strongly by selenium treatment (ie, 1329 [63%] of the 2109 differentially expressed genes in normal epithelial cells were associated with selenium treatment, compared with placebo treatment). In contrast, gene expression in the stromal cells was affected most strongly by vitamin E treatment (ie, 1354 [66%] of the 2051 differentially expressed genes in the stromal cells were associated with vitamin E treatment). Gene expression in tumor cells was most strongly affected by the combination treatment (ie, 329 [56%] of the 587 differentially expressed genes in tumor cells were associated with combination treatment).
Table 4.
Summary of cell types isolated from prostate tissue by treatment group and the corresponding yield of cRNA*
| Treatment group | No. of patients | No. of cells (SD) |
Amplified cRNA, μg (SD) |
||||
| Normal epithelium | Stroma | Tumor | Normal epithelium | Stroma | Tumor | ||
| Selenium | 9 | 6402 (1499) | 7144 (1916) | 1301 (2232) | 32.0 (4.5) | 23.1 (18.2) | 31.2 |
| Vitamin E | 11 | 5582 (414) | 6195 (2044) | 2572 (2797) | 39.1 (30.9) | 16.7 (15.6) | 22.4 (28.6) |
| Combination | 10 | 6205 (694) | 6250 (1111) | 2964 (2730) | 51.1 (31.2) | 26.9 (14.4) | 40.6 (14.7) |
| Placebo | 8 | 7661 (1770) | 6591 (852) | 3338 (3635) | 50.0 (20.4) | 24.5 (14.0) | 30.6 (23.3) |
cRNA was amplified from pooled samples of RNAs isolated from the indicated cell type. cRNA = complementary RNA.
Gene Clustering, Gene Ontology, and Pathway Analysis
We further explored the nature of the biological effect of treatment in the distinct prostate cell types by subjecting the differentially expressed genes to unsupervised hierarchical clustering analysis, which organizes genes into groups according to their similarity in expression patterns across samples. A heatmap is shown in Figure 2 that compares gene expression in tumor and stromal cells from combination treatment and placebo treatment groups. Similar heatmaps were generated for all other treatments and cell types (data not shown).
Figure 2.
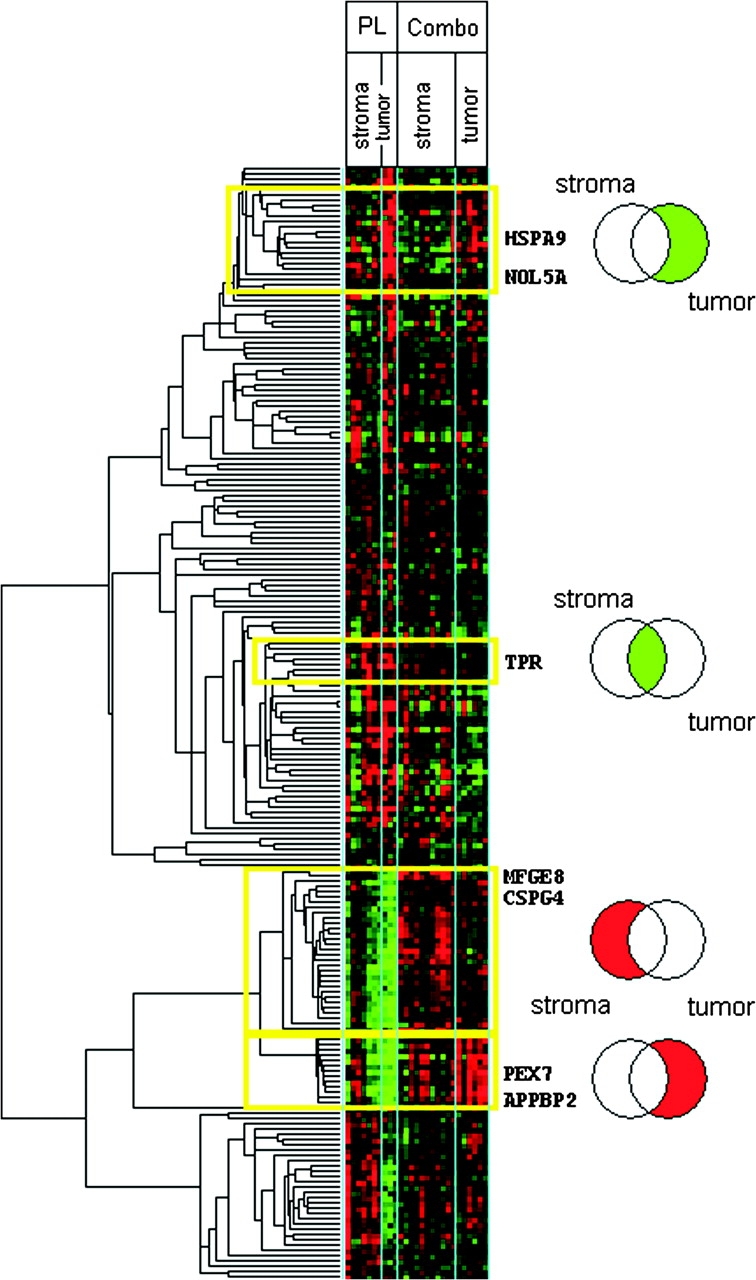
Gene expression values and unsupervised hierarchical clustering of differentially expressed genes in tumor and stromal prostate cells for combination (Combo) vs placebo (PL) treatments. Data are expressed as the logarithm of gene expression values and presented in a heatmap of differentially expressed genes between combination vs placebo treatments for tumor and stromal prostate cells, with a false discovery rate of 2%. In the heatmap, the logarithm of expression levels of genes is shown in colors that reflect expression levels (green = negative log values that represent low expression levels; red = positive log values that represent high expression levels). The Venn diagrams located to the right represent gene groups in select clusters (yellow boxes of the heatmap) that were differentially expressed in stromal cells only (left section of the Venn diagrams), tumor cells only (right section of the Venn diagrams), or both cell types (intersection of the two circles in the Venn diagram). The sections (gene groups) of the Venn diagrams are labeled as red or green, as described for the heatmap. The genes symbols at the right of the heatmap correspond to gene ontology groups shown in Supplementary Figure 2 (available online).
Groups of genes that were differentially expressed in normal, tumor, and stromal cells, in treatment vs placebo groups, were identified and further categorized according to gene ontology attributes by use of Ingenuity Pathways Analysis software (version 5.0; Ingenuity Systems). This analysis identified gene groups that were associated with specific molecular or biological processes (the most representative group is shown in Supplementary Figure 2, available online) (P < .009). The association was determined by Fisher exact test, which compares the number of genes from the gene expression profile that participate in a given biological function with the total number of occurrences of those genes in all functional annotations stored in the software's database. In particular, when differentially expressed genes in normal epithelial cells were compared between treatment and placebo groups, 25 of these genes were associated with RNA modification, six with apoptosis, and nine with cell transformation. When differentially expressed genes in tumor cells were compared between treatment and placebo groups, 11 genes were associated with molecular transport. When differentially expressed genes in stromal cells were compared between treatment and placebo groups, eight genes were associated with the cell cycle and three with invasion.
To narrow our focus to a manageable group of networks, we included only the 213 genes that were differentially expressed in normal (53 genes), tumor (85 genes), or stromal (75 genes) cells between treatment and placebo groups, in both a single treatment group (selenium or vitamin E) and the combination treatment group. Of these genes, the 85 genes in tumor cells are shown in the unsupervised hierarchical clustering in Figure 3. The same clustering approach was applied to the genes in stromal and normal cells (data not shown). We next organized the 213 genes (in tumor, normal, or stromal cells) into networks by use of the Ingenuity Systems’ medical literature database to limit our focus further to 11 networks (Supplementary Figures 3–8, available online). The resulting 11 networks were composed of differentially expressed genes and genes that were not differentially expressed but were connected (in terms of molecular or biological interactions) to many differentially expressed genes. In this way, potential direct and indirect impacts on specific signaling networks may be inferred. Genes with more than 10 network connections that were associated with selenium treatment in normal prostate epithelial cells included TP53, AR, HRAS, JUN, and CASP3; those that were associated with vitamin E treatment in normal epithelial cells included CASP3, TP53, HRAS, NFKB1 and PTEN. Genes with major network connections in stromal cells that were associated with selenium treatment included TNF, TGFB1, and CREB1. Genes with multiple network connections in stromal cells that were associated with vitamin E treatment included TNF and CREB1; those in tumor cells that were associated with selenium treatment included MYC and TP53; and those in tumor cells that were associated with vitamin E treatment included AKT1, GSK3B, TNF, TP53, and MYC.
Figure 3.
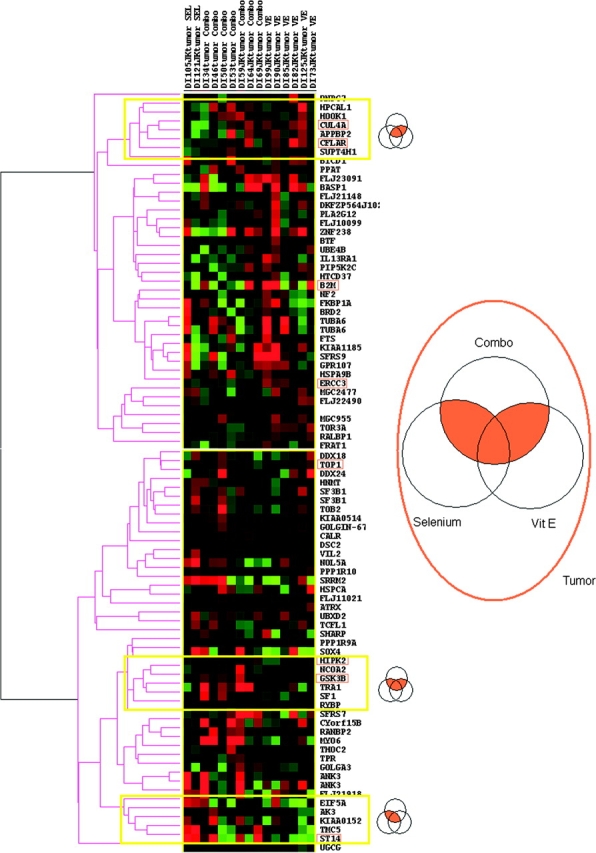
t Scores and unsupervised hierarchical clustering of differentially expressed genes found in combination treatment vs placebo and in at least one single treatment vs placebo group. Differentially expressed genes in both combination compared with placebo treatment and in at least one single treatment group compared with placebo treatment group are shown. In the heatmap, t scores of genes are shown in colors that reflect differences in gene expression levels of treatment (combination, selenium, or vitamin E) compared with placebo (green = decreased expression; red = increased expression). Representative clusters (genes differentially expressed in vitamin E and combination treatment, in all three treatments, and in selenium and combination treatment) are marked with yellow boxes and are presented in the associated Venn diagram. Validated genes (Figure 4) are boxed.
Validation of Differentially Expressed Genes
Pooled RNA was isolated from each cell type (normal epithelial, stromal, and tumor) that had been dissected from frozen sections of prostate tissue for each drug treatment group and then subjected to quantitative RT-PCR to evaluate gene expression levels. Specifically, from each cell type, the expression of representative genes that we identified as being differentially expressed between treatment and placebo groups was evaluated (four genes from normal epithelial cells, nine genes from stromal cells, and eight genes from tumor cells) (Figure 4, A). The differential expression of each gene between treatment and placebo groups was calculated as a ΔΔCt value, and the relationship between the ΔΔCt values and corresponding t scores from the Affymetrix microarray experiment for each gene was assessed by use of the Spearman correlation test. We found that all correlations between the ΔΔCt values and corresponding t scores across all cell types and treatment groups were all statistically significant (eg, for selenium treatment, r = 0.87, 95% CI = 0.53 to 0.99; for vitamin E treatment, r = 0.64, 95% CI = 0.31 to 0.79; and for the combination treatment, r = 0.84, 95% CI = 0.57 to 0.96). Thus, values obtained from RT-PCR and microarrays appear to be consistent with each other.
Figure 4.

Validation of the 21 differentially expressed genes. A) RT-PCR. Data are expressed as the fold change in RNA expression between treatment and placebo groups (ΔΔCt), which is the difference between the normalized RNA expression level (ΔCt, where Ct is the threshold cycle) of the gene in placebo group and that of the gene in the treatment group. The ΔΔCt values represent levels of differential gene expression in tissue from selenium (Sel), vitamin E (VE), or combination (Combo) treatments, compared with placebo treatment. The mRNA expression of ACADS, ANGPT1, ARPC4, CREB1, DNAJ3, EDNRA, MAP3K8, NFKBI, and PRKCI was tested in stromal cells; that of B2M, CFLAR, CUL4A, ERCC3, GSK3B, HIPK2, ST14, and TOP1 was tested in tumor cells; and that of GRN, HRAS, TP53, and PDE8A was tested in normal cells. B) Microarray evaluation. Data are expressed as t scores (levels of differential gene expression in treatment groups compared with the placebo treatment group) for the same genes that were derived from the original microarray experiment. C) Expression of p53 protein in sections of prostatectomy tumor tissue (10× magnification). p53 was stained with anti-p53 antibody and visualized by use of horseradish peroxidase–conjugated secondary anti-rabbit antibody immunoglobulin G and 3,3-diaminobenzine. Left) selenium-treated group. Right) Placebo-treated group. Scale bars = 10 μm. RT-PCR = real-time quantitative polymerase chain reaction.
We also assessed p53 protein levels in tumor tissues from all four treatment groups (selenium, vitamin E, both, or placebo) by immunohistochemistry. The protein p53 was selected because good-quality antibody against p53 was available and because p53 plays a central role in three networks of differentially expressed genes that were identified in the microarray study (diagrams of these networks are presented in Supplementary Figures 4 and 7, network to the right, and Supplementary Figure 8, network in the middle, available online). The mean percentage of p53-positive cells in tumor cells was 26.3% in the selenium treatment group, 17% in the vitamin E treatment group, 9% in the combination treatment group, and 5% in the placebo treatment group. The difference in p53 protein expression between selenium treatment and placebo groups was marginally statistically significant (difference = 21.3%, 95% CI = 0.7 to 41.8%; P = .051) (Figure 4, C) and is consistent with microarray results in tumor cells in which selenium treatment was associated with increased expression of p53 mRNA, compared with placebo treatment (Supplementary Figure 2, available online).
Discussion
We evaluated the prostate cell type– and treatment-specific effects of selenium and vitamin E in a phase IIA randomized, double-blind, placebo-controlled trial. We found that the treatments affected expression levels of several genes, including those in p53-related pathways. The level of p53 protein itself was increased in tumor cells after selenium treatment.
We also demonstrated that it was possible to systematically evaluate the modulation of relevant biomarkers (in this study, by vitamin E and selenium) and that the biological effects of these agents were dependent on cell type and on intervention. Access to the entire prostate gland after surgery and rapid screening were two advantages of our preoperative model, with the former providing data that can be compared with data from biopsy samples and with the latter offering a speedy analysis that is clearly unavailable in trials with many thousands of subjects. In addition, these studies are relatively easy to perform and can provide histological and biological (ie, cellular, molecular, and biochemical) evidence (25). To the best of our knowledge, this study was the first detailed systematic pathological interrogation to be completed in preoperative patients with favorable risk prostate cancer.
Low baseline levels of plasma selenium have been linked to an increased risk of prostate cancer (8). Clark et al. (26) found an inverse relationship between plasma selenium levels at baseline and the strength of the treatment effect. In a study of 249 subjects with prostate cancer who were matched to controls and followed for more than 20 years, Nomura et al. (6) found an inverse relationship between baseline levels of selenium and risk of prostate cancer (ie, the higher the selenium level, the lower the risk), and they also found that this inverse relationship extended to current smokers (n = 76) (odds ratio = 0.2, 95% CI = 0.1 to 0.8; Ptrend = .02, when the quartile with the highest selenium level was compared with the quartile with the lowest). In our study, plasma levels of vitamin E were associated with more statistically significant effects than were selenium levels. Comparisons of pretreatment and posttreatment plasma measures of both alpha-tocopherol (its levels increased) and gamma-tocopherol (its levels decreased) were statistically significantly different in those in the vitamin E–alone arm, alpha-tocopherol (P < .001) and gamma-tocopherol (P < .001) or in the selenium plus vitamin E arm, alpha-tocopherol (P ≤ .001) and gamma-tocopherol (P = .002).
Pretreatment and posttreatment plasma levels of selenium (which increased) were statistically significantly different only in the selenium plus vitamin E arm. Using multiple linear regression to adjust for the low pretreatment level of alpha-tocopherol in one group, we found that both selenium and vitamin E treatments were associated with statistically significant treatment effects. After adjustment for the pretreatment gamma-tocopherol level, we found a statistically significant effect in the vitamin E arm but not in the selenium arm. Spearman rank correlation testing indicated a statistically significant correlation between alpha- and gamma-tocopherol levels in posttreatment samples but not in pretreatment samples, which is consistent with the inverse pharmacodynamic effect of alpha-tocopherol on gamma-tocopherol (27,28).
We found that the peripheral zone was the area most likely to have tumors, as reported previously. Peripheral zone tumors have dominated in other studies; for example, 80% of tumors were in the peripheral zone in the study by Sakai et al. (29), 68% in the study by McNeal et al. (3), and 75% in the study by Augustin et al. (30). The differences between cancers in different zones have been attributed to large differences in the levels of factors that control tumor progression in each zone (31); these results further validate the methodological approach that we have used in this study. With broader and better characterization of these prostate tissue zones, the challenges presented by the multiple tumor foci and pathological zones in prostate cancers (3) can be overcome.
Although we found no statistically significant differences between the apoptotic index or the proliferation index by group in normal peripheral zone cells or in cells from that zone's largest tumor focus, we did find statistically significant differences in the apoptotic index by zone and by logarithm of body mass index and in the proliferation index by treatment group, smoking status, zone, and logarithm of body mass index. Not withstanding the small sample size of this trial (ie, 39 patients), the inverse relationship between body mass index and proliferation index underscores the complex associations between obesity and prostate cancer development and progression (32). We speculate that for patients with early-stage prostate cancer with a nonaggressive nature, obesity may slow disease progression; however, further study is warranted to determine the mechanisms involved. Other investigators (33) have found a statistically significant difference in apoptotic indices in epithelial cells of canine prostate before treatment and after 7 months of selenium treatment. Similar proliferation indices were found in the tumor samples of the placebo and selenium groups. No statistically significant differences were found in proliferation indices between the treatment groups.
We found in microarray studies that combination treatment with selenium and vitamin E appeared to affect p53- and nuclear factor κ B (NFKB)–related pathways. In tumor cells, selenium treatment, in particular, appeared to stimulate the synthesis of both TP53 RNA (Supplementary Figure 2, available online) and protein (Figure 4, C). In normal epithelial cells, vitamin E or combination treatment with vitamin E and selenium induced TP53 expression (a result that was confirmed with RT-PCR) (Figure 4, A), whereas in tumor cells, mRNAs for markers of p53 activation were either induced (ie, CFLAR mRNA by vitamin E treatment and ST14 mRNA by selenium treatment) or repressed (HSP90AA1 mRNA by selenium treatment), consistent with previous reports (34–36). These p53 findings are also consistent with those in proteomic studies (37) of patients’ serum in which combined treatment with selenium and vitamin E induced protein expression patterns that were indicative of being free of prostate cancer.
Changes in the oxidation–reduction balance of cells have been shown to activate an NFKB survival signaling pathway (38,39). NFKB induces expression of antiapoptotic and proinflammatory genes and is constitutively activated in prostate cancer. Antioxidant agents such as selenium have been shown to inhibit NFKB activity in vitro (40) and, in turn, to enhance DNA repair through a p53-related pathway (41). Likewise, in controlled studies, Waters et al. (33) showed that supplementing the diet of the dogs with selenium for 7 months increased epithelial cell apoptosis and decreased DNA damage.
Besides p53 and NFKB, several other factors were also affected. Most important were genes associated with regulation of the androgen receptor. Interestingly, selenium may be involved in the mechanisms that allow prostate tumor cells to become resistant to androgen deprivation therapy. In response, investigators have proposed interventions to target androgen receptor signaling (42). Calreticulin (CALR), an intracellular calcium-binding protein that is able to inhibit binding of the androgen receptor to its DNA element, is responsive to androgen (43). The level of calreticulin RNA was found to be lower in the selenium treatment group than in the placebo treatment group in our study. The expression of HSPA9, a member of the heat shock protein 70 family that enhances androgen receptor activity by inducing BAG1–androgen receptor binding (44), has been found to be reduced in the selenium treatment group, compared with the placebo group. APPBP2 (PAT1), another factor that decreases the transactivation activity of androgen receptor (45), has been found to be induced in the combination group, compared with the placebo group.
The reduced expression in the selenium arm and increased expression in the combination treatment arm of factors that promote androgen receptor function should be viewed in the context of androgen receptor signaling in prostate cancer. In many prostate cancer cell lines, treatment with methylseleninic acid can reduce the expression of prostate-specific antigen and the level of the androgen receptor (46,47); in LAPC-4 and LNCaP cells, selenite can inhibit the expression and activity of the androgen receptor (48). Both selenium forms (selenite and methylseleninic acid) have been shown to reduce the expression of the androgen receptor but by different mechanisms (48).
Pathological studies by others in our group (49) have shown that selenium supplementation results in the accumulation of selenium in the human prostate gland, rather than the seminal vesicles. We extended this observation to the tumor, stromal, and normal prostate epithelial cells. In selenium-treated normal prostate epithelial cells, the expression of TP53 mRNA increased, as well as that of 20 genes that are related to mRNA processing and alternative splicing, processes that are likely to be associated with tumorigenesis or disease progression in prostate cancer (50,51).
Stroma plays an important role in prostate cancer progression because it provides a supportive environment for tumor progression and for tumor cell proliferation at a metastatic site (52). It was therefore important to investigate whether gene expression changes in the stroma can be detected after selenium and vitamin E treatment. Some changes might modulate the environment of the stroma and thereby alter its ability to interact with the epithelium. For example, selenium treatment induced gelsolin (GSN), a gene implicated in actin filament polymerization that is expressed by prostate's stroma and whose expression is decreased in neoplastic lesions of the prostate (53). Induced expression of lactadherin (MFGE8) and CSPG4 by combination treatment may also contribute to an alteration of the stroma milieu with yet unknown consequences because both factors participate in angiogenesis and matrix organization via integrin binding (54,55). Finally, the reduced expression of endothelin receptor α that was observed after combination treatment with selenium and vitamin E indicates that the ability of the stroma to mimic the metastatic site and to “train” cancer cells for their future homing to bone sites may be impaired by treatment. This possibility has also been raised by recent data that endothelin signaling may mediate an interaction between prostate cancer cells and the bone microenvironment (56). This cross talk between epithelial cells and stromal cells plays an important role in prostate cancer progression. Prostate cancer cells, upon metastasis to bone, can activate the stromal cells (ie, osteoblasts), which, in turn, can provide cues to cancer cells for proliferation.
The preprostatectomy model has several limitations, however, including the short duration of the trials, diversity of changes in the cancer field, difficulty in detecting or interpreting biomarkers, and statistical challenges presented by the endpoints. Focally expressed biomarkers or those that lack homogeneous expression may be studied or identified in sections of the prostate, but their expression may not correlate with findings from core biopsy specimens. In addition, biomarkers that are modulated after a short intervention with agents may be time dependent in terms of their expression and may not correlate with a clinical endpoint in large clinical trials. Inasmuch as these studies cannot be carried out to a clinical endpoint, such as all-cause mortality, identifying biomarkers in mechanistic pathways that respond to an agent and that are reliably associated with the efficacy of the intervention is critical. Sampling errors caused by the presence of multifocal tumors and by the diverse nature of the tumors can lead to errors in identification of biomarkers and false-negative findings.
Through a detailed systematic pathological evaluation with a preprostatectomy model, we obtained information about differential biological effects of selenium and vitamin E in the various cell types and zones of the prostate by use of prostatectomy specimens from patients with organ-confined prostate cancer. After a short-term intervention with selenium and vitamin E, we used a microarray platform to identify at least 587 differentially expressed genes in tumor, stromal, and normal epithelial cells. We then linked these differentially expressed genes to molecular processes and characterized the networks to which they belong. This study also provided a proof-of-principle that prostate biopsy specimens can serve as a source of tissue for molecular interrogation. Although the SELECT was a negative study (57), we still have a lot to gain from the molecular studies of cancer risk. Additionally, it would be of interest to compare the expression of genes that are associated with androgen receptor regulation and modulated by selenium with that of genes that are associated with the finasteride response. The fact that a large randomized, double-blind, placebo-controlled trial of finasteride, the Prostate Cancer Prevention Trial, showed a reduction in the incidence, the promotion, and the early progression stages of prostate cancer indicates that androgens may be one of the key players (58). The gene expression changes described in this study need to be validated in studies of SELECT-archived prostate tissue and undergo functional studies to assess their biological significance. Such studies will help determine if they can be used as surrogate biomarkers in other large clinical studies or if their associated genes can be developed as therapy targets.
Funding
National Cancer Institute (R03 CA101072, R21 CA088761, and P50 CA90270).
Supplementary Material
Footnotes
The authors had full responsibility for the design of the study, the collection of the data, the analysis and interpretation of the data, the decision to submit the manuscript for publication, and the writing of the manuscript.
We acknowledge contributions of patients for their participation; David Imperial for his technical expertise in carrying out the laser capture microdissection and RNA preparation; Anh Hoang for his technical expertise in carrying out the immunohistochemical staining; Beth W. Allen, MPH, for editorial assistance; Peiying Yang, PhD, for the analysis of tocopherols; and Keith Johnson and Carlotta Cavazos for patient recruitment.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Guileyardo JM, Johnson WD, Welsh RA, Akazaki K, Correa P. Prevalence of latent prostate carcinoma in two U.S. populations. J Natl Cancer Inst. 1980;65(2):311–316. [PubMed] [Google Scholar]
- 3.McNeal JE, Redwine EA, Freiha FS, Stamey TA. Zonal distribution of prostatic adenocarcinoma. Correlation with histologic pattern and direction of spread. Am J Surg Pathol. 1988;12(12):897–906. doi: 10.1097/00000478-198812000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Yoshizawa K, Willett WC, Morris SJ, et al. Study of prediagnostic selenium level in toenails and the risk of advanced prostate cancer. J Natl Cancer Inst. 1998;90(16):1219–1224. doi: 10.1093/jnci/90.16.1219. [DOI] [PubMed] [Google Scholar]
- 5.Li H, Stampfer MJ, Giovannucci EL, et al. A prospective study of plasma selenium levels and prostate cancer risk. J Natl Cancer Inst. 2004;96(9):696–703. doi: 10.1093/jnci/djh125. [DOI] [PubMed] [Google Scholar]
- 6.Nomura AM, Lee J, Stemmermann GN, Combs GF., Jr Serum selenium and subsequent risk of prostate cancer. Cancer Epidemiol Biomarkers Prev. 2000;9(9):883–887. [PubMed] [Google Scholar]
- 7.Helzlsouer KJ, Huang HY, Alberg AJ, et al. Association between alpha-tocopherol, gamma-tocopherol, selenium, and subsequent prostate cancer. J Natl Cancer Inst. 2000;92(24):2018–2023. doi: 10.1093/jnci/92.24.2018. [DOI] [PubMed] [Google Scholar]
- 8.Brooks JD, Metter EJ, Chan DW, et al. Plasma selenium level before diagnosis and the risk of prostate cancer development. J Urol. 2001;166(6):2034–2038. [PubMed] [Google Scholar]
- 9.Goodman GE, Schaffer S, Omenn GS, Chen C, King I. The association between lung and prostate cancer risk, and serum micronutrients: results and lessons learned from beta-carotene and retinol efficacy trial. Cancer Epidemiol Biomarkers Prev. 2003;12(6):518–526. [PubMed] [Google Scholar]
- 10.van den Brandt PA, Zeegers MP, Bode P, Goldbohm RA. Toenail selenium levels and the subsequent risk of prostate cancer: a prospective cohort study. Cancer Epidemiol Biomarkers Prev. 2003;12(9):866–871. [PubMed] [Google Scholar]
- 11.Clark LC, Combs GF, Jr, Turnbull BW, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996;276(24):1957–1963. [PubMed] [Google Scholar]
- 12.Duffield-Lillico AJ, Dalkin BL, Reid ME, et al. Selenium supplementation, baseline plasma selenium status and incidence of prostate cancer: an analysis of the complete treatment period of the Nutritional Prevention of Cancer Trial. BJU Int. 2003;91(7):608–612. doi: 10.1046/j.1464-410x.2003.04167.x. [DOI] [PubMed] [Google Scholar]
- 13.The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. The Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. N Engl J Med. 1994;330(15):1029–1035. doi: 10.1056/NEJM199404143301501. [DOI] [PubMed] [Google Scholar]
- 14.Heinonen OP, Albanes D, Virtamo J, et al. Prostate cancer and supplementation with alpha-tocopherol and beta-carotene: incidence and mortality in a controlled trial. J Natl Cancer Inst. 1998;90(6):440–446. doi: 10.1093/jnci/90.6.440. [DOI] [PubMed] [Google Scholar]
- 15.Lippman SM, Goodman PJ, Klein EA, et al. Designing the Selenium and Vitamin E Cancer Prevention Trial (SELECT) J Natl Cancer Inst. 2005;97(2):94–102. doi: 10.1093/jnci/dji009. [DOI] [PubMed] [Google Scholar]
- 16.Troncoso P, Babaian RJ, Ro JY, Grignon DJ, von Eschenbach AC, Ayala AG. Prostatic intraepithelial neoplasia and invasive prostatic adenocarcinoma in cystoprostatectomy specimens. Urology. 1989;34(6 suppl):52–56. [PubMed] [Google Scholar]
- 17.Chen ME, Johnston D, Reyes AO, Soto CP, Babaian RJ, Troncoso P. A streamlined three-dimensional volume estimation method accurately classifies prostate tumors by volume. Am J Surg Pathol. 2003;27(10):1291–1301. doi: 10.1097/00000478-200310000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Sinicrope FA, Roddey G, McDonnell TJ, Shen Y, Cleary KR, Stephens LC. Increased apoptosis accompanies neoplastic development in the human colorectum. Clin Cancer Res. 1996;2(12):1999–2006. [PubMed] [Google Scholar]
- 19.Sowell AL, Huff DL, Yeager PR, Caudill SP, Gunter EW. Retinol, alpha-tocopherol, lutein/zeaxanthin, beta-cryptoxanthin, lycopene, alpha-carotene, trans-beta-carotene, and four retinyl esters in serum determined simultaneously by reversed-phase HPLC with multiwavelength detection. Clin Chem. 1994;40(3):411–416. [PubMed] [Google Scholar]
- 20.Zhang L, Miles MF, Aldape KD. A model of molecular interactions on short oligonucleotide microarrays. Nat Biotechnol. 2003;21(7):818–821. doi: 10.1038/nbt836. [DOI] [PubMed] [Google Scholar]
- 21.Cleveland W. Robust locally-weighted regression and smoothing scatterplots. J Am Stat Assoc. 1979;74(368):829–836. [Google Scholar]
- 22.Hocking RR. Methods and Applications of Linear Models. 2nd ed. New York, NY: John Wiley & Sons; 2003. [Google Scholar]
- 23.Venables W, Ripley B. Modern Applied Statistics with SPLUS. 3rd ed. New York, NY: Springer; 1999. [Google Scholar]
- 24.Pounds S, Morris SW. Estimating the occurrence of false positives and false negatives in microarray studies by approximating and partitioning the empirical distribution of p-values. Bioinformatics. 2003;19(10):1236–1242. doi: 10.1093/bioinformatics/btg148. [DOI] [PubMed] [Google Scholar]
- 25.Special report: phase II prostate trials use pre-prostatectomy study design to identify promising prevention agents and biomarkers. NCI Cancer Bulletin. 2004;1(6):1–2. [Google Scholar]
- 26.Clark LC, Dalkin B, Krongrad A, et al. Decreased incidence of prostate cancer with selenium supplementation: results of a double-blind cancer prevention trial. Br J Urol. 1998;81(5):730–734. doi: 10.1046/j.1464-410x.1998.00630.x. [DOI] [PubMed] [Google Scholar]
- 27.Handelman GJ, Machlin LJ, Fitch K, Weiter JJ, Dratz EA. Oral alpha-tocopherol supplements decrease plasma gamma-tocopherol levels in humans. J Nutr. 1985;115(6):807–813. doi: 10.1093/jn/115.6.807. [DOI] [PubMed] [Google Scholar]
- 28.Handelman GJ, Epstein WL, Peerson J, Spiegelman D, Machlin LJ, Dratz EA. Human adipose alpha-tocopherol and gamma-tocopherol kinetics during and after 1 y of alpha-tocopherol supplementation. Am J Clin Nutr. 1994;59(5):1025–1032. doi: 10.1093/ajcn/59.5.1025. [DOI] [PubMed] [Google Scholar]
- 29.Sakai I, Harada K, Kurahashi T, Yamanaka K, Hara I, Miyake H. Analysis of differences in clinicopathological features between prostate cancers located in the transition and peripheral zones. Int J Urol. 2006;13(4):368–372. doi: 10.1111/j.1442-2042.2006.01307.x. [DOI] [PubMed] [Google Scholar]
- 30.Augustin H, Erbersdobler A, Graefen M, et al. Differences in biopsy features between prostate cancers located in the transition and peripheral zone. BJU Int. 2003;91(6):477–481. doi: 10.1046/j.1464-410x.2003.04140.x. [DOI] [PubMed] [Google Scholar]
- 31.Greene DR, Fitzpatrick JM, Scardino PT. Anatomy of the prostate and distribution of early prostate cancer. Semin Surg Oncol. 1995;11(1):9–22. doi: 10.1002/ssu.2980110104. [DOI] [PubMed] [Google Scholar]
- 32.Freedland SJ, Platz EA. Obesity and prostate cancer: making sense out of apparently conflicting data. Epidemiol Rev. 2007;29(1):88–97. doi: 10.1093/epirev/mxm006. [DOI] [PubMed] [Google Scholar]
- 33.Waters D, Shen S, Cooley D, et al. Effects of dietary selenium supplementation on DNA damage and apoptosis in canine prostate. J Natl Cancer Inst. 2003;95(3):237–241. doi: 10.1093/jnci/95.3.237. [DOI] [PubMed] [Google Scholar]
- 34.Bartke T, Siegmund D, Peters N, et al. p53 upregulates cFLIP, inhibits transcription of NF-kappaB-regulated genes and induces caspase-8-independent cell death in DLD-1 cells. Oncogene. 2001;20(5):571–580. doi: 10.1038/sj.onc.1204124. [DOI] [PubMed] [Google Scholar]
- 35.Ceballos E, Munoz-Alonso MJ, Berwanger B, et al. Inhibitory effect of c-Myc on p53-induced apoptosis in leukemia cells. Microarray analysis reveals defective induction of p53 target genes and upregulation of chaperone genes. Oncogene. 2005;24(28):4559–4571. doi: 10.1038/sj.onc.1208652. [DOI] [PubMed] [Google Scholar]
- 36.Amundson SA, Do KT, Vinikoor L, et al. Stress-specific signatures: expression profiling of p53 wild-type and -null human cells. Oncogene. 2005;24(28):4572–4579. doi: 10.1038/sj.onc.1208653. [DOI] [PubMed] [Google Scholar]
- 37.Kim J, Sun P, Lam YW, et al. Changes in serum proteomic patterns by presurgical alpha-tocopherol and L-selenomethionine supplementation in prostate cancer. Cancer Epidemiol Biomarkers Prev. 2005;14(7):1697–1702. doi: 10.1158/1055-9965.EPI-04-0679. [DOI] [PubMed] [Google Scholar]
- 38.Franco AV, Zhang XD, Van Berkel E, et al. The role of NF-kappa B in TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis of melanoma cells. J Immunol. 2001;166(9):5337–5345. doi: 10.4049/jimmunol.166.9.5337. [DOI] [PubMed] [Google Scholar]
- 39.Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192(1):1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 40.Gasparian AV, Yao YJ, Lu J, et al. Selenium compounds inhibit I kappa B kinase (IKK) and nuclear factor-kappa B (NF-kappa B) in prostate cancer cells. Mol Cancer Ther. 2002;1(12):1079–1087. [PubMed] [Google Scholar]
- 41.Combs GF, Jr, Gray WP. Chemopreventive agents: selenium. Pharmacol Ther. 1998;79(3):179–192. doi: 10.1016/s0163-7258(98)00014-x. [DOI] [PubMed] [Google Scholar]
- 42.Nieto M, Finn S, Loda M, Hahn WC. Prostate cancer: Re-focusing on androgen receptor signaling. Int J Biochem Cell Biol. 2007;39(9):1562–1568. doi: 10.1016/j.biocel.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dedhar S, Rennie PS, Shago M, et al. Inhibition of nuclear hormone receptor activity by calreticulin. Nature. 1994;367(6462):480–483. doi: 10.1038/367480a0. [DOI] [PubMed] [Google Scholar]
- 44.Shatkina L, Mink S, Rogatsch H, et al. The cochaperone Bag-1L enhances androgen receptor action via interaction with the NH2-terminal region of the receptor. Mol Cell Biol. 2003;23(20):7189–7197. doi: 10.1128/MCB.23.20.7189-7197.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, Yang Y, Yeh S, Chang C. ARA67/PAT1 functions as a repressor to suppress androgen receptor transactivation. Mol Cell Biol. 2004;24(3):1044–1057. doi: 10.1128/MCB.24.3.1044-1057.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dong Y, Lee SO, Zhang H, Marshall J, Gao AC, Ip C. Prostate specific antigen expression is down-regulated by selenium through disruption of androgen receptor signaling. Cancer Res. 2004;64(1):19–22. doi: 10.1158/0008-5472.can-03-2789. [DOI] [PubMed] [Google Scholar]
- 47.Dong Y, Zhang H, Gao AC, Marshall JR, Ip C. Androgen receptor signaling intensity is a key factor in determining the sensitivity of prostate cancer cells to selenium inhibition of growth and cancer-specific biomarkers. Mol Cancer Ther. 2005;4(7):1047–1055. doi: 10.1158/1535-7163.MCT-05-0124. [DOI] [PubMed] [Google Scholar]
- 48.Husbeck B, Bhattacharyya RS, Feldman D, Knox SJ. Inhibition of androgen receptor signaling by selenite and methylseleninic acid in prostate cancer cells: two distinct mechanisms of action. Mol Cancer Ther. 2006;5(8):2078–2085. doi: 10.1158/1535-7163.MCT-06-0056. [DOI] [PubMed] [Google Scholar]
- 49.Sabichi AL, Lee JJ, Taylor RJ, et al. Selenium accumulation in prostate tissue during a randomized, controlled short-term trial of l-selenomethionine: a Southwest Oncology Group Study. Clin Cancer Res. 2006;12(7 pt 1):2178–2184. doi: 10.1158/1078-0432.CCR-05-0937. [DOI] [PubMed] [Google Scholar]
- 50.Li HR, Wang-Rodriguez J, Nair TM, et al. Two-dimensional transcriptome profiling: identification of messenger RNA isoform signatures in prostate cancer from archived paraffin-embedded cancer specimens. Cancer Res. 2006;66(8):4079–4088. doi: 10.1158/0008-5472.CAN-05-4264. [DOI] [PubMed] [Google Scholar]
- 51.Ozen M, Creighton CJ, Ozdemir M, Ittmann M. Widespread deregulation of microRNA expression in human prostate cancer. Oncogene. 2008;27(12):1788–1793. doi: 10.1038/sj.onc.1210809. [DOI] [PubMed] [Google Scholar]
- 52.Morrissey C, Vessella RL. The role of tumor microenvironment in prostate cancer bone metastasis. J Cell Biochem. 2007;101(4):873–886. doi: 10.1002/jcb.21214. [DOI] [PubMed] [Google Scholar]
- 53.Lee HK, Driscoll D, Asch H, Asch B, Zhang PJ. Downregulated gelsolin expression in hyperplastic and neoplastic lesions of the prostate. Prostate. 1999;40(1):14–19. doi: 10.1002/(sici)1097-0045(19990615)40:1<14::aid-pros2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 54.Fukushi J, Makagiansar IT, Stallcup WB. NG2 proteoglycan promotes endothelial cell motility and angiogenesis via engagement of galectin-3 and alpha3beta1 integrin. Mol Biol Cell. 2004;15(8):3580–3590. doi: 10.1091/mbc.E04-03-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Silvestre JS, Thery C, Hamard G, et al. Lactadherin promotes VEGF-dependent neovascularization. Nat Med. 2005;11(5):499–506. doi: 10.1038/nm1233. [DOI] [PubMed] [Google Scholar]
- 56.Carducci MA, Jimeno A. Targeting bone metastasis in prostate cancer with endothelin receptor antagonists. Clin Cancer Res. 2006;12(20 pt 2):6296s–6300s. doi: 10.1158/1078-0432.CCR-06-0929. [DOI] [PubMed] [Google Scholar]
- 57.Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) [published online ahead of print] JAMA. 2009;301(1):39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349(3):215–224. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.