

Abstract
Epigenetic alterations of the genome such as DNA promoter methylation and chromatin remodeling play an important role in tumorigenesis. Recent findings indicate epigenetic modifications as key factors in breast carcinogenesis. These modifications are quite appealing as targets for preventative care and therapeutics because of their potential for reversal. Future medical care for breast cancer patients will likely depend upon a better understanding of the roles epigenetic modifications play in carcinogenesis. Here, we discuss the importance of epigenetics in breast cancer detection, prognosis, and therapy with an emphasis on mechanisms and epigenetic contributions to field cancerization effects.
Keywords: epigenetics, breast cancer, field cancerization BRCA, ER
1. Introduction and Background
Human cancers arise from a multi-step process characterized by tumor initiation and progression. Much of the research focus on this process has investigated the role of direct changes or mutations to DNA sequences. Both inherited and de novo mutations are well established in carcinogenesis. These changes lead to activation of oncogenes and inactivation of tumor suppressor genes. Studies have begun to evaluate the role of epigenetics in tumor development. Epigenetics is the inheritance of information on the basis of gene expression rather then direct changes to sequence composition. Epigenetic alterations include methylation of CpG dinucletides in promoters and changes in chromatin structure that may lead to silencing of tumor suppressor genes. Activation of oncogenes may also be a result of epigenetic changes through post-translational modifications in histone acetylation or DNA conformation.
There are distinct mechanisms that initiate and sustain epigenetic modifications(1–3). Of these DNA methylation and post-translational modifications of histone proteins are the best understood. DNA methylation is a covalent addition of a methyl group to DNA, usually to a cytosine located 5′ to guanosine (CpG dinucleotide). CpG dinucleotides are under-represented in the genome except for small clusters, referred to as CpG islands, located in or near the promoter of greater than 70% of all genes(4–6). Promoter methylation is known to participate in reorganizing chromatin structure and also plays a role in transcriptional inactivation. It is believed that the chromatin surrounding an active promoter containing an unmethylated CpG island is “open” and allows for the access of transcription factors and other coactivators. An inactive promoter containing methylated CpG dinucleotides is associated with a “closed” chromatin configuration and results in transcription factors unable to access the promoter. In addition to promoter methylation, chromatin modification may also contribute to silencing genes in cancer cells. Post-translational modifications to histone proteins occur after translation primarily in the NH2 terminal tail of histones and include acetylation, phosphorylation, ubiquitination, or methylation. Other epigenetic modifiers have been identified, including the Polycomb group (PcG) proteins and small non-coding RNAs. PcG repressors serve as a docking platform for DNA methyltransferases and target a gene for permanent silencing by methylation of hisone H3 on lysine 27 (Figure 1). Reversal of permanent silencing is only overcome by de-differentiation processes in the germline. Small non-coding RNA molecules, such as microRNAs, regulate gene expression by targeting RNA degradation (2). These RNAs have also been found to also target gene promoters and result in transcriptional gene silencing (4, 7).
Figure 1.
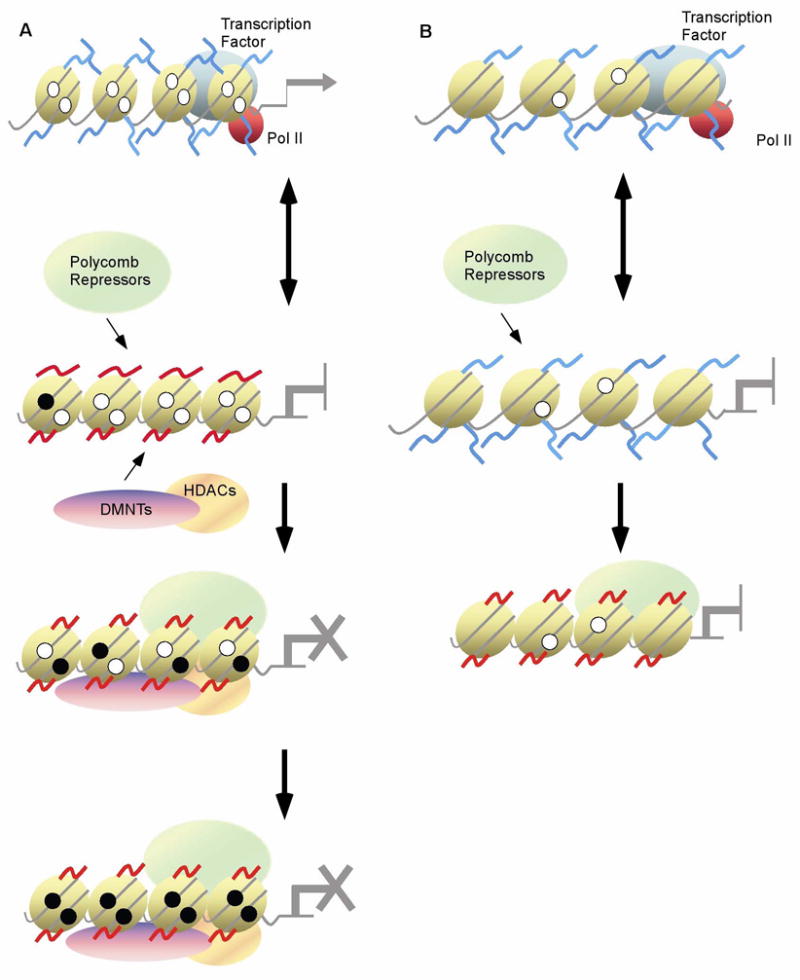
Proposed models for epigenetic long-term silencing of a gene. A. Active transcription of a gene is dependent on transcription factor (TF) binding. Upon removal of the TF through down-regulation, gene silencing or other means, down-regulation of the targeted gene occurs. In the absence of activating transcription factors, PcGs, HDACs, and DNMTs are recruited to the promoter and initiate long-term transcriptional repression. CpG sites adjacent to the promoter are then methylated (represented by filled circles) and eventually a heterochromatin state of long-term silencing is established. B. As in panel A transcription of a gene is dependent upon TF binding. In the absence of TF binding, PcG is recruited to a promoter and initiates long-term transcriptional repression through modification of histones. This can occur in the absence of CpG islands or methylation.
Breast cancer is the most common cancer among women in the United States. Approximately 200,000 women and 2,000 men are diagnosed with breast cancer each year (http://www.cancer.org). There are well understood genetic alterations associated with breast carcinogenesis, including specific gene amplifications, deletions, point mutations, chromosome rearrangements, and aneuploidy. In addition to these highly characterized mutations, epigenetic alterations resulting in aberrant gene expression are key contributors to breast tumorigenesis (8–15).
In this review we will discuss the role and mechanisms of epigenetic alterations in breast cancer for detection, prognosis, and treatment. We will also focus on the effect of epigenetics on field cancerization and triple negative breast cancer. Unlike germline mutations, epigenetic modifications can potentially be reversed making them very appealing for preventative care and therapeutics. Targeting epigenetic alterations represents an active area for breast cancer drug investigation and treatment targets.
2.0 BIOMARKERS/DETECTION
2.1 Methods to identify epigenetic alterations
Gene specific epigenetic changes for breast cancer are likely to occur early in tumorigenesis and have the potential to be used for early detection and prevention (1). DNA methylation as a biomarker for early detection of breast cancer has several advantages over sequence mutations. First, incidences of aberrant methylation of specific CpG islands are higher than those of mutations and methylation can be measured by genome-wide screening. Secondly, aberrant methylation patterns can be detected even when they are embedded in an excess amount of normal DNA molecules. Third, techniques for the detection of methylation patterns are relatively simple(16). As epigenetic modifications become used as biomarkers for individualized breast cancer treatment and therapeutic intervention, it is important to understand the many different techniques available for detecting the presence of methylation, histone modifications, and microRNAs (4).
New promising high-throughput methylation detection methods are available which allow researchers and clinicians to identify an “epigenetic signature” specific to breast cancer. Currently, breast cancer detection relies on various screening methods such as MRI and mammography. Criteria for diagnosis and characterization of breast cancer status include abnormal biopsy, tumor size, histological grade, hormone receptor status, and HER2/Neu amplification. Cytology is the main standard for identification of abnormalities typical of cellular transformation. Future means to diagnosis breast cancer could incorporate the presence of aberrantly methylated genes. Quantitative multiplex methylation specific PCR (QM-MSP) is a highly sensitive method to quantitate cumulative gene promoter hypermethylation in samples in which there is limited available DNA (17). Using QM-MSP, a panel of nine genes shown to be commonly methylated in early breast cancer has been tested for its sensitivity and specificity to detecting premalignant changes. Breast cells were isolated using ductal lavage, nipple aspiration fluids, and fine needle aspirates. Using this detection panel, the rate of detection of breast cancer cells rose from 43% sensitivity with cytologic examination alone to 71.4% (17). Hypermethylation of genes commonly methylated in breast cancer in sera in breast cancer patients has also been used to detect early malignant changes. Sera methylation can be detected using methylation-specific polymerase chain reaction (MS-PCR). This technique utilizes primers designed for methylated or unmethylated bisulfite-modified DNA (18), and early studies indicate that MS-PCR is a promising approach to screen putative cancer patients(19). For example, RASSF1A gene hypermethylation has been detected using MS-PCR in sera of ovarian cancer patients with 100% specificity (19).
Whereas epigenetic modifications involving chromatin are not yet used clinically for breast cancer detection, future panels of epigenetic chromatin modifications may be incorporated into standard tests. One method used to detect chromatin remodeling is a combination of chromatin immunoprecipitation (ChIP) and PCR, which allows for quantification of the amount of protein binding to a specific region of DNA. A large collection of antibodies for specific forms of methylated and acetylated chromatin exist. ChIP-chip combines chromatin immunoprecipitation with array technology allowing for interrogation of thousands of promoter elements. The newest technology ChIP-seq, which combines ChIP with new generation sequencing, is highly quantitative and not biased by which features are on an array. Results from ChIP-seq-based studies are already leading to the identification of new genomic elements, showing epigenetic regulation in cancer(20).
2.2 RASSF1A: A breast cancer biomarker
Allelic losses of 3p, including a critical region at 3p21.3, are frequently detected in many cancers including breast cancer. The Ras-associated domain family member 1 gene (RASSF1) maps to the region of frequent loss. It is comprised of eight exons and through different promoter usage and alternative splicing generates seven unique transcripts, RASSF1A-G. RASSF1A is transcribed from a CpG island promoter region, and is one of the most frequently hypermethylated genes thus far described in human cancer. The CpG island of RASSF1A is hypermethylated in 60–77% of breast cancers [13,22] resulting in gene silencing in cancer cell lines and primary tissues. Its diverse functions include regulation of apoptosis, growth regulation, and microtubule dynamics during mitotic progression. Specifically, RASSF1A is a Ras effector and induces apoptosis through its interactions with pro-apoptotic kinase MST1. When cells lacking RASSF1A expression are treated with a DNA methyltransferase, such as 5-aza-2′-deoxycytidine, expression can be reactivated(19). Mouse knockout studies show that RASSF1A−/− mice are prone to spontaneous development of lung adenomas, lymphomas and breast adenocarcinomas. These mice are prone to early spontaneous tumorigenesis and show a severe tumor susceptibility phenotype compared to that of littermate wild-type mice (19).
There are two main reasons RASSF1A methylation is a good biomarker for breast cancer. First, RASSF1A methylation is rare in normal tissue providing a marker with high specificity. Second, the frequency of methylation is observed in 60 to 77% of cells from a tumor which provides a high frequency of diagnostic coverage(8, 21). In addition to breast tumors, hypermethylation of RASSF1A can be detected in non-malignant breast cells and patient sera. In one study, hypermethylation of sera in breast cancer patients was detected in six out of 26 cases (19). Promoter methylation of RASSF1A was observed in 70% of samples from women at high-risk of developing breast cancer versus only 29% of samples from women at low-risk. Women with a previous history of benign breast growths are statistically more likely to have RASSF1A methylation (22). Thus, hypermethylation of RASSFIA could be used as a form of breast cancer screening to detect breast cancer at its earliest stages.
3. Field Cancerization/Microenvironment
In breast cancer, multiple genes are hypermethylated compared to non-cancerous tissue (23). These include genes involved in evasion of apoptosis (RASSF1A, HOXA5, TWIST1), limitless replication potential (CCND2, p16, BRCA1, RARβ), growth (ERα, PGR), and tissue invasion and metastasis (CDH1)(3, 17, 24, 25). These genes are not only hypermethylated in tumor cells, but show increased epigenetic silencing in normal epithelium surrounding the tumor site. The first observations of this phenomenon were in oral cancer. Slaughter et al (1953) was the first group to use the term “field cancerization” which refers to the presence of cancer causing changes in apparently normal tissue surrounding a neoplasm. They theorized the existence of (pre-) neoplastic processes at multiple sites, with the unproven assumption that these have developed independently(26). In subsequent years, the presence of field cancerization has been described in head and neck squamous cell carcinoma, lung, esophagus, vulva, cervix, colon, bladder, skin, and breast cancers(27). Studies have demonstrated that normal adjacent cells to tumors frequently harbor loss of heterozygosity, microsatellite and chromosome instability, and gene mutations(28). Recently DNA methylation has been added to list as hypermethylated normal tissue immediately adjacent to tumor sites has been found (29).
Epigenetic modifications are believed to be early events in cancer development (30). It is thought that once epigenetic alterations are established in premalignant tissues, the extent of modifications will accumulate as the disease progresses. Varying theories have been proposed on how this field defect arises. One theory is based on the self-metastasis model and the idea that the primary tumor is composed of multiple self-metastases that form around a seed from the tumor to itself (31). A second theory has been seen in gastric cancers and is based on cell methylation profiles influencing H. pylori infection which leads to additional methylation of promoters in gastric mucosal cells and accompanying increases in risk for gastric cancer (32). Another theory has supportive evidence in breast cancer and is based on the idea that early epigenetic changes are associated with a large area of pre-malignant changes, and the “epicenter” appears to accumulate additional epigenetic changes (27).
The detection of epigenetic events resulting in field cancerization effects will be important for breast cancer therapy. Before adjuvant radiotherapy, recurrence rates for cancer in the ipsilateral breast could be as high as forty percent despite apparent negative pathologic margins. Adjuvant radiotherapy reduces this to less than or equal to ten percent. The high recurrence rate in breast cancer strongly suggests that residual “normal” breast is in fact at risk for harboring occult precancerous cells. Yan et al. (2006) identified epigenetic biomarkers frequently hypermethylated in normal breast tissue immediately adjacent to tumor sites. They examined four zones of normal breast tissue in the ipsilateral and contralateral breasts of patients with invasive breast cancer. Using differential methylation hybridization, they observed hypermethylation of the promoter and first exon of RASSF1A in the primary tumor and adjacent breast ducts. Overall levels of RASSF1 methylation in adjacent ducts were lower than primary tumor, but significantly higher than levels in breast of healthy individuals. In a subset of patients they observed a gradient pattern of RASSF1A methylation in which a higher degree of methylation was seen in tissue closest to the tumor compared to tissue two to four centimeters away. Other genes show this effect. When CYP26A1, KCNAB1, and SNCA were hypermethylated in primary tumors, the paired normal adjacent tissues showed methylation 70% of the time (27).
Preliminary evidence exists that epigenetically mediated gene silencing in the epithelial genome can be directed by neighboring fibroblasts (33). One study placed an immortalized breast epithelial cell line in different settings of primary fibroblasts isolated from breast cancer patients or cancer-free women. The methylation status of various loci was subsequently shown to be epigenetically altered when fibroblasts came from breast cancer patients. The findings suggest that breast cancer patient microenvironments induced epigenetic changes in this immortalized breast cell line(33).
A better understanding of how field cancerization occurs has practical implications for predicting the future risk of local recurrence in breast cancer patients undergoing lumpectomy. Current evaluations rely on pathologic examinations of margins free of gross tumor tissue. The use of epigenetic markers will be an important adjunct for predicting local recurrence in histologically normal margins.
4. PROGNOSIS
Breast cancer is heterogeneous. With the availability of an increasing number of therapy options, it is important to identify ways to predict individual tumor response to a given therapy. It is also crucial to streamline treatment and spare patients from receiving often toxic and expensive therapies that are not likely to be effective. The methylation status of many genes and microRNAs are likely to be important for prognosis. Here, we review two genes, RASSF1A and BRCA1 which are good indicators of prognosis.
4.1 RASSF1A
As described here, RASSF1A methylation is important in breast cancer detection. Methylated RASSF1A in breast serum from breast cancer patients is also strongly associated with metastasis, tumor size, and increased relative risk for death (21). RASSF1A methylation in serum of breast cancer patients has been identified as a marker for response to adjuvant Tamoxifen treatment. Methylation post surgery indicates a resistance to Tamoxifen and loss of methylation indicates a response (34).
4.2 BRCA1
Breast Cancer Gene 1 (BRCA1) is a tumor suppressor genes for both breast and ovarian cancer(8). It encodes a multifunctional protein with roles in DNA repair, cell cycle check point control, protein ubiquitization, and chromatin remodeling (10). In vitro experiments showed that decreased BRCA1 expression in cells led to increased levels of tumor growth, while increased expression of BRCA1 led to growth arrest and apoptosis. Recent studies indicate that BRCA1 methylation is an important marker for prognosis. The magnitude of the decrease of functional BRCA1 protein correlates with disease prognosis(10, 13). Tumors with BRCA1 mutations are usually more likely to be higher-grade, poorly differentiated, highly proliferative, estrogen receptor (ER) negative, and progesterone receptor (PR) negative, and harbor p53 mutations. BRCA1 mutated breast cancers are also associated with poor survival in some studies(35–37). Phenotypically, BRCA1-methylated tumors are similar to tumors from carriers of germline BRCA1 mutations.
BRCA1 is thought to be a classical tumor suppressor gene for which Knudson’s two-hit hypothesis holds true. About 20% of individuals with a strong personal and family history of breast and ovarian cancer carry germline mutations in the BRCA1 gene (38, 39). A second hit is thought to be required in the wild-type BRCA1 allele for the development of BRCA-associated cancer (40–42). However, about 20% of all tumors from BRCA mutation carriers do not show LOH of the wildtype BRCA1 (40, 43). A handful of studies have looked at the rate of BRCA1 methylation in germline carriers. In one study, BRCA1 promoter hypermethylation was observed in one of two tumors from BRCA1 carriers lacking LOH(44). In a second study of population-based ovarian tumors, two of eight tumors with germline BRCA1 mutations showed neither LOH nor promoter methylation(45). Another more systematic study of 47 breast tumors from hereditary breast cancer families identified three BRCA1 carriers of which two showed BRCA1 promoter methylation in their tumors(39). These studies suggest that methylation of BRCA1 may be serve as a second hit in tumors from a subset of BRCA1 mutation carriers.
Xu et al. (2008) were one of the first groups to conduct an epidemiological study on the prognostic value of BRCA1 methylation. They found BRCA1 promoter methylation was more frequent in invasive than in situ carcinoma. In a subset of their population they found no correlation between BRCA1 promoter methylation and ER/PR status. However, they also found a higher prevalence of BRCA1 promoter methylation in cases with at least one node involved and with tumor size greater than 2cm. Based on their findings higher methylation levels may correlate with more advanced tumor stage at diagnosis. They also observed a 45% increase in mortality of individuals with BRCA1 methylation positive tumors compared to those who had unmethylated BRCA1 promoters(46). Honrado et al (2007) conducted a familial breast cancer based study and found contradicting results. They observed no overall correlation of ER, PR, or grade with hypermethylation of BRCA1 in the tumors from BRCA1 mutation negative families. However, seven individuals had both promoter hypermethylation and LOH; the majority of these tumors had a basal-like phenotype and were triple negative (47).
4.3 The role of epigenetic alterations in triple negative breast cancer
The presence or absence of ER expression is another important prognostic indicator for survival(8, 9). ER negative tumors are unresponsive to antiestrogens, more likely to have a more aggressive clinical course, more likely to be poorly differentiated, have a higher histological grade and are associated with a higher recurrence rate and decreased overall survival. Along with other genes, the estrogen receptors, ERα and ERβ, have been implicated in breast cancer development. ERα is encoded by ESR1, and when estrogen activated, it stimulates cell proliferation. ERβ is encoded by ESR2 and is known to inhibit the proliferation and invasion of breast cancer cells. ERα and ERβ both have promoter associated CpG islands that can be abnormally methylated in breast cancer, but ESR2 methylation is less well studied (8, 9). Estrogen and its receptors play key roles in normal development and reproduction. Upon estrogen binding, ERα functions as a transcription factor by binding to DNA targets or tethering to other transcription factors. This activity controls the activity of ERα downstream genes important to breast epithelium development. There is evidence implicating ERα and its ligand, estrogen, in the pathogenesis, progression, and treatment of breast cancer. Recent studies have created a compelling case for dynamic regulation of ERα function in breast cancers through post-translational modification by acetylation and epigenetic signaling cascades. Almost all breast cancers show some degree of DNA methylation of the ESR1 gene promoter, but this methylation is only associated with gene repression in ~30% of tumors (8, 9). Approximately 66% of breast cancers express ERα. A fraction of breast cancers that are initially ERα-positive lose ER expression during tumor progression, but it is unclear if this is due to methylation or other causes (9).
The combination of ER, PR, and Her-2/Neu status has been recognized as more informative then ER status alone for prognosis and prediction for the response to treatment with endocrine therapy. Triple negative breast cancer (negative ER, PR, and absent Her-2/Neu amplification) accounts for 10–17% of all breast carcinomas. Triple negative tumors frequently affect younger patients (<50 years) and are more prevalent in African-American women. They often present as interval cancers and are significantly more aggressive (48). DNA methylation of certain genes has been reported to be associated with hormone receptor status. Feng et al. investigated methylation profiles of 12 genes identified as hypermethylated using genome-wide methylated CpG island amplification in 90 pairs of breast cancer and normal tissue. They found that ER status was positively associated with high HIN-1 and RASSF1A methylation but was negatively correlated to RIL (PDLIM4) methylation levels. (PR status was positively associated with high HIN-1 methylation levels and negatively associated with high CDH13 methylation levels(49).) Fifty-eight percent of triple negative cancers exhibit positive correlation to methylation of RIL/CDH13 and 41% of triple negative breast cancers failed to methylate HIN-1/RASSF1A(49). These results suggest the existence of an interaction between DNA methylation and hormone receptor biology.
4.4. Mechanism of epigenetic silencing in triple negative breast cancers
The epigenetic process is complex and the molecular sequence leading to the establishment of epigenetic gene silencing is not well established. One model suggests that histone modifications are the primary initiating event in transient repression while a different model suggests that DNA methylation can actually specify unique histone codes for maintaining the silenced state of a gene (30). To establish DNA methylation in a subset of genes, polycomb protein EZH2 must associate with DNMTs (50). It is thought that polycomb proteins could collaborate with DNMTs by recruiting them to silenced promoters to establish long-term silencing (51). Leu et al (2004) investigated whether the removal of ERα signaling could cause changes in DNA methylation and chromatin structure of ERα target promoters. They used RNAi to transiently disable ERα in breast cancer cells and found that polycomb repressors and histone deacetylases assemble in the promoter of an ERα target gene. Accumulation of DNA methylation in these silenced targets like the PR promoter region then occurs and can be stably transmitted to cell progeny for long-term silencing (Figure 1). Both ERα expression and DNA demethylation appear to be required to restore PR expression. They also observed a trend that more ERα negative tumors had more methylated loci than ERα positive tumors (30). This indicates that dysregulation of normal signaling in cancer cells may result in stable silencing of downstream targets maintained by epigenetic machinery.
Other post-translational modifications of ERα such as phosphorylation, ubiquitination, glycosylation, and acetylation are believed to play a role in breast cancer promotion. ERα is modified by p300 on two lysine residues (302 and 303) located in the hinge region (between DNA- and ligand binding domains). When these lysine residues are mutated, ERα had increased hormone sensitivity. Thirty-four percent of atypical breast hyperplasia samples have mutations of the lysine at 303 (K303R) of the ERα (52–54) suggesting a functional role of these mutations in breast cancer promotion. A better understanding of the mechanism of ERα acetylation and deacetylation may lead to better therapies for ER negative and triple negative breast cancers.
5. TREATMENT/THERAPY
Breast cancer prevention, treatments, and diagnostics are being developed to target epigenetic changes leading to breast cancer. Treatments for breast cancer currently being evaluated focus on reversing aberrant DNA methylation and histone acetylation of tumor suppressor genes and genes involved in therapeutic response. Combinations of epigenetic targeted therapies with conventional chemotherapeutic agents may provide a way to resensitize drug-resistant tumors (reviewed in (55)). Also, combinations of epigenetic drug treatments could potentially work synergistically in increasing therapeutic affects. Examples of epigenetic drug treatments currently in clinical use include: 5-Azacytide, Procainamide, and Hydralazine(55).
ER-positive breast cancers can be treated with anti-estrogenic drugs, like Tamoxifen and Fluvestrant. ER-negative cells are no longer responsive to estrogen, therefore, anti-estrogenic drugs have no effect (9). A therapy that could restore gene expression of ERS1 (for ER-negative cancer patients) could reestablish cancer cell growth regulation through estrogen. After re-expression of ERS1, anti-estrogenic drugs could subsequently be used. A number of drugs including DNMT inhibitors and HDAC inhibitors are being used to reactivate ER expression. DNMTs and HDACs work synergistically to silence gene expression of ER. One problem found in treatments that target DNMTs and HDACs is that alone they may not be enough to reverse ER promoter hypermethylation. Fan et al. investigated whether long-term treatment with Tamoxifen or fulvestrant could case changes in DNA methylation. They studied DNA methylation profiles of Tamoxifen- and Fluvestrant-resistant MCF7 human breast cancer cell derivatives. Sixteen commonly hypermethylated/hypomethylated promoters were found in both resistant cell lines. These data suggest that distinct promoters are targeted for epigenetic modification by each drug, however more promoter hypomethylation was prevalent in antiestrogen-resistant sublines than MCF7 cells(56).
Tamoxifen is a commonly used drug for hormone-dependent (ERα positive) breast cancers. Initially, 70% of breast cancer patients with ERα positive lesions benefit from this antiestrogen therapy. Unfortunately, acquired resistance of Tamoxifen occurs in nearly 40% of patients. There have been numerous proposed mechanisms for why most patients eventually relapse with tumors resistant to Tamoxifen. Chang et al. (2005) investigated the proposed mechanism of the role of methylation of ERα and ERβ effect on Tamoxifen resistance in breast cancer. They found CpG methylation rate of ERβ gene is lower in Tamoxifen-resistant tumors than controls. Among the methylated tumors the Tamoxifen-resistant tumors showed denser methylation of the ERα and ERβ genes than controls. This result suggests that hypermethylation of the ERβ gene is involved in the development of Tamoxifen-resistance(57). It is important to identify patients most likely to respond to Tamoxifen and to identify individuals likely to acquire resistance. One promising biomarker for Tamoxifen response is WWOX. Preliminary studies show that WWOX expression levels predict Tamoxifen resistance better than the two previously known biomarkers, PR and HER2 (24). WWOX appears to mediate Tamoxifen sensitivity, and its expression is reduced in a large fraction (63.2%) of breast cancers (58). The primary mechanism of down-regulation of WWOX is through DNA hypermethylation of its regulatory region.
6. CONCLUSION
Epigenetic alterations are clearly involved in breast cancer initiation and progression. Early studies focused on single genes important in prognosis and prediction, but newer genome-wide methods are identifying many genes whose regulation is epigenetically altered during breast cancer progression. Detection of hypermethylation in specific genes like RASSF1A could be used as a form of surveillance to detect early stage breast cancer, however future studies may find that the addition of multiple genes and the inclusion of histone alterations to predictive panels may improve sensitivity and specificity. In addition to the use of epigenetic alterations as a means of screening, epigenetic alterations in a tumor or adjacent tissues may also help clinicians in determining prognosis and treatment in breast cancer patients. Analysis of histologically normal tumor margins for epigenetic alterations and field cancerization will increase the ability to remove all “pre-cancerous” tissues and decrease local recurrences. As we understand specific epigenetic alterations contributing to breast tumorigenesis and prognosis, these discoveries will lead to significant advances for breast cancer treatment. Therapeutics that target methylation and histone modifications in breast cancer already exist. Newer versions of these drugs are likely to play an important role in future clinical treatment. Since epigenetic modifications can also be used as biomarkers, targeted therapies may some day be used as preventative measures.
Acknowledgments
This work was funded in part by the American Cancer Society and the National Institutes of Health.
Footnotes
Conflict of interest: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Balch C, Montgomery JS, Paik HI, et al. New anti-cancer strategies: epigenetic therapies and biomarkers. Front Biosci. 2005;10:1897–931. doi: 10.2741/1668. [DOI] [PubMed] [Google Scholar]
- 2.Luczak MW, Jagodzinski PP. The role of DNA methylation in cancer development. Folia Histochem Cytobiol. 2006;44:143–54. [PubMed] [Google Scholar]
- 3.Widschwendter M, Jones PA. DNA methylation and breast carcinogenesis. Oncogene. 2002;21:5462–82. doi: 10.1038/sj.onc.1205606. [DOI] [PubMed] [Google Scholar]
- 4.Balch C, Huang THM, Nephew KP. High-Throughput Assessments of Epigenetics in Human Disease. In: Kim S, Mardis ER, Tang H, editors. Advances in Genome Sequencing Technology and Algorithms. Artech House Publishers, Inc; 2007. [Google Scholar]
- 5.Brena RM, Huang TH, Plass C. Quantitative assessment of DNA methylation: Potential applications for disease diagnosis, classification, and prognosis in clinical settings. J Mol Med. 2006;84:365–77. doi: 10.1007/s00109-005-0034-0. [DOI] [PubMed] [Google Scholar]
- 6.Hellebrekers DM, Griffioen AW, van Engeland M. Dual targeting of epigenetic therapy in cancer. Biochim Biophys Acta. 2007;1775:76–91. doi: 10.1016/j.bbcan.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Han J, Kim D, Morris KV. Promoter-associated RNA is required for RNA-directed transcriptional gene silencing in human cells. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12422–7. doi: 10.1073/pnas.0701635104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campan M, Weisenberger DJ, Laird PW. DNA methylation profiles of female steroid hormone-driven human malignancies. Current topics in microbiology and immunology. 2006;310:141–78. doi: 10.1007/3-540-31181-5_8. [DOI] [PubMed] [Google Scholar]
- 9.Giacinti L, Claudio PP, Lopez M, Giordano A. Epigenetic information and estrogen receptor alpha expression in breast cancer. Oncologist. 2006;11:1–8. doi: 10.1634/theoncologist.11-1-1. [DOI] [PubMed] [Google Scholar]
- 10.Mirza S, Sharma G, Prasad CP, et al. Promoter hypermethylation of TMS1, BRCA1, ERalpha and PRB in serum and tumor DNA of invasive ductal breast carcinoma patients. Life Sci. 2007;81:280–7. doi: 10.1016/j.lfs.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 11.Sharma D, Blum J, Yang X, Beaulieu N, Macleod AR, Davidson NE. Release of methyl CpG binding proteins and histone deacetylase 1 from the Estrogen receptor alpha (ER) promoter upon reactivation in ER-negative human breast cancer cells. Mol Endocrinol. 2005;19:1740–51. doi: 10.1210/me.2004-0011. [DOI] [PubMed] [Google Scholar]
- 12.Sui M, Huang Y, Park BH, Davidson NE, Fan W. Estrogen receptor alpha mediates breast cancer cell resistance to paclitaxel through inhibition of apoptotic cell death. Cancer Res. 2007;67:5337–44. doi: 10.1158/0008-5472.CAN-06-4582. [DOI] [PubMed] [Google Scholar]
- 13.Vincent-Salomon A, Ganem-Elbaz C, Manie E, et al. X inactive-specific transcript RNA coating and genetic instability of the X chromosome in BRCA1 breast tumors. Cancer Res. 2007;67:5134–40. doi: 10.1158/0008-5472.CAN-07-0465. [DOI] [PubMed] [Google Scholar]
- 14.Visvanathan K, Sukumar S, Davidson NE. Epigenetic biomarkers and breast cancer: cause for optimism. Clin Cancer Res. 2006;12:6591–3. doi: 10.1158/1078-0432.CCR-06-2001. [DOI] [PubMed] [Google Scholar]
- 15.Zhou Q, Davidson NE. Silencing estrogen receptor alpha in breast cancer cells. Cancer Biol Ther. 2006;5:848–9. doi: 10.4161/cbt.5.7.3205. [DOI] [PubMed] [Google Scholar]
- 16.Wajed SA, Laird PW, DeMeester TR. DNA methylation: an alternative pathway to cancer. Annals of surgery. 2001;234:10–20. doi: 10.1097/00000658-200107000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fackler MJ, Malone K, Zhang Z, et al. Quantitative multiplex methylation-specific PCR analysis doubles detection of tumor cells in breast ductal fluid. Clin Cancer Res. 2006;12:3306–10. doi: 10.1158/1078-0432.CCR-05-2733. [DOI] [PubMed] [Google Scholar]
- 18.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfeifer GP, Dammann R. Methylation of the tumor suppressor gene RASSF1A in human tumors. Biochemistry. 2005;70:576–83. doi: 10.1007/s10541-005-0151-y. [DOI] [PubMed] [Google Scholar]
- 20.Feng W, Liu Y, Wu J, Nephew KP, Huang TH, Li L. A Poisson mixture model to identify changes in RNA polymerase II binding quantity using high-throughput sequencing technology. BMC genomics. 2008;9 (Suppl 2):S23. doi: 10.1186/1471-2164-9-S2-S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muller HM, Widschwendter A, Fiegl H, et al. DNA methylation in serum of breast cancer patients: an independent prognostic marker. Cancer Res. 2003;63:7641–5. [PubMed] [Google Scholar]
- 22.Lewis CM, Cler LR, Bu DW, et al. Promoter hypermethylation in benign breast epithelium in relation to predicted breast cancer risk. Clin Cancer Res. 2005;11:166–72. [PubMed] [Google Scholar]
- 23.Agrawal A, Murphy RF, Agrawal DK. DNA methylation in breast and colorectal cancers. Mod Pathol. 2007;20:711–21. doi: 10.1038/modpathol.3800822. [DOI] [PubMed] [Google Scholar]
- 24.Guler G, Iliopoulos D, Guler N, Himmetoglu C, Hayran M, Huebner K. Wwox and Ap2γ Expression Levels Predict Tamoxifen Response. Clin Cancer Res. 2007:13. doi: 10.1158/1078-0432.CCR-07-1282. [DOI] [PubMed] [Google Scholar]
- 25.Yan PS, Chen CM, Shi H, et al. Dissecting complex epigenetic alterations in breast cancer using CpG island microarrays. Cancer Res. 2001;61:8375–80. [PubMed] [Google Scholar]
- 26.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963–8. doi: 10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 27.Yan PS, Venkataramu C, Ibrahim A, et al. Mapping geographic zones of cancer risk with epigenetic biomarkers in normal breast tissue. Clin Cancer Res. 2006;12:6626–36. doi: 10.1158/1078-0432.CCR-06-0467. [DOI] [PubMed] [Google Scholar]
- 28.Braakhuis BJ, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727–30. [PubMed] [Google Scholar]
- 29.Ushijima T. Epigenetic field for cancerization. Journal of biochemistry and molecular biology. 2007;40:142–50. doi: 10.5483/bmbrep.2007.40.2.142. [DOI] [PubMed] [Google Scholar]
- 30.Leu YW, Yan PS, Fan M, et al. Loss of estrogen receptor signaling triggers epigenetic silencing of downstream targets in breast cancer. Cancer Res. 2004;64:8184–92. doi: 10.1158/0008-5472.CAN-04-2045. [DOI] [PubMed] [Google Scholar]
- 31.Norton L. Conceptual and practical implications of breast tissue geometry: toward a more effective, less toxic therapy. Oncologist. 2005;10:370–81. doi: 10.1634/theoncologist.10-6-370. [DOI] [PubMed] [Google Scholar]
- 32.Maekita T, Nakazawa K, Mihara M, et al. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res. 2006;12:989–95. doi: 10.1158/1078-0432.CCR-05-2096. [DOI] [PubMed] [Google Scholar]
- 33.Huey-Jen L, Lin TZ, Lin Ching-Hung, Kuo Chieh Ti, Liyanarachchi Sandya, Sun Shuying, Shen Rulong, Deatherage Daniel E, Potter Dustin, Asamoto Lisa, Lin Shili, Yan Pearlly S, Cheng Ann-Lii, Ostrowski Michael C, Huang Tim HM. Breast Cancer-Associated Fibroblasts Confer AKT1-Mediated Epigenetic Silencing of Cystatin M in Epithelial Cells. Cancer Research. 2008:68. doi: 10.1158/0008-5472.CAN-08-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fiegl H, Millinger S, Mueller-Holzner E, et al. Circulating tumor-specific DNA: a marker for monitoring efficacy of adjuvant therapy in cancer patients. Cancer Res. 2005;65:1141–5. doi: 10.1158/0008-5472.CAN-04-2438. [DOI] [PubMed] [Google Scholar]
- 35.Chappuis PO, Kapusta L, Begin LR, et al. Germline BRCA1/2 mutations and p27(Kip1) protein levels independently predict outcome after breast cancer. J Clin Oncol. 2000;18:4045–52. doi: 10.1200/JCO.2000.18.24.4045. [DOI] [PubMed] [Google Scholar]
- 36.Robson ME, Chappuis PO, Satagopan J, et al. A combined analysis of outcome following breast cancer: differences in survival based on BRCA1/BRCA2 mutation status and administration of adjuvant treatment. Breast Cancer Res. 2004;6:R8–R17. doi: 10.1186/bcr658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stoppa-Lyonnet D, Ansquer Y, Dreyfus H, et al. Familial invasive breast cancers: worse outcome related to BRCA1 mutations. J Clin Oncol. 2000;18:4053–9. doi: 10.1200/JCO.2000.18.24.4053. [DOI] [PubMed] [Google Scholar]
- 38.Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res. 2006;8:R38. doi: 10.1186/bcr1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tapia T, Smalley SV, Kohen P, et al. Promoter hypermethylation of BRCA1 correlates with absence of expression in hereditary breast cancer tumors. Epigenetics. 2008;3:157–63. doi: 10.4161/epi.3.3.6387. [DOI] [PubMed] [Google Scholar]
- 40.Chenevix-Trench G, Healey S, Lakhani S, et al. Genetic and histopathologic evaluation of BRCA1 and BRCA2 DNA sequence variants of unknown clinical significance. Cancer Res. 2006;66:2019–27. doi: 10.1158/0008-5472.CAN-05-3546. [DOI] [PubMed] [Google Scholar]
- 41.Osorio A, de la Hoya M, Rodriguez-Lopez R, et al. Loss of heterozygosity analysis at the BRCA loci in tumor samples from patients with familial breast cancer. International journal of cancer. 2002;99:305–9. doi: 10.1002/ijc.10337. [DOI] [PubMed] [Google Scholar]
- 42.Osorio A, Milne RL, Honrado E, et al. Classification of missense variants of unknown significance in BRCA1 based on clinical and tumor information. Human mutation. 2007;28:477–85. doi: 10.1002/humu.20470. [DOI] [PubMed] [Google Scholar]
- 43.Meric-Bernstam F. Heterogenic loss of BRCA in breast cancer: the “two-hit” hypothesis takes a hit. Annals of surgical oncology. 2007;14:2428–9. doi: 10.1245/s10434-007-9379-7. [DOI] [PubMed] [Google Scholar]
- 44.Esteller M, Fraga MF, Guo M, et al. DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Human molecular genetics. 2001;10:3001–7. doi: 10.1093/hmg/10.26.3001. [DOI] [PubMed] [Google Scholar]
- 45.Press JZ, De Luca A, Boyd N, et al. Ovarian carcinomas with genetic and epigenetic BRCA1 loss have distinct molecular abnormalities. BMC cancer. 2008;8:17. doi: 10.1186/1471-2407-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu X, Gammon MD, Zhang Y, et al. BRCA1 promoter methylation is associated with increased mortality among women with breast cancer. Breast cancer research and treatment. 2008 doi: 10.1007/s10549-008-0075-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Honrado E, Osorio A, Milne RL, et al. Immunohistochemical classification of non-BRCA1/2 tumors identifies different groups that demonstrate the heterogeneity of BRCAX families. Mod Pathol. 2007;20:1298–306. doi: 10.1038/modpathol.3800969. [DOI] [PubMed] [Google Scholar]
- 48.Reis-Filho JS, Tutt AN. Triple negative tumours: a critical review. Histopathology. 2008;52:108–18. doi: 10.1111/j.1365-2559.2007.02889.x. [DOI] [PubMed] [Google Scholar]
- 49.Feng W, Shen L, Wen S, et al. Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res. 2007;9:R57. doi: 10.1186/bcr1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Human molecular genetics. 2007;16:R50–9. doi: 10.1093/hmg/ddm018. Spec No 1. [DOI] [PubMed] [Google Scholar]
- 51.Matarazzo MR, De Bonis ML, Strazzullo M, et al. Multiple binding of methyl-CpG and polycomb proteins in long-term gene silencing events. Journal of cellular physiology. 2007;210:711–9. doi: 10.1002/jcp.20879. [DOI] [PubMed] [Google Scholar]
- 52.Margueron R, Duong V, Castet A, Cavailles V. Histone deacetylase inhibition and estrogen signalling in human breast cancer cells. Biochemical pharmacology. 2004;68:1239–46. doi: 10.1016/j.bcp.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 53.Wang C, Fu M, Angeletti RH, et al. Direct acetylation of the estrogen receptor alpha hinge region by p300 regulates transactivation and hormone sensitivity. The Journal of biological chemistry. 2001;276:18375–83. doi: 10.1074/jbc.M100800200. [DOI] [PubMed] [Google Scholar]
- 54.Popov VM, Wang C, Shirley LA, et al. The functional significance of nuclear receptor acetylation. Steroids. 2007;72:221–30. doi: 10.1016/j.steroids.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dworkin A, Huang THM, Toland AE. The role of Epigenetics in Breast Cancer: Implications for Diagnosis, Prognosis, and Treatment. In: Leyland-Jones B, editor. Pharmacogenetics of Breast Cancer. New York: Informa Healthcare USA; 2008. pp. 45–59. [Google Scholar]
- 56.Fan M, Yan PS, Hartman-Frey C, et al. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res. 2006;66:11954–66. doi: 10.1158/0008-5472.CAN-06-1666. [DOI] [PubMed] [Google Scholar]
- 57.Chang HG, Kim SJ, Chung KW, et al. Tamoxifen-resistant breast cancers show less frequent methylation of the estrogen receptor beta but not the estrogen receptor alpha gene. J Mol Med. 2005;83:132–9. doi: 10.1007/s00109-004-0596-2. [DOI] [PubMed] [Google Scholar]
- 58.Guler G, Uner A, Guler N, et al. The fragile genes FHIT and WWOX are inactivated coordinately in invasive breast carcinoma. Cancer. 2004;100:1605–14. doi: 10.1002/cncr.20137. [DOI] [PubMed] [Google Scholar]