

Abstract
Objective
Vascular NADPH oxidases (Noxes) have been implicated in cardiovascular diseases; however, the importance of individual Nox homologues remains unclear. Here, the role of the vascular smooth muscle cell (VSMC) Nox1 in neointima formation was studied using genetically modified animal models.
Methods and results
Wire injury-induced neointima formation in the femoral artery, along with proliferation and apoptosis, was reduced in Nox1y/- mice, but there was little difference in TgSMCnox1 mice compared with wild type (WT) mice. Proliferation and migration were reduced in cultured Nox1y/- VSMCs and increased in TgSMCnox1 cells. TgSMCnox1 cells exhibited increased fibronectin secretion, but neither collagen I production nor cell adhesion was affected by alteration of Nox1. Using antibody microarray and Western blotting analysis, increased cofilin phosphorylation and mDia1 expression and decreased PAK1 expression were detected in Nox1y/- cells. Overexpression of S3A, a constitutively active cofilin mutant, partially recovered reduced migration of Nox1y/- cells, suggesting that reduction in cofilin activity contributes to impaired migration of Nox1y/- VSMCs.
Conclusions
These results indicate that Nox1 plays a critical role in neointima formation by mediating VSMC migration, proliferation and extracellular matrix production, and that cofilin is a major effector of Nox1-mediated migration. Inhibition of Nox1 may be an efficient strategy to suppress neointimal formation.
Introduction
The abnormal intimal growth of blood vessels as a `response to injury' is key in the development of vascular occlusive diseases such as in-stent stenosis, intimal proliferation following vein grafts and atherosclerosis; hence, it is the major limitation for the efficacy of corrective surgery.1 Vascular smooth muscle cells (VSMCs) are a main constituent of the neointima in these lesions. Following injury, VSMCs migrate to the damaged area, proliferate and elaborate extracellular matrix (ECM), largely in response to platelet-derived growth factor (PDGF) stimulation.2 The molecular mechanisms underlying these events are poorly understood.
Reactive oxygen species (ROS) such as superoxide and hydrogen peroxide mediate signal transduction pathways that contribute to the pathophysiological responses of VSMCs including migration, proliferation, apoptosis, phenotypic modulation, and hypertrophy.3 Major sources of ROS in VSMCs, especially in pathological conditions, are the NADPH oxidase (Nox) family of enzymes. VSMCs from conduit arteries express Nox1 and Nox4,4 while those from resistance arteries express Nox2 and Nox4.5 These oxidases serve different functions within the cells,6 presumably owing to their distinct intracellular compartmentalization and different mechanism of regulation and activation.7 Of interest, studies have linked Nox1 to VSMC phenotypic changes including angiotensin II-induced hypertrophy8, serum-induced proliferation4, ECM production9 and basic fibroblast growth factor (bFGF)-induced migration.10
In vivo studies of the role of specific Nox homologues in vascular lesions are limited. However, enhanced generation of superoxide and increased NADPH oxidase expression or activity are observed in rat balloon-injured carotid11 and coronary12 arteries and vein grafts,13. In accordance with these findings, antioxidant treatment with tempol or N-acetyl-cysteine reduces injury-induced restenosis.14 Furthermore, superoxide production is prominent in neointimal and medial SMCs after carotid injury,11 and intimal SMCs are predominantly responsible for the elevated NADPH oxidase activity in venous bypass graft intimal hyperplasia.13 The expression of Nox1 increases early in the restenotic response and remains elevated during the growth phase of the lesion.11
Based on these observations, we hypothesized that Nox1-derived ROS participate in neointimal formation by mediating PDGF-induced signaling. We tested this by subjecting Nox1 knockout (KO) and smooth muscle-specific, Nox1 overexpressing (OE) mice to wire injury of the femoral artery. Our data show that deletion of Nox1 indeed impairs the response to injury, support a role for Nox1 in proliferation, migration and extracellular matrix secretion, and provide insight into the signaling that regulates such responses. Understanding the role of specific NADPH oxidases such as Nox1 will permit better design of therapies targeted to reducing oxidative stress in vascular disease.
Materials and Methods
An expanded Materials and Methods section is available in the online data supplement at http://atvb.ahajournals.org.
Reagents
All reagents and antibodies used here were purchased from standard suppliers. The coding sequence for the S3A cofilin mutant in pcDNA3 expression vector was kindly provided by Dr. J. S. Condeelis (Albert Einstein College of Medicine, Bronx, New York).15 Anti-Nox4 rabbit polyclonal antibody was prepared as previously described.7
Animals
Nox1y/- mice were generated by Dr. K. H. Krause.9, 16 TgSMCnox1 mice, transgenic mice overexpressing Nox1 in smooth muscle were also previously described.8 All mice are fully backcrossed onto a C57Bl/6 background.
Mouse femoral artery injury model
Transluminal mechanical injury of bilateral femoral arteries was induced by introducing a large wire as previously reported.17 At 21 days, the mice were sacrificed and pressure-perfused at 100 mm Hg with 0.9% sodium chloride, followed by pressure-fixation with 10% formalin. Arteries were then carefully excised and embedded in paraffin. To assess the early apoptotic response to injury, arteries were obtained 2 hrs after injury induction, as described previously.17
Histological analysis of neointima
Hematoxylin and eosin (H&E) staining was used to assess morphological analysis. Proliferating cell nuclear antigen (PCNA) and TUNEL staining were performed to identify proliferating and apoptotic cells, respectively. Fibronectin and collagen were measured to determine matrix accumulation. Mac-3 was used to detect macrophages. Images were acquired with an Axioskop microscope and Axiocam CCD camera, and analyzed using NIH ImageJ or MetaMorph (Molecular Devices) software. Percent stenosis was calculated from the ratio of intimal area to the area inside the internal elastic lamina x 100.
Cell culture
VSMCs were isolated from mouse aorta by enzymatic digestion18 and used between passages 3 to 10.
RNA extraction and reverse transcriptase-polymerase chain reaction (RT-PCR)
Total RNA was extracted with the RNeasy kit (Qiagen). cDNA was synthesized using random primers and Superscript II (Invitrogen)4. For PCR, primers (forward: 5'-CTGAGGGGCACCTGCTCATT-3', backward: 5'-CTGGAATTTGTACCAGATAGATTTCAAG-3') were designed to amplify both mouse and human Nox1 cDNA, but not DNA from KO cells that lack the annealing site for the forward primer.
NADPH oxidase activity
NADPH oxidase activity was assayed in membrane preparations.19 Superoxide was quantified by NADPH-dependent, SOD-inhibitable formation of 3-carboxy-proxyl radical (CP•) from 1-hydroxy-3-carboxy-pyrrolidine (CPH), using ESR.
Cell proliferation assay
Proliferation was assessed by cell counting as previously described.20
Migration assay
Migration was measured using a modified Boyden chamber assay. Migration was stimulated by 10 ng/ml PDGF for 3 hrs. Migrated cells were stained with DAPI. Four (x20) fields were visualized using Zeiss Axioskop and quantified with NIH ImageJ.
Cell transfection
VSMCs were transfected with pcDNA3/S3Acofilin or pcDNA3 by electroporation using a Nucleofector (Amaxa Biosystems). The transfection was made 12 hrs before a 48 h serum deprivation prior to the experiments.
Extracellular matrix (ECM) production
ECM production was assessed by measuring collagen I and fibronectin in the culture media. Serum-deprived VSMCs were maintained in media containing 50 μg/ml ascorbic acid and 50 μg/ml β-aminopropionitrile (β-APN) with or without 10 ng/ml PDGF. After 72 hrs, media was collected and subjected to Western blot analysis. To correct for possible differences in ECM protein generation caused by differences in cell growth rate, gel loading was adjusted for total protein concentration of the media.
Cell adhesion assay
VSMCs were plated and allowed to adhere for 30, 60 and 90 min. Attached cells were fixed/stained with 0.5% crystal violet. Dye was extracted with 0.5 ml of 1% SDS, and absorbance was measured at 590 nm.
Antibody microarray
Characterization of expression and phosphorylation of signaling molecules was performed by Kinexus (Vancouver, Canada) using the Kinex Antibody Microarray. All changes reported in Results were confirmed by Western analysis.
Western blotting
VSMCs lysates were prepared and subjected to SDS-PAGE.21 Immunoreactive proteins were detected by chemiluminescence. Band intensity was quantified by densitometry using NIH ImageJ.
Statistical Analysis
Data are reported as mean±SE. Statistical significance was assessed by ANOVA on untransformed data, followed by contrast analysis. A p<0.05 was considered statistically significant.
Results
Analysis of injury-induced neointima formation and histological characteristics in Nox1y/- and TgSMCnox1 mice
To assess the role of Nox1 in neointimal formation, a wire-injury model of the mouse femoral artery was employed. Three weeks after injury, morphological analysis of the injured artery was performed. We confirmed that there was no increased intimal or medial thickness in sham-operated mice. As shown in Figure 1, both intimal thickness and cross-sectional area of femoral arteries were significantly reduced in Nox1y/- mice, but there was minimal change in TgSMCnox1 mice. No significant difference was found in medial thickness or area among WT, Nox1y/- and TgSMCnox1 mice. Accordingly, the intima/media ratio and % stenosis were diminished in Nox1y/- mice.
Figure 1.

Analysis of neointima formation in WT, Nox1y/- and TgSMCnox1 mice. Injury was induced by insertion of a guide wire into the left femoral artery, and arteries were harvested after 21 days. A. Representative images of arterial cross-sections from WT, Nox1y/- and TgSMCnox1 mice. B. Assessment of medial and intimal area, intima/media ratio, and %stenosis. Areas of media and intima were averaged from duplicate cross-sections, from which intima/media ratio and %stenosis were calculated. Values are means±SE of 13 (WT) and 6 (Nox1y/- and TgSMCnox1) animals. *Significantly different from WT control (p < 0.05). Scale bar=50 μm.
Cellular responses related with neointimal growth, proliferation, apoptosis, matrix accumulation were examined with histological methods such as PCNA, TUNEL, fibronectin and collagen staining. Low levels of PCNA expression and TUNEL positive cells were observed in neointima of Nox1y/- arteries, whereas TgSMCnox1 arteries were not different from WT arteries (Figure 2A and B). Expression of fibronectin was lower in Nox1y/- arteries; however, there was no significant difference in collagen among groups (Figure 2C). To investigate the involvement of inflammation in this injury model, we stained for macrophages using Mac3-conjugated quantum dots (Figure S1). Macrophage distribution was not detected in the neointima, consistent with previous reports,17 and no difference was observed among groups. These results suggest that Nox1 is necessary for neointimal formation, but overexpression in smooth muscle itself does not augment the response to injury.
Figure 2.

Histological analysis of proliferation, apoptosis, and extracellular matrix distribution in arterial cross-sections obtained from WT, Nox1y/- and TgSMCnox1 mice. Arterial sections obtained on day 21 after injury induction were stained for proliferating cell nuclear antigen (PCNA) (A), fibronectin (C, top panels) and collagen (C, bottom panels). TUNEL staining was performed after 2 hrs of injury induction (B). PCNA and TUNEL positive cells were stained red and dark brown, respectively (A and B). Red or orange color indicates fibronectin or collagen, respectively (C). Background images were obtained from WT arteries omitting primary antibody staining (A and C upper panel), and nuclei were counterstained with hematoxylin (C upper panel). Arrowhead indicates positive staining of PCNA (A), and neointimal area analyzed for fibronectin is indicated with a solid line in images (C upper panel). Images for PCNA, TUNEL, collagen and fibronectin staining are representative of sections from at least 3 animals of each genotype. Mean±SE are provided to the right of each image. `M' and `N' denote media and neointima (A). Scale bar=30 (A), 20 (C upper panel), 50 (B and C lower panel) μm.
Characterization of VSMCs from Nox1y/- and TgSMCnox1 mice
To determine how Nox1 influences the cellular functions of smooth muscle that contribute to neointimal formation, we prepared VSMCs from Nox1y/- or TgSMCnox1 animals. Cell identity was confirmed with smooth muscle α-actin and calponin staining (Figure S2), and nox1 expression was detected by RT-PCR. As predicted, nox1 expression was undetectable in Nox1y/- cells, while TgSMCnox1 cells exhibited a marked increase (Figure 3A). Because rodent aortic SMCs exclusively express Nox1 and Nox4 among gp91 homologues,4 we investigated a possible compensatory Nox4 upregulation by measuring Nox4 expression in Nox1y/- and TgSMCnox1 cells. However, consistent with previous studies,8, 16 we found no change in Nox4 basal expression (Figure 3B). This excludes a differential effect of Nox4 in the following cell function studies employing Nox1y/- and TgSMCnox1 cells.
Figure 3.

Characterization of primary-cultured aortic smooth muscle cells from Nox1y/- and TgSMCnox1 mice. A. PCR was conducted with primer pairs that can distinguish cDNA of Nox1y/- cells from that of WT or TgSMCnox1 cells. PCR products were detected on agarose gel electrophoresis. A representative gel image is presented. B. Nox4 protein expression was measured by Western blotting. CDK4 was measured as a loading control. Representative images are shown in upper panels. Values are means±SE from 3 independent experiments. C. Serum-starved VSMCs were treated with 25 ng/ml PDGF for 4 h, which corresponds to maximal activation of NADPH oxidase. Membrane fractions were prepared, and NADPH-dependent, SOD-inhibitable superoxide generation was measured with ESR using CPH. Values shown are means±SE from 5 independent experiments. *Significantly different from corresponding control (p < 0.05).
To test whether alteration of Nox1 expression reflects a functional change in NADPH oxidase activity, NADPH oxidase-dependent superoxide production was measured in basal and PDGF-activated conditions. As shown in Figure 3C, WT superoxide production was increased by PDGF, which is consistent with previous reports demonstrating that Nox1 is PDGF-inducible.4 However, such an increase was not observed in Nox1y/- cells, whereas TgSMCnox1 exhibited higher levels of superoxide compared with WT cells both basally and after treatment. Compared with Nox4, basal expression of Nox1 is remarkably low;4 therefore, basal production of superoxide was not different between Nox1y/- and WT cells. These results confirm that manipulations of Nox1 expression are in fact functional.
Effect of Nox1 on VSMC proliferation, migration, adhesion and extracellular matrix production
VSMC proliferation, adhesion, extracellular matrix (ECM) production and migration are major contributors to neointimal formation.22-24 Hence, the effect of Nox1 on these cellular functions was tested in Nox1-modified VSMCs. Nox1y/- cells had a slightly, but significantly, reduced proliferative activity, while the growth rate of TgSMCnox1 cells was higher than that of WT cells (Figure 4A). Cellular migration was modified to a greater extent than proliferation. Compared to that in WT cells, PDGF-induced migration was inhibited by 35 ± 8% in Nox1y/- cells, and both basal and PDGF-stimulated migration was significantly increased in TgSMCnox1 cells (180 ± 17% and 180 ± 23% of WT cells, respectively, Figure 4B). This was not due to alterations in cell adhesion, since altering Nox1 expression did not induce any difference in adhesion capacity (Figure S3). These results suggest that Nox1 contributes to the proliferative and migratory responses of VSMCs.
Figure 4.

Effect of Nox1 expression on proliferation, migration and extracellular matrix (ECM) production. A. Growth curves were obtained from WT, Nox1y/- and OE cells. B. Cell migration was assessed using the modified Boyden chamber assay. Quiescent VSMCs were treated with 10 ng/ml PDGF for 3 h. C. Measurement of collagen I and fibronectin in culture media. Serum-starved VSMCs were maintained in the presence of 50 μg/mL ascorbic acid and β-aminopropionitrile with or without 10 ng/ml PDGF for 72 h. Media was collected and subjected to Western blotting. Representative immunoblots are shown in C. Values are means±SE of 6 (A) and 3 (B and C) independent observations. * Significantly different from corresponding control (p < 0.05).
The effect of Nox1 on ECM deposition was assessed by measuring the most abundant proteins present in ECM, collagen I and fibronectin. 22, 25, 26 Both basal and PDGF-stimulated fibronectin production were significantly enhanced in TgSMCnox1 compared with WT cells, while Nox1 knockout had no effect (Figure 4C). Neither Nox1y/- nor TgSMCnox1 cells exhibited significant changes in collagen I production. It thus appears that Nox1 is capable of inducing fibronectin production, but may not be required for this response.
Mechanism of Nox1-mediated VSMC migration
The ROS-sensitive mechanisms underlying Nox1-mediated proliferation have been well studied,6, 27, 28 but those regulating migration are less understood. To clarify these mechanisms, we examined potential signaling molecules responsible for Nox1-mediated functional alterations of VSMCs. The goal was to identify targets that satisfied two criteria: relation with migration or proliferation, and regulation by ROS. Initially, we tested whether the activation of PDGF receptor itself is altered in Nox1y/- or TgSMCnox1 cells. Both the total expression and site-specific phosphorylation at Tyr716, an indicator of intrinsic receptor tyrosine kinase activity, were examined. Neither expressed protein nor activation by PDGF was changed by differential expression of Nox1 (Figure S4). Antibody microarray analysis was then performed with 4 samples, WT-untreated cell lysate, and lysates from WT, Nox1y/- and TgSMCnox1 cells treated with 10 ng/ml PDGF for 5 min. Cogent results from the antibody microarray were confirmed with Western blotting. No significant difference was detected in total protein and/or phosphorylated levels of Src, PLCγ1, Erk1/2, p38, PRK1, p85S6K, PKCδ, PP2A, or vinculin (Table S1). However, significant changes in protein expression or activation were detected in the cytoskeletal-associated proteins cofilin (a regulator of actin depolymerization), mDia1 (a RhoA adapter protein), and PAK1 (a serine/threonine kinase that promotes cytoskeletal reorganization) (Table S1 and Figure 5). Nox1y/- cells exhibited more phosphorylated cofilin both basally and after PDGF stimulation, without alteration of cofilin expression. Expression of mDia1 was increased and PAK1 was decreased in Nox1y/- cells. Taken together, these data suggest that Nox1 may regulate migration by modulating the actin cytoskeleton.
Figure 5.

Alteration of cofilin, mDia and PAK1 expression or activity in WT, Nox1y/- and TgSMCnox1 VSMCs. Lysates were prepared from serum-deprived, untreated cells for total protein measurement. To investigate phosphocofilin levels, serum-starved VSMCs were treated with or without 10 ng/ml PDGF for 15 min, and cell lysates were subjected to Western blotting. Band density of each protein was normalized to that of CDK4 (cofilin and PAK1), cofilin (phospho-cofilin), or β-tubulin (mDia1), and the relative fold-changes over WT groups were calculated. Values are means ± SE of 4 (cofilin) or 3 (phospho-cofilin, mDia1, PAK1) independent experiments. * Significantly different from corresponding control (p < 0.05).
Cofilin is known to be primarily involved in cytoskeletal reorganization by depolymerizing actin, which influences cell migration and growth.29 Phosphorylation of cofilin at Ser3 inhibits its activity.15 The observed increase in cofilin phosphorylation suggests that persistent inactivation of cofilin may explain the impaired migration in Nox1y/- cells. To examine this hypothesis, we attempted to reverse the reduced migration in Nox1y/- cells by expressing a constitutively active cofilin mutant, S3A.15 As shown in Figure 6, S3A recovered the impaired migration of Nox1y/- cells to the control level. This result suggests that decreased cofilin activity is mainly responsible for reduced migration of Nox1y/- cells.
Figure 6.
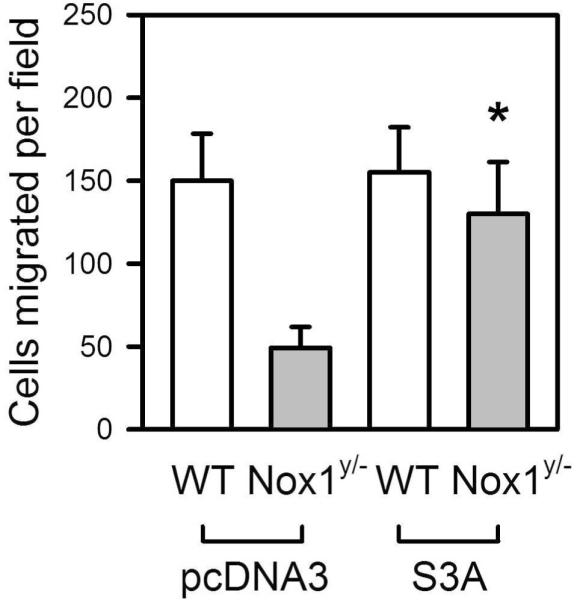
Recovery of impaired migration in Nox1y/- VSMCs by expression of constitutively active cofilin, S3A. pcDNA3/S3A or pcDNA3 was introduced into Nox1y/- or WT VSMCs by electroporation. After 12 h, serum was removed for 48 h, and migration induced by PDGF (10 ng/ml) was measured. Values are means±SE of 3 independent observations. *Significantly different from corresponding control, Nox1y/- cells transfected with pcDNA (p < 0.05).
Discussion
The results of this study support a role for Nox1 in the process of neointima formation induced by injury. Nox1 is required for the formation of neointima, but overexpression in VSMC above the normal upregulation of Nox1 after injury does not significantly enhance injury-induced neointimal formation. In vitro studies reveal that both migration and growth of VSMCs are dependent on expression of Nox1. Regulation of cytoskeletal dynamics is presumably responsible for the effect of Nox1 on migration, and in particular, cofilin serves as Nox1 effector. Thus, suppression of Nox1 may be a promising strategy to treat vascular diseases associated with neointimal formation.
The process of neointimal formation is complex, involving endothelial injury, thrombosis, phenotypic transformation followed by migration and proliferation of VSMCs, elaboration of ECM, redifferentiation of VSMCs and regrowth of the endothelium. For this reason, we studied the overall effect of Nox1y/- or TgSMCnox1 in vivo, and used in vitro assays to probe those events related to VSMCs. The reduced neointima in Nox1y/- mice indicates that Nox1 is necessary for the neointimal response. In contrast, its overexpression does not induce a further response, since neointimal formation in TgSMCnox1 mice was similar to WT. In other words, additional activity of Nox1 does not evoke stronger signals, perhaps because Nox1 is already upregulated in injured WT arteries.11 It is also possible that because Nox1 mediates early proliferative and migratory responses, potential differences between TgSMCnox1 and WT mice were obscured by 3 weeks after injury. This view is well correlated with the finding that, in these experimental conditions, there was no prominent increase in proliferating cells in TgSMCnox1 (Figure 2A). Finally, non-VSMC components contribute to neointimal formation in vivo, making it difficult to quantify enhanced smooth-muscle specific responses in this setting. For these reasons, the reduction in neointimal formation in Nox1y/- animals gives a clearer picture of the importance of Nox1 in this response.
Our studies suggest several mechanisms by which Nox1 can affect lesion formation and healing in vivo. Previous studies showed that Nox1 mediates cell proliferation.20, 27, 28 Similarly, we found that overexpression of Nox1 enhanced, while knockout of Nox1 inhibited, proliferation. A similar pattern was seen for Nox1 in migration. Nox1 knockout also resulted in reduced apoptosis after injury, which is consistent with previous reports demonstrating the involvement of ROS signaling in apoptotic signals.30 The role of apoptosis in neointima formation is still controversial, but early apoptosis after injury is generally believed to induce a greater wound healing process.31 Therefore, the observed reduction of the apoptotic response in Nox1y/- arteries is consistent with subsequent decreased neointimal growth. However, we saw a limited effect of Nox1 on ECM production. Alteration of Nox1 had no effect on collagen in culture or in vivo. Fibronectin accumulation was reduced in the neointima of Nox1y/- arteries and augmented in TgSMCnox1 VSMCs. This suggests that Nox1 may differentially regulate matrix components, leading to enrichment of fibronectin. Taken together, our results indicate that Nox1 mediates multiple processes associated with the de-differentiated, synthetic VSMC phenotype.
Because others have delineated many of the growth-related signaling pathways associated with Nox1,27, 28 we focused here on understanding the mechanisms by which Nox1 regulates cell migration. The Src/phosphoinositide-dependent kinase-1 (PDK1)/PAK signaling axis has been implicated as a major mediator of PDGF-induced, ROS- and NADPH oxidase-dependent migration of VSMCs,32, 33 while thrombin-induced migration appears to depend on ROS-regulated p38 MAPK activation,34 and c-Jun-N-terminal kinase (JNK) regulates bFGF-induced, Nox1-dependent migration of VSMCs.10 However, we found no changes in Src, p38 MAPK (Table S1), or JNK (in antibody microarray, not shown), suggesting that migratory signaling may be agonist- and Nox-specific.
Migration is an integrated, dynamic, and cyclical process, dependent upon a well-orchestrated regulation of the actin cytoskeleton.35 Important regulators of the actin cytoskeleton include cofilin, mDia1 and PAK1, all of which are effectors of the Rho GTPase.36 Cofilin is capable of disassembling and severing actin filaments.29 The activity of cofilin is controlled by its phosphorylation state: Enhancement of cofilin activity by dephosphorylation accelerates filamentous actin turnover and migration, while its phosphorylation impairs actin turnover.37 Our data revealed that Nox1y/- cells possess less active cofilin than WT cells in both basal and PDGF-activated conditions. In addition, rescue of cofilin activity with a constitutively active mutant restored impaired migration of Nox1y/- cells, suggesting that cofilin is responsible for the impaired migration of Nox1y/- cells. These results are consistent with a previous report demonstrating that Nox1 mediates Ras-induced cofilin activation (dephosphorylation), and silencing of Nox1 by siRNA recovers phosphocofilin levels in normal rat kidney fibroblasts.38
The reason for the increase in cofilin phosphorylation in Nox1y/- cells is unclear. PAK1 is upstream of cofilin, and activates the cofilin kinase LIMK,36 so one would expect its expression/activity to be increased, rather than decreased. The observed decrease in PAK expression may thus represent an attempt by the cell to compensate for persistent cofilin inactivation. In support of a compensatory effect, we also found an increase in mDia1 in Nox1y/- cells. mDia contributes to cytoskeletal remodeling by regulating actin polymerization through profilin interaction, and promoting the stabilization and polarization of microtubules.39, 40 Loss of mDia is associated with impaired T-lymphocyte trafficking,41 so increased mDia expression is unlikely to account for impaired migration in the Nox1y/- cells. One other possibility is that Nox1 may influence slingshot (SSH) phosphatase, which mediates PDGF-induced cofilin phosphorylation.42 The redox-sensitivity of SSH is unknown; however, Nox1 may affect cofilin dephosphorylation by regulating SSH activation. Nonetheless, it is clear that Nox1 has a strong influence on cytoskeletal dynamics that will require further investigation.
In summary, Nox1 is a critical element of neointimal formation after vascular injury. It apparently exerts its effects in part by modulating VSMC growth and migration, and by influencing matrix accumulation. Because neointima formation is a complex and multistage process in which diverse cell types participate,43-47 the clear reduction in neointima in Nox1y/- mice suggests that the role of Nox1 should be investigated in other vascular cells as well. Meanwhile, these results identify Nox1 as a potentially novel target for therapy aimed at reduction of intimal hyperplasia.
Acknowledgments
This work was supported by NIH grants HL38206, HL093115 and HL058863, and American Heart Association fellowship #0525465B to ASM. We are grateful to Dr. John Condeelis for providing the S3A plasmid.
Footnotes
Disclosures None.
References
- 1.Zargham R. Preventing restenosis after angioplasty: a multistage approach. Clin Sci (Lond) 2008;114:257–264. doi: 10.1042/CS20070228. [DOI] [PubMed] [Google Scholar]
- 2.Ferns GA, Raines EW, Sprugel KH, Motani AS, Reidy MA, Ross R. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science. 1991;253:1129–1132. doi: 10.1126/science.1653454. [DOI] [PubMed] [Google Scholar]
- 3.Clempus RE, Griendling KK. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc Res. 2006;71:216–225. doi: 10.1016/j.cardiores.2006.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lassègue B, Sorescu D, Szöcs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91phox homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 5.Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, Schiffrin EL. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ Res. 2002;90:1205–1213. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 6.Lyle AN, Griendling KK. Modulation of vascular smooth muscle signaling by reactive oxygen species. Physiology (Bethesda) 2006;21:269–280. doi: 10.1152/physiol.00004.2006. [DOI] [PubMed] [Google Scholar]
- 7.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:677–683. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- 8.Dikalova A, Clempus R, Lassègue B, Cheng G, McCoy J, Dikalov S, San Martin A, Lyle A, Weber DS, Weiss D, Taylor WR, Schmidt HHW, Owens GK, Lambeth JD, Griendling KK. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–2676. doi: 10.1161/CIRCULATIONAHA.105.538934. [DOI] [PubMed] [Google Scholar]
- 9.Gavazzi G, Deffert C, Trocme C, Schappi M, Herrmann FR, Krause KH. NOX1 deficiency protects from aortic dissection in response to angiotensin II. Hypertension. 2007;50:189–196. doi: 10.1161/HYPERTENSIONAHA.107.089706. [DOI] [PubMed] [Google Scholar]
- 10.Schroder K, Helmcke I, Palfi K, Krause KH, Busse R, Brandes RP. Nox1 mediates basic fibroblast growth factor-induced migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2007;27:1736–1743. doi: 10.1161/ATVBAHA.107.142117. [DOI] [PubMed] [Google Scholar]
- 11.Szöcs K, Lassègue B, Sorescu D, Hilenski LL, Valppu L, Couse TL, Wilcox JN, Quinn MT, Lambeth JD, Griendling KK. Upregulation of Nox-based NAD(P)H oxidases in restenosis after carotid injury. Arterioscler Thromb Vasc Biol. 2002;22:21–27. doi: 10.1161/hq0102.102189. [DOI] [PubMed] [Google Scholar]
- 12.Shi Y, Niculescu R, Wang D, Patel S, Davenpeck KL, Zalewski A. Increased NAD(P)H oxidase and reactive oxygen species in coronary arteries after balloon injury. Arterioscler Thromb Vasc Biol. 2001;21:739–745. doi: 10.1161/01.atv.21.5.739. [DOI] [PubMed] [Google Scholar]
- 13.West N, Guzik T, Black E, Channon K. Enhanced superoxide production in experimental venous bypass graft intimal hyperplasia: role of NAD(P)H oxidase. Arterioscler Thromb Vasc Biol. 2001;21:189–194. doi: 10.1161/01.atv.21.2.189. [DOI] [PubMed] [Google Scholar]
- 14.Kappert K, Sparwel J, Sandin A, Seiler A, Siebolts U, Leppanen O, Rosenkranz S, Ostman A. Antioxidants relieve phosphatase inhibition and reduce PDGF signaling in cultured VSMCs and in restenosis. Arterioscler Thromb Vasc Biol. 2006;26:2644–2651. doi: 10.1161/01.ATV.0000246777.30819.85. [DOI] [PubMed] [Google Scholar]
- 15.Zebda N, Bernard O, Bailly M, Welti S, Lawrence DS, Condeelis JS. Phosphorylation of ADF/cofilin abolishes EGF-induced actin nucleation at the leading edge and subsequent lamellipod extension. J Cell Biol. 2000;151:1119–1128. doi: 10.1083/jcb.151.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, Krause KH. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006;580:497–504. doi: 10.1016/j.febslet.2005.12.049. [DOI] [PubMed] [Google Scholar]
- 17.Sata M, Maejima Y, Adachi F, Fukino K, Saiura A, Sugiura S, Aoyagi T, Imai Y, Kurihara H, Kimura K, Omata M, Makuuchi M, Hirata Y, Nagai R. A mouse model of vascular injury that induces rapid onset of medial cell apoptosis followed by reproducible neointimal hyperplasia. J Mol Cell Cardiol. 2000;32:2097–2104. doi: 10.1006/jmcc.2000.1238. [DOI] [PubMed] [Google Scholar]
- 18.Ohmi K, Masuda T, Yamaguchi H, Sakurai T, Kudo Y, Katsuki M, Nonomura Y. A novel aortic smooth muscle cell line obtained from p53 knock out mice expresses several differentiation characteristics. Biochem Biophys Res Commun. 1997;238:154–158. doi: 10.1006/bbrc.1997.7218. [DOI] [PubMed] [Google Scholar]
- 19.Hanna IR, Dikalova A, Hilenski L, Quinn MT, Griendling KK. Nox 1 binds p22phox to form a functional oxidase in vascular smooth muscle cells (VSMCs) Circulation. 2002;106:II–164. [Google Scholar]
- 20.Suh Y, Arnold RS, Lassègue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 21.Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, Schmidt HH, Lassegue B, Griendling KK. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2007;27:42–48. doi: 10.1161/01.ATV.0000251500.94478.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farb A, Kolodgie FD, Hwang JY, Burke AP, Tefera K, Weber DK, Wight TN, Virmani R. Extracellular matrix changes in stented human coronary arteries. Circulation. 2004;110:940–947. doi: 10.1161/01.CIR.0000139337.56084.30. [DOI] [PubMed] [Google Scholar]
- 23.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz SM. Perspectives series: cell adhesion in vascular biology. Smooth muscle migration in atherosclerosis and restenosis. J Clin Invest. 1997;99:2814–2816. doi: 10.1172/JCI119472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Magnusson MK, Mosher DF. Fibronectin: structure, assembly, and cardiovascular implications. Arterioscler Thromb Vasc Biol. 1998;18:1363–1370. doi: 10.1161/01.atv.18.9.1363. [DOI] [PubMed] [Google Scholar]
- 26.Raines EW. The extracellular matrix can regulate vascular cell migration, proliferation, and survival: relationships to vascular disease. Int J Exp Pathol. 2000;81:173–182. doi: 10.1046/j.1365-2613.2000.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arnold RS, Shi J, Murad E, Whalen AM, Sun CQ, Polavarapu R, Parthasarathy S, Petros JA, Lambeth JD. Hydrogen peroxide mediates the cell growth and transformation caused by the mitogenic oxidase Nox1. Proc Natl Acad Sci U S A. 2001;98:5550–5555. doi: 10.1073/pnas.101505898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ranjan P, Anathy V, Burch PM, Weirather K, Lambeth JD, Heintz NH. Redox-dependent expression of cyclin D1 and cell proliferation by Nox1 in mouse lung epithelial cells. Antioxid Redox Signal. 2006;8:1447–1459. doi: 10.1089/ars.2006.8.1447. [DOI] [PubMed] [Google Scholar]
- 29.Chen H, Bernstein BW, Bamburg JR. Regulating actin-filament dynamics in vivo. Trends Biochem Sci. 2000;25:19–23. doi: 10.1016/s0968-0004(99)01511-x. [DOI] [PubMed] [Google Scholar]
- 30.Pollman MJ, Hall JL, Gibbons GH. Determinants of vascular smooth muscle cell apoptosis after balloon angioplasty injury. Influence of redox state and cell phenotype. Circ Res. 1999;84:113–121. doi: 10.1161/01.res.84.1.113. [DOI] [PubMed] [Google Scholar]
- 31.Mayr M, Xu Q. Smooth muscle cell apoptosis in arteriosclerosis. Exp Gerontol. 2001;36:969–987. doi: 10.1016/s0531-5565(01)00090-0. [DOI] [PubMed] [Google Scholar]
- 32.ten Freyhaus H, Huntgeburth M, Wingler K, Schnitker J, Baumer AT, Vantler M, Bekhite MM, Wartenberg M, Sauer H, Rosenkranz S. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc Res. 2006;71:331–341. doi: 10.1016/j.cardiores.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 33.Weber DS, Taniyama Y, Rocic P, Seshiah PN, Dechert MA, Gerthoffer WT, Griendling KK. Phosphoinositide-dependent kinase 1 and p21-activated protein kinase mediate reactive oxygen species-dependent regulation of platelet-derived growth factor-induced smooth muscle cell migration. Circ Res. 2004;94:1219–1226. doi: 10.1161/01.RES.0000126848.54740.4A. [DOI] [PubMed] [Google Scholar]
- 34.Wang Z, Castresana MR, Newman WH. Reactive oxygen species-sensitive p38 MAPK controls thrombin-induced migration of vascular smooth muscle cells. J Mol Cell Cardiol. 2004;36:49–56. doi: 10.1016/j.yjmcc.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 35.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 36.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 37.Wang W, Mouneimne G, Sidani M, Wyckoff J, Chen X, Makris A, Goswami S, Bresnick AR, Condeelis JS. The activity status of cofilin is directly related to invasion, intravasation, and metastasis of mammary tumors. J Cell Biol. 2006;173:395–404. doi: 10.1083/jcb.200510115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shinohara M, Shang WH, Kubodera M, Harada S, Mitsushita J, Kato M, Miyazaki H, Sumimoto H, Kamata T. Nox1 redox signaling mediates oncogenic Ras-induced disruption of stress fibers and focal adhesions by down-regulating Rho. J Biol Chem. 2007;282:17640–17648. doi: 10.1074/jbc.M609450200. [DOI] [PubMed] [Google Scholar]
- 39.Li F, Higgs HN. The mouse Formin mDia1 is a potent actin nucleation factor regulated by autoinhibition. Curr Biol. 2003;13:1335–1340. doi: 10.1016/s0960-9822(03)00540-2. [DOI] [PubMed] [Google Scholar]
- 40.Wen Y, Eng CH, Schmoranzer J, Cabrera-Poch N, Morris EJ, Chen M, Wallar BJ, Alberts AS, Gundersen GG. EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat Cell Biol. 2004;6:820–830. doi: 10.1038/ncb1160. [DOI] [PubMed] [Google Scholar]
- 41.Sakata D, Taniguchi H, Yasuda S, Adachi-Morishima A, Hamazaki Y, Nakayama R, Miki T, Minato N, Narumiya S. Impaired T lymphocyte trafficking in mice deficient in an actin-nucleating protein, mDia1. J Exp Med. 2007;204:2031–2038. doi: 10.1084/jem.20062647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.San Martin A, Lee MY, Williams HC, Mizuno K, Lassègue B, Griendling KK. Dual regulation of cofilin activity by LIMK and slingshot 1L phosphatase controls PDGF-induced migration of human aortic smooth muscle cells. Circ Res. 2007 doi: 10.1161/CIRCRESAHA.107.158923. in press. [DOI] [PubMed] [Google Scholar]
- 43.Chandrasekar B, Tanguay JF. Platelets and restenosis. J Am Coll Cardiol. 2000;35:555–562. doi: 10.1016/s0735-1097(99)00596-3. [DOI] [PubMed] [Google Scholar]
- 44.Irani K. Oxidant signaling in vascular cell growth, death, and survival: a review of the roles of reactive oxygen species in smooth muscle and endothelial cell mitogenic and apoptotic signaling. Circ Res. 2000;87:179–183. doi: 10.1161/01.res.87.3.179. [DOI] [PubMed] [Google Scholar]
- 45.Li G, Chen SJ, Oparil S, Chen YF, Thompson JA. Direct in vivo evidence demonstrating neointimal migration of adventitial fibroblasts after balloon injury of rat carotid arteries. Circulation. 2000;101:1362–1365. doi: 10.1161/01.cir.101.12.1362. [DOI] [PubMed] [Google Scholar]
- 46.Welt FG, Rogers C. Inflammation and restenosis in the stent era. Arterioscler Thromb Vasc Biol. 2002;22:1769–1776. doi: 10.1161/01.atv.0000037100.44766.5b. [DOI] [PubMed] [Google Scholar]
- 47.Tanaka K, Sata M, Natori T, Kim-Kaneyama JR, Nose K, Shibanuma M, Hirata Y, Nagai R. Circulating progenitor cells contribute to neointimal formation in nonirradiated chimeric mice. Faseb J. 2007 doi: 10.1096/fj.06-6884com. [DOI] [PubMed] [Google Scholar]