

Abstract
The present study extends our previous work on characterizing the autistic behavior profile of boys with fragile X syndrome (FXS) who meet Diagnostic and Statistical Manual for Mental Disorders, 4th Edition criteria for autism spectrum disorder (ASD) into a longitudinal evaluation of ASD in FXS (FXS+ASD). Specifically, we aimed to determine the stability of the diagnosis and profile of ASD in FXS over time. Through regression models, we also evaluated which autistic and social behaviors and skills were correlates of diagnosis and autistic behavior severity (i.e., Autism Diagnostic Interview-Revised total scores). Finally, we assessed the evolution of cognitive parameters in FXS+ASD. A population of 56 boys (30–88 months at baseline) with FXS was evaluated using measures of autistic, social, and cognitive behaviors and skills at three yearly evaluations. We found that the diagnosis of ASD in FXS was relatively stable over time. Further emphasizing this stability, we found a set of behaviors and skills, particularly those related to peer relationships and adaptive socialization, that differentiated FXS+ASD from the rest of the FXS cohort (FXS+None) and contributed to autistic severity at all time points. Nevertheless, the general improvement in autistic behavior observed in FXS+ASD coupled with the concurrent worsening in FXS+None resulted in less differentiation between the groups over time. Surprisingly, FXS+ASD IQ scores were stable while FXS+None non-verbal IQ scores declined. Our findings indicate that ASD is a distinctive subphenotype in FXS characterized by deficits in complex social interaction, with similarities to ASD in the general population.
Keywords: Fragile X, autism, longitudinal, Autism Diagnostic Interview, adaptive socialization
INTRODUCTION
Fragile X syndrome (FXS) is the most common cause of inherited mental retardation [Cohen et al., 2005], affecting approximately 1:4,000 males and 1:6,000 females [Sherman, 2002]. FXS is associated with an unstable expansion of a CGG trinucleotide repeat within the 5’-untranslated region of the FMR1 gene, located in the X chromosome [Kaufmann and Reiss, 1999]. According to the number of CGG repeats [Maddalena et al., 2001], alleles are classified as normal (5–40 repeats), intermediate or gray zone (~45–54 repeats), premutation (~55–200 repeats), or full mutation (>200 repeats), the latter corresponding to the clinical diagnosis of FXS [Hagerman et al., in press]. A mixed pattern of full mutation and premutation alleles is termed size mosaicism [Rousseau et al. 1994; Kaufmann et al, 1999], and it is also included under the FXS label. Full mutation leads to hypermethylation of the FMR1 gene which, in turn, results in the silencing of the gene, expressed phenotypically in males by cognitive impairment and behavioral abnormalities [Hagerman et al., in press]. Premutation alleles are associated with milder cognitive impairment than full mutation alleles [Hagerman et al., 1994; Merenstein et al., 1996; Kaufmann and Reiss, 1999; Kaufmann et al., 1999], and better adaptive skills development [Cohen at al., 1996]. FXS affects both males and females; however, males, with only one X chromosome, are typically more severely impaired.
Autism (Aut) is one of the most severe behavioral abnormalities observed in FXS [Hagerman et al., 1986; Baumgardner et al., 1995; Cohen, 1995; Bailey et al., 1998; Kaufmann and Reiss, 1999; Bailey et al., 2001; Hagerman, 2002; Kau et al., 2004; Kaufmann et al., 2004; Hatton et al., 2006; Clifford et al., 2007; Hall et al., 2008], with prevalence rates ranging from 15–47% [Hagerman et al., 1986; Bregman et al., 1987; Dykens and Volkmar, 1997; Feinstein and Reiss, 1998; Bailey et al., 1998; Bailey et al., 2001; Rogers et al., 2001] and from 25–52% in males in more recent studies [Kaufmann et al., 2004; Philofsky et al., 2004; Hatton et al., 2006; Clifford et al., 2007; García-Nonell et al., 2008; Hall et al., 2008], depending on the criteria and methods used for diagnosis. Importantly, up to 90% of males with FXS display some form of atypical behavior characteristic of Aut. This includes atypical social interaction (e.g., avoidant eye contact, social withdrawal, social anxiety) as well as repetitive or stereotyped behaviors (e.g., perseveration, hand flapping, self-injury) [Merenstein et al., 1996; Bailey et al., 1998; Hatton et al., 1999; Hagerman, 2002; Kau et al., 2004; Kaufmann et al., 2004; Budimirovic et al., 2006; Hall et al., 2008]. Since Rogers et al. [2001] described the distinctive profile of very young boys with FXS and Aut (FXS+Aut) and its striking global similarities to that of boys with idiopathic Aut, specifically in terms of performance on the Autism Diagnostic Interview-Revised (ADI-R) [Lord et al., 1994] and the Autism Diagnostic Observation Schedule-Generic (ADOS-G) [Lord et al., 1999], several cross-sectional studies have concentrated on the differentiation and characterization of the FXS+Aut subphenotype. In addition to confirming the general resemblance between FXS+Aut and idiopathic Aut [Kau et al., 2004], we also demonstrated that the diagnosis of autism spectrum disorder [ASD, including both the Aut and the milder Pervasive Developmental Disorder-Not Otherwise Specified (PDD) diagnoses] in boys with FXS reflects an impairment in complex social interaction rather than in the nonverbal social behaviors typically described in FXS (e.g., eye gaze avoidance). We also found that boys with both FXS and ASD (FXS+ASD) display overall greater impairment in cognition and adaptive behavior and more severe aberrant behavior than FXS boys without ASD (FXS+None) [Kau et al., 2004; Kaufmann et al., 2004] and that, in line with this, delayed socialization skills and severity of social withdrawal are primary determinants of ASD status [Kaufmann et al., 2004; Budimirovic et al., 2006].
Few studies have examined the longitudinal evolution of Aut in FXS. Bailey and colleagues [Bailey et al., 1998; Hatton et al., 1999; Bailey et al., 2000; Bailey et al., 2001; Hatton et al., 2003; Hatton et al., 2006] have significantly contributed to our current understanding of autistic behavior in FXS. Hatton et al. [2006] examined Childhood Autism Rating Scale (CARS) scores [Schopler et al., 1988] in children with FXS over time. They found that the CARS-based classification of Aut (e.g., not autistic, autistic) was stable and that CARS total scores increased (i.e., worsened) slowly, but significantly, over time. However, strong conclusions could not be drawn because CARS only classifies individuals into categories of autistic behavior severity, not diagnostic categories in line with the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-IV) [American Psychiatric Association, 1994] criteria. Moreover, the study did not report changes over time separately for the autistic and not autistic subgroups. Sabaratnam et al. [2003] reported on a 10-year follow-up study assessing the autistic-like behavior profile in older males with FXS (mean age at baseline 35.8 ± 18.8 years). They found that autistic-like behavior, as measured by the Brief Disability Assessment Schedule (B-DAS) [Holmes et al., 1982] and the Handicaps, Behaviour and Skills Schedule (HBS) [Wing, 1980], were stable over time in FXS [Sabaratnam et al. 2003]. This finding must be taken with caution, as well, since the B-DAS and HBS are not primary measures of autistic behavior and none of the subjects met DSM, 3rd Edition Revised [American Psychiatric Association, 1987] criteria for autism at any evaluation. Further, the age range was considerably wide, from 6–76 years.
The present investigation is a continuation of our previous work on characterizing the autistic and general social behavior profiles in boys with FXS+ASD [Kau et al., 2004; Kaufmann et al., 2004; Budimirovic et al., 2006] and intends to analyze the phenomenon of ASD in FXS over time. Based on our hypotheses that FXS+ASD is a distinctive subphenotype of FXS and that the core elements of ASD in FXS will be present over time, our first goal was to determine the longitudinal changes in the diagnosis and severity of ASD in FXS, as determined by DSM-IV and ADI-R criteria. Our second goal was to examine whether the deficits in complex social interaction we described [Kaufmann et al., 2004], as well as the social behavior determinants of ASD diagnosis [Budimirovic et al., 2006], were stable over time. To our knowledge, this is the first study to evaluate diagnosis and features of ASD in FXS in DSM-IV-classified subjects over time. In a cohort of FXS boys, between 30–88 months at baseline, assessed yearly over a three-year period, we attempted to answer the following specific questions:
Is the diagnosis and severity of ASD in FXS stable over time?
Is the autistic behavior profile of ASD in FXS consistent over time and what behaviors are correlates of severity and diagnosis?
What is the relationship between social skills and other abnormal social behaviors and autistic behavior and diagnosis over time?
What is the longitudinal progression of cognitive parameters, namely IQ and language skills, in boys with FXS+ASD?
MATERIALS AND METHODS
Subjects
The present study includes a total of 56 boys with FXS (mean age at baseline 56.6 ± 13.7 months, mean full-scale IQ at baseline 55.9 ± 17.0) both with (FXS+ASD) and without (FXS+None) ASD, recruited as part of a longitudinal study of autistic and social behaviors and cognitive skills in young males with FXS at the Kennedy Krieger Institute (Baltimore, MD). Diagnosis of FXS was determined by standard Southern blotting techniques [Rousseau et al., 1991], complemented by clinical examination. 14 (25%) of the subjects evaluated at baseline (T1) exhibited allele size mosaicism for the FMR1 mutation (combination of full mutation and premutation) and 1 subject (~2%) was mosaic for methylation (mixture of completely and partially methylated full mutation alleles) [Maddalena et al., 2001]. The remaining 41 subjects (~73%) had typical full mutation. The distribution of the mutations was relatively equal across diagnostic groups, which were based upon DSM-IV criteria and confirmed by the ADI-R. Eighteen (~44%) of the 41 full mutation subjects and six (40%) of the 15 mosaic subjects, or approximately 43% of the entire cohort, met criteria for ASD diagnosis at T1. Due to the approximate equal distribution of the two patterns of FMR1 mutation across the FXS+None and FXS+ASD groups, it was not necessary to divide the cohort according to mutation type. The ASD category encompassed 10 subjects (~18%) diagnosed with the milder PDD and 14 subjects (25%) diagnosed with Aut. Based upon the developmental and cognitive profiles, no subject met DSM-IV criteria for the diagnosis of Asperger syndrome. The ethnic composition of the sample at T1 was predominantly white (~95%), with approximately 3% Hispanic and 2% Black. The majority of the families were middle class and ~35% were of low socioeconomic class. A substantial proportion of the mothers of the FXS subjects had premutation; however, mean parental (primarily maternal) IQ scores at T1 were within the normal range (105.77 ± 15.1). Of the 56 subjects recruited at T1, 44 (~79%) returned for a one-year follow up (T2), and 34 (~61%) returned for a two-year follow up (T3). One subject returned at T3 but not at T2. The average number of assessments per subject was 2.4. A summary of the distribution and characteristics of the subjects in this study is shown in Table I. This study was approved by the Johns Hopkins Medical Institutions’ Institutional Review Board and written informed consent was obtained from all parents or legal guardians of the subjects after the procedures were fully explained.
Table I.
Prevalence of ASD and Autism Over Time
| T1 | T2 | T3 | ||||
|---|---|---|---|---|---|---|
| Baseline (T1) Dx | T1 N (% of cohort) |
Age (months) mean (SD) |
T2 N (% of cohort) |
Age (months) mean (SD) |
T3 N (% of cohort) |
Age (months) mean (SD) |
| Fragile X | 56 (100%) | 56.6 (13.7) | 44 (100%) | 69.1 (13.2) | 34 (100%) | 82.3 (13.2) |
| +None | 32 (57.1%) | 56.8 (13.0) | 26 (56.1%) | 70.2 (12.8) | 21 (64.7%) | 83.0 (12.2) |
| +ASD | 24 (42.9%) | 56.4 (14.8) | 18 (43.9%) | 67.6 (14.0) | 13 (35.3%) | 81.2 (15.1) |
| +PDD | 10 (17.9%) | 53.7 (13.3) | 6 (19.5%) | 59.7 (2.2) | 3 (11.8%) | 73.3 (3.5) |
| +Aut | 14 (25.0%) | 58.3 (16.0) | 12 (24.4%) | 71.6 (15.8) | 10 (23.5%) | 83.6 (16.5) |
Dx, diagnosis; ASD, Autism Spectrum Disorder; PDD, Pervasive Developmental Disorder; Aut, autism; SD, standard deviation.
Instrumentation
Autistic Behavior
The diagnosis of ASD was established by DSM-IV criteria [American Psychiatric Association, 1994] and confirmed by the ADI-R [Lord et al., 1994], which was administered at each time point to assess autistic features in our FXS cohort. The ADI-R is a highly standardized semi-structured interview conducted with the child’s caregiver in order to obtain detailed descriptions of behavioral symptoms associated with the criteria required for a DSM-IV diagnosis of ASD. The ADI-R relies on descriptions of behaviors that demonstrate developmental deviance rather than developmental delay, and higher scores indicate greater impairment. This assessment provides a total score as well as separate scores in three distinct areas, or domains: Reciprocal Social Interaction (Recs), Communication (Comm), and Repetitive Behaviors and Stereotyped Patterns (Reps). Each domain has a threshold score, with that number and any number higher signifying meeting criteria for the DSM-IV diagnosis of Aut in that domain: the Recs threshold is 11, the Comm threshold is 9 for “verbal” subjects or 8 for “non-verbal” subjects, and the Reps threshold is 3. There is no threshold for total score; rather, a diagnosis of Aut is given, according to DSM-IV and ADI-R criteria, if the participant meets the cut-off score for each of the three domains and the developmental deviance occurred before 3 years of age. A diagnosis of PDD is given if the participant meets the cut-off criteria for Recs (i.e., the core domain) and one of the other two domains. In order to fully appreciate the range of abnormal behaviors, as reported [Kaufmann et al., 2004], scores of 3 (indicating the highest level of abnormal behavior) on individual ADI-R items were not converted to scores of 2, as ADI-R scoring protocol suggests for diagnostic purposes. This approach, however, did not affect the diagnosis for any of the subjects at any time point, yet provided the widest spectrum of scores for analytical purposes. The ADI-R was administered to the participant’s caregiver by two trained interviewers, who were blind to group membership. At each assessment, “current” functioning was evaluated rather than “most abnormal” or “ever” behavior. Reliability of total scores across items for the three domains was established between the psychologist trainer and the two interviewers. Reliability intraclass correlation coefficients ranged between 0.88 and 0.94.
The ADOS-G was also administered to a proportion (~39%) of our FXS cohort at T1, but the ASD diagnoses and autistic behavior profiles yielded by this assessment were not consistent with the DSM-IV/ADI-R diagnoses (the latter in complete agreement). As reported in autism of unknown cause [Tomanik et al., 2007], when there is a discrepancy between ADI-R and ADOS-G diagnoses, the Vineland Adaptive Behavior Scales is a valid tool for settling this disagreement. An analysis comparing adaptive behavior in True ASD (i.e., DSM-IV/ADI-R and ADOS-G diagnoses agree) and False ASD (i.e., DSM-V/ADI-R yields a negative ASD diagnosis while ADOS-G yields a positive diagnosis) showed that, as expected, the True ASD group (n=12) had significantly lower Socialization and Composite mean scores than the False ASD group (n=9), further supporting the “true” and “false” nature of the diagnoses. The high false positivity rate of the ADOS-G is further illustrated by the fact that, at T1, the ADOS-G diagnosed 21 (~95%) out of the 22 subjects who were administered the measure with ASD. For all these reasons, we feel we were justified in excluding data from the ADOS-G in our analyses. Nonetheless, we considered it important to mention our ADOS-G testing to illustrate the comprehensive nature of the autistic behavior assessments used in this study. In future studies, we will attempt to identify the factors that determined the high false positivity rate of the ADOS-G.
Cognitive function
The Stanford Binet, 4th Edition (SB-IV) [Thorndike et al., 1986] or the Bayley Scales of Infant Development, 2nd Edition, Mental Scales (BSID-II) [Bayley, 1993] were administered at each time point to assess the cognitive abilities of the subjects. The SB-IV was used for all subjects who were able to establish a true basal [i.e., 3 consecutive correct items; 34 subjects (~61%) at T1, 32 subjects (~73%) at T2, and 28 subjects (~85%) at T3]. The remaining subjects, 22 (~39%) at T1, 12 (~27%) at T2, and 5 (~15%) at T3, were evaluated by the BSID-II in order to obtain the Mental Developmental Index (MDI). If the child’s chronological age was greater than 42 months, a BSID-II-estimated IQ was calculated by dividing the mental age-equivalent by the chronological age and multiplying the ratio by 100 (i.e., DQ). These scores, including the test composite scores from the SB-IV, the MDI, and the BSID-II-estimated IQ, all of which represent levels of overall cognitive abilities, were labeled as “full-scale IQ” (FSIQ) for data analysis purposes. Non-verbal (NVIQ) and verbal (VIQ) IQ scores were also calculated according to Sattler [1988].
Adaptive behavior
Adaptive behavior was assessed using the Vineland Adaptive Behavior Scales, Survey Form, Interview Edition (VABS) [Sparrow et al., 1984]. The VABS, a semi-structured interview with the parent, provides a general assessment of developmentally adaptive behavior in a variety of areas, and is appropriate for infancy through 18-year-old age groups. The VABS yields standard and age-equivalent scores on global function (i.e., Adaptive Behavior Composite) and on four individual domains: Communication skills, Daily Living skills, Socialization skills, and Motor skills. Lower scores indicate greater impairment. Standard scores were used for our analyses.
Language skills
Language skills were assessed using the Preschool Language Scale, 3rd Edition (PLS-3) [Zimmerman et al., 1992] or the Clinical Evaluation of Language Fundamentals, 3rd Edition (CELF-3) [Semel et al., 1995]. The PLS-3 is a standardized measure used to evaluate the semantics and language structure of children from birth through age 6. Like many children with FXS, a majority of our sample demonstrated limited verbalization. Therefore, since the PLS-3 is designed primarily for pre-verbal children and both the Auditory Comprehension and Expressive Communication subscales require limited verbalization, the assessment was well suited for our sample. The Auditory Comprehension and Expressive Communication subscales yield both raw and standardized scores.
The CELF-3 is a similar measure of language skills that can be applied to older populations, aged 6 to 21 years, and includes Receptive Language and Expressive Language subscales. For data analysis purposes, the Receptive Language subscale of the CELF-3 and the Auditory Comprehension subscale of the PLS-3 were labeled as “receptive language”, while the Expressive subscales of the two language assessments were grouped into one “expressive language” label. Children who were above age 6 were administered the CELF-3, while children up to 6 years of age were administered the PLS-3. A few subjects above 6 years of age, 2 (~4%) at T1, 2 (~5%) at T2, and 13 (~38%) at T3, were too low functioning to score on the CELF-3 and were instead administered the PLS-3; however, these subjects were too old for the conversion of subscale raw scores to standardized scores, and, because standardized scores were used in our analysis of language skills, were excluded from our analyses of language skills.
Problem/aberrant behavior
Problem/aberrant behaviors were assessed using the Child Behavior Checklist (CBCL) [Achenbach, 1991, 1992] and the Aberrant Behavior Checklist-Community (ABC-C) [Aman and Singh, 1986]. The CBCL is a widely used parent/teacher report for assessing behavioral and emotional problems in children. Either the 2–3 years version [Achenbach, 1992] or the 4–18 years version [Achenbach, 1991] was used. There are six subscales in the 2–3 years version and eight subscales in the 4–18 years version. These subscales are primarily grouped into Internalizing or Externalizing domains. Separate Sleep and Somatic problems subscales and Social, Thought, and Attention problems subscales are also part of the 2–3 years and 4–18 years versions, respectively. T scores are generated for each individual syndrome subscale and for both the Internalizing and Externalizing behavior domain composites. A total T score, combining all subscales, is also calculated. This study focused primarily on CBCL measures related to the previously described social behavior determinants of FXS+ASD, a series of parameters of adaptive and problem/aberrant behavior linked to autistic features in FXS [Kau et al., 2004; Kaufmann et al., 2004], which are mainly included in the Withdrawn (CBCLw) subscale [Budimirovic et al., 2006].
The ABC-C is another parent report measure, which assesses the prevalence of inappropriate and maladaptive behaviors in individuals between 3 and 18 years with developmental disabilities. It is composed of five subscales: Irritability, Lethargy/Social Withdrawal (ABCsw), Stereotypic Behaviors, Hyperactivity, and Inappropriate Speech. Like the CBCL, higher ABC-C scores indicate greater impairment. The ABC-C only yields raw scores for each subscale. As for the CBCL, we focused on ABC measures previously found to be related to FXS+ASD, specifically those in the ABCsw subscale [Budimirovic et al., 2006].
Study Design and Data Analysis
Based on their T1 DSM-IV/ADI-R diagnoses, for analytical purposes, subjects were divided into two groups (e.g., FXS+ASD vs. FXS+None). The rationale for an ASD versus None comparison includes the conceptualization of PDD as a milder form of Aut, which we have previously demonstrated [Kaufmann et al., 2004], particularly when defined as a condition with core impairment in social interaction, and the need for a larger statistical sample with a wider range of impairments for our regression analyses. Furthermore, ASD has become an umbrella term accepted in the field that encompasses the diagnoses of Aut and PDD. Subjects were also divided into three groups (e.g., FXS+Aut vs. FXS+PDD vs. FXS+None) only for analyzing diagnostic trends over time. The latter were investigated by contingency tables employing the McNemar Test and Chi-Squared Test for Trend [Maxwell, 1961] for the FXS+None/FXS+ASD analysis and by the Stuart-Maxwell Test for the FXS+None/FXS+PDD/FXS+Aut analysis. These tests mainly evaluated whether there were significant differences in diagnostic stability between the diagnostic groups.
Since the behavioral profile of FXS+ASD not only refers to a particular pattern of autistic behaviors (e.g., ADI-R profile) but also to a specific set of delays or impairments in communication and socialization skills, we examined the evolution of other cognitive and behavioral parameters, such as the VABS, CBCL, and ABC-C. Moreover, in addition to providing a more complete picture of ASD in FXS, these parameters contribute a DSM-independent behavioral framework for understanding the meaning of ASD in FXS. Several statistical approaches were used for data analysis, as reported in our previous studies on the subject [Kau et al., 2004; Kaufmann et al., 2004; Budimirovic et al., 2006]. Differences between the FXS+ASD and FXS+None cohorts on most measures of autistic behavior, skills, and aberrant behaviors were determined by non-parametric tests because of the relatively small sample size and lack of significant age differences between the FXS+ASD and FXS+None groups requiring an analysis of covariance (ANCOVA)-like approach. Relationships between ADI-R total scores (i.e., autistic behavior severity), as well as diagnostic categories (i.e., ASD classification), and measures of autistic behaviors, aberrant behaviors, and skills were investigated through various linear regression models, including multivariate, stepwise, and logistic regressions. Changes over time in the abovementioned assessments within the FXS+ASD and FXS+None groups were analyzed using repeated measures analyses of variance (ANOVA) and confirmed with longitudinal regression models, using generalized estimating equations (GEE) models to predict the longitudinal courses of our FXS+ASD and FXS+None subcohorts as reported [Kraut et al., 2004]. GEE allowed us to assess the mean response at each time point as if the outcome measures were handled as cross-sectional data, but also allowed us to estimate more accurate standard errors and confidence intervals around these means that take into account the correlations of measured responses within subject. For all the regression analyses, F values ≥ 3.96–4.00 corresponding to P values ≤ 0.05 were considered significant. Corrections for multiple comparisons, by the Bonferroni method, were conducted where appropriate.
RESULTS
Prevalence and Stability of ASD Diagnosis in FXS
Contingency table analyses were performed to determine diagnostic stability over time. The prevalence of ASD was approximately equal at T1 and T2 and lowest at T3 (Table I). The McNemar Test revealed no significant shift in diagnoses between any time point, demonstrating that the stability of the FXS+ASD and FXS+None diagnoses were not significantly different from one another and that the diagnoses changed at relatively similar rates over time. Quantitatively, we found that more subjects remained within the diagnostic category of ASD from T1 to T2 than from T2 to T3 or T1 to T3 (~82% vs. 60% and ~57%, respectively). Despite the drop in agreement in the latter two time points, we found that the Chi-Squared Test for Trend [Maxwell, 1961] was not significant. Across all time points, there was an average agreement in ASD diagnosis of ~68% (Table II).
Table II.
Stability of ASD and Autism Diagnosis Over Time
| T1 | T2 | T3 | |||||
|---|---|---|---|---|---|---|---|
| Aut Dx | N (with T2 data, with T3 data) |
None | PDD | Aut | None | PDD | Aut |
| None | 32 (24, 20) | 20 (83.3%) | 3 (12.5%) | 1 (4.2%) | 16 (80.0%) | 2 (10.0%) | 2 (10.0%) |
| PDD | 10 (6, 4) | 2 (33.3%) | 2 (33.3%) | 2 (33.3%) | 3 (75.0%) | 0 (0.0%) | 1 (25.0%) |
| Aut | 14 (11, 10) | 1 (9.1%) | 3 (27.3%) | 7 (63.6%) | 3 (30.0%) | 2 (20.0%) | 5 (50.0%) |
Dx, diagnosis; PDD, Pervasive Developmental Disorder; Aut, autism.
Three-group diagnoses (i.e., FXS+None, FXS+PDD, and FXS+Aut) analyses were used to quantify the stability of the specific ASD diagnoses and to determine whether improvement or worsening within the ASD was gradual (e.g., change from FXS+PDD to FXS+None) or swift (e.g., change from FXS+Aut to FXS+None). We observed about equal prevalence of Aut at each time point (Table I). As expected, the diagnosis of PDD was found to be unstable, with average agreement across time points of only ~21%. The diagnosis of Aut demonstrated relative stability, with average agreement over time of ~59%. Despite these quantitative differences in stability between FXS+Aut and FXS+PDD, there were not significant differences in their stability between any time point according to the Stuart-Maxwell Test. Although some improvement was observed within the FXS+Aut group, ~78% of subjects remained within the autism spectrum across time points (i.e., either retained the Aut diagnosis or improved only slightly to PDD) (Table II), emphasizing the stability of ASD within the FXS population.
Profiles of Autistic Behavior Over Time in FXS
We previously defined a distinctive profile of deficits in autistic behaviors, as measured by the ADI-R, in boys with FXS and ASD. This pattern mainly involved behaviors within the Recs domain of the ADI-R, which represented deficits in complex social interactions [Kaufmann et al., 2004]. In order to assess the stability of this profile of autistic behavior in FXS, we first determined which behaviors were significantly more impaired in FXS+ASD subjects than in FXS+None subjects at each time point and were thus able to distinguish the FXS+ASD group from the rest of the FXS cohort. Through various regression models (logistic, stepwise, and standard multivariate), we then determined which behaviors were correlated to ASD diagnosis/status (i.e., classification) and/or ADI-R scores (i.e., severity) at each time point. In our 2004 cross-sectional study [Kaufmann et al., 2004], the foundation on which the current longitudinal study is based, we presented stepwise regression models which demonstrated that the Recs domain was the most significant ADI-R determinant of autistic behavior severity. In the present study, we performed similar analyses at each time point to assess if this trend continued over time, and also to serve as a means of comparison to the 2004 publication (T1 of the current study). These regressions are presented in Table III. In addition, parallel standard multivariate regressions yielded the same results as the stepwise regressions. Since Recs is the core domain of the ADI-R, and its impairment is necessary for the diagnosis of ASD, we expected Recs to continue to be the most important factor in the distinction of ASD within FXS. We found that the FXS+ASD group averaged significantly higher scores (i.e., greater impairment) than the FXS+None group on all ADI-R domains at both T1 and T2; however, Recs was the only domain that continued to be significantly higher in the FXS+ASD group at T3 and was thus the only domain to differentiate FXS+ASD at all time points. Further, Recs was the most significant correlate of both ASD diagnosis and autistic behavior severity at each time point (Table III). Within the Recs domain, significant impairments in subdomains Recs-2 and Recs-4, representing Failure to develop peer relationships and Lack of socioemotional reciprocity, respectively, which reproduced our initially reported pattern [Kaufmann et al., 2004], were maintained in FXS+ASD subjects over time, but only Recs-2 consistently influenced Recs and ADI-R total scores at all three time points (Table III). Recs-2 also predicted ASD diagnosis significantly at T1 and T2 and at trend level at T3.
Table IV.
Effects of ADI-R Subdomains on ADI-R Total Scores Over Time
| Variable/parameter | T1 | T2 | T3 | |||||
|---|---|---|---|---|---|---|---|---|
| R squared | 0.829 | 0.788 | 0.885 | |||||
| Adj. R squared | 0.816 | 0.771 | 0.878 | |||||
| DF regression | 4 | 3 | 2 | |||||
| F regression | 62.020 | 45.898 | 119.399 | |||||
| P regression | <.0001 | <.0001 | <.0001 | |||||
| Coefficient | F | Coefficient | F | Coefficient | F | |||
| Recs-1 | 1.387 | 9.993 (R) | 0.272 | 2.876 (E) | 0.019 | 0.011 (E) | ||
| Recs-2 | 1.613 | 30.348 (R) | 2.004 | 23.678 (R) | 2.417 | 52.037 (R) | ||
| Recs-3 | 1.212 | 13.088 (R) | 1.269 | 4.663 (R) | 2.522 | 47.822 (R) | ||
| Recs-4 | 1.562 | 27.988 (R) | 1.795 | 13.097 (R) | 0.239 | 1.813 (E) | ||
| R squared | 0.730 | 0.711 | 0.776 | |||||
| Adj. R squared | 0.719 | 0.696 | 0.762 | |||||
| DF regression | 2 | 2 | 2 | |||||
| F regression | 71.493 | 46.746 | 53.847 | |||||
| P regression | <.0001 | <.0001 | <.0001 | |||||
| Coefficient | F | Coefficient | F | Coefficient | F | |||
| Comm-1 | 2.474 | 45.084 (R) | 2.459 | 27.941 (R) | 1.530 | 5.013 (R) | ||
| Comm-4 | 1.726 | 13.708 (R) | 2.024 | 16.687 (R) | 3.682 | 50.286 (R) | ||
| R squared | 0.371 | 0.540 | 0.204 | |||||
| Adj. R squared | 0.347 | 0.515 | 0.179 | |||||
| DF regression | 2 | 2 | 1 | |||||
| F regression | 15.623 | 22.266 | 8.212 | |||||
| P regression | <.0001 | <.0001 | 0.0073 | |||||
| Coefficient | F | Coefficient | F | Coefficient | F | |||
| Reps-1 | 0.090 | 0.426 (E) | 6.130 | 10.276 (R) | 0.302 | 3.116 (E) | ||
| Reps-2 | 0.158 | 1.328 (E) | 0.127 | 0.602 (E) | 0.301 | 3.094 (E) | ||
| Reps-3 | 2.872 | 4.956 (R) | 0.012 | 0.005 (E) | 0.220 | 1.580 (E) | ||
| Reps-4 | 7.273 | 17.929 (R) | 5.306 | 9.016 (R) | 6.900 | 8.212 (R) | ||
R, regression coefficient; Adj., adjusted; DF, degrees of freedom; F, F value; P, P value; (E), F-to-enter (not significant in forward model); (R), F-to-remove (significant in forward model). F values ≥ 3.96–4.00 are the equivalent of P ≤ 0.05.
Since impairments in both the Comm and the Reps domains are not necessarily required for ASD diagnosis, we expected scores on these domains to differentiate the FXS+ASD group to a lesser extent than Recs. Again, although FXS+ASD subjects showed significantly greater impairment than FXS+None subjects in all ADI-R domains at T1 and T2, the groups did not have significantly different mean scores on Comm or Reps at T3. The Comm domain, however, was a significant correlate of autistic behavior severity at all time points (Table III) but only predicted ASD classification at T3. Although Comm did not differentiate the groups at T3, mean scores on the non-verbal communication (CommNV) domain, comprised of subdomains Comm-1 and Comm-4, representing Lack of, or delay in, spoken language and failure to compensate through gesture and Lack of varied spontaneous make-believe or social imitative play, respectively, were trend-level higher in the FXS+ASD group at T3 and significantly higher at both T1 and T2 when compared to mean scores of the FXS+None group. Within CommNV, FXS+ASD subjects showed significantly greater deficits in Comm-1 at T1 and T2 but not at T3, while significant impairments were maintained in Comm-4 at all time points. At T1 and T2, Comm-1 had more influence than Comm-4 on both Comm and ADI-R total scores as well as ASD diagnosis, but at T3 Comm-4 became the major correlate of both ADI-R scores (Table III) and ASD diagnosis. Verbal communication (CommV), comprised of subdomains Comm-2 and Comm-3, was excluded from our analyses since this domain and its subdomains were only applicable to a relatively small subset of “verbal” FXS subjects.
Reps did not contribute to severity of autistic behavior at any time point (Table III), but did predict ASD status at T1 and T2. Of the subdomains, Reps-3 and Reps-4, representing Stereotyped and repetitive motor mannerisms and Preoccupations with parts of objects, respectively, were significantly more impaired in FXS+ASD subjects than in FXS+None subjects at both T1 and T2, but none of the four subdomains could differentiate the two groups at T3. Reps-1, representing Preoccupation or circumscribed pattern of interest, contributed to Reps scores at all time points but only influenced ADI-R total scores at T2. Reps-4, on the other hand, contributed to ADI-R total scores at all time points but only influenced Reps scores at T2 (Table III). No Reps subdomain consistently predicted ASD diagnosis.
Altogether, we found the autistic behavior profiles in FXS to be almost identical at T1 and T2, but different at T3, with less distinction of the FXS+ASD group from the rest of the FXS cohort. In order to ascertain what underlies this difference and how the autistic behavior profile changes over time, repeated measures ANOVAs were performed on measures of the ADI-R and then confirmed by longitudinal regression models (GEE). We found a clear difference in the nature of the change of autistic behavior over time between the groups, by which FXS+ASD subjects generally improved (i.e., decreased scores) while FXS+None subjects generally worsened (i.e., increased scores). Within the FXS+ASD subcohort, all domains improved, particularly CommNV and Recs, which showed significant and trend level improvement, respectively. At the subdomain level, Recs-4 and Comm-1 improved significantly over time. Within the FXS+None subcohort, all domains worsened, particularly Recs, which showed significant worsening. Recs-2 was the only subdomain to significantly worsen over time. Neither the FXS+ASD subjects nor the FXS+None subjects showed any significant changes over time in Reps or any of its subdomains. Figure 1 depicts the changes in Recs over time.
Figure 1.
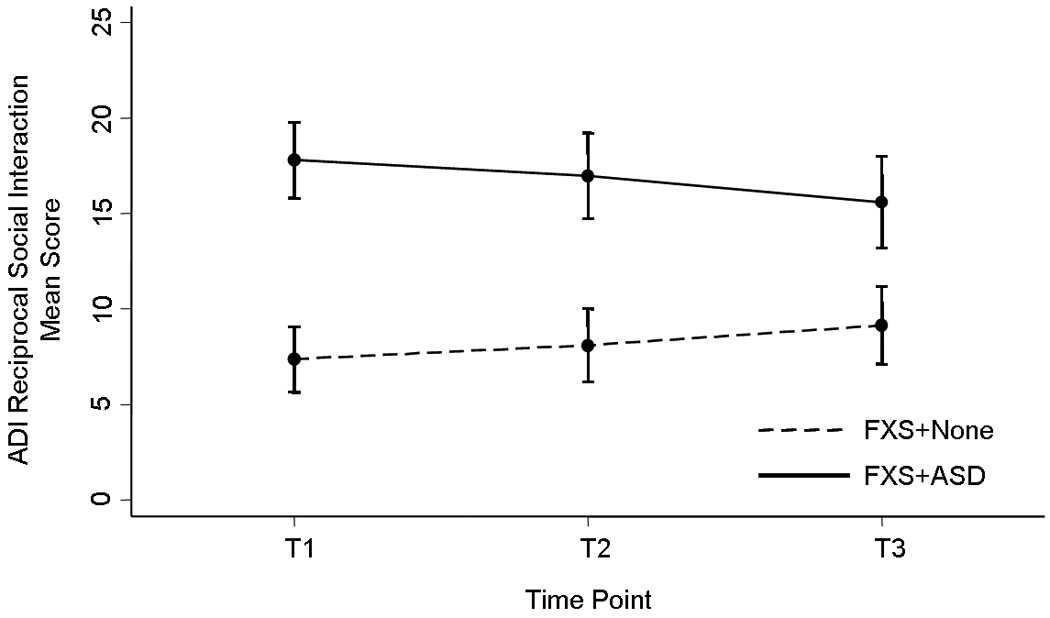
Social Behavior Profile Over Time in FXS Boys with ASD
We have characterized a specific group of communication and social behavior impairments that are closely associated with autistic behavior proper in FXS boys. This profile in FXS+ASD includes several measures of skills and problem/aberrant behavior, such as the VABS, ABC-C, and CBCL, and reflects a core deficit in socialization skills, as opposed to a primary impairment in communication skills [Kau et al., 2004; Kaufmann et al., 2004], associated with social withdrawal [Budimirovic et al., 2006]. In the current study, we found that although adaptive socialization (VABSsoc) mean scores were significantly lower (i.e., more impaired) in the FXS+ASD subcohort than in the FXS+None subcohort at T1 and T2, there was no significant difference in scores between the groups at T3. In contrast, mean scores on adaptive communication (VABScom) remained significantly more impaired in FXS+ASD subjects at all time points. Despite finding the ABC-C Lethargy/Social Withdrawal (ABCsw) subscale to be more informative of ASD status than the CBCL Withdrawn (CBCLw) subscale [Budimirovic et al., 2006], our results showed that CBCLw was the only other measure that was able to differentiate the FXS+ASD group from the rest of the cohort at all time points. The FXS+ASD group had significantly greater impairment (i.e., higher scores) on ABCsw and the ABC-C Stereotypic Behaviors (ABCstereo) subscale than the FXS+None group at T1 and T2, but not at T3.
In parallel to our longitudinal findings on the autistic behavior profile mentioned above, the social behavior profile in our FXS cohort was nearly identical at T1 and T2 but again differed at T3, with much less distinction between groups. Analyses were performed to determine the longitudinal progression of these social behavior determinants of FXS+ASD; we found that the FXS+ASD group remained relatively stable over time, showing significant worsening in only one parameter, VABScom, which showed markedly declining scores (p<0.0001). In contrast, FXS+None subjects worsened over time, demonstrating significant score declines in VABScom and particularly in VABSsoc. This group also showed worsening (i.e., increase) in ABCsw scores.
In our 2004 cross-sectional investigation [Kaufmann et al., 2004], from which the current longitudinal investigation extends, we used an integrated stepwise regression model incorporating social behaviors and skills, specifically measures of communication (receptive language, expressive language and VABScom) and socialization (VABSsoc), and showed that VABSsoc was the most significant correlate of autistic behavior severity and of ASD diagnosis. We also identified, among aberrant behaviors, that social withdrawal behaviors were the most informative of ASD diagnosis. When compared with the other four ABC-C subscales and other CBCL subscales with high scores, ABCsw and CBCLw, respectively, were the only significant correlates of ASD diagnosis, and ABCsw was found to be the stronger correlate of the two. However, when ABCsw was paired with VABSsoc, VABSsoc was shown to be the more important predictor of ASD status [Budimirovic et al., 2006]. In the present study, similar stepwise regression models were repeated at all time points to evaluate if socialization skills continued to be more influential than both communication skills and social withdrawal, again as a means of comparison to our previous publication (T1 of the present study). These regression models are presented in Table IV. Parallel standard multivariate regressions were also performed, and, as was the case in the regression analysis of the ADI-R, yielded the same results as the stepwise regressions. We found that, indeed, VABSsoc remained the most important correlate of ADI-R total scores at all time points. VABSsoc also predicted ASD diagnosis at T1 and T2; however, it had no influence at T3. No other measures influenced ADI-R total scores or ASD diagnosis at more than one time point.
Table IV.
Effects of Integrated Skills and Social Behaviors on ADI-R Total Scores
| Variable/Parameter | T1 | T2 | T3 | |||
|---|---|---|---|---|---|---|
| R.Squared | 0.527 | 0.453 | 0.506 | |||
| Adj. R Squared | 0.518 | 0.438 | 0.481 | |||
| DF Regression | 1 | 1 | 1 | |||
| F Regression | 57.948 | 30.651 | 19.499 | |||
| P Regression | <.0001 | <.0001 | 0.0003 | |||
| Coefficient | F | Coefficient | F | Coefficient | F | |
| Rec language | −0.127 | 0.830 (E) | −0.188 | 1.319 (E) | −0.197 | 0.729 (E) |
| Exp language | 0.068 | 0.238 (E) | 0.052 | 0.096 (E) | −0.239 | 1.092 (E) |
| VABScom | 0.091 | 0.425 (E) | −0.024 | 0.021 (E) | −0.197 | 0.726 (E) |
| VABSsoc | −0.548 | 57.948 (R) | −0.465 | 30.651 (R) | −0.624 | 19.499 (R) |
R, regression coefficient; Adj., adjusted; DF, degrees of freedom; F, F value; P, P value; (E), F-to-enter (not significant in forward model); (R), F-to-remove (significant in forward model). F values ≥ 3.96–4.00 are the equivalent of P ≤ 0.05.
Evolution of Cognitive Skills in Boys with FXS+ASD
Analyses were also performed in order to explore the evolution of cognitive parameters in FXS+ASD, namely IQ and language skills. To be certain the BSID-II-based IQ estimates were not skewing our results, a separate analysis was performed including only subjects who were administered the SB-IV. The findings were strikingly similar to the combined SB-IV/BSID-II analysis, particularly in terms of the longitudinal progression of IQ, and so we report the results from the combined SB-IV/BSID-II cohort. As expected, FXS+ASD subjects had significantly lower (i.e., more impaired) full-scale IQ (FSIQ) than FXS+None subjects at T1 and T2. However, while the FXS+None group showed a markedly significant decline (p<0.0001) in FSIQ scores over time, the FXS+ASD group was surprisingly stable, which led to a lack of significant difference in FSIQ between the two groups at T3. Upon further analysis, we found that this drop in FSIQ within the FXS+None cohort was due to worsening in non-verbal IQ (NVIQ) scores, which markedly declined (p<0.0001) over time. Similar to FSIQ, NVIQ in the FXS+ASD group was stable, and FXS+ASD subjects had significantly greater deficits in NVIQ than FXS+None subjects at T1 and T2 but not at T3 (Fig 2). Verbal IQ (VIQ) also differentiated between groups at T1 and T2 but not at T3; however, no significant changes over time were observed in either group. Although both FSIQ and NVIQ standard scores decreased over time in the FXS+None group, raw scores either remained stable or increased slightly. Similarly, raw scores for VABScom and VABSsoc did not decrease in the FXS+None group. This suggests FXS+None subjects are not losing skills, but rather developing at a much slower rate than normal children. Consistent with the pattern seen in the autistic and social behavior profiles, the IQ profiles at T1 and T2 were nearly identical but different from T3, at which point there were no significant differences in any measure of IQ.
Figure 2.
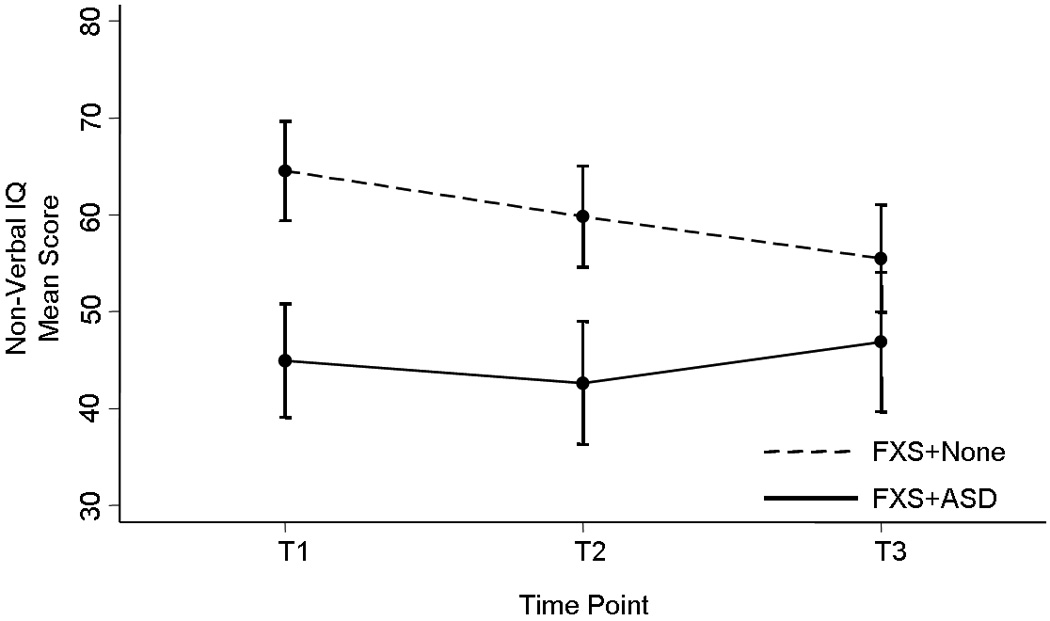
In regards to language skills, we found receptive language distinguished the FXS+ASD group from the rest of the FXS cohort at all time points, with significantly lower mean scores (i.e., greater impairment) in the FXS+ASD subcohort. Over time, receptive language worsened significantly and similarly within both groups, and therefore the significant deficit in the FXS+ASD group was maintained. There was no significant difference in expressive language between the groups at T1, but because the FXS+ASD group’s scores declined significantly and the FXS+None group showed slight (non-significant) improvement over time, expressive language was able to differentiate the groups at T2 and T3, with significantly greater impairment in the FXS+ASD group. Again, the decline in standard scores did not represent a loss of skills, as the raw scores either remained stable or increased slightly over time, but rather slow development. An interesting note is that expressive language was always more impaired than receptive language within the FXS+None cohort, which is a characteristic of FXS that has been reported by other studies [Philofsky et al., 2004; Lewis et al., 2006] and our results now suggest to be a stable pattern over time.
DISCUSSION
The present study builds upon the findings of our previous investigations that characterized the autistic and related social behavior profiles of boys with FXS who meet DSM-IV criteria for ASD [Kau et al., 2004; Kaufmann et al., 2004; Budimirovic et al., 2006], by extending into a longitudinal evaluation of ASD in FXS. Our results demonstrated that ASD, and in particular Aut, is a stable diagnosis, with approximately 70% diagnostic agreement over time. FXS+ASD also seems to be a distinct FXS subphenotype, since, for the most part, the same profile of autistic behaviors and other abnormalities in social behaviors contributed to either differentiation between the FXS+ASD and FXS+None groups or autistic behavior severity over the three-year period: relatively greater impairment in complex social interactions, predominantly peer relationships; deficits in socially-relevant communication, mainly make-believe or social imitative play; severity of social withdrawal; and delays in adaptive communication and receptive language skills. In addition, in terms of skills, delay in adaptive socialization was the most consistent correlate of autistic behavior severity. Despite this overall stability, autistic behavior scores decreased in FXS+ASD boys across time points, which, in conjunction with the worsening of autistic behaviors and social withdrawal in FXS+None boys, led to less differentiation between the two subcohorts at the last time point (T3) than at baseline (T1) and the midpoint of assessment (T2). Scores on several cognitive parameters changed over time within the FXS+None group but were mainly stable in FXS+ASD, contributing to less group distinction at T3. Prominent among these parameters were adaptive socialization and non-verbal IQ. Although some social behaviors improved in the FXS+ASD group (e.g., socioemotional reciprocity), acquisition of communication skills did not progress at a steady rate, leading to a decrease in receptive and expressive language skills over time. Consequently, the last two parameters differentiated FXS+ASD from FXS+None at T3. The finding that the two groups, FXS+ASD and FXS+None, have clearly different developmental trajectories further supports the conclusion of FXS+ASD as a unique entity in FXS.
At present, FXS is one of the main genetic causes of ASD [Muhle et al., 2004; Schaefer et al., 2008]. The controversy about the meaning of meeting DSM-IV criteria for an individual with a well-delineated genetic disorder, such as FXS, will probably remain well beyond the time when current diagnostic guidelines are replaced in 2012 [Kaufmann et al., 2008]. Therefore, in-depth characterizations of FXS+ASD are needed in order to develop adequate diagnostic and therapeutic strategies for this substantial subset of mainly male FXS subjects [Hagerman et al., in press]. Stability of a diagnostic label is an important criterion for its validity. In longitudinal studies of idiopathic Aut utilizing the ADI-R, among other measures, stability of Aut diagnosis over time was reported to be between 67–80% [Charman et al., 2005; Moss et al., 2008; Kleinman et al., 2008]. However, no studies, to our knowledge, have examined whether the DSM-IV classification of ASD is a stable diagnosis in the FXS population. Our data indicate that, in FXS boys aged approximately 3–8 years, the diagnosis of ASD is relatively stable. Approximately 80% of boys with FXS+Aut remained within the ASD spectrum, with about 60% maintaining the Aut diagnosis. This contrasts with the approximate 20% diagnostic agreement over time for PDD. Between T1 and T3, 30% of boys with FXS+Aut improved out of the ASD category while the proportion in boys with FXS+PDD was 75%. On the other hand, 20% of boys with FXS+None worsened into the ASD category. These figures reflect the relative improvement in autistic behavior over time in FXS+ASD and the opposite in FXS+None, which led to a slight decrease in the overall proportion of FXS+ASD subjects during the three-year observation period (43% at T1 vs. 35% at T3). The marked stability of the Aut diagnostic label in this study further supports its consideration as a distinctive entity within the FXS phenotype. Conversely, the instability of the PDD diagnosis probably represents the influence of communication and other deficits in young children that can mimic mild autistic impairment. To some extent, the situation in FXS is not substantially different from that in the general population, in which young children receive the PDD diagnosis early but are found to be out of the spectrum later in life [Turner and Stone, 2007; Kleinman et al., 2008]. Considering the trend towards improvement in a variety of abnormal behaviors but stagnation in the acquisition of language skills in our FXS+ASD group, a longer follow up is needed in order to fully appreciate the significance of ASD diagnosis in males with FXS.
One of the main outcomes of our previous research on ASD in FXS was the demonstration of a distinctive profile of autistic behavior in FXS+ASD [Kaufmann et al., 2004], as well as the identification of other social behaviors, both social skills and aberrant behaviors, that are major correlates of ASD in FXS [Kau et al., 2004; Budimirovic et al., 2006]. The present investigation demonstrated that the profile of relatively more prominent deficits in peer relationships and socioemotional reciprocity in FXS+ASD [Kaufmann et al., 2004] is indeed a core feature, since it was found to be the main correlate of autistic behavior severity and a distinguishing factor for ASD status at all time points. Only impairment in socioemotional reciprocity was not a determinant of ADI-R total scores or of ASD status at the last observation, because of a relative improvement in scores of FXS+ASD boys in this area. Contrary to some assumptions about the FXS neurobehavioral phenotype [Baumgartner et al., 1995; Hagerman, 2002], basic nonverbal social behaviors (e.g., eye gaze avoidance) and stereotypic and repetitive behaviors do not have major influence on diagnosis or severity of autistic behavior. Other than social interaction, only deficits in communication, as measured by the ADI-R, contributed to autistic severity over time. Examination of skills confirmed that only adaptive socialization, and not communication skills, consistently influenced severity of autistic behavior at all time points, and ASD status, as we reported earlier [Kaufmann et al., 2004; Budimirovic et al., 2006], at the first two time points but not at T3. Social withdrawal was a parameter that distinguished FXS+ASD from FXS+None over time, but not a determinant of ASD status per se. Similarly, adaptive communication and receptive language skills longitudinally differentiated the FXS+ASD group but were not determinants of ASD status.
These data support the conclusions of two of our previous studies [Kaufmann et al., 2004; Budimirovic et al., 2006], that FXS+ASD is characterized by a primary impairment in socialization skills and deficits in communication skills are a secondary component, as well as that severe social withdrawal is an important but not obligatory attribute. Data on adaptive socialization in idiopathic ASD and in FXS highlight the importance of this parameter in understanding and diagnosing FXS+ASD [Kraijer, 2000; Fisch et al., 2002; Fenton et al., 2003; Hatton et al., 2003; Budimirovic et al., 2006]. While our current findings on the longitudinal ADI-R profiles are consistent with studies of autistic behavior in idiopathic ASD, which report significant improvement in scores in ADI-R’s Recs and Comm domains and less drastic, or lack of, change in ADI-R’s Reps domain [Piven et al., 1996; Fecteau et al., 2003; Seltzer et al., 2003; Charman et al., 2005; Shattuck et al., 2007; Moss et al., 2008], to our knowledge, no other study on ADI-R profiles in FXS equivalent to the present one has been published to date. Therefore, the significance of our findings in FXS for ASD of other causes is still unknown. Nevertheless, our longitudinal data in conjunction with the few cross-sectional studies of FXS+ASD [Rogers et al., 2001; Kau et al., 2004], examining a variety of cognitive and behavioral parameters, support the notion of FXS+ASD as a distinctive subphenotype in FXS that shares many features with idiopathic ASD.
A secondary set of findings in this study involved the evolution of cognitive parameters in boys with FXS. Fisch and colleagues [2002] demonstrated that in children with FXS+None or idiopathic Aut, declines in IQ scores were observed during the younger years in both groups, but as children aged, the idiopathic Aut group attained a plateau while the FXS+None group continued to decline. Importantly, however, the authors point out the decline in scores was not representative of loss of skills, but rather of slow development. This is in line with our current findings of marked declines in FSIQ standard scores over time, particularly in NVIQ, in our FXS+None subcohort, reflective of delayed development as raw scores either improved slightly or remained stable. In contrast, the FXS+ASD group remained cognitively stable. This unexpected profile contributed to the diminished cognitive and behavioral distinction between the FXS+ASD and FXS+None groups at T3. The findings in the FXS+ASD group are in limited agreement with the literature on idiopathic ASD, which indicates that the initial relative greater delay in VIQ (vs. NVIQ) tends to disappear by adolescence [Werner et al., 2005]. In our FXS+ASD sample, both VIQ and NVIQ tended to remain stable and, if anything, verbal skills appeared to be more impaired in late childhood. Since the mean ages in our cohort were about 5–7 years from T1 to T3, perhaps declines in IQ within the FXS+ASD subcohort occurred prior to enrollment in our study and at a younger age than in FXS boys without ASD, who may reach a plateau at an age outside the present study’s range. Wright-Talamante and colleagues [1996] also studied longitudinal changes in IQ in FXS children. After comparing FXS boys with full methylation with those with either allele size or methylation mosaic patterns, they concluded that mosaic subjects, with a higher mean IQ, may be at greater risk for decline since their initial cognitive abilities were higher than the full mutation subjects. These findings are in correspondence with our findings of IQ score decline in our higher functioning FXS+None subcohort. However, in the Wright-Talamante et al. study the mean baseline age of the full mutation group was well above the mean baseline age of our cohort, while the mean baseline age of the mosaic subjects was in the middle of our cohort’s age range. Therefore, further longitudinal studies of younger and older populations with different behavioral profiles will be needed to better understand IQ changes in FXS over time.
Similar to Philofsky et al. [2004], we found receptive language was a relative strength in our FXS+None subcohort, showing higher scores than in expressive language at all time points. Receptive and expressive language scores were almost identical in our FXS+ASD subcohort at each time point, strengthening the already established thought that early deficits in receptive language are an early warning sign for ASD [Philofsky et al., 2004], although language skills did not influence autistic behavior severity or ASD status.
The present study is, to our knowledge, the first attempt at evaluating ASD in FXS over time using DSM-IV criteria. A recent investigation [Hatton et al., 2006] of autistic behavior in FXS applying the Childhood Autism Rating Scale (CARS) found a slight, but significant, increase in CARS scores over time. However, this report did not differentiate autistic and non-autistic groups; therefore, the results may be concordant with the increase in ADI-R scores in our FXS+None group. No other investigation has addressed the ASD phenomenon in FXS children from a longitudinal perspective. In spite of the strength of our analytical approach and the use of DSM-IV criteria, we recognize our study presented several limitations. Our sample size and diagnostic composition of the ASD cohort (i.e., low N for PDD and Aut by T3) were limiting factors. The use of two different measures of cognitive function, and the incorporation of an IQ-like DQ ratio for a subset of subjects, as well as two measures of language skills, were other limitations inherent to the study of young children with lower cognitive function. A direct measure of non-verbal communication should also be incorporated in future studies, since many children in this population are non-verbal and deficits in communication were shown to have some influence on autistic behavior severity and ASD classification. Based on our results, interventions targeting the behaviors and skills found to remain impaired or delayed over time in the FXS+ASD subcohort, such as peer relationships and adaptive socialization, could be a more effective strategy in the treatment of children with FXS and ASD. Further, our data show there is general improvement in autistic behavior in FXS+ASD subjects over time and perhaps with intervention this improvement could be further enhanced. Similarly, early intervention in skills that worsened over time in FXS+None subjects, such as NVIQ and behaviors related to peer relationships, may prove useful to help prevent this decline. The strength of our data indicates FXS+ASD is clearly a distinctive and significant entity, with implications for ASD in general, which warrants further research efforts using behavioral and other approaches (e.g., neuroimaging). Of concern is the wide range of prevalence rates of Aut in FXS due to several different methods and measures employed by the various research groups, which can be misleading. We stress the caution that must be exercised when using the ADOS-G for primary or confirmatory diagnosis of ASD in children with FXS due to its susceptibility for false positives. The high prevalence of social anxiety in the FXS population [Budimirovic et al., 2006; Sullivan et al., 2007; Hagerman et al., in press] may be a contributing factor to these false positives; however, this issue has not been formally studied. Therefore, we suggest a standard method for diagnosing ASD in FXS that adheres to DSM-IV criteria using both the ADI-R and the ADOS-G, and, if necessary, the utilization of the VABS to settle diagnostic disagreements between the two assessments, as reported [Tomanik et al., 2007]. Although ADI-R items were included in our analyses, due to the length and complexity of this paper, we chose to exclude these results. However, examination of impairment in specific behaviors (e.g., “play” behaviors) in FXS+ASD and their contributions to ASD status is an important aspect that should be investigated further.
ACKNOWLEDGMENTS
We thank Lisa Freund, Alice Kau, and William Trescher for assistance with subject recruitment and behavioral data collection. We are also extremely grateful to the families who participate in the Young Males with Fragile X Project at the Center for Genetic Disorders of Cognition & Behavior, Kennedy Krieger Institute, Baltimore, Maryland.
Grant sponsor: National Institutes of Health; Grant numbers: HD33175, MH67092.
REFERENCES
- Achenbach TM. Manual for the Child Behavior Checklist/4‘18 and 1991 Profile. Burlington, VT: University of Vermont; 1991. [Google Scholar]
- Achenbach TM. Manual for the Child Behavior Checklist/2–3 and 1992 Profile. Burlington, VT: University of Vermont; 1992. [Google Scholar]
- Aman MG, Singh NN. Aberrant Behavior Checklist-Community Manual. E Aurora, NY: Slosson Educational publications; 1986. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 3rd Edition Revised. Washington, DC: American Psychiatric Association; 1987. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th Edition. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- Bailey DB, Jr, Hatton DD, Mesibov G, Ament N, Skinner M. Early development, temperament, and functional impairment in autism and fragile X syndrome. J Autism Dev Disord. 2000;30:49–59. doi: 10.1023/a:1005412111706. [DOI] [PubMed] [Google Scholar]
- Bailey DB, Jr, Hatton DD, Skinner M, Mesibov G. Autistic behavior, FMR1 protein, and developmental trajectories in young males with fragile X syndrome. J Autism Dev Disord. 2001;31:165–174. doi: 10.1023/a:1010747131386. [DOI] [PubMed] [Google Scholar]
- Bailey DB, Jr, Mesibov GB, Hatton DD, Clarke RD, Roberts JE, Mayhew L. Autistic behavior in young boys with fragile X syndrome. J Autism Dev Disord. 1998;28:499–508. doi: 10.1023/a:1026048027397. [DOI] [PubMed] [Google Scholar]
- Baumgardner TL, Reiss AL, Freund LS, Abrams MT. Specification of the neurobehavioral phenotype in males with fragile X syndrome. Pediatrics. 1995;95:744–752. [PubMed] [Google Scholar]
- Bayley N. Bayley Sales of Infant Development: Second Edition Manual. San Antonio, TX: Psychological Corporation; 1993. [Google Scholar]
- Bregman JD, Dykens E, Watson M, Ort SI, Leckman JF. Fragile X syndrome: Variability of phenotypic expression. J Am Acad Child Adolesc Psychiatry. 1987;26:463–471. doi: 10.1097/00004583-198707000-00001. [DOI] [PubMed] [Google Scholar]
- Budimirovic DB, Bukelis I, Cox C, Gray RM, Tierney E, Kaufmann WE. Autism spectrum disorder in fragile X syndrome: Differential contribution of adaptive socialization and social withdrawal. Am J Med Genet Part A. 2006;140A:1814–1826. doi: 10.1002/ajmg.a.31405. [DOI] [PubMed] [Google Scholar]
- Charman T, Taylor E, Drew A, Cockerill H, Brown JA, Baird G. Outcome at 7 years of children diagnosed with autism at age 2: Predictive validity of assessments conducted at 2 and 3 years of age and pattern of symptom change over time. J Child Psychol Psychiatry. 2005;46:500–513. doi: 10.1111/j.1469-7610.2004.00377.x. [DOI] [PubMed] [Google Scholar]
- Clifford S, Dissanayake C, Bui QM, Huggins R, Taylor AK, Loesch DZ. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord. 2007;37:738–747. doi: 10.1007/s10803-006-0205-z. [DOI] [PubMed] [Google Scholar]
- Cohen D, Pichard N, Tordjman S, Baumann C, Burglen L, Excoffier E, Lazar G, Mazet P, Pinquier C, Verloes A, Heron D. Specific genetic disorders and autism: Clinical contribution toward their identification. J Autism Dev Disord. 2005;35:103–116. doi: 10.1007/s10803-004-1038-2. [DOI] [PubMed] [Google Scholar]
- Cohen IL. Behavioral profiles of autistic and nonautistic fragile X males. Dev Brain Dysfunction. 1995;8:252–269. [Google Scholar]
- Cohen IL, Nolin SL, Sudhalter V, Ding SH, Dobkin CS, Brown WT. Mosaicism for the FMR1 gene influences adaptive skills development in fragile X-affected males. Am J Med Genet. 1996;64:365–369. doi: 10.1002/(SICI)1096-8628(19960809)64:2<365::AID-AJMG26>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Dykens EM, Volkmar FR. Medical conditions associated with autism. In: Cohen DJ, Volkmar FR, editors. Handbook of autism and pervasive developmental disorder. New York: Wiley; 1997. pp. 388–410. [Google Scholar]
- Fecteau S, Mottron L, Berthiaume C, Burack JA. Developmental changes of autistic symptoms. Autism. 2003;7:255–268. doi: 10.1177/1362361303007003003. [DOI] [PubMed] [Google Scholar]
- Feinstein C, Reiss AL. Autism: The point of view from fragile X studies. J Autism Dev Disord. 1998;28:393–405. doi: 10.1023/a:1026000404855. [DOI] [PubMed] [Google Scholar]
- Fenton G, D’Ardia C, Valente D, Del Vecchio I, Fabrizi A, Bernabei P. Vineland adaptive behavior profiles in children with autism and moderate to severe developmental delay. Autism. 2003;7:269–287. doi: 10.1177/1362361303007003004. [DOI] [PubMed] [Google Scholar]
- Fisch GS, Simensen RJ, Schroer RJ. Longitudinal changes in cognitive and adaptive behavior scores in children and adolescents with the fragile x mutation or autism. J Autism Dev Disord. 2002;32:107–114. doi: 10.1023/a:1014888505185. [DOI] [PubMed] [Google Scholar]
- García-Nonell C, Ratera ER, Harris S, Hessl D, Ono MY, Tartaglia N, Marvin E, Tassone F, Hagerman RJ. Secondary medical diagnosis in fragile X syndrome with and without autism spectrum disorder. Am J Med Genet Part A. 2008;146A:1911–1916. doi: 10.1002/ajmg.a.32290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ. The physical and behavioral phenotype. In: Hagerman RJ, Cronister A, editors. Fragile X syndrome: Diagnosis, treatment, and research. Baltimore, MD: The Johns Hopkins University Press; 2002. pp. 3–109. [Google Scholar]
- Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A, Kronk R, Delahunty C, Hessl D, Visootsak J, Picker J, Gane L, Tranfaglia M. Advances in the treatment of fragile X syndrome. Pediatrics. 2008 doi: 10.1542/peds.2008-0317. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Hull CE, Safanda JF, Carpenter I, Staley LW, O’Connor RA, Seydel C, Mazzocco MM, Snow K, Thibodeau SN, Kuhl D, Nelson DL, Caskey ST, Taylor AK. High functioning fragile X males: Demonstration of an unmethylated fully expanded FMR-1 mutation associated with protein expression. Am J Med Genet. 1994;51:298–308. doi: 10.1002/ajmg.1320510404. [DOI] [PubMed] [Google Scholar]
- Hagerman RJ, Jackson AW, III, Levitas A, Rimland B, Braden M. An analysis of autism in fifty males with fragile X syndrome. Am J Med Genet. 1986;23:359–374. doi: 10.1002/ajmg.1320230128. [DOI] [PubMed] [Google Scholar]
- Hall SS, Lightbody AA, Reiss AL. Compulsive, self-injurious, and autistic behavior in children and adolescents with fragile X syndrome. Am J Ment Retard. 2008;113:44–53. doi: 10.1352/0895-8017(2008)113[44:CSAABI]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Hatton DD, Bailey DB, Jr, Hargett-Beck MO, Skinner M, Clark RD. Behavioral style of young boys with fragile X Syndrome. Dev Med Child Neurol. 1999;41:625–632. doi: 10.1017/s0012162299001280. [DOI] [PubMed] [Google Scholar]
- Hatton DD, Sideris J, Skinner M, Mankowski J, Bailey DB, Jr, Roberts J, Mirrett P. Autistic behavior in children with fragile X syndrome: Prevalence, stability, and the impact of FMRP. Am J Med Genet Part A. 2006;140A:1804–1813. doi: 10.1002/ajmg.a.31286. [DOI] [PubMed] [Google Scholar]
- Hatton DD, Wheeler AC, Skinner ML, Bailey DB, Sullivan KM, Robert JE, Mirrett P, Clark RD. Adaptive behavior in children with fragile X syndrome. Am J Ment Retard. 2003;108:373–390. doi: 10.1352/0895-8017(2003)108<373:ABICWF>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Holmes N, Shah A, Wing L. The Disability Assessment Schedule: A brief screening device for use with the mentally retarded. Psychol Med. 1982;12:879–890. doi: 10.1017/s0033291700049175. [DOI] [PubMed] [Google Scholar]
- Kau ASM, Tierney E, Bukelis I, Stump MH, Kates, Trescher WH, Kaufmann WE. Social behavior profile in young males with fragile X syndrome: Characteristics and specificity. Am J Med Genet Part A. 2004;126A:9–17. doi: 10.1002/ajmg.a.20218. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Abrams MT, Chen W, Reiss AL. Genotype, molecular phenotype, and cognitive phenotype: Correlations in fragile X syndrome. Am J Med Genet. 1999;83:286–295. [PubMed] [Google Scholar]
- Kaufmann WE, Capone GT, Clarke M, Budimirovic DB. Autism: Current Theories and Evidence. In: Zimmerman AW, editor. Autism in genetic intellectual disability: Insights into idiopathic autism. Totowa, NJ: The Humana Press Inc; 2008. pp. 81–108. [Google Scholar]
- Kaufmann WE, Cortell R, Kau AS, Bukelis I, Tierney E, Gray RM, Cox C, Capone GT, Stanard P. Autism spectrum disorder in fragile X syndrome: Communication, social interaction, and specific behaviors. Am J Med Genet Part A. 2004;129A:225–234. doi: 10.1002/ajmg.a.30229. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Reiss AL. Molecular and cellular genetics of fragile X syndrome. Am J Med Genet. 1999;88:11–24. doi: 10.1002/(sici)1096-8628(19990205)88:1<11::aid-ajmg3>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Kleinman JM, Vontola PE, Pandey J, Verbalis AD, Barton M, Hodgson S, Green J, Dumont-Mathieu T, Robins DL, Fein D. Diagnostic stability in very young children with autism spectrum disorders. J Autism Dev Disord. 2008;38:606–615. doi: 10.1007/s10803-007-0427-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraijer D. Review of adaptive behavior studies in mentally retarded persons with autism/pervasive developmental disorder. J Autism Dev Disord. 2000;30:39–47. doi: 10.1023/a:1005460027636. [DOI] [PubMed] [Google Scholar]
- Kraut MA, Gerring JP, Cooper KL, Thompson RE, Denckla MB, Kaufmann WE. Longitudinal evolution of unidentified bright objects in children with neurofibromatosis-1. Am J Med Genet Part A. 2004;129A:113–119. doi: 10.1002/ajmg.a.20656. [DOI] [PubMed] [Google Scholar]
- Lewis P, Abbeduto L, Murphy M, Richmond E, Giles N, Bruno L, Schroeder S. Cognitive, language and social-cognitive skills of individuals with fragile X syndrome with and without autism. J Intellect Disabil Res. 2006;50:532–545. doi: 10.1111/j.1365-2788.2006.00803.x. [DOI] [PubMed] [Google Scholar]
- Lord C, Risi S, Lambrecht L, Cook EH, Jr, Leventhal BL, DiLavore PC, Pickles A, Rutter M. The Autism Diagnostic Observation Schedule-Generic: A standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord. 2000;30:205–223. [PubMed] [Google Scholar]
- Lord C, Rutter M, DiLavore P, Risi S. Autism Diagnostic Observation Schedule-WPS Edition. Los Angeles, CA: Western Psychological Services; 1999. [Google Scholar]
- Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised. A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- Maddalena A, Richards CS, McGinniss MJ, Brothman A, Desnick RJ, Grier RE, Hirsch B, Jacky P, Mcdowell GA, Popovisch B, Watson M, Wolff DJ. Technical standards and guidelines for fragile X: The first of a series of disease-specific supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics. Quality Assurance Subcommittee of the Laboratory Practice Committee. Genet Med. 2001;3:200–205. doi: 10.1097/00125817-200105000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell AE. Analyzing Qualitative Data. London: Methuen; 1961. [Google Scholar]
- Merenstein SA, Sobesky WE, Taylor AK, Riddle JE, Tran HX, Hagerman RJ. Molecular-clinical correlations in males with an expanded FMR1 mutation. Am J Med Genet. 1996;64:388–394. doi: 10.1002/(SICI)1096-8628(19960809)64:2<388::AID-AJMG31>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Moss J, Magiati I, Charman T, Howlin P. Stability of the autism diagnostic interview-revised from pre-school to elementary school age in children with autism spectrum disorders. J Autism Dev Disord. 2008;38:1081–1091. doi: 10.1007/s10803-007-0487-9. [DOI] [PubMed] [Google Scholar]
- Muhle R, Trentacoste SV, Rapin I. The genetics of autism. Pediatrics. 2004;113:e472–e486. doi: 10.1542/peds.113.5.e472. [DOI] [PubMed] [Google Scholar]
- Philofsky A, Hepburn SL, Hayes A, Hagerman R, Rogers SJ. Linguistic and cognitive functioning and autism symptoms in young children with fragile X syndrome. Am J Ment Retard. 2004;109:208–218. doi: 10.1352/0895-8017(2004)109<208:LACFAA>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Piven J, Harper J, Palmer P, Arndt S. Course of behavioral change in autism: A retrospective study of high-IQ adolescents and adults. J Am Acad Child Adolsec Psychiatry. 1996;35:523–529. doi: 10.1097/00004583-199604000-00019. [DOI] [PubMed] [Google Scholar]
- Rogers SJ, Wehner DE, Hagerman R. The behavioral phenotype in fragile X: Symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J Dev Behav Pediatr. 2001;22:409–417. doi: 10.1097/00004703-200112000-00008. [DOI] [PubMed] [Google Scholar]
- Rousseau F, Heitz D, Biancalana V, Blumenfeld S, Kretz C, Boue J, Tommerup N. Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N Engl J Med. 1991;325:1673–1681. doi: 10.1056/NEJM199112123252401. [DOI] [PubMed] [Google Scholar]
- Rousseau F, Heitz D, Tarleton J, MacPherson J, Malmgren H, Dahl N, Barnicoat A, Mathew C, Mornet E, Tejada I, Maddalena A, Spiegel R, Schinzel A, Marcos JAG, Schorderet DF, Schaap T, Maccioni L, Russo S, Jacobs PA, Schwartz C, Mandel JL. A multicenter study on genotype-phenotype correlations in the fragile X syndrome, using direct diagnosis with probe StB12.3: The first 2,253 cases. Am J Hum Genet. 1994;55:225–237. [PMC free article] [PubMed] [Google Scholar]
- Sabaratnam M, Murthy NV, Wijeratne A, Buckingham A, Payne S. Autistic-like behaviour profile and psychiatric morbidity in fragile X syndrome: A prospective ten-year follow-up study. Eur Child & Adolesc Psychiatry. 2003;12:172–177. doi: 10.1007/s00787-003-0333-3. [DOI] [PubMed] [Google Scholar]
- Sattler JM. Assessment of Children. 3rd edition. San Diego, CA: J.M. Sattler; 1988. [Google Scholar]
- Schaefer GB, Mendelsohn NJ. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders. Genet Med. 2008;10:464. doi: 10.1097/GIM.0b013e31816b5cc9. Professional Practice and Guidelines Committee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopler E, Reichler RJ, Renner BR. Childhood Autism Rating Scale. Los Angeles: Western Psychological Services; 1988. [Google Scholar]
- Seltzer MM, Krauss MW, Shattuck PT, Orsmond G, Swe A, Lord C. The symptoms of autism spectrum disorders in adolescence and adulthood. J Autism Dev Disord. 2003;33:565–581. doi: 10.1023/b:jadd.0000005995.02453.0b. [DOI] [PubMed] [Google Scholar]
- Semel E, Wiig EH, Secord WA. Clinical Evaluation of Language Fundamentals. 3rd Edition. San Antonio, TX: Psychological Corporation; 1995. [Google Scholar]
- Shattuck PT, Seltzer MM, Greenberg JS, Orsmond GI, Bolt D, Kring S, Lounds J, Lord C. Change in autism symptoms and maladaptive behaviors in adolescents and adults with an autism spectrum disorder. J Autism Dev Disord. 2007;37:1735–1747. doi: 10.1007/s10803-006-0307-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman S. Epidemiology. In: Hagerman RJ, Hagerman PJ, editors. Fragile X syndrome: Diagnosis, treatment, and research. 3rd edition. Baltimore, MD: Johns Hopkins University Press; 2002. pp. 136–139. [Google Scholar]
- Sparrow SS, Balla DA, Cicchetti DV. Vineland Adaptive Behavior Scales. Circle Pines, MN: American Guidance Service; 1984. [Google Scholar]
- Sullivan K, Hooper S, Hatton D. Behavioural equivalents of anxiety in children with fragile X syndrome: parent and teacher report. J Intellect Disabil Res. 2007;51:54–65. doi: 10.1111/j.1365-2788.2006.00899.x. [DOI] [PubMed] [Google Scholar]
- Thorndike R, Hagen E, Sattler J. Guide for administering and scoring the Stanford-Binet Intelligence Scale. 4th edition. Chicago, IL: The Riverside Publishing Company; 1986. [Google Scholar]
- Tomanik SS, Pearson DA, Loveland KA, Lane DM, Shaw JB. Improving the reliability of autism diagnoses: Examining the utility of adaptive behavior. J Autism Dev Disord. 2007;37:921–928. doi: 10.1007/s10803-006-0227-6. [DOI] [PubMed] [Google Scholar]
- Turner LM, Stone WL. Variability in outcome for children with an ASD diagnosis at age 2. J Child Psychol Psychiatry. 2007;48:793–802. doi: 10.1111/j.1469-7610.2007.01744.x. [DOI] [PubMed] [Google Scholar]
- Werner E, Dawson G, Munson J, Osterling J. Variation in early developmental course in autism and its relations with behavioral outcome at 3–4 years of age. J Autism Dev Disord. 2005;35:337–350. doi: 10.1007/s10803-005-3301-6. [DOI] [PubMed] [Google Scholar]
- Wing L. The MRC handicaps, behaviour and skills schedule. Aca Psychiatr Scand. 1980;62:241–248. [Google Scholar]
- Wright-Talamante C, Cheema A, Riddle JE, Luckey DW, Taylor AK, Hagerman RJ. A controlled study of longitudinal IQ changes in females and males with fragile X syndrome. Am J Med Genet. 1996;64:350–355. doi: 10.1002/(SICI)1096-8628(19960809)64:2<350::AID-AJMG23>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Zimmermann IL, Steiner VG, Pond RE. English Edition. 3rd Edition. San Antonio, TX: The Psychological Corporation; 1992. Preschool Language Scale. [Google Scholar]