

Abstract
Technologies to synthesize and transplant a complete genome into a cell have opened limitless potential to redesign organisms for complex, specialized tasks. However, large-scale re-engineering of a biological circuit will require systems-level optimization that will come from a deep understanding of operational relationships among all the constituent parts of a cell. The integrated framework necessary for conducting such complex bioengineering requires the convergence of systems and synthetic biology. Here, we review the status of these rapidly developing interdisciplinary fields of biology and provide a perspective on plausible venues for their merger.
Multidisciplinary approaches in science have always resulted in new ideas, broader and deeper levels of understanding, and of course, controversies. The inception of many modern transformative products and technologies, including drug synthesis1 and recombinant microbial enzyme factories2 can be attributed to the merging of, until then, distinct disciplines. With recent advances to deconstruct biological function at a systems scale3,4 and design biological subcircuits with defined properties5 we can begin to envision spectacular solutions that combine unique functions that have evolved in organisms from diverse environments. In other words, the stage is set for yet another revolutionary merger of two powerful interdisciplinary fields with fundamentally different but complementary outlooks — synthetic biology and systems biology (FIG. 1).
Figure 1. Timeline of events that resulted in the development of systems biology and synthetic biology.

Key milestones within the fields of systems biology and synthetic biology are highlighted. For brevity and clarity, we have only included key advances in molecular systems biology and have not included important events related to other macroscale systems biology efforts; for example, human physiological systems and ecological systems. Although it is difficult to differentiate the specific impact some events have had on either of the two fields, we have attempted to do so for consistency. Particularly noteworthy are the events in which complete organism sequences were made available to the public and paired with gene regulatory and metabolic reconstruction models. Next generation sequencing refers to pyrosequencing on beads, polony sequencing and sequencing by synthesis. Pivotal technologies that produced new paradigms for systems and synthetic research are also important; for example, the use of mass spectrometry (MS) for proteomic analysis gave rise to quantitative proteomics and eventually metabolomics, multilayer soft lithography and microfluidic devices, and next-generation sequencing technologies enabled deep metagenomics studies. ChIP–chip, chromatin immunoprecipitation with microarray hybridization; ChIP–Seq, chromatin immunoprecipitation with sequencing; COBRA, constraints-based reconstruction and analysis; EGRIN, environment and gene regulatory influence network; ESI, electrospray ionization; FDA, Food and Drug Administration; GFP, green fluorescent protein; GRN, gene regulatory network; GMO, genetically modified organism; GSMR, genome-scale metabolic reconstruction; HGP, Human Genome Project; ICAT, isotopic coded affinity tag; iTRAQ, isobaric tags for relative and absolute quantitation; MALDI, matrix-assisted laser desorption ionization; MLS, multi-layer soft lithography; qPCR, quantitative PCR; SILAC, stable isotope labelling with amino acid in cell culture; TOF, time of flight; YAC, yeast artificial chromosome.
A systems biologist aims to model and understand an entire organism by characterizing dynamic environment-dependent functional interrelationships between its constituent parts (for example, genes, RNAs, proteins and metabolites). A synthetic biologist, however, uses well characterized parts that are shaped by natural evolution to construct artificial systems that perform new tasks. These fields are on trajectories that are bound to cross paths and even merge as they begin to inform one another. We envision that systems biology will provide both the parts and wiring diagrams for entire cells to guide complex circuit design for synthetic biology approaches. In this Opinion article, we focus on recent advances in the design of synthetic circuits and predictive modelling of prokaryotes, and the convergence of these two disciplines for rational biological systems re-engineering. Given that both fields are rapidly progressing on multiple fronts, we acknowledge that despite every attempt to make this article comprehensive, it is by no means complete.
Why use systems re-engineering?
By rewiring regulatory circuits to couple new enzymes, sensors and transporters, a cell can be redesigned to execute novel and complex processes. Although carefully designed genomic reconfiguration and reduction has been shown to be feasible under strictly controlled laboratory conditions6, such re-engineering can reduce the fitness of an organism, especially in dynamic and complex environments, by introducing taxes on the limited energy and biochemical resources of the cell7. A distinguishing aspect of systems re-engineering will be the integration of compensatory regulatory and metabolic adjustments to optimize resource management and restore homeostasis in the newly engineered system. Such balanced re-engineering will enable complex biological circuit designs driven by specific spatio-temporal control that have the potential to help solve such diverse and pressing problems as environmental pollution, energy production and complex diseases.
Take bioremediation as an example. Isolated parts of a desired system exist in a diverse collection of organisms that withstand radioactivity (for example, Deinococcus radiodurans8), thrive in high salt concentrations (Halobacterium salinarum3) or high temperatures (Pyrococcus furiosus9), or detoxify metals (species of Geobacter and Shewanella10). Using these independently evolved parts, a plausible design would be a self-sustaining integrated system (for example, within single microorganisms or as microbial consortia11) that is capable of sensing and relocating towards pollutants, producing detoxification agents and self-destructing on cue to avoid biological contamination. Given the drawbacks of current technologies, the successful implementation of such a system is incredibly ambitious. There are numerous challenges to be addressed on a case-by-case basis, such as the incompatibility of heterologous gene products within a selected host system. Nonetheless, similar but simplified strategies, such as the use of microorganisms in the production of biofuels12, bioremediation strategies13 and drug delivery14, have proven successful. These strategies constitute concept models for re-engineering biological systems that could be developed and optimized further by a combination of systems and synthetic biology. Control, robustness, reliability and predictability of such re-engineered systems would present advantages over natural organisms that may not possess the features necessary to perform such specific tasks. This exciting journey will lead to new technologies and provide a deeper understanding of biological systems, but also raises important ethical, legal and environmental concerns that warrant careful consideration15,16.
Programming a cell: synthetic biology
At its core, synthetic biology is the practice of applying engineering principles to rationally redesign biological subsystems with new and useful dynamic properties (BOX 1). This process typically begins with a mathematical model of interactions with desired dynamic properties. These models are often based on known circuits from laboratory ‘workhorses’, such as Escherichia coli and Saccharomyces cerevisiae (budding yeast), and, in most cases, are limited to a dozen or so interacting elements. The synthetic system is further refined iteratively through computational simulation and experimental validation to resolve inconsistencies.
Box 1 | Synthetic biology approaches to the design of circuits
Parts
Individual components that make up gene expression machinery. These components are specific DNA sequences that code for gene promoters and upstream regulatory sites, ribosome binding sites, gene or protein coding regions, and mRNA translation termination sites. Sequences for RNA-only machinery, such as small interfering RNA and ribozyme coding sequences, are also used.
Devices
Assemblages of parts (promoters and genes) that carry out specific functions. There are several basic devices, including reporters, inverters and devices that carry out signalling and protein generation. Each device includes one or two promoter–ribosome binding site–gene terminator constructs. Subsequently, these basic devices can be combined into ‘composite devices’ to achieve more complex behaviour.
Systems
Collections of specific devices that enable individual parts to be quantified. Generally, a subset of the composite devices is used to characterize the strength and efficiency of promoters under non-repressive conditions.
Chassis
The host cell in which the systems and devices described above are assembled and used. To date, these cells have been primarily from Escherichia coli strains, although a few devices and systems have been tested in yeast and mammalian cells. A more exotic chassis includes cell-free systems that use DNA transcription and mRNA translation chemistry derived from whole-cell extracts and encapsulated in artificial membranes.
Systems Biology Mark-up Language (SBML)
SBML is an XML (Extensible Markup Language)-like language used to encode reaction sets that constitute a typical biological model. These include cell signalling, metabolic pathways, biochemical reactions and gene regulation kinetics. SBML is a computable format: it incorporates all the information about a model and can be directly used to perform simulations. Such information includes the definition of mathematical functions, units, species types and compartmentalization, initial values, constraints, species rules and reaction rates. There is no ‘one-size-fits-all’ encoding language, owing to the many modelling approaches that are available to systems and synthetic biologists. SBML is therefore intended to be a common intermediate format, allowing for the transfer of essential components of a model between diverse computational analysis and simulation tools. The list of SBML compatible programmes is comprehensive and continually expanding (see Further information for a link to the SBML Software Matrix).
See the MIT Registry of Standard Biological Parts for further information.
The first and most common approach in synthetic biology is the incorporation of exogenous or synthetic gene constructs into a host system. Synthetic biology differs dramatically from biotechnology17,18 in the complexity of re-engineering, especially in the synthetically designed regulatory network for controlling gene expression. This approach has benefited from the development of natural or synthetic mobile regulatory circuits (orthogonal systems) that can function in a context-independent manner without host interference19.
In a second approach, deleting genes to leave behind a skeletal architecture for processing nutrients, protein production and cellular reproduction simplifies an existing biological network. This minimal biological system is then used as a basic and easily manipulated framework on which to build novel regulatory circuitry20. An important limitation of this approach is the potential instability of such minimal systems to complex environmental changes21.
A third, and perhaps most ambitious, approach is the complete de novo synthesis of an organism. Synthetic genomes of increasing complexity can now be generated22,23. The feasibility of transplanting genomes between organisms, as a means of ‘rebooting’, has been recently demonstrated24. However, this approach is still in its infancy and functional gene expression from such entirely synthetic systems is short-lived (~6–8 hours), limiting the use of this approach to simple regulatory systems25. Moreover, incorporation of all the necessary components for basic cellular function is an understated challenge.
Although synthetic biology has mainly been applied to the fundamental sciences26, a number of approaches have potentially useful applications. Examples related to human health include the efficient production of antimalarial drugs and ‘smart’ bio-agent-based cancer therapies. Artemisinic acid, a precursor to a potent antimalarial drug, can now be produced in engineered bacteria27 and yeast28 cultures more efficiently than previous methods that relied on chemical extraction from rare plants. Notably, this effort was far more complex than the development of Humulin (human insulin), trastuzumab (Herceptin) and other recombinant therapeutics, which relied on the overexpression of a single gene2,17. Instead, Martin et al.27 and Ro et al.28 combined a microbial mevalonate pathway for terpenoid production with an engineered amorphadiene synthase and a heterologously expressed cytochrome p450 monooxygenase from the plant Artemisia annua. In another example, controlled invasion of cancer cells was accomplished by re-engineering E. coli to express invasion proteins and cancerostatic agents (colicin E3, bacteriocin, RNA-based therapeutics29 or other prodrugs) after encountering tumour-environment characteristics14. In this case, the synthetic circuit components originated from diverse organisms, including invasion machinery from Yersinia pseudotuberculosis and quorum sensors from Vibrio fischeri.
Synthetic biology may also help address the renewed concerns regarding the availability and environmental impact of fossil fuels. The production of biofuels by engineered microorganisms is an alternative that is gaining much interest12. production of alcohols that contain higher energy, such as butanol, is possible by modifying amino acid biosynthetic pathways in E. coli30.
The fundamental synthetic biology tenet illustrated by these examples is that knowledge of component dynamics and mechanisms leads to predictable behaviour in artificially constructed circuits of increasing complexity31. This approach, coupled to complementary experimental techniques, has enormous potential for understanding fundamental regulatory dynamics of both uncharacterized and well-known systems32. Importantly, such efforts have produced regulatory circuits that exhibit precise behaviours, including autoregulatory positive feedback loops33, toggle switches34 and oscillatory systems35,36. These successes have also brought to light the importance of noise in biological systems. Expression measurements in single cells can be noisy, but result in smooth and deterministic behaviour at the population level, affording adaptability to the overall system37. The desired re-engineered system must therefore be able to either attenuate or take advantage of such noise with an objective goal of robust, predictable circuit behaviour38.
However, despite these impressive achievements, classic engineering design principles, such as abstraction, modularization and standardization, have yet to be consistently and robustly implemented for sophisticated synthetic biology re-engineering. The circuits discussed above were not based on standardized (for example, easily interchangeable) parts, and therefore require specific modifications to achieve the desired dynamic behaviours. In other words, this laborious ad hoc process does not scale well — the leap from creating discrete modules of defined function to re-engineering an entire cell or organism and incorporating highly interconnected systems with homeostasis is not straightforward.
Standard parts list and vocabulary
An important requirement for the construction of complex circuits is a standardized language and lists of parts with detailed specifications. There are two notable examples of such resources: the freely accessible MIT Registry of Standard Biological parts (sensors, genetic circuits and actuators)39 and the standardized open source Systems Biology Markup language (SBML)40 for representing biological circuit models (see Further information) (BOX 1). Mathematical design rules emerging from these efforts will eventually unite the well established engineering framework of standardization, decoupling and abstraction with biological regulatory circuits39. This union will produce a library of reusable parts and devices with defined data sheets41, easing the production of more complex systems. However, a key specification, the systems-level operational relationships of various parts and subcircuits, is missing; this cannot be ignored if the eventual goal is to restore homeostasis in the engineered system. We posit that systems biology approaches will help pave the way to the production of more complex systems by defining the global rules that govern complex cellular circuit behaviours.
Systems biology: modelling a cell
By iteratively driving developments in biology, computation and technology, systems biology tackles the reverse engineering problem of deconstructing integrated biological circuits into holistic and predictive models42. These circuits have assembled over 3.5 billion years of evolution to sense environmental signals, turn genes on and off, control enzyme activities and transporters, and coordinate these processes with structural changes in the cell, DNA replication, segregation and reproduction.
There are two complementary approaches to deconstruct a biological circuit43: ‘bottom-up’ and ‘top-down’ approaches. In the traditional approach, information gathered through detailed characterization of individual parts and their interactions is used to assemble circuits bottom-up44. However, unlike the architectures of electronic circuits, which are constant in time and space, biological circuits assemble dynamically through environment-dependent synthesis, reuse and sharing of parts. A circuit assembled purely from a bottom-up approach is therefore unable to capture the global dynamics of a system.
A top-down approach takes advantage of high-throughput technologies (BOX 2) to investigate the dynamic operational interrelationships of all constituent parts of a system simultaneously. Although this has sacrificed the resolution at which biological properties are characterized, new high-throughput sequencing and high-density array technologies, as well as mathematical techniques that extract quantitative kinetic parameters from sparse time-course data, are minimizing the loss of detailed information45.
Box 2 | Experimental approaches used in systems biology
Sequencing technologies
Sequencing efforts were greatly advanced and accelerated by the Human Genome Project. Randomly generated genomic fragments are analysed by a capillary-based Sanger method on sequencing platforms developed by Applied Biosystems91. Massively higher throughput is achieved by the use of new technologies, such as 454 pyrosequencing, SOLiD (sequencing by ligation) and Solexa (sequencing by synthesis)66,67,92. Quantitative transcriptome profiling, metagenomics of complex microbial environments and sequencing of multiple prokaryotic strains are being driven by such technologies51.
DNA microarrays and high-throughput sequencing
Slides containing spatially organized DNA probes with known sequences are used as a platform for the identification and quantification of fluorescently labelled nucleic acids samples. Applications of this technology include, but are not limited to, gene expression studies (RNA and cDNA hybridizations)93, comparative genomics (comparative genomic hybridization)94, identification of protein–DNA binding sites (ChIP–chip; chromatin immunoprecipitation with microarray hybridization)95 and DNA methylation studies96.
High-throughput sequencing is offering alternative solutions to a number of array-based technologies, including gene expression and protein–DNA interaction measurements. For example, DNA fragments in enriched transcription complexes can be localized at higher resolution by this approach (ChIP–Seq; chromatin immunoprecipitation with sequencing)69.
Proteomics technologies
The study of all the proteins in a cell, including their dynamics, modifications, interactions and structure. High-throughput analysis of proteins has been driven by the development of mass spectrometry (MS)-based approaches for the identification, quantitation and analysis of modifications with increasing sensitivity97. The development of array-based platforms, such as protein or antibody microarrays, in addition to two-hybrid approaches, are allowing higher throughput of biochemical assays to assess protein functions98. Tagging green fluorescent protein and fluorescence resonance energy transfer analysis allow in vivo study by systematic analysis of protein localization in the cell98. Structural proteomics is also being developed using X-ray crystallography and nuclear magnetic resonance, electron microscopy and electron tomography of protein complexes99.
Metabolomics technologies
The study of metabolites, the pathways they affect (or are affected by) and their interplay with cellular regulation at the gene level. Powerful and accurately predictive approaches have been successfully applied to assess the phenotypic behaviours of Escherichia coli, yeast and red blood cells79. Collecting the data required for these models can be common bench-scale assays to high-throughput methods, such as automated high-performance liquid chromatography and MS-based approaches100.
A systems approach begins with the systematic perturbation of a complex process, a single cell, a multicellular organism or a community of organisms. Although this step is not always hypothesis driven in the first iteration, subsequent iterations tend to be, as they build on prior knowledge and data. Molecular and phenotypic responses are observed globally, documented and integrated with prior information, including data from bottom-up approaches, to produce a quantitative model that recapitulates prior observations and predicts behaviours in new environments (BOX 3). Given the context-dependent dynamics of biological responses, careful and controlled experiments are needed to provide accurate data to model the system46. experimental and in silico testing of systems-generated hypotheses resolves inconsistencies and feeds new information into model revisions. Although this is similar to the synthetic biology approach, the global measurements and modelling approaches used in the iterative cycle of systems biology (especially the top-down approach) can delineate principles governing assembly of complex biological circuits, including most of the cellular components3,4.
Box 3 | network models for systems biology
Network models101 that integrate a myriad of information have been developed to describe and predict properties of complex biological systems. The balance between data availability and the complexity of the model is an important factor to be considered102: the choice of model must fit with the parameters and generating predictions. Once the validation step is performed, the model can be improved iteratively and interactively. All these steps can be a combination of approaches; for example, simultaneously using gene expression levels (continuous) and discretized protein–protein interaction information. Three examples of successful modelling approaches are provided in the figure and the table. Other diverse models have been used in eukaryotic organisms58 and can be adapted for the system of interest.
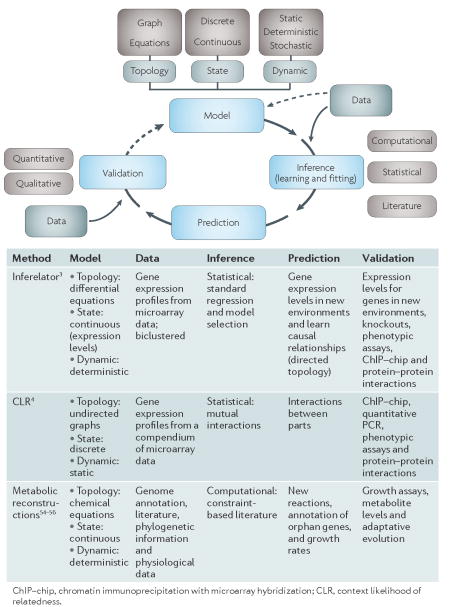
In general, the top-down approach is regarded as the paradigm for systems biology. This methodology has roots in whole genome sequencing, microarray technology, proteomics and other high-throughput methods that can be used to observe numerous system elements simultaneously (BOX 2). Although the varying limitations of these technologies yield data with different levels of uncertainty, these often tend to be systematic and specific to a given approach. By integrating the various global data using a cross-validation procedure (for example, by using a gold standard data set, such as membership in gene ontology function categories or an objective function that evaluates a metric of relatedness, such as co-expression), it is possible to find meaningful information that is supported by multiple sources of evidence. This is illustrated by the integration of expression data, comparative genomics and physical interactions to discover conditionally co-regulated genes that are co-expressed, share promoter motif signatures, are functionally associated and/or physically interact47. Therefore, although the richness and depth of the data are initially overwhelming, the top-down approach is equally useful and can be integrated with the bottom-up approach. Even well studied model organisms and macromolecular processes benefit from the merger of holistic and reductionist scientific approaches. Notable examples include the study of cell cycle regulation in Caulobacter crescentus44, bacterial chemotaxis48 and subcellular organization of proteins and DNA in bacterial cells49. To date, the study of transcriptional regulatory networks (TRNs) are examples of the best-characterized systems with genome-scale predictive models. Even the most studied prokaryotic model organism, E. coli, has benefited from systems analysis of these networks. Curated resources, such as RegulondB50 (see Further information), house many experimental and in silico data that are common entry points for further biological exploration of this organism. This has enabled rapid development and verification of predictive global models that integrate known and novel regulatory interactions to broaden our view of transcriptional regulation of diverse biological processes4.
Systems-level inquiries empower rapid characterization of poorly studied organisms, especially owing to the current wealth of newly sequenced, but poorly annotated, microbial genomes51. In our own work, using a top-down approach of biclustering, coupled with statistical network inference, it was possible to integrate diverse, system-wide data for the halophilic archaeon H. salinarum NRC-1 to construct an environmental and gene regulatory influence network model that reliably predicted transcriptional changes in 80% of all its genes to new environmental and genetic perturbations3. Comparable systems-level models of environmentally important organisms, such as the metabolically versatile Shewanella oneidensis MR-1 (REF. 52), and pathogenic bacterial systems53 are also well underway.
Substantial advances have also been made in the mathematical reconstruction of metabolism in different prokaryotic organisms54,55; the metabolic model for E. coli, for example, now contains 48% of all genes with experimentally determined functions. These models have been used to design strains for metabolite production, identify putative genes for orphan reactions, and for physiological and evolutionary studies56. Importantly, approaches for reconstructing metabolic networks can be extended to complex eukaryotic systems, such as yeast, Aspergillus nidulans57, Caenorhabditis elegans58 and cell lines that model human physiology59–61. Such bottom-up reconstruction, integrated with high-throughput global analysis (especially metabolome and fluxome data), is moving the field forwards towards systems-scale kinetic models62.
Although construction of systems-scale integrated models of TRNs and metabolism is admittedly challenging, recent successes in simultaneous high-throughput analyses of biological processes at different levels offer promising solutions to help decipher some of these interrelationships63,64.
Examples of poorly represented information in systems-scale models include translation efficiencies, interference by non-coding RNAs, protein modifications, structural information and experimentally unexplored regions of metabolic maps65. In other words, there is still work to be done to incorporate such missing information to make systems-scale models mechanistically accurate and quantitatively predictive — a fundamental basis for re-engineering circuits. There is evidence of some progress on this front through deep sequencing of transcripts66,67, precise localization of genomic binding sites for transcription factors68 and chromatin remodelling proteins69, transcription start site mapping for accurate promoter discovery70 and ultra-high-accuracy mass spectrometry for metabolome mapping65.
In the future, in addition to comparisons based on genes, genome organization and protein sequences, organisms will be compared based on the network architecture of interacting genes, proteins, metabolites and RNAs71,72. The comparison of network topologies in the context of phylogeny will lead to a broader and deeper understanding of the evolution of complex biological systems.
Convergence: global network rewiring
Biological systems have evolved in a constrained environmental framework of reproducible physicochemical relationships73. Consequently, they have learned these relationships to modulate various cellular processes appropriately in response to immediate challenges and in anticipation of future environmental changes74. This ability to reallocate biochemical and energy stores dynamically to various cellular processes lends a fitness and competitive advantage to a cell in a resource-limited environment. By ignoring such relationships while redesigning a biological circuit, we risk decoupling cellular processes from the environmental network, and as such, rendering them less fit or unstable. Fortunately, biological systems are resilient and have some capability to accommodate small changes both in the external environmental network74 and their own networks75. However, they are not equipped to deal with large changes to their circuitry, including the introduction of entirely new processes, which could shift the internal balance of energy and resources. It will therefore become necessary to understand the coupling of various cellular processes, so that additional compensatory changes can be introduced to push the redesigned biological circuit towards the desired optimal state. Knowledge of biological noise in single cells and populations and its role in providing robustness to cellular responses will also be crucial in this re-engineering effort. To this end, directed laboratory evolution will be indispensable for completely optimizing a rationally redesigned circuit76.
Unsurprisingly, current models in both synthetic and systems biology highlight the interwoven relationship between environmental influences and the responses of biological networks. However, these models operate at different scales, and to realize the new paradigm of rational systems re-engineering, synthetic and systems biology fields must collaborate closely and eventually merge. Notably, the strengths and weaknesses of both fields are highly complementary.
Synthetic biology and bottom-up systems biology methods extract discrete, accurate, quantitative, kinetic and mechanistic details of regulatory subcircuits. The models generated from these approaches provide an explicit mathematical foundation that can ultimately be used in systems redesign and re-engineering. However, these approaches are confounded by high dimensionality, non-linearity and poor prior knowledge of key dynamic parameters77 when scaled to large systems of hundreds to thousands of unique interacting components. As a result, modular subnetwork characterization is performed under the unrealistic assumption that the network is isolated from the rest of the host system.
The top-down systems biology approach leverages data-rich high-throughput experimental analyses that catalogue the entire set of components within a system. Although these measures retain high statistical confidence, they are usually qualitative or semi-quantitative; for example, gene expression profiles are quantized categorically as up, down or not changed relative to other genes of interest or a specific reference condition. Models of global systems are similarly qualitative, tending toward algorithmic descriptions of component interactions78. Such models are amenable to the experimental data used to develop them, but usually sacrifice the finer kinetic and mechanistic details of the molecular components involved79. Thus, accurate quantitative and mechanistic prediction of how the entire system would operate in new environments is currently beyond the means of such analyses.
How we can bridge the divide between the synthetic and systems views is being actively discussed, and several potential solutions have been suggested80,81. A common suggestion is the use of multi-scale approaches: those that translate the dynamics quantified at the component level into the inferred, logical interactions at the global network level. Such approaches would represent an ideal interface for the union of synthetic biology and systems biology, but would require a detailed description of the global network and environmental influences on its operation and modular interconnections, as well as careful characterization of several (if not all) of its component subcircuits. It is therefore inevitable that we will have to leverage strengths of both fields. The time is ripe to do so, as new technologies, such as deep sequencing, multiplexed tiling arrays and high-resolution mass spectrometers (BOX 2), are capturing both kinetic and mechanistic details of genetic information processing and metabolic processes at a genome scale, albeit with limited spatio-temporal resolution.
Furthermore, new approaches, such as those based on microfluidic technologies for cell culturing and multiplexed high-resolution measurement of biological responses, are enabling unprecedented detailed characterization of subcircuits82,83. Together, these new technological advances will provide a much needed impedance match for interweaving global systems models with detailed subcircuit models.
We propose two dovetailed tandem cycles of systems biology and synthetic biology that will drive the re-engineering process through simultaneous global modelling and subsequent detailed characterization of subcircuit dynamics (FIG. 2). Weaknesses of both fields are immediately resolved: subsystem connections are provided by the global analysis to the synthetic biology approach, whereas quantitative parameters and mechanistic details are introduced to the systems approach.
Figure 2. convergence of systems and synthetic biology.

The systems biology cycle begins with a specific hypothesis that is tested by systematic perturbations through targeted environmental and genetic changes. Molecular changes are measured globally at multiple levels (for example, transcription, translation and physical interactions) using high-throughput technologies. This produces large and diverse data sets that drive the development of algorithms to process raw signal and integrate all available information to infer predictive models of how the inputs (environmental and genetic perturbations) were converted to outputs (for example, phenotypes, transcriptional changes and interactions)102. Owing to the complexity of these models, their exploration requires a framework that enables the integration and interoperation of diverse databases and applications for visualizing and analysing the data used to construct the models103. The exploration of systems models enables a biologist to design experiments to test model predictions using classical genetics, biochemistry and molecular biology approaches. This helps define subcircuits and feeds the next iterations of the systems and synthetic biology cycles. In the synthetic biology cycle, the hypothesis begins as a specific network topology, regulatory subcircuit or set of molecular interactions. The system is mathematically formulated as systems of ordinary differential equations (ODEs), stochastic differential equations (SDEs) or a stochastic reaction network to produce a quantitative and kinetic representative model that is fit for computational simulation and testing. Key parameters of the system (synthesis and degradation rates, binding cooperativities and association or dissociation constants) are derived based on estimates from other models of well known systems. The system is analysed by focusing on exploring the parameter space and testing the kinetic properties of the system (for example, through frequency response analysis) to locate the regimes of desired dynamic behaviour and define their limits. Simulations elucidate revisions in the topology of the system to produce the desired output or enhance unexpected, but desirable, dynamic characteristics. Once these characteristics are well defined computationally, they are verified experimentally through system construction and implementation. Experimental exploration of the parameter space is performed using flow cytometry and microfluidics83-based assays, which provide high-throughput measurements at population and single cell scales simultaneously. Finally, the experimental implementation incorporates revisions, leading to another iteration of computational modelling to validate the dynamics of the system. New challenges to construct hybrid models that link detailed ODE or SDE models of subcircuits to the statistically learned systems models will emerge. We suggest that such a tandem ‘top-down’ and ‘bottom-up’ approach will be essential to precisely manipulate a biological circuit and accurately predict its system-level outcomes. E represents an effector (either global regulatory influences or environmental factors), r represents a regulator gene, a represents gene A and b represents gene B.
The systems biology cycle starts with a specific hypothesis regarding properties of a particular system, and this hypothesis is then tested through systematic perturbations. Large and diverse data sets are analysed and integrated to infer predictive models, which are tested using classical genetics, biochemistry and molecular biology approaches. This helps define subcircuits and feeds subsequent iterations of the systems and the synthetic biology cycles. In the synthetic biology cycle, the systems-scale model provides a hypothesis regarding a specific network topology of a regulatory subcircuit. Quantitative and kinetic representative models are formulated, key parameters are estimated and the subcircuit is iteratively refined through simulation and experimental testing. This refined mechanistic detail is eventually fed into the systems biology cycle.
Continued iterations of these tandem cycles will lead to the development of a framework that accurately predicts dynamic cellular behaviour through multiple scales. In addition to the production of well defined system behaviour, this will also enable the abstraction and standardization of principles for re-engineering biology84. It is clear that this will bring us closer to rationally redesigning a cell, but given the enormous complexity of biological systems, we anticipate that some aspects of system behaviour will not be fully understood for some time. However, it should be possible to account for these poorly understood mechanisms through the application of evolutionary processes (random mutagenesis coupled to appropriate selective pressures) to select for robust re-engineered circuits76,85 in which homeostasis is completely restored.
This framework will enable the re-engineering of cross-scale systems with desired and robust behaviours, from nanoscale engineered systems to interspecies communities86, bringing our theoretical closed-loop microbial system for bioremediation closer to reality. The merging of systems and synthetic biology will also contribute to a new generation of in vitro applications87, including nanoscale constructions that will eventually lead to entirely artificial cells, in which every component is synthesized bottom-up88.
The emergence of systems re-engineering will undoubtedly bring a dramatic shift in the way we understand and responsibly use biology, be it studies of individual cells89, communities11, ecologies90 or infectious diseases53. In just a few years, we will see a dramatic increase in the complexity of engineered biosystems. Although this will raise new challenges in bioethics, it will also provide greater control and predictability over biological processes77,85. Gone will be the days of simple recombinant overexpression of a few molecules and the painstaking task of optimizing the process. Instead, in its place, will be a foundation of mathematics and engineering principles for constructing biological circuits, which will solve many complex problems that affect humankind.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIH; P50GM076547 and 1R01GM077398-01A2), Department of Environment (DoE; DE-FG02-07ER64327 and DE-FG02-07ER64327), National Science Foundation (NSF; EF-0313754, EIA-0220153, MCB-0425825 and DBI-0640950) and NASA (National Aeronautics and Space Administration; NNG05GN58G) to N.S.B. The authors thank D. Reiss and A. Schmid for critical reading of the manuscript and R. Vencio for helpful discussions.
Footnotes
DATABASES
Entrez Genome Project: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=genomeprj
Aspergillus nidulans | Caulobacter crescentus |Caenorhabditis elegans | Deinococcus radiodurans |Escherichia coli | Halobacterium salinarum Nrc-1 |Pyrococcus furiosus | Saccharomyces cerevisiae | Shewanella oneidensis MR.1 | Vibrio fischeri | Yersinia pseudotuberculosis
FURTHER INFORMATION
Halobacterium systems Biology: http://baliga.systemsbiology.net/
Mit registry of standard Biological Parts: http://parts.mit.edu/
RegulonDB: http://regulondb.ccg.unam.mx/
SBML software Matrix: http://sbml.org/sBML_software_Guide/sBML_software_Matrix
Synthetic Biology community: http://syntheticbiology.org/
References
- 1.Yeh BJ, Lim WA. Synthetic biology: lessons from the history of synthetic organic chemistry. Nature Chem Biol. 2007;3:521–525. doi: 10.1038/nchembio0907-521. [DOI] [PubMed] [Google Scholar]
- 2.Williams DC, Frank RMV, Muth WL, Burnett JP. Cytoplasmic inclusion bodies in Escherichia coli producing biosynthetic human insulin proteins. Science. 1982;215:687–689. doi: 10.1126/science.7036343. [DOI] [PubMed] [Google Scholar]
- 3.Bonneau R, et al. A predictive model for transcriptional control of physiology in a free living cell. Cell. 2007;131:1354–1365. doi: 10.1016/j.cell.2007.10.053. [DOI] [PubMed] [Google Scholar]
- 4.Faith JJ, et al. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5:e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sprinzak D, Elowitz MB. Reconstruction of genetic circuits. Nature. 2005;438:443–448. doi: 10.1038/nature04335. [DOI] [PubMed] [Google Scholar]
- 6.Sharma SS, Blattner FR, Harcum SW. Recombinant protein production in an Escherichia coli reduced genome strain. Metab Eng. 2007;9:133–141. doi: 10.1016/j.ymben.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grilly C, Stricker J, Pang WL, Bennett MR, Hasty J. A synthetic gene network for tuning protein degradation in Saccharomyces cerevisiae. Mol Syst Biol. 2007;3:127. doi: 10.1038/msb4100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cox MM, Battista JR. Deinococcus radiodurans — the consummate survivor. Nature Rev Microbiol. 2005;3:882–892. doi: 10.1038/nrmicro1264. [DOI] [PubMed] [Google Scholar]
- 9.Laksanalamai P, Whitehead TA, Robb FT. Minimal protein-folding systems in hyperthermophilic archaea. Nature Rev Microbiol. 2004;2:315–324. doi: 10.1038/nrmicro866. [DOI] [PubMed] [Google Scholar]
- 10.Shi L, Squier TC, Zachara JM, Fredrickson JK. Respiration of metal (hydr)oxides by Shewanella and Geobacter: a key role for multihaem c-type cytochromes. Mol Microbiol. 2007;65:12–20. doi: 10.1111/j.1365-2958.2007.05783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brenner K, You L, Arnold FH. Engineering microbial consortia: a new frontier in synthetic biology. Trends Biotechnol. 2008;26:483–489. doi: 10.1016/j.tibtech.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 12.Mukhopadhyay A, Redding AM, Rutherford BJ, Keasling JD. Importance of systems biology in engineering microbes for biofuel production. Curr Opin Biotechnol. 2008;19:228–234. doi: 10.1016/j.copbio.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 13.de Lorenzo V. Systems biology approaches to bioremediation. Curr Opin Biotechnol. 2008;19:579–589. doi: 10.1016/j.copbio.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 14.Anderson JC, Clarke EJ, Arkin AP, Voigt CA. Environmentally controlled invasion of cancer cells by engineered bacteria. J Mol Biol. 2006;355:619–627. doi: 10.1016/j.jmb.2005.10.076. [DOI] [PubMed] [Google Scholar]
- 15.Boldt J, Muller O. Newtons of the leaves of grass. Nature Biotechnol. 2008;26:387–389. doi: 10.1038/nbt0408-387. [DOI] [PubMed] [Google Scholar]
- 16.Rai A, Boyle J. Synthetic biology: caught between property rights, the public domain, and the commons. PLoS Biol. 2007;5:e58. doi: 10.1371/journal.pbio.0050058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dingermann T. Recombinant therapeutic proteins: production platforms and challenges. Biotechnol J. 2008;3:90–97. doi: 10.1002/biot.200700214. [DOI] [PubMed] [Google Scholar]
- 18.Shewry PR, Jones HD, Halford NG. Plant biotechnology: transgenic crops. Adv Biochem Eng Biotechnol. 2008;111:149–186. doi: 10.1007/10_2008_095. [DOI] [PubMed] [Google Scholar]
- 19.Wang K, Neumann H, Peak-Chew SY, Chin JW. Evolved orthogonal ribosomes enhance the efficiency of synthetic genetic code expansion. Nature Biotechnol. 2007;25:770–777. doi: 10.1038/nbt1314. [DOI] [PubMed] [Google Scholar]
- 20.Pósfai G, et al. Emergent properties of reduced-genome Escherichia coli. Science. 2006;312:1044–1046. doi: 10.1126/science.1126439. [DOI] [PubMed] [Google Scholar]
- 21.Kafri R, Levy M, Pilpel Y. The regulatory utilization of genetic redundancy through responsive backup circuits. Proc Natl Acad Sci USA. 2006;103:11653–11658. doi: 10.1073/pnas.0604883103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith HO, Hutchison CA, Pfannkoch C, Venter JC. Generating a synthetic genome by whole genome assembly: phiX174 bacteriophage from synthetic oligonucleotides. Proc Natl Acad Sci USA. 2003;100:15440–15445. doi: 10.1073/pnas.2237126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibson DG, et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science. 2008;319:1215–1220. doi: 10.1126/science.1151721. [DOI] [PubMed] [Google Scholar]
- 24.Lartigue C, et al. Genome transplantation in bacteria: changing one species to another. Science. 2007;317:632–638. doi: 10.1126/science.1144622. [DOI] [PubMed] [Google Scholar]
- 25.Noireaux V, Bar-Ziv R, Godefroy J, Salman H, Libchaber A. Toward an artificial cell based on gene expression in vesicles. Phys Biol. 2005;2:P1–P8. doi: 10.1088/1478-3975/2/3/P01. [DOI] [PubMed] [Google Scholar]
- 26.Serrano L. Synthetic biology: promises and challenges. Mol Syst Biol. 2007;3:158. doi: 10.1038/msb4100202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin VJ, Pitera DJ, Withers ST, Newman JD, Keasling JD. Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nature Biotechnol. 2003;21:796–802. doi: 10.1038/nbt833. [DOI] [PubMed] [Google Scholar]
- 28.Ro DK, et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440:940–943. doi: 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- 29.Wei MQ, Mengesha A, Good D, Anné J. Bacterial targeted tumour therapy-dawn of a new era. Cancer Lett. 2008;259:16–27. doi: 10.1016/j.canlet.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 30.Atsumi S, Hanai T, Liao JC. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature. 2008;451:86–89. doi: 10.1038/nature06450. [DOI] [PubMed] [Google Scholar]
- 31.Guido NJ, et al. A bottom-up approach to gene regulation. Nature. 2006;439:856–860. doi: 10.1038/nature04473. [DOI] [PubMed] [Google Scholar]
- 32.Bennett MR, et al. Metabolic gene regulation in a dynamically changing environment. Nature. 2008;454:1119–1122. doi: 10.1038/nature07211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Isaacs FJ, Hasty J, Cantor CR, Collins JJ. Prediction and measurement of an autoregulatory genetic module. Proc Natl Acad Sci USA. 2003;100:7714–7719. doi: 10.1073/pnas.1332628100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000;403:339–342. doi: 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
- 35.Elowitz MB, Leibler S. A synthetic oscillatory network of transcriptional regulators. Nature. 2000;403:335–338. doi: 10.1038/35002125. [DOI] [PubMed] [Google Scholar]
- 36.Fung E, et al. A synthetic gene-metabolic oscillator. Nature. 2005;435:118–122. doi: 10.1038/nature03508. [DOI] [PubMed] [Google Scholar]
- 37.Maheshri N, O’Shea EK. Living with noisy genes: how cells function reliably with inherent variability in gene expression. Annu Rev Biophys Biomol Struct. 2007;36:413–434. doi: 10.1146/annurev.biophys.36.040306.132705. [DOI] [PubMed] [Google Scholar]
- 38.López-Maury L, Marguerat S, Bähler J. Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nature Rev Genet. 2008;9:583–593. doi: 10.1038/nrg2398. [DOI] [PubMed] [Google Scholar]
- 39.Endy D. Foundations for engineering biology. Nature. 2005;438:449–453. doi: 10.1038/nature04342. [DOI] [PubMed] [Google Scholar]
- 40.Hucka M, et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics. 2003;19:524–531. doi: 10.1093/bioinformatics/btg015. [DOI] [PubMed] [Google Scholar]
- 41.Canton B, Labno A, Endy D. Refinement and standardization of synthetic biological parts and devices. Nature Biotechnol. 2008;26:787–793. doi: 10.1038/nbt1413. [DOI] [PubMed] [Google Scholar]
- 42.Aderem A. Systems biology: its practice and challenges. Cell. 2005;121:511–513. doi: 10.1016/j.cell.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 43.Schmid AK, Baliga NS. In: Systems Biology. Al-Rubeai M, Fussenegger M, editors. Vol. 5. Springer; New York: 2007. pp. 395–423. [Google Scholar]
- 44.Laub MT, Shapiro L, McAdams HH. Systems biology of Caulobacter. Annu Rev Genet. 2007;41:429–441. doi: 10.1146/annurev.genet.41.110306.130346. [DOI] [PubMed] [Google Scholar]
- 45.Wang X, Wu M, Li Z, Chan C. Short time-series microarray analysis: methods and challenges. BMC Syst Biol. 2008;2:58. doi: 10.1186/1752-0509-2-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Facciotti MT, Bonneau R, Hood L, Baliga NS. Systems biology experimental design: considerations for building predictive gene regulatory network models for prokaryotic systems. Curr Genomics. 2004;5:527–544. [Google Scholar]
- 47.Reiss DJ, Baliga NS, Bonneau R. Integrated biclustering of heterogeneous genome-wide datasets for the inference of global regulatory networks. BMC Bioinformatics. 2006;7:280. doi: 10.1186/1471-2105-7-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baker MD, Wolanin PM, Stock JB. Systems biology of bacterial chemotaxis. Curr Opin Microbiol. 2006;9:187–192. doi: 10.1016/j.mib.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 49.Thanbichler M, Shapiro L. Getting organized — how bacterial cells move proteins and DNA. Nature Rev Microbiol. 2008;6:28–40. doi: 10.1038/nrmicro1795. [DOI] [PubMed] [Google Scholar]
- 50.Gama-Castro S, et al. Regulon DB (version 6.0): gene regulation model of Escherichia coli K-12 beyond transcription, active (experimental) annotated promoters and textpresso navigation. Nucleic Acids Res. 2008;36:D120–D124. doi: 10.1093/nar/gkm994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Medini D, et al. Microbiology in the post-genomic era. Nature Rev Microbiol. 2008;6:419–430. doi: 10.1038/nrmicro1901. [DOI] [PubMed] [Google Scholar]
- 52.Fredrickson JK, et al. Towards environmental systems biology of Shewanella. Nature Rev Microbiol. 2008;6:592–603. doi: 10.1038/nrmicro1947. [DOI] [PubMed] [Google Scholar]
- 53.Young D, Stark J, Kirschner D. Systems biology of persistent infection: tuberculosis as a case study. Nature Rev Microbiol. 2008;6:520–528. doi: 10.1038/nrmicro1919. [DOI] [PubMed] [Google Scholar]
- 54.Price ND, Reed JL, Palsson BØ. Genome-scale models of microbial cells: evaluating the consequences of constraints. Nature Rev Microbiol. 2004;2:886–897. doi: 10.1038/nrmicro1023. [DOI] [PubMed] [Google Scholar]
- 55.Reed JL, Famili I, Thiele I, Palsson BØ. Towards multidimensional genome annotation. Nature Rev Genet. 2006;7:130–141. doi: 10.1038/nrg1769. [DOI] [PubMed] [Google Scholar]
- 56.Feist AM, Palsson BØ. The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nature Biotechnol. 2008;26:659–667. doi: 10.1038/nbt1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.David H, Ozçelik IS, Hofmann G, Nielsen J. Analysis of Aspergillus nidulans metabolism at the genome-scale. BMC Genomics. 2008;9:163. doi: 10.1186/1471-2164-9-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee I, et al. A single gene network accurately predicts phenotypic effects of gene perturbation in Caenorhabditis elegans. Nature Genet. 2008;40:181–188. doi: 10.1038/ng.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zak DE, Aderem A. Systems biology of innate immunity. Immunol Rev. 2009;227:264–282. doi: 10.1111/j.1600-065X.2008.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lusis AJ, Attie AD, Reue K. Metabolic syndrome: from epidemiology to systems biology. Nature Rev Genet. 2008;9:819–830. doi: 10.1038/nrg2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zahn JM, Kim SK. Systems biology of aging in four species. Curr Opin Biotechnol. 2007;18:355–359. doi: 10.1016/j.copbio.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jamshidi N, Palsson BØ. Formulating genome-scale kinetic models in the post-genome era. Mol Syst Biol. 2008;4:171. doi: 10.1038/msb.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ishii N, et al. Multiple high-throughput analyses monitor the response of E. coli to perturbations. Science. 2007;316:593–597. doi: 10.1126/science.1132067. [DOI] [PubMed] [Google Scholar]
- 64.Barrett CL, Herring CD, Reed JL, Palsson BØ. The global transcriptional regulatory network for metabolism in Escherichia coli exhibits few dominant functional states. Proc Natl Acad Sci USA. 2005;102:19103–19108. doi: 10.1073/pnas.0505231102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Breitling R, Vitkup D, Barrett MP. New surveyor tools for charting microbial metabolic maps. Nature Rev Microbiol. 2008;6:156–161. doi: 10.1038/nrmicro1797. [DOI] [PubMed] [Google Scholar]
- 66.Shendure J, et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science. 2005;309:1728–1732. doi: 10.1126/science.1117389. [DOI] [PubMed] [Google Scholar]
- 67.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reiss DJ, Facciotti MT, Baliga NS. Model-based deconvolution of genome-wide DNA binding. Bioinformatics. 2008;24:396–403. doi: 10.1093/bioinformatics/btm592. [DOI] [PubMed] [Google Scholar]
- 69.Robertson G, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nature Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 70.McGrath PT, et al. High-throughput identification of transcription start sites, conserved promoter motifs and predicted regulons. Nature Biotechnol. 2007;25:584–592. doi: 10.1038/nbt1294. [DOI] [PubMed] [Google Scholar]
- 71.Sharan R, Ideker T. Modeling cellular machinery through biological network comparison. Nature Biotechnol. 2006;24:427–433. doi: 10.1038/nbt1196. [DOI] [PubMed] [Google Scholar]
- 72.Kelley BP, et al. Pathblast: a tool for alignment of protein interaction networks. Nucleic Acids Res. 2004;32:W83–W88. doi: 10.1093/nar/gkh411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baliga NS. Systems biology: the scale of prediction. Science. 2008;320:1297–1298. doi: 10.1126/science.1159485. [DOI] [PubMed] [Google Scholar]
- 74.Tagkopoulos I, Liu YC, Tavazoie S. Predictive behavior within microbial genetic networks. Science. 2008;320:1313–1317. doi: 10.1126/science.1154456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Isalan M, et al. Evolvability and hierarchy in rewired bacterial gene networks. Nature. 2008;452:840–845. doi: 10.1038/nature06847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barrett CL, Kim TY, Kim HU, Palsson BØ, Lee SY. Systems biology as a foundation for genome-scale synthetic biology. Curr Opin Biotechnol. 2006;17:488–492. doi: 10.1016/j.copbio.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 77.Fisher J, Henzinger TA. Executable cell biology. Nature Biotechnol. 2007;25:1239–1249. doi: 10.1038/nbt1356. [DOI] [PubMed] [Google Scholar]
- 78.Schlitt T, Brazma A. Current approaches to gene regulatory network modelling. BMC Bioinformatics. 2007;8(Suppl 6):S9. doi: 10.1186/1471-2105-8-S6-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Price ND, Shmulevich I. Biochemical and statistical network models for systems biology. Curr Opin Biotechnol. 2007;18:365–370. doi: 10.1016/j.copbio.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stein M, Gabdoulline RR, Wade RC. Bridging from molecular simulation to biochemical networks. Curr Opin Struct Biol. 2007;17:166–172. doi: 10.1016/j.sbi.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 81.Ayton GS, Noid WG, Voth GA. Multiscale modeling of biomolecular systems: in serial and in parallel. Curr Opin Struct Biol. 2007;17:192–198. doi: 10.1016/j.sbi.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 82.Whitesides GM. The origins and the future of microfluidics. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 83.Melin J, Quake SR. Microfluidic large-scale integration: the evolution of design rules for biological automation. Annu Rev Biophys Biomol Struct. 2007;36:213–231. doi: 10.1146/annurev.biophys.36.040306.132646. [DOI] [PubMed] [Google Scholar]
- 84.Kremling A, Saez-Rodriguez J. Systems biology — an engineering perspective. J Biotechnol. 2007;129:329–351. doi: 10.1016/j.jbiotec.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 85.Burgard AP, Pharkya P, Maranas CD. Optknock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol Bioeng. 2003;84:647–657. doi: 10.1002/bit.10803. [DOI] [PubMed] [Google Scholar]
- 86.Follows MJ, Dutkiewicz S, Grant S, Chisholm SW. Emergent biogeography of microbial communities in a model ocean. Science. 2007;315:1843–1846. doi: 10.1126/science.1138544. [DOI] [PubMed] [Google Scholar]
- 87.Meyer A, Pellaux R, Panke S. Bioengineering novel in vitro metabolic pathways using synthetic biology. Curr Opin Microbiol. 2007;10:246–253. doi: 10.1016/j.mib.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 88.Doktycz MJ, Simpson ML. Nano-enabled synthetic biology. Mol Syst Biol. 2007;3:125. doi: 10.1038/msb4100165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Longo D, Hasty J. Dynamics of single-cell gene expression. Mol Syst Biol. 2006;2:64. doi: 10.1038/msb4100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Raes J, Bork P. Molecular eco-systems biology: towards an understanding of community function. Nature Rev Microbiol. 2008;6:693–699. doi: 10.1038/nrmicro1935. [DOI] [PubMed] [Google Scholar]
- 91.Hood L, Galas D. The digital code of DNA. Nature. 2003;421:444–448. doi: 10.1038/nature01410. [DOI] [PubMed] [Google Scholar]
- 92.Bennett ST, Barnes C, Cox A, Davies L, Brown C. Toward the 1,000 dollars human genome. Pharmacogenomics. 2005;6:373–382. doi: 10.1517/14622416.6.4.373. [DOI] [PubMed] [Google Scholar]
- 93.Hoheisel JD. Microarray technology: beyond transcript profiling and genotype analysis. Nature Rev Genet. 2006;7:200–210. doi: 10.1038/nrg1809. [DOI] [PubMed] [Google Scholar]
- 94.Gresham D, Dunham MJ, Botstein D. Comparing whole genomes using DNA microarrays. Nature Rev Genet. 2008;9:291–302. doi: 10.1038/nrg2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ren B, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 96.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nature Rev Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 97.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 98.Phizicky E, Bastiaens PI, Zhu H, Snyder M, Fields S. Protein analysis on a proteomic scale. Nature. 2003;422:208–215. doi: 10.1038/nature01512. [DOI] [PubMed] [Google Scholar]
- 99.Sali A, Glaeser R, Earnest T, Baumeister W. From words to literature in structural proteomics. Nature. 2003;422:216–225. doi: 10.1038/nature01513. [DOI] [PubMed] [Google Scholar]
- 100.Want EJ, Nordstrom A, Morita H, Siuzdak G. From exogenous to endogenous: the inevitable imprint of mass spectrometry in metabolomics. J Proteome Res. 2007;6:459–468. doi: 10.1021/pr060505+. [DOI] [PubMed] [Google Scholar]
- 101.Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nature Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 102.Karlebach G, Shamir R. Modelling and analysis of gene regulatory networks. Nature Rev Mol Cell Biol. 2008;9:770–780. doi: 10.1038/nrm2503. [DOI] [PubMed] [Google Scholar]
- 103.Shannon PT, Reiss DJ, Bonneau R, Baliga NS. The gaggle: an open-source software system for integrating bioinformatics software and data sources. BMC Bioinformatics. 2006;7:176. doi: 10.1186/1471-2105-7-176. [DOI] [PMC free article] [PubMed] [Google Scholar]