

Abstract
Background:
Variation in APOE genotype is a determinant of Alzheimer disease (AD), but the risk associated with variation in plasma apoE levels has yet to be determined. Here, we studied offspring with and without a parental history of AD to identify the effect of plasma apoE levels at middle age on the risk of late-onset AD.
Methods:
Some 203 offspring from 92 families with a parental history of AD were compared with 197 offspring from 97 families without a parental history of AD. APOE genotypes and plasma apoE levels were assessed in all offspring. Difference in plasma apoE level between subjects with and without a parental history of AD was calculated using robust linear regression, both stratified and adjusted for APOE genotype.
Results:
Offspring with a parental history of AD were more likely to be an APOE ɛ4 allele carrier (46% vs 21%, p < 0.001) than offspring without such a parental history. Mean plasma apoE levels strongly decreased from ɛ2 to ɛ3ɛ3 to ɛ4 carriers (p < 0.001). Offspring with a parental history of AD had lower plasma apoE levels than subjects without such a history, both in analyses adjusted for APOE genotype (difference: −0.21 mg/dL, p = 0.02) and when using standardized Z scores, when stratified for APOE genotype (difference: −0.22, p = 0.009).
Conclusions:
Our findings suggest that lower plasma apoE levels in middle age could be a risk factor for Alzheimer disease in old age, independent of APOE genotype.
GLOSSARY
- AD
= Alzheimer disease;
- BMI
= body mass index;
- CI
= confidence interval;
- CVD
= cardiovascular disease;
- EOAD
= early-onset Alzheimer disease;
- HDL
= high-density lipoprotein;
- LDL
= low-density lipoprotein;
- LOAD
= late-onset Alzheimer disease;
- MMSE
= Mini-Mental State Examination;
- VaD
= vascular dementia.
Variation in the apolipoprotein E (APOE) gene is the strongest genetic risk factor of late-onset Alzheimer disease (AD). Two common nucleotide polymorphisms constitute the ɛ2/ɛ3/ɛ4 alleles that encode the functionally and structurally different apoE2, apoE3, and apoE4 isoforms. Carriers of the ɛ4 allele are at an increased risk of AD.1 Although the exact biologic mechanism explaining the risk differences is unclear, structural variation in the apoE protein has been shown to play a role in numerous processes contributing to AD, such as cholesterol transport and cellular uptake, clearance of oxidative products, and inflammation.2–4
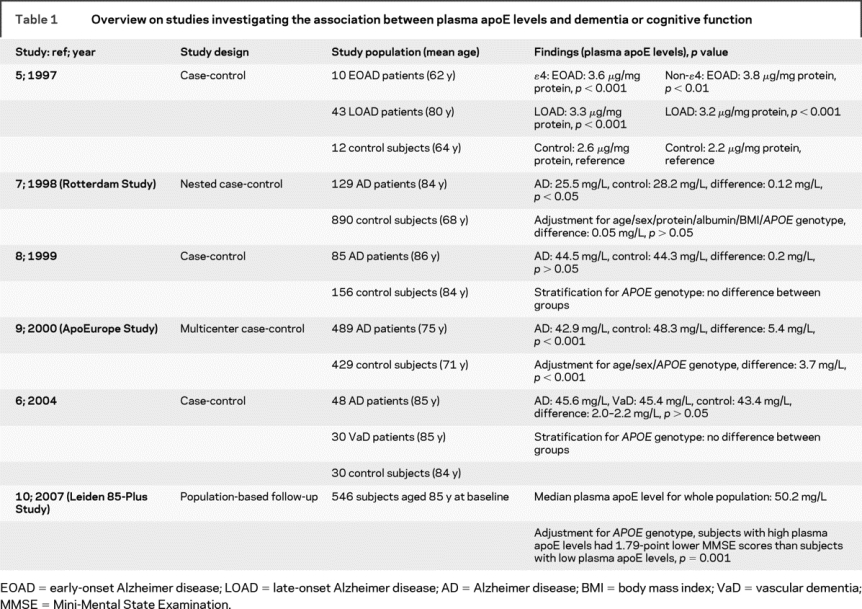
Several studies have reported on the association between plasma apoE levels and AD or cognitive impairment, but with opposing results (table 1).5–10 In some cross-sectional studies, plasma apoE levels were similar in patients with AD and controls,6–8 whereas others found lower plasma apoE levels in patients with AD.9 On the contrary, an earlier study reported higher plasma apoE levels in patients with AD.5 In the Leiden 85-Plus Study, we recently found that ɛ3ɛ3 and ɛ3ɛ4 carriers with high plasma apoE levels had worse cognitive function during a 5-year follow-up period.10 The mechanisms that underlie these divergent results are unknown, but possibly disease state influences plasma apoE levels.
Table 1 Overview on studies investigating the association between plasma apoE levels and dementia or cognitive function
To overcome the possible distorting effect of disease on the apoE phenotype, we used a family study design comparing middle-aged offspring with and without a parental history of AD to identify and quantify whether variation in plasma apoE levels is associated with an increased risk of AD. Additionally, adjustment for lipid levels was performed to study the association, independent of the lipid profile. All offspring were characterized for APOE genotype, and plasma apoE levels were determined.
METHODS
Study design and participants.
From 2006 to 2007, we initiated a family study to investigate midlife factors that are associated with an increased risk of late-life AD in subjects with and subjects without a parental history of AD. Ninety-two consecutive patients aged 70 years and older with a diagnosis of probable AD according to National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association criteria (mean age 82 years) were recruited from the memory clinic of the Alzheimer Center of the Vrije Universiteit Medical Center and affiliated nursing homes. Subjects with mixed-type dementia or vascular dementia were excluded. Ninety-seven married couples, aged 70 years and older, who were free from dementia, were also recruited (mean age 82.6 years). At least 1 spouse participated in either the Longitudinal Aging Study Amsterdam or the Leiden 85-Plus Study, 2 Dutch prospective population-based studies. Subjects were classified as free from dementia when having a Mini-Mental State Examination score greater than 27 points. When one of the spouses was deceased (n = 55), a history on cognitive function from the surviving spouse was obtained. Children from the patients with AD (n = 203) and the married couples without AD (n = 197) were invited to participate in the study. All measurements were confined to the offspring of patients with AD and offspring of couples with good cognitive function, hereafter described as “offspring with or without a parental history of AD.”
Standard protocol approvals, registrations, and patient consents.
We received approval to perform this study from the Medical Ethical Committee for Mental Health Care of The Netherlands. Consent for participation in the study was given by all married couples or the legal guardian of eligible patients with AD.
APOE genotyping.
For genotyping, 2 TaqMan assays (Applied Biosystems, Foster City, CA) were used, which has been described in detail elsewhere.11
Plasma measurements.
Nonfasting blood samples were collected early in the morning. Plasma levels of total cholesterol, high-density lipoprotein cholesterol, and triglycerides were analyzed at baseline on fully automated computerized analyzers (Modular P 800 analyzer, Roche, Almere, The Netherlands). The level of low-density lipoprotein cholesterol was estimated by the Friedewald equation.12
Plasma apoE levels were determined using a human apoE-specific sandwich ELISA as described in detail elsewhere.11
Cardiovascular status.
A history of cardiovascular disease was obtained through a questionnaire, filled out by participating subjects. The ankle-arm index was determined by calculating the ratio between ankle and arm systolic blood pressure. An ankle-arm index <1.0 was considered to represent the presence of peripheral artery disease.
Statistical analysis.
First, the difference in distribution of APOE genotypes between subjects with and subjects without a parental history of AD was assessed with the χ2 test.
Second, the difference in mean plasma apoE level between offspring with and offspring without a parental history of AD was analyzed with linear regression analysis, using robust standard errors to correct for familial aggregation. The robust character of the linear regression model adequately handles the multiple observations (subjects) per family. All models were additionally adjusted for sex, age, and plasma lipids. Analyses were stratified for APOE ɛ2/ɛ3ɛ3/ɛ4 carriership, with ɛ2ɛ2 and ɛ2ɛ4 carriers excluded from the analyses.
Third, the difference in plasma apoE level between offspring with and offspring without a parental history of AD was calculated for all subjects combined, using robust linear regression analysis with Z scores of plasma apoE level as determinant. Z scores of plasma apoE level were calculated per APOE genotype group separately.
Finally, broad heritability of plasma apoE levels was estimated with the following formula: heritability = 2 * (between-families variance)/(between-families variance + within-families variance), under the assumption of shared environment by siblings.
Calculations were performed using SPSS software (version 14.0.1, SPSS Inc., Chicago, IL) and STATA statistical software (version SE 9.0, StataCorp LP, College Station, TX).
RESULTS
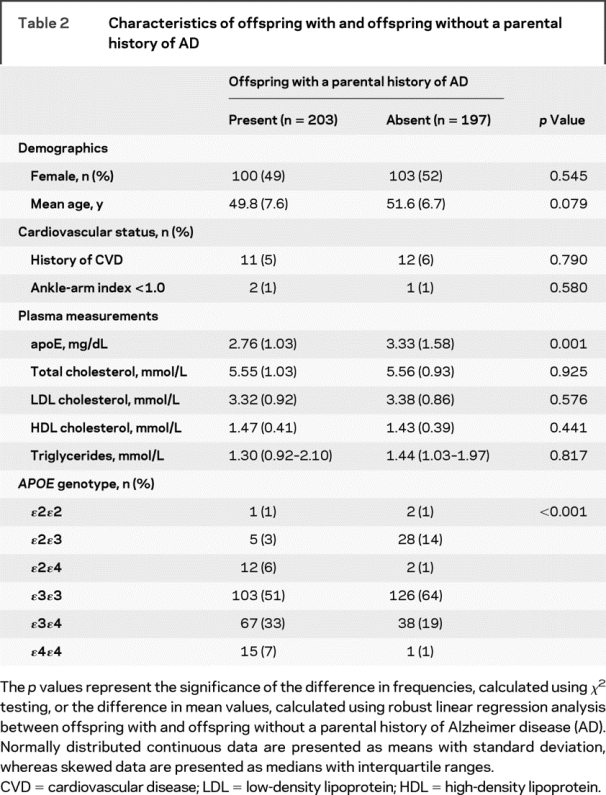
Plasma apoE measurement failed in 2 subjects. Table 2 shows that among the remaining 398 subjects, without correcting for APOE genotype, a parental history of AD was associated with a 0.57-mg/dL lower plasma apoE level (p = 0.001). This finding stands out because most other factors, most notably plasma lipid levels, were similar regardless of parental history of AD. The APOE allele frequencies for all offspring were ɛ2, 0.07; ɛ3, 0.75; and ɛ4, 0.19, and the genotypes were in Hardy-Weinberg equilibrium. We noted that, expectedly, offspring with a parental history of AD were more likely to carry the APOE ɛ4 allele compared with subjects without such a history (46% vs 21% ɛ4 allele carriership, p < 0.001), giving rise to the possibility of confounding by the APOE genotype.
Table 2 Characteristics of offspring with and offspring without a parental history of AD
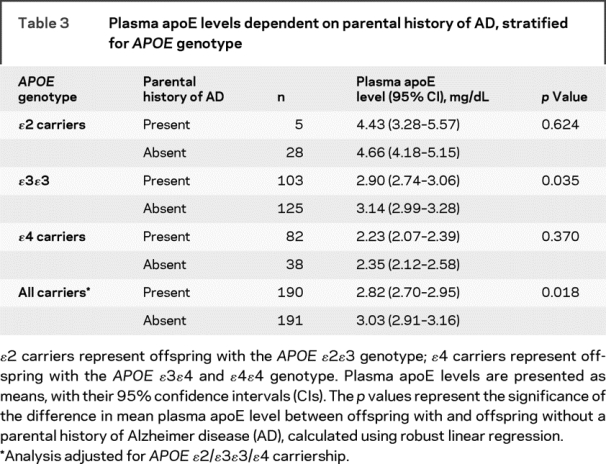
Thus, we accounted for this possibility by comparing plasma apoE levels according to APOE genotype. There were large differences in plasma apoE levels according to APOE genotype with decreasing levels from ɛ2 to ɛ3ɛ3 to ɛ4 carriers (table 3; p < 0.001). However, for each APOE genotype there was approximately a 0.20-mg/dL lower plasma apoE level among offspring with compared with offspring without a parental history of AD. After adjustment for APOE genotype, sex, age, and lipid levels in a linear model, overall, offspring with a parental history of AD had a 0.21-mg/dL lower plasma apoE level (p = 0.005).
Table 3 Plasma apoE levels dependent on parental history of AD, stratified for APOE genotype
Taking an alternative approach, we standardized plasma apoE levels for each APOE genotype, creating Z scores as an outcome variable for use in linear models. Taking this approach, a parental history of AD was associated with a 0.22-point lower Z score (figure; p = 0.009). Adjustment for factors related to cardiovascular disease did not materially alter this estimate (difference in Z score: 0.20 points, p = 0.02). Broad heritability of plasma apoE levels was calculated as 0.24 when using Z scores, whereas it was 0.61 when using nonstandardized plasma apoE levels.
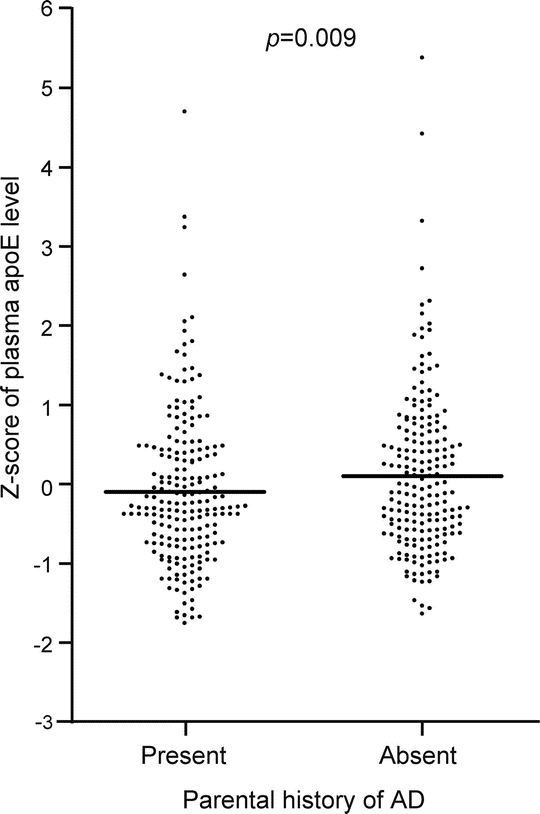
Figure Association between plasma apolipoprotein E levels and parental history of AD
Dots represent Z scores of plasma apoE levels, calculated for each APOE genotype separately. The p value represents the significance of the difference in mean Z score between offspring with and offspring without a parental history of Alzheimer disease (AD), calculated with robust linear regression model, adjusted for sex, age, and levels of plasma total cholesterol, low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, and triglycerides.
DISCUSSION
The main finding of this study is that middle-aged offspring with a parental history of AD have lower plasma apoE levels when compared with offspring without such a parental history of AD. This association was also observed after adjustment for APOE genotype and when using plasma apoE levels standardized for APOE genotype. Our findings suggest that low plasma apoE levels in midlife are associated with an increased risk of late-life AD, independent of APOE genotype.
The present study was designed to identify heritable risk factors in midlife that contribute to the development of late-onset AD. From twin studies, heritability of AD has been estimated to be as high as 0.77,13 in line with the observation that first-degree relatives of patients with AD have an increased risk of developing AD.14 Therefore, in the present study, we have compared offspring without a parental history of AD with offspring with a parental history of AD who are enriched for risk factors of AD but are not likely to have the disease yet. The increased risk is reflected in an overrepresentation of the APOE ɛ4 allele among offspring with a parental history of AD. Because circulating levels of apoE are under tight genetic control,15 variation in plasma apoE levels is highly heritable. The lower plasma apoE levels in middle-aged offspring with a parental history of AD might therefore be a risk factor for AD in late life.
Possible pathophysiologic pathways, explaining the association between lower plasma apoE levels and the risk of AD, can be found in the numerous actions of apoE. First, apoE plays an important role in lipid transportation, clearance, and metabolism.2 Higher levels of plasma apoE may protect against atherosclerosis, because a deficiency in apoE has been shown to result in the accumulation of very-low-density lipoprotein and remnant particles and subsequent atherosclerosis in mice. Second, apoE has been shown to have antioxidant effects, mainly by functioning as a scavenger of lipid peroxidation products.3 Third, although apoE produced by macrophages has been shown lately to be involved in lipid antigen presentation,16 numerous studies showed immunosuppressive properties of apoE, which may result in decreased inflammation.4 A decrease in atherosclerosis burden, the prevention of oxidative stress, and decreased inflammation may all protect against AD. These protective effects are generally weakest for apoE4 and strongest for apoE2. Therefore, ɛ4 allele carriers may be at an increased risk of AD, because besides the less functional protective properties of the structural variant apoE4, ɛ4 allele carriers also have the lowest plasma apoE levels.
The presented results are in accordance with the ApoEurope Study, in which lower plasma apoE levels were found in patients with AD.9 On the contrary, in a prospective population-based study, we found previously that lower plasma apoE levels were associated with better cognitive function,10 whereas others found no association between plasma apoE levels and cognitive function.7,8 A possible explanation for the opposing results could be the difference in study populations. In the current study, we studied middle-aged offspring with and without a parental history of AD. All other studies were performed in subjects who were selected on the presence of dementia, were age-matched controls, or were selected on old age. Possibly, the effect of plasma apoE levels on the risk of dementia is prone to change with increasing age, as has been suggested for other risk factors for dementia, such as blood pressure and cholesterol levels.17,18 In old age, high plasma apoE levels may be the result of decreased clearance of lipids,11 or could be the result of an adaptive response to systemic damage, such as cardiovascular disease and dementia, which could up-regulate the expression of apoE. A recently published study showed that an inflammatory response in mice led to a decreased clearance of circulating apoE, which resulted in increased plasma apoE levels, despite the down-regulation of apoE gene expression.19 Therefore, we hypothesize that high plasma apoE levels in late life may reflect ill health, whereas high plasma apoE levels in midlife may reflect higher innate apoE expression, which subsequently may be protective for AD. Indeed, in another family study, which we performed to study determinants of longevity, plasma apoE levels were higher in the old-aged parental generation compared with the middle-aged offspring generation (p < 0.001, unpublished results). Unfortunately, plasma apoE levels were not determined in the parental generation in the current study.
As hypothesized, lower plasma apoE levels may be the result of decreased innate production, which could be explained by variation in gene regulatory regions. Previous studies have indeed shown that polymorphisms in the APOE gene regulatory region and the hepatic control region are associated with plasma apoE levels.20 Three polymorphisms in the promoter region of the APOE gene have been studied extensively. The -219 G/T, -427 C/T, and -491 A/T polymorphisms have been shown to be associated with plasma apoE levels.21–23 These polymorphisms have also been shown to be associated with the risk of AD, although results are not consistent throughout all studies.22,24,25 Because these polymorphisms are in strong linkage with the common APOE polymorphisms, this may explain some part of the inconsistencies between studies. The observed lower plasma apoE levels in ɛ4 carriers and the higher plasma apoE levels in ɛ2 carriers, which is in accordance with the results from other studies,5–8,10 are not likely explained by a difference in innate production. Evidence points in the direction of different clearance of the various isoforms as the explanation for the genotype dependent plasma apoE levels. ApoE4 has the highest affinity for the low-density lipoprotein receptor and is cleared fastest, whereas apoE2 has the lowest affinity for the low-density lipoprotein receptor and is cleared at a much lower rate, explaining the variation in plasma apoE levels.26,27
Besides the influence of genetic variation on plasma apoE levels, phenotypic variation, such as dietary intake, has also been shown to influence plasma apoE levels. Several studies have reported increasing plasma apoE levels after the administration of diets containing saturated fatty acids, or fish and vegetable oil,28,29 although others did not report such an association.30,31 In the current study, the collection of blood samples was performed in nonfasted subjects. Although this could have introduced some increased random variation in the plasma apoE level measurements, this is unlikely to be a confounder in our study, because feeding state is not likely to be different between offspring with and offspring without a parental history of AD. Other studies reporting on plasma apoE levels and dementia or cognitive function could have been confounded by measuring in nonfasted blood samples, because eating patterns of patients with dementia have been shown to be different from healthy subjects, e.g., due to decreased food-seeking behavior. However, the studies reporting on nonfasted plasma apoE levels showed higher plasma apoE levels to be associated with dementia or worse cognitive function, making confounding as an explanation for the findings unlikely.5,10
An important strength of this study is that the family study design allowed for the identification and quantification of the effect of plasma apoE levels measured in middle age on an increased risk of AD later in life, without the possible distorting effect of disease on plasma levels of apoE. A limitation of this study is that, although subjects with a parental history of AD are likely to have an increased risk of having AD in late life, it is uncertain which subjects will eventually experience dementia. Therefore, associations could only be made based on a likely increased risk for the group of subjects with a parental history of AD and not on an individual level. Another limitation is that plasma apoE levels represent systemically produced apoE and do not reflect intracerebral apoE, which is locally produced. Brain autopsy studies have shown apoE levels to be lower in hippocampal and frontal cortical regions.32,33 Moreover, some studies have shown CSF apoE levels to be lower in patients with AD compared with controls,34,35 although other studies did not find this association36,37 or even showed an association in the opposite direction.38,39 However, all proposed pathophysiologic pathways, explaining the observed association in this study, may play an important part in the development of AD and do not involve intracerebral apoE.
AUTHOR CONTRIBUTIONS
Statistical analyses were performed by P. van Vliet.
ACKNOWLEDGMENT
The authors thank Anna Paauw, Anna Westerveld, Inge Mooijekind, Chris Hettinga, Ellen Vollebregt, Marja Kersbergen, and Margo van Schie for their technical assistance. The Alzheimer Center of the VU University Center and the Zonnehuis Groep are gratefully acknowledged for their help in contacting the offspring with a parental history of AD. The Longitudinal Aging Study Amsterdam and the Leiden 85-Plus Study are gratefully acknowledged for assistance in contacting offspring without a parental history of AD.
DISCLOSURE
Dr. van Vliet, Dr. Eikelenboom, Dr. Comijs, Dr. Frolich, Dr. Bakker, and Dr. van der Flier report no disclosures. Dr. Westendorp serves as editor of Experimental Gerontology and on the editorial board of Ageing Reviews; receives/has received research support from the Netherlands Genomics Initiatives [NGI 050-040-200 (PI)], Lifespan Network of Excellence [FP6-2005-036894 (PI)], IOP Genomics [IGE05007A (PI)], and the Netherlands Organization for Scientific Research [NWO MaGW 051-14-050 (PI) and NWO WOTRO W 93-467 (PI)]; and serves as executive director of Leyden Academy. Dr. van Exel receives research support from the NIA [R03 AG 276163-01 (pilot grant)] and the Internationale Stichting Alzheimer Onderzoek.
Address correspondence and reprint requests to Dr. P. van Vliet, Leiden University Medical Center, Department of Gerontology and Geriatrics (C2-R), PO Box 9600, 2300 RC, Leiden, The Netherlands p.van_vliet@lumc.nl
Supported by grants from the National Institute of Aging, USA (R03 AG 276163-01), the EU project LifeSpan (LSHG-CT-2007-036894), and the Internationale Stichting Alzheimer Onderzoek (International Foundation for Alzheimer Research, The Netherlands).
Disclosure: Author disclosures are provided at the end of the article.
Received March 5, 2009. Accepted in final form June 5, 2009.
REFERENCES
- 1.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993;261:921–923. [DOI] [PubMed] [Google Scholar]
- 2.Eichner JE, Dunn ST, Perveen G, Thompson DM, Stewart KE, Stroehla BC. Apolipoprotein E polymorphism and cardiovascular disease: a HuGE review. Am J Epidemiol 2002;155:487–495. [DOI] [PubMed] [Google Scholar]
- 3.Pedersen WA, Chan SL, Mattson MP. A mechanism for the neuroprotective effect of apolipoprotein E: isoform-specific modification by the lipid peroxidation product 4-hydroxynonenal. J Neurochem 2000;74:1426–1433. [DOI] [PubMed] [Google Scholar]
- 4.Jofre-Monseny L, Minihane AM, Rimbach G. Impact of apoE genotype on oxidative stress, inflammation and disease risk. Mol Nutr Food Res 2008;52:131–145. [DOI] [PubMed] [Google Scholar]
- 5.Taddei K, Clarnette R, Gandy SE, Martins RN. Increased plasma apolipoprotein E (apoE) levels in Alzheimer’s disease. Neurosci Lett 1997;223:29–32. [DOI] [PubMed] [Google Scholar]
- 6.Folin M, Baiguera S, Conconi MT, et al. Apolipoprotein E as vascular risk factor in neurodegenerative dementia. Int J Mol Med 2004;14:609–613. [PubMed] [Google Scholar]
- 7.Slooter AJ, de Knijff P, Hofman A, et al. Serum apolipoprotein E level is not increased in Alzheimer’s disease: the Rotterdam study. Neurosci Lett 1998;248:21–24. [DOI] [PubMed] [Google Scholar]
- 8.Scacchi R, Gambina G, Ruggeri M, et al. Plasma levels of apolipoprotein E and genetic markers in elderly patients with Alzheimer’s disease. Neurosci Lett 1999;259:33–36. [DOI] [PubMed] [Google Scholar]
- 9.Siest G, Bertrand P, Qin B, et al. Apolipoprotein E polymorphism and serum concentration in Alzheimer’s disease in nine European centres: the ApoEurope study. ApoEurope group. Clin Chem Lab Med 2000;38:721–730. [DOI] [PubMed] [Google Scholar]
- 10.Mooijaart SP, van Vliet P, van Heemst D, et al. Plasma levels of apolipoprotein E and cognitive function in old age. Ann NY Acad Sci 2007;1100:148–161. [DOI] [PubMed] [Google Scholar]
- 11.Mooijaart SP, Berbee JF, van Heemst D, et al. ApoE plasma levels and risk of cardiovascular mortality in old age. PLoS Med 2006;3:e176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 1972;18:499–502. [PubMed] [Google Scholar]
- 13.Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 2006;63:168–174. [DOI] [PubMed] [Google Scholar]
- 14.van Duijn CM, Clayton D, Chandra V, et al. Familial aggregation of Alzheimer’s disease and related disorders: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol 1991;20(suppl 2):S13–S20. [DOI] [PubMed] [Google Scholar]
- 15.Beekman M, Heijmans BT, Martin NG, et al. Heritabilities of apolipoprotein and lipid levels in three countries. Twin Res 2002;5:87–97. [DOI] [PubMed] [Google Scholar]
- 16.van den Elzen P, Garg S, Leon L, et al. Apolipoprotein-mediated pathways of lipid antigen presentation. Nature 2005;437:906–910. [DOI] [PubMed] [Google Scholar]
- 17.Qiu C, Winblad B, Fratiglioni L. The age-dependent relation of blood pressure to cognitive function and dementia. Lancet Neurol 2005;4:487–499. [DOI] [PubMed] [Google Scholar]
- 18.van Vliet P, van de Water W, de Craen AJ, Westendorp RG. The influence of age on the association between cholesterol and cognitive function. Exp Gerontol 2009;44:112–122. [DOI] [PubMed] [Google Scholar]
- 19.Li L, Thompson PA, Kitchens RL. Infection induces a positive acute phase apolipoprotein E response from a negative acute phase gene: role of hepatic LDL receptors. J Lipid Res 2008;49:1782–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klos K, Shimmin L, Ballantyne C, et al. APOE/C1/C4/C2 hepatic control region polymorphism influences plasma apoE and LDL cholesterol levels. Hum Mol Genet 2008;17:2039–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lambert JC, Brousseau T, Defosse V, et al. Independent association of an APOE gene promoter polymorphism with increased risk of myocardial infarction and decreased APOE plasma concentrations: the ECTIM study. Hum Mol Genet 2000;9:57–61. [DOI] [PubMed] [Google Scholar]
- 22.Scacchi R, Gambina G, Martini MC, et al. Polymorphisms of the apolipoprotein E gene regulatory region and of the LDL receptor gene in late-onset Alzheimer’s disease in relation to the plasma lipidic pattern. Dement Geriatr Cogn Disord 2001;12:63–68. [DOI] [PubMed] [Google Scholar]
- 23.Laws SM, Taddei K, Martins G, et al. The -491AA polymorphism in the APOE gene is associated with increased plasma apoE levels in Alzheimer’s disease. Neuroreport 1999;10:879–882. [DOI] [PubMed] [Google Scholar]
- 24.Lambert JC, Araria-Goumidi L, Myllykangas L, et al. Contribution of APOE promoter polymorphisms to Alzheimer’s disease risk. Neurology 2002;59:59–66. [DOI] [PubMed] [Google Scholar]
- 25.Artiga MJ, Bullido MJ, Frank A, et al. Risk for Alzheimer’s disease correlates with transcriptional activity of the APOE gene. Hum Mol Genet 1998;7:1887–1892. [DOI] [PubMed] [Google Scholar]
- 26.Chou CY, Jen WP, Hsieh YH, Shiao MS, Chang GG. Structural and functional variations in human apolipoprotein E3 and E4. J Biol Chem 2006;281:13333–13344. [DOI] [PubMed] [Google Scholar]
- 27.Lund-Katz S, Wehrli S, Zaiou M, Newhouse Y, Weisgraber KH, Phillips MC. Effects of polymorphism on the microenvironment of the LDL receptor-binding region of human apoE. J Lipid Res 2001;42:894–901. [PubMed] [Google Scholar]
- 28.Jackson KG, Wolstencroft EJ, Bateman PA, Yaqoob P, Williams CM. Acute effects of meal fatty acids on postprandial NEFA, glucose and apo E response: implications for insulin sensitivity and lipoprotein regulation? Br J Nutr 2005;93:693–700. [DOI] [PubMed] [Google Scholar]
- 29.Georgopoulos A, Applebaum-Bowden D, Margolis S. Improved glycemic control lowers plasma apoprotein E and triglyceride levels following ingestion of a fat load in insulin-dependent diabetic subjects. Metabolism 1988;37:837–843. [DOI] [PubMed] [Google Scholar]
- 30.Blum CB. Dynamics of apolipoprotein E metabolism in humans. J Lipid Res 1982;23:1308–1316. [PubMed] [Google Scholar]
- 31.Moreno JA, Lopez-Miranda J, Marin C, et al. The influence of the apolipoprotein E gene promoter (-219G/T) polymorphism on postprandial lipoprotein metabolism in young normolipemic males. J Lipid Res 2003;44:2059–2064. [DOI] [PubMed] [Google Scholar]
- 32.Pirttila T, Soininen H, Heinonen O, et al. Apolipoprotein E (apoE) levels in brains from Alzheimer disease patients and controls. Brain Res 1996;722:71–77. [DOI] [PubMed] [Google Scholar]
- 33.Beffert U, Cohn JS, Petit-Turcotte C, et al. Apolipoprotein E and beta-amyloid levels in the hippocampus and frontal cortex of Alzheimer’s disease subjects are disease-related and apolipoprotein E genotype dependent. Brain Res 1999;843:87–94. [DOI] [PubMed] [Google Scholar]
- 34.Landen M, Hesse C, Fredman P, Regland B, Wallin A, Blennow K. Apolipoprotein E in cerebrospinal fluid from patients with Alzheimer’s disease and other forms of dementia is reduced but without any correlation to the apoE4 isoform. Dementia 1996;7:273–278. [DOI] [PubMed] [Google Scholar]
- 35.Song H, Saito K, Seishima M, Noma A, Urakami K, Nakashima K. Cerebrospinal fluid apo E and apo A-I concentrations in early- and late-onset Alzheimer’s disease. Neurosci Lett 1997;231:175–178. [DOI] [PubMed] [Google Scholar]
- 36.Lefranc D, Vermersch P, Dallongeville J, Daems-Monpeurt C, Petit H, Delacourte A. Relevance of the quantification of apolipoprotein E in the cerebrospinal fluid in Alzheimer’s disease. Neurosci Lett 1996;212:91–94. [DOI] [PubMed] [Google Scholar]
- 37.Hahne S, Nordstedt C, Ahlin A, Nyback H. Levels of cerebrospinal fluid apolipoprotein E in patients with Alzheimer’s disease and healthy controls. Neurosci Lett 1997;224:99–102. [DOI] [PubMed] [Google Scholar]
- 38.Lindh M, Blomberg M, Jensen M, et al. Cerebrospinal fluid apolipoprotein E (apoE) levels in Alzheimer’s disease patients are increased at follow up and show a correlation with levels of tau protein. Neurosci Lett 1997;229:85–88. [DOI] [PubMed] [Google Scholar]
- 39.Merched A, Blain H, Visvikis S, Herbeth B, Jeandel C, Siest G. Cerebrospinal fluid apolipoprotein E level is increased in late-onset Alzheimer’s disease. J Neurol Sci 1997;145:33–39. [DOI] [PubMed] [Google Scholar]