

Abstract
Deficiency in plasminogen activator inhibitor-1 (PAI-1) gene expression is known to promote growth factor activation and regeneration in a number of hepatotoxicity models. To evaluate if PAI-1 has similar effects in acetaminophen (APAP) hepatotoxicity, wild-type (WT) and PAI-1 gene knockout mice (PAI-KO) were treated with 200 mg/kg APAP and liver injury and its repair were assessed. In WT animals, plasma alanine aminotransferase (ALT) activities increased during the first 12 h and then returned to baseline within 48 h. The area of necrosis increased in parallel to the ALT values, peaked between 12 and 24 h and was completely resolved by 96 h. The regenerative response of cells outside the necrotic area, as indicated by proliferating cell nuclear antigen protein and cyclin D1 gene expression, was observed within 24 h, peaked at 48 h and then declined but remained elevated until 96 h. Liver injury in response to APAP was similar in PAI-KO as in WT animals during the first 12 h. However, plasma ALT values and the area of necrosis further increased during the following 12 h with development of massive intrahepatic hemorrhage. Approximately, 50% of the PAI-KO animals did not survive. Although liver injury of the surviving animals was repaired, the regeneration process was delayed until 48 h. A potential reason for this delay may have been due to the more severe injury and/or the increased expression of the cell cycle inhibitor p21. Our data indicate that PAI activation limits liver injury and mortality during APAP hepatotoxicity by preventing excessive hemorrhage and thereby facilitating tissue repair.
Keywords: acetaminophen, hepatotoxicity, regeneration
Acetaminophen (APAP) hepatotoxicity is the leading cause of drug-induced liver failure in the United States (Larson et al., 2005) and other industrialized nations (Björnsson et al., 2005). The mechanism of toxicity includes the formation of a reactive metabolite (N-acetyl-p-benzoquinoneimine; NAPQI), which is initially detoxified by cellular glutathione (Nelson, 1990). However, after the GSH levels are exhausted, NAPQI binds to cellular proteins, especially mitochondrial proteins (Nelson, 1990). This results in mitochondrial dysfunction with inhibition of mitochondrial respiration, reactive oxygen, and peroxynitrite formation, and declining ATP levels (Jaeschke and Bajt, 2006). The mitochondrial dysfunction eventually leads to the mitochondrial membrane permeability transition pore opening and collapse of the mitochondrial membrane potential (Kon et al., 2004). In addition, the mitochondrial release of intermembrane proteins initiates nuclear DNA fragmentation (Bajt et al., 2006, 2008), which contributes to necrotic cell death (Shen et al., 1992). However, there is increasing evidence that the fate of a cell is not solely determined by injury mechanisms but also by an opposing repair/regenerative response (Mehendale, 2005). In fact, a number of diverse interventions have been shown to stimulate regeneration and limit liver injury including treatment with various hepatotoxic chemicals (Chanda et al., 1995; Dalhoff et al., 2001), antioxidants (Bajt et al., 2003), or growth factors (Donahower et al., 2006).
A substantial upregulation of plasminogen activator inhibitor-1 (PAI-1) has been shown in a variety of liver injury models including hemorrhagic shock (Lagoa et al., 2005), bile duct ligation (Bergheim et al., 2006a; Wang et al., 2005, 2007b), APAP hepatotoxicity (Ganey et al., 2007; Reilly et al., 2001) and alcohol-induced liver injury (Bergheim et al., 2006b). In addition, increased plasma PAI-1 protein levels have been observed in mice (Ganey et al., 2007) and in patients with APAP overdose (Leiper et al., 1994). PAI-1 is an inhibitor of tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA). Both PAs convert plasminogen to the active serine protease plasmin, which breaks down fibrin clots (Eddy, 2002). Because PAI-1 inhibits tPA-mediated plasmin generation, it prevents fibrinolysis in blood vessels. In the extracellular space, PAI-1 inhibits uPA-mediated extracellular matrix turnover (Eddy, 2002).
In addition to their role in blood coagulation, both PAs are able to cleave the inactive single chain hepatocyte growth factor (HGF) to its active two-chain form (Mars et al., 1993). Thus, PAI-1 deficiency can lead to elevated uPA and tPA activities with increased mature HGF levels and activated survival pathways and regeneration (Lagoa et al., 2005; Shimizu et al., 2001; Wang et al., 2005). Reduced tissue injury after bile duct ligation or hemorrhagic shock in PAI-1 gene knockout (PAI-KO) mice was thought to be caused by the pro-regenerative effect of HGF activation (Bergheim et al., 2006a; Lagoa et al., 2005; Wang et al., 2005, 2007a). Given the extensive beneficial effects of PAI-1 inhibition in various models of acute liver injury and the fact that PAI-1 mRNA is upregulated early after APAP overdose (Reilly et al., 2001), we hypothesized that PAI-1 may also play an important role in APAP hepatotoxicity and tissue repair.
MATERIALS AND METHODS
Experimental protocol.
PAI-KO mice and the respective age-matched WT animals from the colony (C57BL/6J background) were purchased from Jackson Laboratories (Bar Harbor, ME). All animals were housed in an environmentally controlled room with 12-h light/dark cycle and allowed free access to food (certified rodent diet no. 8640, Harlan Teklad, Indianapolis, IN) and water. The experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Arizona and the University of Kansas Medical Center and followed the recommendations of the National Research Council for the care and use of laboratory animals in research. All animals were fasted overnight before the experiments. Animals received an intraperitoneal injection of 200 mg/kg APAP (Sigma Chemical Co., St Louis, MO) between 8 and 9 A.M. APAP was dissolved in warm (37–39°C) saline (15 mg/ml).
At selected times after APAP treatment, animals were killed by cervical dislocation under isoflurane (Abbott Laboratories, North Chicago, IL) anesthesia. Blood was drawn from the vena cava into heparinized syringes and centrifuged. The plasma was used for determination of ALT activities (assayed with kinetic test kit 68-B, Biotron Diagnostics, Inc., Hernet, CA). Immediately after collecting the blood, the livers were excised and rinsed in saline. A section from each liver was placed in 10% phosphate buffered formalin to be used for immunohistochemical analyses. The remaining liver was frozen in liquid nitrogen and stored at −80°C.
Histology and immunohistochemistry.
Formalin-fixed tissue samples were embedded in paraffin and 5-μm sections were cut. Replicate sections were stained with hematoxylin and eosin (H&E) for evaluation of necrosis (Gujral et al., 2002). The percent of necrosis was estimated by evaluating the number of microscopic fields with necrosis compared with the entire cross section. In general, necrosis was estimated at low power (×100); questionable areas were evaluated at higher magnification (×200 or ×400). The pathologist (A.F.) evaluated all histological sections in a blinded fashion. For the terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling (TUNEL) assay, sections of liver were stained with the In Situ Cell Death Detection Kit, AP (Roche Diagnostics, Indianapolis, IN) as described in the manufacturer's instructions (Gujral et al., 2002). Sections were also stained for proliferating cell nuclear antigen (PCNA) using a rabbit polyclonal anti-PCNA antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and standard immunohistochemical procedures (Bajt et al., 2003).
Western blotting and glutathione measurements.
Western blotting for PCNA was described in detail (Bajt et al., 2003). The primary antibody was a mouse monoclonal anti-PCNA antibody and the secondary antibody was a horseradish peroxidase-coupled anti-mouse IgG (both were from Santa Cruz Biotechnology Santa Cruz, CA). Proteins were visualized by enhanced chemiluminescence (Amersham Pharmacia Biotech. Inc., Piscataway, NJ) according to the manufacturer's instructions. Hepatic glutathione (GSH + GSSG) levels were determined with the Tietze assay as described in detail (Knight et al., 2001).
Reverse transcription and quantitative real-time polymerase chain reaction.
Total RNA was extracted from liver tissue using TRI reagent (Sigma, St Louis, MO), reversed transcribed into cDNA using random primers and M-MLV reverse transcriptase (Invitrogen, Carlsbad, CA) at 0.1 μg/μl. The cDNA was diluted 1/10 and 5 μl were used as a template in each PCR reaction. Real-time PCR was used to determine the mRNA expression levels of PAI, cyclin D1, and p21. Power SYBR green (Applied Biosystems, Foster City, CA) was applied as the detector. Beta-actin was chosen as an internal standard. All samples were run in triplicate in 96-well reaction plates (Applied Biosystems), using corresponding primers, on the 7300 Real-Time PCR System (Applied Biosystems). The primer sequences (Table 1) were provided by Dr Jie Liu (NIEHS, Research Triangle Park, NC). The delta Ct value was obtained with each sample by subtracting the corresponding internal controls. Results are expressed as fold changes based on controls.
TABLE 1.
Real-time PCR Primers for Analysis
| Gene | Genebank accession no. | Direction (5′–3′) | Sequence | Amplicon size (bp) |
| Beta-actin | M12481 | Forward | GTATGACTCCACTCACGGCAAA | 101 |
| Reverse | GGTCTCGCTCCTGGAAGATG | |||
| p21 | NM007669.3 | Forward | GGTGGGCCCGGAACAT | 72 |
| Reverse | GCGCTTGGAGTGATAGAAATCTG | |||
| PAI | M33960 | Forward | TGCATCGCCTGCCATTG | 85 |
| Reverse | GGACATTTCCACAGTGGACCTT | |||
| Cyclin D1 | M64403 | Forward | GGGCACCTGGATTGTTCT | 102 |
| Reverse | CACCGGAGACTCAGAGCA |
Statistics.
Data are expressed as means ± SE. Comparison between two groups were performed with Student's t-test or one-way ANOVA followed by Bonferroni t-test for multiple groups. If the data were not normally distributed, the Mann-Whitney test was applied for comparison of two groups and the Kruskal-Wallis test (nonparametric ANOVA) followed by Dunn's multiple comparisons test for multiple groups. p < 0.05 was considered significant.
RESULTS
Administration of a moderate overdose of APAP (200 mg/kg) resulted in an increase in PAI mRNA levels of 290% compared with baseline in WT animals at 6 h (Fig. 1). However, there was a further dramatic increase of PAI gene expression by 24 h followed by a decline to baseline levels by 72 h (Fig. 1). As expected, no mRNA of PAI was detected in PAI-KO mice (Fig. 1). To assess if there is a difference in the capacity to metabolically activate APAP between WT and PAI-KO mice, hepatic glutathione (GSH + GSSG) levels were determined before and 20 min after APAP administration (Fig. 2). APAP caused an 88% depletion of hepatic glutathione levels within 20 min, which reflects the formation of NAPQI and its conjugation with GSH. However, there was no significant difference between WT and PAI-KO mice indicating a similar capacity to metabolically activate APAP (Fig. 2).
FIG. 1.

Time course of PAI-1 mRNA expression in WT and PAI-KO mice before and after 200 mg/kg APAP treatment. Data are expressed as the PAI-to-actin mRNA ratio; the value of WT control animals was set as 100%. The mean ± SE of n = 4–6 animals per time point is shown. *p < 0.05 (compared with PAI-KO mice).
FIG. 2.

Hepatic glutathione levels were determined in animals 20 min after treatment with either the vehicle saline (S) or 200 mg/kg APAP. WT and PAI-KO mice were compared. Data represent the mean ± SE of n = 4 animals per group. *p < 0.05 (compared with the respective vehicle group).
To evaluate liver injury after 200 mg/kg APAP, plasma ALT activities were measured over 4 days (Fig. 3A). In WT animals, plasma ALT activities were significantly increased at 12 and 24 h and declined to baseline by 48 h. These results indicate that acute cell injury occurred mainly during the first 12 h after APAP overdose. In PAI-KO mice, slightly higher ALT levels were observed at 12 h. However, in contrast to the declining ALT levels in WT animals, there was a substantial further increase of plasma ALT activities by 24 h (Fig. 3A). Similar to WT animals, plasma ALT activities declined to baseline by 48 h. Blinded quantitation of the area of necrosis confirmed these findings. At 12 h after APAP treatment the area of necrosis was similar in WT and PAI-KO mice (Fig. 3B). The area of necrosis remained constant in WT animals at 24 h and then declined during the next 2 days. However, there was a clear expansion of the necrotic area in KO mice between 12 and 24 h (Fig. 3B). The area of necrosis was reduced at 48 h and beyond. The substantial decline between 24 and 48 h was mainly caused by the death of the most severely injured animals of the 48-h group (Fig. 3B). The 48-h mortality rate was 50% for the PAI-KO group compared with 0% of WT animals. Interestingly, the aggravation of liver injury between 24 and 48 h in the PAI-KO mice was correlated with massive hemorrhage (Fig. 4B), which was not observed in WT animals (Fig. 4A). Hemorrhage in PAI-KO mice was still detectable at 48 h (Figs. 4C and 4D) but then disappeared over the next 2 days.
FIG. 3.

Plasma ALT activities (A) and the area of necrosis (B) were measured as indicators for APAP-induced liver injury in WT and in PAI-KO animals. Animals received 200 mg/kg APAP at t = 0. Data represent means ± SE of n = 4–6 animals per group. *p < 0.05 (compared with WT mice).
FIG. 4.
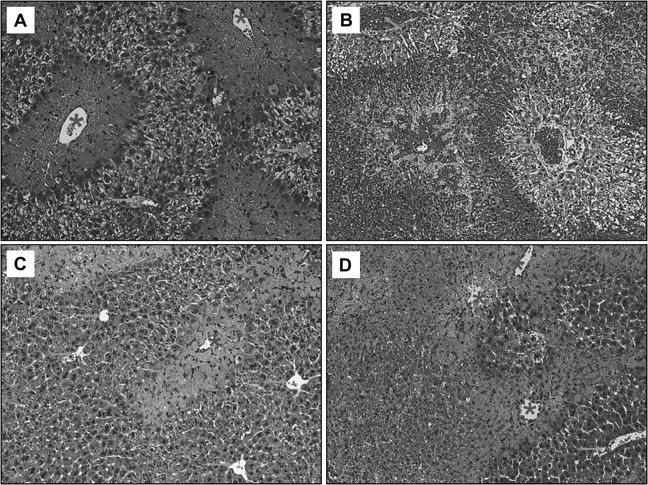
Representative liver sections stained with H&E from APAP-treated animals (200 mg/kg). Sections were taken from the 24 h (A, B) and the 48 h (C, D) groups of WT (A, C) and PAI-KO mice (B, D). Sections show extensive necrosis around the centrilobular areas (*) of WT and KO mice and severe hemorrhage only in PAI-KO mice. Arrows indicate periportal areas (×100).
DNA fragmention is a hallmark of APAP toxicity and an additional measure of cell damage (Shen et al., 1992). APAP-induced DNA damage can be detected with the TUNEL assay (Lawson et al., 1999). Control WT and PAI-KO mice showed only very few individual TUNEL-positive cells randomly scattered throughout the tissue (data not shown). However, 12 and 24 h after APAP overdose, large numbers of hepatocytes around the centrilobular areas were TUNEL-positive (Figs. 5A and 5C). Similar to the area of necrosis, there was no relevant further increase of TUNEL-positive cells in WT animals between 12 and 24 h. In contrast, the number of TUNEL-positive cells further increased in PAI-KO mice during this time frame (Figs. 5B and 5D). This increase of the number of cells with DNA damage correlated with the expanding area of necrosis in PAI-KO mice.
FIG. 5.
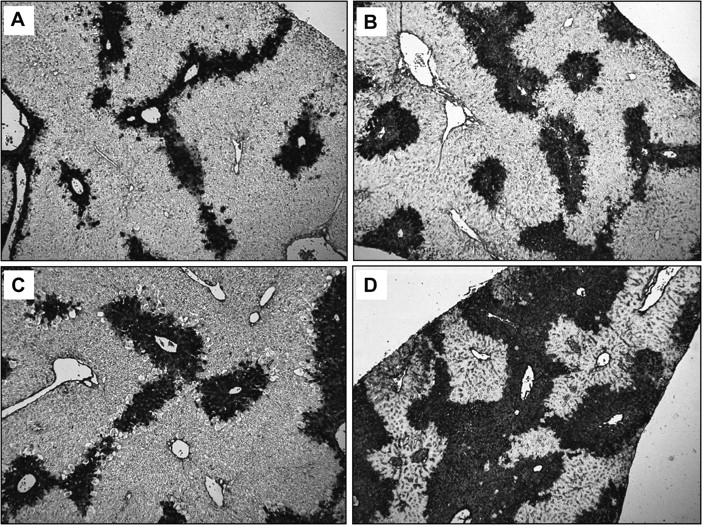
DNA fragmentation was assessed by the TUNEL assay in WT (A, C) and in PAI-KO mice (B, D) at 12 h (A, B) and 24 h (C, D) after APAP (200 mg/kg) treatment. The panels show TUNEL-positive cells around the centrilobular areas (×50).
The timely onset of tissue repair processes can limit liver injury and promote regeneration of lost tissue mass (Mehendale, 2005). To evaluate cell cycle activation, PCNA protein expression was determined after APAP treatment (Fig. 6). Hepatic PCNA protein levels, as determined by Western blotting, showed a significant increase at 24 h (Fig. 6A) and at 48 h (Fig. 6B). The peak expression at 48 h was followed by a decline to levels, which remained significantly above baseline at 72 and 96 h (Fig. 6C). Immunohistochemical localization of cells with PCNA-positive nuclei indicated that most nonnecrotic cells stained positive for PCNA (Fig. 7A). However, the most intense staining was observed in cells adjacent to necrotic cells at 24 h (Fig. 7A) and at 48 h (Fig. 7C). In PAI-KO mice, the time course of PCNA staining was very similar to WT animals (Fig. 6C) with one exception. The onset of PCNA staining was clearly delayed. There was little to no staining in PAI-KO mice at 24 h after APAP treatment (Figs. 6A and 7B), however, at 48 h and beyond, overall PCNA protein expression (Figs. 6B and 6C) and cellular staining (Fig. 7D) was not significantly different between WT and KO mice. The findings were confirmed with a different parameter of cell cycle activation, cyclin D1 gene expression (Fig. 8A). Cyclin D1 mRNA level changes followed closely PCNA expression levels in both WT and PAI gene KO mice. However, the most significant difference between WT and KO mice was the substantial delay of cyclin D1 expression at 24 h (Fig. 8A). To evaluate a potential reason for this effect, expression of p21, a cell cycle inhibitor, was measured. Although there was a significant increase of p21 gene expression in WT animals at 24 h, p21 gene expression in PAI-KO mice was three times higher (Fig. 8B). On the other hand, p21 mRNA levels dropped below WT values at 48 h (Fig. 8B), which may have allowed the regenerative process to catch up in PAI-KO mice.
FIG. 6.

Western blot analysis of PCNA expression in livers of controls or animals treated with 200 mg/kg APAP for 24 h (A) or 48 h (B). Livers were obtained from WT animals and PAI-KO mice. One to three representative samples are shown from each group. (C) Densitometric analysis of PCNA western blots from controls and APAP-treated WT and PAI-KO mice. Data of control animals were set as 100%. All data represent means ± SE of n = 4–6 animals per time point. *p < 0.05 (compared with controls), #p < 0.05 (compared with WT).
FIG. 7.
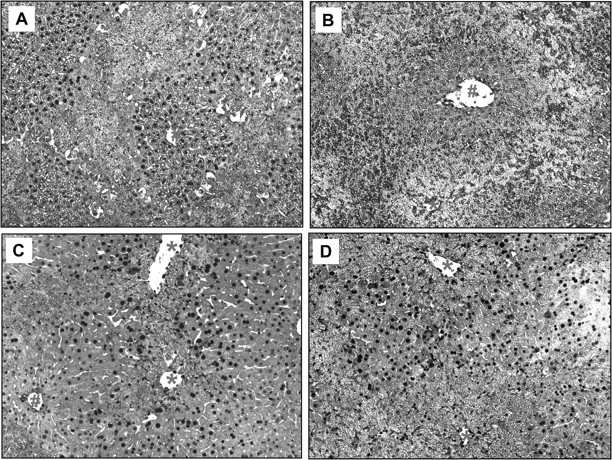
Representative liver sections obtained from APAP-treated animals (200 mg/kg) were stained for PCNA. Sections were taken from the 24 h (A, B) and the 48 h (C, D) groups of WT (A, C) and PAI-KO mice (B, D). Sections show extensive PCNA staining (dark brown nuclei) outside the areas of centrilobular necrosis (*) of WT (24, 48 h) and KO mice (48 h only). #Indicates periportal areas (×100).
FIG. 8.
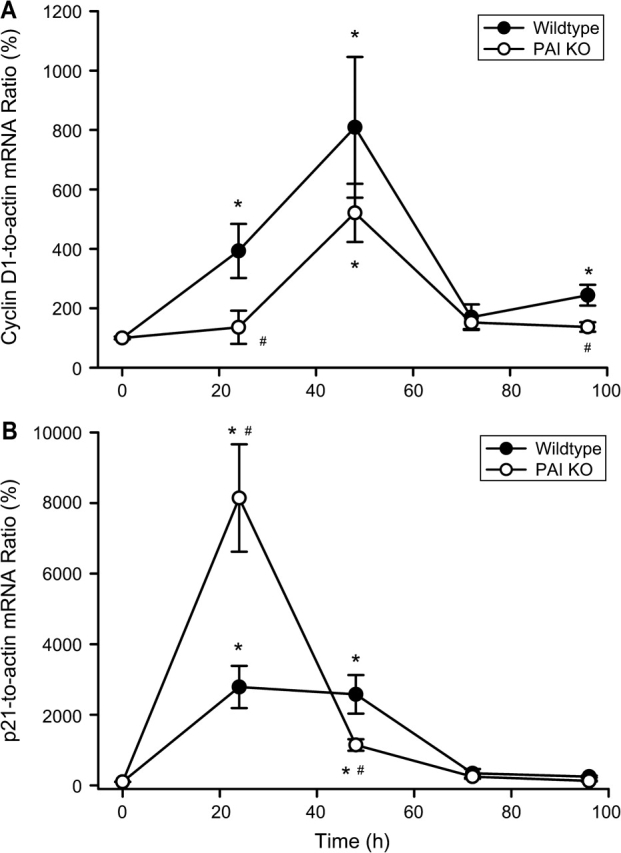
Time course of mRNA expression of cyclin D1 (A) and p21 (B) before and after treatment with 200 mg/kg APAP in WT animals and PAI-KO mice. Cyclin D1 and p21 mRNA levels were compared with the housekeeping gene β-actin and the gene-to-actin ratio is reported as percent of control animals (t =0 h). All data represent means ± SE of n = 4–6 animals per time point. *p < 0.05 (compared with controls), #p < 0.05 (compared with WT).
DISCUSSION
The main objective of this study was to evaluate the functional significance of PAI in the pathogenesis of APAP-induced liver cell injury. Our data indicate that in the absence of PAI gene expression APAP caused massive hemorrhage during the first 24 h, which may have been the main reason for the aggravated tissue injury between 12 and 24 h and the increased mortality rate in the KO animals. In addition, cell cycle activation and the regenerative response were also delayed. These findings were clearly not caused by a difference in the capacity to metabolically activate APAP, that is, form the reactive metabolite NAPQI, in WT and PAI-KO mice. The exponential decline of hepatic GSH levels during the first 30 min after APAP exposure directly reflects the actual formation of NAPQI and its conjugation with GSH in vivo (Jaeschke, 1990). Because there was no difference in the rapid loss of GSH between WT and PAI-KO mice, it can be concluded that both WT and PAI-KO mice generated the same amount of NAPQI, which is the critical determinant of the initial toxicity (Nelson, 1990).
Blood Coagulation and APAP Hepatotoxicity
A recent report demonstrated activation of the blood coagulation cascade and fibrin deposition around the centrilobular area during the first 6 h after APAP overdose (Ganey et al., 2007). The molecular mechanisms of this effect remained unclear. However, sinusoidal endothelial cell damage due to metabolic activation of APAP (DeLeve et al., 1997) and peroxynitrite formation in endothelial cells (Knight et al., 2001) occurs early during APAP-induced liver injury (Ito et al., 2003). In case of endothelial cell damage, tissue factor (TF) expressed on extravascular cells can trigger the extrinsic pathway of the blood coagulation cascade resulting in formation of fibrin clots (Mackman, 2005). Although the constitutive expression of TF on liver cells has not been demonstrated, TF mRNA is induced during APAP-induced liver injury (Welch et al., 2006). In addition, mice with low TF expression or wild-type (WT) animals treated with anticoagulants had less fibrin deposits and reduced liver injury 6 h after APAP overdose (Ganey et al., 2007). These fibrin deposits were still present at 24 h and it was suggested that a further progression of liver injury between 6 and 24 h was at least in part caused by these fibrin clots (Ganey et al., 2007). However, our results suggest that these fibrin deposits may also be beneficial. The massive expression of PAI in WT animals between 6 and 24 h most likely inhibited the formation of plasmin by plasminogen activators and therefore prevented fibrinolysis. Because the absence of PAI gene expression in PAI-KO mice during APAP toxicity resulted in massive hemorrhage and aggravation of tissue necrosis, in particular between 12 and 24 h after APAP administration, it can be concluded that a premature lysis of these fibrin deposits might have triggered these effects. Thus, activation of the coagulation cascade after APAP overdose appears to have a dual function; fibrin deposits can aggravate the early injury but a too rapid fibrinolysis can cause hemorrhage in the liver, which can lead to enhanced tissue damage. The massive accumulation of blood in the extravascular space of the liver impairs the microcirculation and reduces oxygen supply to hepatocytes (Ito et al., 2005). In addition, the loss of blood from the circulation will cause hypotension and reduce blood flow in all organs including the liver, which will further reduce oxygen delivery and can result in ischemic necrosis. The ischemic necrosis may have been the main reason for the expansion of tissue injury in PAI-KO mice between 12 and 24 h and the hypovolemic shock most likely caused the increased mortality in these animals. The hemorrhage and hypovolemic shock observed in PAI-KO mice after APAP overdose are very similar to events occurring in mice treated with galactosamine/endotoxin (Ito et al., 2006) and after treatment with an anti-Fas antibody (Bajt et al., 2000). In these models, the breakdown of the vascular barrier in sinusoids appears to be either caused by direct endothelial cell apoptosis (Bajt et al., 2000) or initial endothelial gap formation and damage by extravasating neutrophils (Ito et al., 2006; Jaeschke et al., 1998). Once this occurs, massive blood accumulation in the liver, similar to the APAP model, leads to hypovolemic shock and death of the animals. Gaps within the endothelium and injury to sinusoidal endothelial cells occur early in all models (Bajt et al., 2000; Deleve et al., 1997; Ito et al., 2003, 2006). However, it appears that fibrin deposits may limit the hemorrhage after APAP overdose. Although it was postulated that these fibrin deposits may be detrimental (Ganey et al., 2007), our data suggest that these deposits may also prevent hemorrhage and can be beneficial. Thus, any pharmacological intervention strategy targeting the blood coagulation cascade in APAP overdose needs to consider these opposing effects.
PAI and Liver Regeneration during APAP Hepatotoxicity
In previous studies it was shown that lack of PAI attenuated liver injury induced by hemorrhagic shock (Lagoa et al., 2005), bile duct ligation (Wang et al., 2005), and alcohol-induced liver injury (Bergheim et al., 2006b). The protection was correlated with activation of survival pathways and improved regeneration (Bergheim et al., 2006a; Lagoa et al., 2005; Wang et al., 2005, 2007a). The molecular background for the improved regeneration in PAI-KO mice was thought to be the enhanced formation of active HGF by tissue plasminogen activators in the absence of PAI activation. However, our results show the opposite effect of PAI in APAP hepatotoxicity, that is, a delayed regenerative response as indicated by the delayed expression of both PCNA and cyclin D1. For hepatocytes to replicate they have to leave the resting phase (G0) and enter the cell cycle with DNA synthesis (S phase) and cell division (M phase) (Albrecht and Hansen, 1999). Progression in the cell cycle is controlled by a number of checkpoints. The expression of cyclin D1 signals that cells have passed the G1 restriction point of the cell cycle and are committed to DNA synthesis (G1/S phase transition) (Fausto, 2000). In addition, PCNA is expressed in the G1 and S phase, which causes the nuclear staining of proliferating cells (Bravo and MacDonald-Bravo, 1987). Thus, the induction of both genes in WT animals at 24 h signals the beginning of the regeneration phase, which ends in the complete restoration of the tissue after 96 h. It is well established that cell cycle inhibitors such as p21 and p27 are also expressed during cell cycle activation (Fausto, 2000). The simultaneous expression of cell cycle activators and inhibitors allows for a sensitive regulation of regeneration. However, in PAI-KO mice, there was a substantial overexpression of p21 at 24 h, which might have been the reason for the delay in cyclin D1 and PCNA expression, that is, a delay in cell cycle activation, in these animals. Interestingly, p21 gene expression levels declined rapidly between 24 and 48 h, which allowed regeneration to proceed also in PAI-KO mice. In fact, beyond the 24 h time point, there was no significant difference in PCNA expression and tissue recovery between PAI-KO and WT animals. However, one has to keep in mind that some of the animals with the most severe hemorrhage and injury died and are not included in the recovery phase. Thus, the 48 to 96 h groups of the PAI-KO animals contain only the more robust survivors. Because it was repeatedly shown that absence of PAI in liver injury models leads to enhanced growth factor activation, the delayed activation of the regeneration process in PAI-KO mice was most likely an indirect effect caused by the higher injury during the time of excessive hemorrhage. Nevertheless, the data indicate that once the animals survive the injury phase without severe hemorrhage, both WT and PAI-KO animals eventually repair the tissue injury equally well.
In summary, our study showed extensive PAI activation during APAP-induced liver injury with peak levels at 24 h. In the absence of PAI, APAP caused severe hemorrhage with increased liver damage between 12 and 24 h resulting in the death of some of the animals. During the aggravation of liver injury due to the hemorrhage, there was a delay in cell cycle activation in PAI-KO mice compared with WT animals. However, the surviving PAI-KO mice showed a similar regenerative response and complete repair of the tissue damage during the following days. The data indicate that PAI activation during APAP hepatotoxicity functions to limit liver injury and death by preventing excessive hemorrhage.
FUNDING
National Institutes of Health grants (R01 DK070195 and R01 AA12916), and National Center for Research Resources grants (P20 RR016475 and P20 RR 021940), a component of the National Institutes of Health.
Acknowledgments
We thank Dr Jie Liu (NIEHS) for providing the primer sequences for real-time RT-PCR.
References
- Albrecht JH, Hansen LK. Cyclin D1 promotes mitogen-independent cell cycle progression in hepatocytes. Cell Growth Differ. 1999;10:397–404. [PubMed] [Google Scholar]
- Bajt ML, Cover C, Lemasters JJ, Jaeschke H. Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver injury. Toxicol. Sci. 2006;94:217–225. doi: 10.1093/toxsci/kfl077. [DOI] [PubMed] [Google Scholar]
- Bajt ML, Farhood A, Lemasters JJ, Jaeschke H. Mitochondrial bax translocation accelerates DNA fragmentation and cell necrosis in a murine model of acetaminophen hepatotoxicity. J. Pharmacol. Exp. Ther. 2008;324:8–14. doi: 10.1124/jpet.107.129445. [DOI] [PubMed] [Google Scholar]
- Bajt ML, Knight TR, Farhood A, Jaeschke H. Scavenging peroxynitrite with glutathione promotes regeneration and enhances survival during acetaminophen-induced liver injury in mice. J. Pharmacol. Exp. Ther. 2003;307:67–73. doi: 10.1124/jpet.103.052506. [DOI] [PubMed] [Google Scholar]
- Bajt ML, Lawson JA, Vonderfecht SL, Gujral JS, Jaeschke H. Protection against Fas receptor-mediated apoptosis in hepatocytes and nonparenchymal cells by a caspase-8 inhibitor in vivo: Evidence for postmitochondrial processing of caspase-8. Toxicol. Sci. 2000;58:109–117. doi: 10.1093/toxsci/58.1.109. [DOI] [PubMed] [Google Scholar]
- Bergheim I, Guo L, Davis MA, Duveau I, Arteel GE. Critical role of plasminogen activator inhibitor-1 in cholestatic liver injury and fibrosis. J. Pharmacol. Exp. Ther. 2006a;316:592–600. doi: 10.1124/jpet.105.095042. [DOI] [PubMed] [Google Scholar]
- Bergheim I, Guo L, Davis MA, Lambert JC, Beier JI, Duveau I, Luyendyk JP, Roth RA, Arteel GE. Metformin prevents alcohol-induced liver injury in the mouse: Critical role of plasminogen activator inhibitor-1. Gastroenterology. 2006b;130:2099–2112. doi: 10.1053/j.gastro.2006.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björnsson E, Jerlstadm P, Bergqvistm A, Olsson R. Fulminant drug-induced hepatic failure leading to death or liver transplantation in Sweden. Scand. J. Gastroenterol. 2005;40:1095–1101. doi: 10.1080/00365520510023846. [DOI] [PubMed] [Google Scholar]
- Bravo R, Macdonald-Bravo H. Existence of two populations of cyclin/proliferating cell nuclear antigen during the cell cycle: Association with DNA replication sites. J. Cell Biol. 1987;105:1549–1554. doi: 10.1083/jcb.105.4.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda S, Mangipudy RS, Warbritton A, Bucci TJ, Mehendale HM. Stimulated hepatic tissue repair underlies heteroprotection by thioacetamide against acetaminophen-induced lethality. Hepatology. 1995;21:477–486. [PubMed] [Google Scholar]
- Dalhoff K, Laursen H, Bangert K, Poulsen HE, Anderson ME, Grunnet N, Tygstrup N. Autoprotection in acetaminophen intoxication in rats: The role of liver regeneration. Pharmacol. Toxicol. 2001;88:135–141. doi: 10.1034/j.1600-0773.2001.d01-94.x. [DOI] [PubMed] [Google Scholar]
- DeLeve LD, Wang X, Kaplowitz N, Shulman HM, Bart JA, van der Hoek A. Sinusoidal endothelial cells as a target for acetaminophen toxicity. Direct action versus requirement for hepatocyte activation in different mouse strains. Biochem. Pharmacol. 1997;9:1339–1345. doi: 10.1016/s0006-2952(97)00048-8. [DOI] [PubMed] [Google Scholar]
- Donahower B, McCullough SS, Kurten R, Lamps LW, Simpson P, Hinson JA, James LP. Vascular endothelial growth factor and hepatocyte regeneration in acetaminophen toxicity. Am. J. Physiol. Gastrointest. Liver Physiol. 2006;291:G102–G109. doi: 10.1152/ajpgi.00575.2005. [DOI] [PubMed] [Google Scholar]
- Eddy AA. Plasminogen activator inhibitor-1 and the kidney. Am. J. Physiol. Renal Physiol. 2002;283:F209–F220. doi: 10.1152/ajprenal.00032.2002. [DOI] [PubMed] [Google Scholar]
- Fausto N. Liver regeneration. J. Hepatol. 2000;32(Suppl. 1):19–31. doi: 10.1016/s0168-8278(00)80412-2. [DOI] [PubMed] [Google Scholar]
- Ganey PE, Luyendyk JP, Newport SW, Eagle TM, Maddox JF, Mackman N, Roth RA. Role of the coagulation system in acetaminophen-induced hepatotoxicity in mice. Hepatology. 2007;46:1177–1186. doi: 10.1002/hep.21779. [DOI] [PubMed] [Google Scholar]
- Gujral JS, Knight TR, Farhood A, Bajt ML, Jaeschke H. Mode of cell death after acetaminophen overdose in mice: Apoptosis or oncotic necrosis? Toxicol. Sci. 2002;67:322–328. doi: 10.1093/toxsci/67.2.322. [DOI] [PubMed] [Google Scholar]
- Ito Y, Abril ER, Bethea NW, McCuskey RS. Inhibition of matrix metalloproteinases minimizes hepatic microvascular injury in response to acetaminophen in mice. Toxicol. Sci. 2005;83:190–196. doi: 10.1093/toxsci/kfh291. [DOI] [PubMed] [Google Scholar]
- Ito Y, Abril ER, Bethea NW, McCuskey MK, Cover C, Jaeschke H, McCuskey RS. Mechanisms and pathophysiological implications of sinusoidal endothelial cell gap formation following treatment with galactosamine/endotoxin in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2006;291:G211–G218. doi: 10.1152/ajpgi.00312.2005. [DOI] [PubMed] [Google Scholar]
- Ito Y, Bethea NW, Abril ER, McCuskey RS. Early hepatic microvascular injury in response to acetaminophen toxicity. Microcirculation. 2003;10:391–400. doi: 10.1038/sj.mn.7800204. [DOI] [PubMed] [Google Scholar]
- Jaeschke H. Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: The protective effect of allopurinol. J. Pharmacol. Exp. Ther. 1990;255:935–941. [PubMed] [Google Scholar]
- Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol. Sci. 2006;89:31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- Jaeschke H, Fisher MA, Lawson JA, Simmons CA, Farhood A, Jones DA. Activation of caspase 3 (CPP32)-like proteases is essential for TNF-alpha-induced hepatic parenchymal cell apoptosis and neutrophil-mediated necrosis in a murine endotoxin shock model. J. Immunol. 1998;160:3480–3486. [PubMed] [Google Scholar]
- Knight TR, Kurtz A, Bajt ML, Hinson JA, Jaeschke H. Vascular and hepatocellular peroxynitrite formation during acetaminophen-induced liver injury: Role of mitochondrial oxidant stress. Toxicol. Sci. 2001;62:212–220. doi: 10.1093/toxsci/62.2.212. [DOI] [PubMed] [Google Scholar]
- Kon K, Kim JS, Jaeschke H, Lemasters JJ. Mitochondrial permeability transition in acetaminophen-induced necrotic and apoptotic cell death to cultured mouse hepatocytes. Hepatology. 2004;40:1170–1179. doi: 10.1002/hep.20437. [DOI] [PubMed] [Google Scholar]
- Lagoa CE, Vodovotz Y, Stolz DB, Lhuillier F, McCloskey C, Gallo D, Yang R, Ustinova E, Fink MP, Billiar TR, et al. The role of hepatic type 1 plasminogen activator inhibitor (PAI-1) during murine hemorrhagic shock. Hepatology. 2005;42:390–399. doi: 10.1002/hep.20797. [DOI] [PubMed] [Google Scholar]
- Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, Reisch JS, Schiodt FV, Ostapowicz G, Shakil AO, et al. Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: Results of a United States multicenter, prospective study. Hepatology. 2005;42:1364–1372. doi: 10.1002/hep.20948. [DOI] [PubMed] [Google Scholar]
- Lawson JA, Fisher MA, Simmons CA, Farhood A, Jaeschke H. Inhibition of Fas receptor (CD95)-induced hepatic caspase activation and apoptosis by acetaminophen in mice. Toxicol. Appl. Pharmacol. 1999;156:179–186. doi: 10.1006/taap.1999.8635. [DOI] [PubMed] [Google Scholar]
- Leiper K, Croll A, Booth NA, Moore NR, Sinclair T, Bennett B. Tissue plasminogen activator, plasminogen activator inhibitors, and activator-inhibitor complex in liver disease. J. Clin. Pathol. 1994;47:214–217. doi: 10.1136/jcp.47.3.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackman N. Tissue-specific hemostasis in mice. Arterioscler. Thromb. Vasc. Biol. 2005;25:2273–2281. doi: 10.1161/01.ATV.0000183884.06371.52. [DOI] [PubMed] [Google Scholar]
- Mars WM, Zarnegar R, Michalopoulos GK. Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am. J. Pathol. 1993;143:949–958. [PMC free article] [PubMed] [Google Scholar]
- Mehendale HM. Tissue repair: An important determinant of final outcome of toxicant-induced injury. Toxicol. Pathol. 2005;33:41–51. doi: 10.1080/01926230590881808. [DOI] [PubMed] [Google Scholar]
- Nelson SD. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin. Liver Dis. 1990;10:267–278. doi: 10.1055/s-2008-1040482. [DOI] [PubMed] [Google Scholar]
- Reilly TP, Bourdi M, Brady JN, Pise-Masison CA, Radonovich MF, George JW, Pohl LR. Expression profiling of acetaminophen liver toxicity in mice using microarray technology. Biochem. Biophys. Res. Commun. 2001;282:321–328. doi: 10.1006/bbrc.2001.4576. [DOI] [PubMed] [Google Scholar]
- Shen W, Kamendulis LM, Ray SD, Corcoran GB. Acetaminophen-induced cytotoxicity in cultured mouse hepatocytes: Effects of Ca(2+)-endonuclease, DNA repair, and glutathione depletion inhibitors on DNA fragmentation and cell death. Toxicol. Appl. Pharmacol. 1992;112:32–40. doi: 10.1016/0041-008x(92)90276-x. [DOI] [PubMed] [Google Scholar]
- Shimizu M, Hara A, Okuno M, Matsuno H, Okada K, Ueshima S, Matsuo O, Niwa M, Akita K, Yamada Y, et al. Mechanism of retarded liver regeneration in plasminogen activator-deficient mice: Impaired activation of hepatocyte growth factor after Fas-mediated massive hepatic apoptosis. Hepatology. 2001;33:569–576. doi: 10.1053/jhep.2001.22650. [DOI] [PubMed] [Google Scholar]
- Wang H, Vohra BP, Zhang Y, Heuckeroth RO. Transcriptional profiling after bile duct ligation identifies PAI-1 as a contributor to cholestatic injury in mice. Hepatology. 2005;42:1099–1108. doi: 10.1002/hep.20903. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhang Y, Heuckeroth RO. Tissue-type plasminogen activator deficiency exacerbates cholestatic liver injury in mice. Hepatology. 2007a;45:1527–1537. doi: 10.1002/hep.21613. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhang Y, Heuckeroth RO. PAI-1 deficiency reduces liver fibrosis after bile duct ligation in mice through activation of tPA. FEBS Lett. 2007b;581:3098–3104. doi: 10.1016/j.febslet.2007.05.049. [DOI] [PubMed] [Google Scholar]
- Welch KD, Reilly TP, Bourdi M, Hays T, Pise-Masison CA, Radonovich MF, Brady JN, Dix DJ, Pohl LR. Genomic identification of potential risk factors during acetaminophen-induced liver disease in susceptible and resistant strains of mice. Chem. Res. Toxicol. 2006;19:223–233. doi: 10.1021/tx050285z. [DOI] [PubMed] [Google Scholar]