

Abstract
Motor neuron axonopathy in diseases such as amyotrophic lateral sclerosis can be modeled and probed with neurotoxic chemicals that induce similar patterns of pathology, such as axonal spheroids that represent focal accumulation of anterogradely transported neurofilaments (NFs). The aromatic γ-diketone–like 1,2-diacetylbenzene (1,2-DAB), but not its 1,3-DAB isomer, reacts with ϵ-amino- or sulfyhydryl groups of (neuro)proteins, forms adducts, and causes NFs to accumulate at proximal sites of elongate motor axons. We exploit the protein-reactive properties of neurotoxic 1,2-DAB versus the nonprotein-reactive properties of non-neurotoxic 1,3-DAB to unveil proteomic changes associated with this type of pathology. We used two-dimensional differential in-gel electrophoresis (2D-DIGE), matrix-assisted laser desorption/ionization time-of-flight tandem mass spectrometry to analyze the lumbosacral spinal cord proteome of adult Sprague-Dawley rats treated systemically with 20 mg/kg/day 1,2-DAB, equimolar dose of 1,3-DAB, or equivalent volume of vehicle (saline containing 2% acetone), 5 days a week, for 2 weeks. 1,2-DAB significantly altered the expression of protein disulfide isomerase, an enzyme involved in protein folding, and gelsolin, an actin-capping and -severing protein. Modifications of these two proteins have been incriminated in the pathogenesis of nerve fiber degeneration. Protein-reactive and neurotoxic 1,2-DAB appears to be excellent tool to dissect mechanisms of nerve fiber (axon) degeneration.
Keywords: axonal swellings, γ-diketones, gelsolin, protein disulfide isomerase, proteomics, solvent neuropathy
Molecular mechanisms underlying motor neuron axonopathy in diseases such as amyotrophic lateral sclerosis (ALS) have yet to be understood. Pathological changes often occur in axons before nerve cell bodies suggesting the existence of axon-specific targets and/or mechanisms associated with neurodegeneration. For instance, an early pathological sign in ALS consists of intraspinal axonal spheroids filled with neurofilaments (NFs) and abnormal protein aggregates, whereas the full-blown picture of pathological changes (dying back-axons, inclusions in nerve cell bodies, and cell death) is seen late in the course of the disease (Kato, 2008). Focal accumulation of axonal NFs is also seen in rodent models of axonopathy induced by certain neurotoxic compounds, such as aliphatic γ-diketone 2,5-hexanedione (2,5-HD) or its aromatic cousin 1,2-diacetylbenzene (1,2-DAB) (Spencer et al., 2002; Tshala-Katumbay et al., 2005). Direct application of 2,5-HD to nerve fibers induces within minutes a reorganization of axoplasmic structures comparable to that seen in systemic intoxication with 1,2-DAB or 2,5-HD (Tshala-Katumbay et al., 2005; Zagoren et al., 1983), which suggests the targets of these agents reside within the axon.
We and others have suggested previously that aromatic 1,2-DAB and aliphatic 2,5-HD, but not their respective 1,3-DAB or 2,4-HD isomers, react with ϵ-amino- or sulfyhydryl (SH)- groups of (neuro)proteins, form adducts, and promote protein cross-linking and polymerization (DeCaprio, 1986, 1987; Kim et al., 2002). We exploit the protein-reactive property of 1,2-DAB versus the nonprotein-reactive property of 1,3-DAB to unveil proteomic changes associated with axonopathy. We used a high-throughput proteomic methodology consisting of two-dimensional differential in-gel electrophoresis (2D-DIGE) followed by protein identification (ID) by matrix-assisted laser desorption ionization time-of-flight/tandem mass spectrometry (MALDI-TOF/MS-MS) to analyze the lumbar spinal cord proteome of adult male Sprague-Dawley rats systemically treated with 1,2-DAB. We show that 1,2-DAB modifies the expression of proteins and/or enzymes involved in protein folding, maintaining axonal shape and dimensions, detoxification and redox mechanisms, and in supporting energy metabolism. Our approach has identified novel actors in the play performed by 1,2-DAB, including protein disulfide isomerase (PDI), an enzyme that modulates protein folding, and gelsolin, an actin-capping and -severing protein.
METHODS
Chemicals
1,2-DAB (99%) and its isomer 1,3-DAB (97%) were purchased from Aldrich Chemical Co. (Madison, WI) and desiccated at room temperature (1,2-DAB) or stored at 4°C (1,3-DAB). Stock samples were checked by gas chromatography–mass spectrometry to confirm the purity of the chemicals. Impurities could not be readily identified, but 1,3-DAB was found to be free of 1,2-DAB and vice versa.
Animals
Experiments were conducted with male Sprague-Dawley rats (Charles Rivers, CA) weighing 175–225 g upon arrival. Animals were individually caged and given rodent chow (PMI Nutrition International, NJ) and water ad libitum. They were acclimated for 5 days in a room maintained at 20°C on a 12-h/12-h light-dark cycle prior to the commencement of the study.
Treatment of Animals
Rats were treated with 20 mg/kg/day 1,2-DAB (n = 5), 1,3-DAB (n = 5), or an equivalent volume of vehicle (saline containing 2% acetone) (n = 5), 5 days a week, for 2 weeks. Agents were administered intraperitoneally with a 1-ml disposable plastic syringe equipped with a 27-gauge needle. Injection sites were rotated around the abdomen and care was taken to minimize leakage and discomfort. Animals were observed daily for changes in behavior, physical appearance, and locomotion in an open field before and after treatment. They were also weighed before treatment to track changes in body weight and adjust the daily dose of the chemicals.
Tissue Preparation
At study termination, rats (n = 3 per treatment group) were anesthetized with 4% isofluorane (0.7 l oxygen/min), their blood collected via cardiac puncture, and then decapitated prior to the removal of the spinal cord. The spinal cord was removed by pressurized saline extrusion, divided into two halves (cranial and caudal halves), and flash-frozen in liquid nitrogen and stored at –80°C prior to proteomic studies.
For morphological studies, the remaining animals were anesthetized as described above, the abdomen and chest opened, and the heart injected with 50 μl of heparin solution (1000 United States Pharmacopea units/ml). Immediately thereafter, the left atrium and ventricular apex were excised, a cannula spurting perfusate introduced and clamped into the ascending aorta, and the animal perfused with cold 4% paraformaldehyde followed by 5% glutaraldehyde, each in 0.2M sodium cacodylate buffer (pH 7.4). The lumbosacral spinal cord and sciatic nerve were sampled, postfixed in excess cacodylate-buffered 1% osmium tetroxide, dehydrated, and embedded in epoxy resin. Cross-sections (∼900 nm) were stained with 1% toluidine blue and screened by bright-field microscopy. Thin sections (∼90 nm) of regions of interest were stained with 2% uranyl acetate followed by 1% lead citrate for examination by transmission electron microscopy.
Proteomic Studies
Sample preparation.
Flash-frozen rat lumbosacral spinal cords were shipped on dry ice to Applied Biomics (Hayward, CA) for proteomic studies. Tissues were washed three times in buffer containing 10mM Tris-HCl and 5mM magnesium acetate (pH 8.0) to remove contaminating blood prior to the commencement of proteomic experiments that were conducted independently three times. Thereafter, proteins were dissolved in 2-D lysis buffer (30mM Tris-HCl, pH 8.8, containing 7M urea, 2M thiourea, and 4% 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS)) and incubated at room temperature for 30 min on a shaker. The lysed tissue samples were centrifuged for 30 min at 16,000 × g. The supernatants were collected and the protein quantity was measured using the Bio-Rad protein assay kit (Bio-Rad, CA).
CyDye labeling.
To label proteins, 30 μg of each protein samples were incubated with 0.7 ml of diluted CyDye (1:5 diluted in dimethylformamide from 1 nmol/ml stock, GE Healthcare, NJ) at 4°C for 30 min. The labeling was stopped by adding 0.7 ml of 10mM L-lysine and incubating at 4°C for 15 min. The labeled samples were mixed together, and equal volume of 2× 2-D sample buffer (8M urea, 4% CHAPS, 20 mg/ml DTT, 2% pharmalytes and trace amount of bromophenol blue) and 100 ml of destreak solution (GE Healthcare) were added. The total sample volumes were adjusted to 260 ml by adding rehydration buffer (7M urea, 2M thiourea, 4% CHAPS, 20 mg/ml dithiotreitol [DTT], 1% pharmalytes and trace amount of bromophenol blue). The samples were incubated at room temperature for 10 min on a shaker and centrifuged for 10 min at 16,000 × g. The supernatants were loaded onto a 13-cm immobilized pH gradient gel (IPG) strip holder (GE Healthcare) prior to isoelectric focusing (IEF) and sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE).
IEF and SDS-PAGE.
Thirteen-centimeter IPG strips (pH 3–10) were put on the loaded samples and 1 ml of mineral oil added on top of the strips. IEF was run according to the protocol provided by the manufacturer (GE Healthcare). Upon completion of the IEF, the IPG strips were incubated in freshly made equilibration buffer 1 (50mM Tris-HCl, pH 8.8, containing 6M urea, 30% glycerol, 2% SDS, trace amount of bromophenol blue, and 10 mg/ml DTT) for 15 min with gentle shaking. Thereafter, they were rinsed in the equilibration buffer 2 (50mM Tris-HCl, pH 8.8, containing 6M urea, 30% glycerol, 2% SDS, trace amount of bromophenol blue, and 45 mg/ml DTT) for 10 min with gentle shaking, and 1× in the SDS-gel running buffer prior to their transfer onto gradient SDS-gels (9–12%) prepared using low fluorescent glass plates and sealed with 0.5% (wt/vol) agarose solution (in SDS-gel running buffer). The SDS-gels were run at 15°C and stopped when the front dye had run out of the gels.
Image scan and software analysis.
Gel scanning was carried out immediately after the SDS-PAGE using Typhoon TRIO (Amersham Biosciences, NJ). Scanned images were analyzed by Image Quant software (version 5.0, Amersham Biosciences) and subjected to in-gel analysis and cross-gel analysis using the DeCyder software (version 6.0, GE Healthcare) with a detection limit of 0.2 ng of protein per spot. The ratio change for differentially expressed protein spots was obtained from the in-gel DeCyder analysis.
Preparative gel and spot picking for mass spectrometry.
A total of 600–700 μg of unlabeled proteins was run in a preparative gel and stained with Deep Purple total protein stain (GE Healthcare) prior to mass spectrometry analysis. Twenty-two protein spots that were consistently differentially expressed in 1,2-DAB- versus vehicle- and 1,3-DAB-treated samples across a minimum of two gels were picked up by the Ettan Spot Picker (GE Healthcare) and subjected to in-gel trypsin digestion, peptide extraction, and desalting prior to MALDI-TOF/MS-MS.
In-gel digestion, mass spectrometry, and database search.
Proteins of interest were digested in-gel with modified porcine trypsin protease (Trypsin Gold, Promega, WI). The digested tryptic peptides were desalted by Zip-tip C18 (Millipore, CA). Peptides were eluted from the Zip-tip with 0.5 μl of matrix solution (α-cyano-4-hydroxycinnamic acid (5 mg/ml in 50% acetonitrile, 0.1% trifluoroacetic acid, 25mM ammonium bicarbonate) and spotted on the MALDI plate (model ABI 01-192-6-AB). MALDI-TOF MS and TOF/TOF tandem MS/MS were performed on an ABI 4700 mass spectrometer (Applied Biosystems, CA). MALDI-TOF mass spectra were acquired in reflectron positive ion mode, averaging 4000 laser shots per spectrum. TOF/TOF tandem MS fragmentation spectra were acquired for each protein, averaging 4000 laser shots per fragmentation spectrum on each of the 10 most abundant ions present in each sample (excluding trypsin autolytic peptides and other known background ions). Both the resulting peptide mass and the associated fragmentation spectra were submitted to GPS Explorer workstation equipped with MASCOT search engine (Matrix science, MA) to search the National Center for Biotechnology Information nonredundant databases. Searches were performed without constraining protein molecular weight or isoelectric point, with variable carbamidomethylation of cysteine and oxidation of methionine residues, and with one missed cleavage also allowed in the search parameters. Candidates with protein- and ion-confidence score(s) greater than 95% were considered significant.
Statistical Analysis
Each animal's body weight was measured five times per week for 2 weeks and was summarized by fitting a linear trend to the series of body weight measurements. The estimated slope coefficient from each fit reflects the change in body weight per day. Nonparametric procedures (Kruskall-Wallis, Wilcoxon rank-sum) were used to determine whether the slope coefficients differed among the three groups. All computations were performed using R version 2.6.1 (R Development Core Team, 2007).
Changes in number of protein spots that were differentially expressed were analyzed using log-linear models with an allowance made for overdispersion.
RESULTS
Physical and Neuropathological Changes
Rats treated with 1,2-DAB, but not with 1,3-DAB or vehicle, developed neuromuscular hind limb weakness together with postural and gait abnormalities. These signs occurred after ∼1 week of treatment with 1,2-DAB. Daily changes in body weight significantly differed among the three groups of animals treated with 1,2-DAB, or 1,3-DAB, or vehicle (χ2 (2df) = 9.42, p = 0.01; Kruskal-Wallis test). There was no difference in body weight changes between 1,3-DAB- and vehicle-treated animals (W = 14, p = 0.84). These two groups typically gained 9.9 g/day (W = 55, p = 0.002; 95% CI: 8.7–11.2 g/day). Relative to animals treated with either 1,3-DAB or vehicle, those given 1,2-DAB lost approximately 14 g/day more weight (W = 50, p < 0.001; 95% CI: 10.8–19.1 g/day more weight lost) (Table 1).
TABLE 1.
Estimated Change in Body Weight (g) per Day (i.e., Slope Coefficient) for Each of the Five Animals in each Treatment Group
| Vehicle-treated animals | 1,3-DAB-treated animals | 1,2-DAB-treated animals |
| 7.35 | 7.68 | −10.28 |
| 8.79 | 9.08 | −6.69 |
| 10.27 | 9.75 | −3.14 |
| 12.18 | 10.85 | −2.16 |
| 12.41 | 10.94 | −0.93 |
Only animals treated with 1,2-DAB showed nerve fiber damage. Light microscopy of the lumbar spinal cord revealed 10-nm NF-filled swollen axons in spinal cord anterior horns and proximal ventral roots. Neurons in the adjacent dorsal horns were unremarkable. Electron microscopy of enlarged axons revealed disorganization of the cytoskeleton in the form of clustering of microtubules and organelles and NF segregation. In some instances, NF accumulated inside the nerve cell bodies (Fig. 1).
FIG. 1.
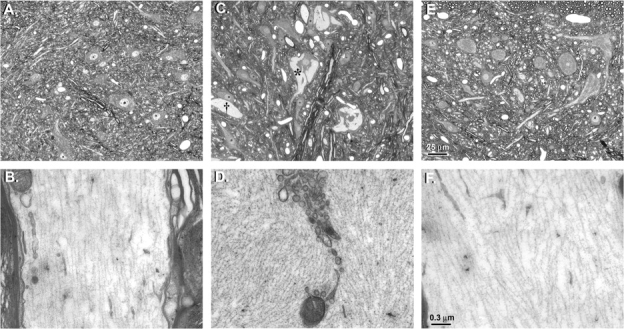
Cross-sections of lumbar spinal cords of rats treated i.p. with vehicle (A and B), 20 mg/kg/day 1,2-DAB (C and D), or 1,3-DAB (E and F), 5 days a week, for 2 weeks. The upper panel shows light-microscope images and the lower panel are the corresponding electron microscopy images. Hypochromic zones (*) corresponding to NF accumulation in anterior horn cells and giant axonal swellings (†) were evident in animals treated with 1,2-DAB (C). Electron microscopy of this region revealed disorganization of the cytoskeleton and clustering of organelles in axonal swellings that were densely packed with NF (D). Spinal cords from animals treated with vehicle (A, B) or 1,3-DAB (E, F) were unremarkable. Cross-sections (∼900 nm) for light microscopy were stained with 1% toluidine blue. Thin sections (∼90 nm) of regions of interest were stained with 2% uranyl acetate followed by 1% lead citrate for transmission electron microscopy.
Proteomic Changes
The Decyder analysis of protein expression across the proteomic experiments revealed that both 1,2-DAB and 1,3-DAB induced changes in expression levels of proteins. However, axonopathic1,2-DAB induced greater change in protein expression relative to 1,3-DAB and/or vehicle. For example, relative to 1,3-DAB, the effect of 1,2-DAB was to significantly increase the number of protein spots showing both “increased” and “decreased” protein expression (p < 0.001). The number of spots with high or low expression levels was 3.2 times higher (95% CI: 1.6–6.5 times higher) or 2.4 times higher (95% CI: 1.4–4.2 times higher), respectively, in 1,2-DAB- versus vehicle-treated relative to 1,3-DAB- versus vehicle-treated animals (Table 2, Fig. 2). The observed changes in protein expression were consistent across the gels (p = 0.82) suggesting reliable conditions and results across the proteomic experiments.
TABLE 2.
Number of Protein Spots Identified by the Decyder Software as being Differentially Expressed in 1,2-DAB or 1,3-DAB Relative to Vehicle-Treated Samples
| 1,2-DAB versus vehicle |
1,3-DAB versus vehicle |
|||||
| Protein expression | Gel 1 | Gel 2 | Gel 3 | Gel 1 | Gel 2 | Gel 3 |
| Decreased | 134 | 90 | 114 | 28 | 35 | 77 |
| Increased | 99 | 90 | 58 | 16 | 42 | 19 |
| Unchanged | 1246 | 1268 | 1223 | 1435 | 1371 | 1299 |
FIG. 2.

Average proportion of protein spots differentially expressed in 1,3-DAB- versus vehicle-treated samples (left) and 1,2-DAB- versus vehicle-treated samples (right). Relative to 1,3-DAB, 1,2-DAB significantly altered (increased or decreased) the expression levels of proteins.
After visual inspection of fluorescent images of analytical gels, 22 protein spots were classified independently by two experienced neuroscientists blind to the experimental conditions as being markedly altered by treatment with 1,2-DAB relative to 1,3-DAB and vehicle (spots 1–22, Fig. 3 and Table 3). Mass spectrometry ID of these 22 protein spots revealed proteins and/or enzymes that are involved in protein folding (e.g., PDI), maintaining axonal shape and dimensions (e.g., NF-L or gelsolin), detoxification and redox mechanisms (e.g., glutathione-S transferase), and supporting energy metabolism (e.g., ATP citrate lyase). Novel findings in the pathogenesis of γ-diketone axonopathy included the downregulation of gelsolin and PDI (spots 5 and 9 in Fig. 3 and Table 3, Fig. 4). We studied the mRNA expression of NF-L, gelsolin, and PDI and showed no transcriptional changes. To validate our proteomic findings, we have confirmed changes in the expression level of a selected protein, that is, PDI using Western blot (data not shown).
FIG. 3.

Representative 2D-DIGE patterns of lumbar spinal cords from animals treated with 20 mg/kg/day 1,2-DAB versus 1,3-DAB, 5 days a week, for 2 weeks. The 22 differentially expressed protein spots (circles 1–22) that were differentially modulated by 1,2-DAB versus vehicle (left) and 1,2-DAB versus 1,3-DAB (right) were subjected to MALDI-TOF/MS-MS for protein ID.
TABLE 3.
List of Proteins Differentially Affected by 1,2-DAB (100% Confidence Score for Protein ID by MALDI-TOF/MS-MS)
| Spot number | Protein name | Symbols | Accession number | Molecular weight | Isoelectric point | Peptides matched | Function | Fold changea |
| 1 | Fatty acid synthase | Fasn | 204099 | 272,493.9 | 5.96 | 27 | Metabolism of lipids | −1.65 |
| 2 | ATP citrate lyase | Acly | 8392839 | 120,558.7 | 6.96 | 23 | Energy metabolism | −1.57 |
| 3 | Endoplasmin | 51858886 | 74,161.6 | 5.02 | 26 | Heat shock protein | −1.44 | |
| 4 | Formyltetrahydrofolate dehydrogenase | Fthfd | 57921067 | 98,811.5 | 5.64 | 34 | Detoxification | −2.52 |
| 5 | Gelsolin | Gsn | 51854227 | 86,014.1 | 5.76 | 16 | Protein binding/structural protein | −2.42 |
| 6 | Hydroxyacyl-coenzyme A dehydrogenase/3-ketoacyl-coenzyme A thiolase/enoyl-coenzyme A hydratase (TFP), alpha subunit | Hadha | 60688124 | 82612.5 | 9.16 | 22 | Energy metabolism | −1.69 |
| 7 | Dihydrolipoamide acetyltransferase | Odp2 | 34863356 | 67,123.4 | 8.76 | 12 | Energy metabolism | −1.63 |
| 8 | Pyruvate kinase muscle | Pkm2 | 56929 | 57,780.9 | 6.63 | 18 | Energy metabolism | +2.02 |
| 9 | PDI related 3 | Pdi | 38382858 | 56,587.7 | 5.88 | 20 | Protein folding | −2.86 |
| 10 | 3-Hydroxy-3-methylglutaryl-coenzyme A synthase 1 (Hmgcs1) | 8393538 | 57,397.4 | 5.58 | 16 | Energy metabolism | −2.87 | |
| 11 | GDP dissociation inhibitor 2 | Gdi2 | 40254781 | 50,504.7 | 5.93 | 18 | Catalytic activity/vesicle trafficking | −1.19 |
| 12 | Aldolase A | Aldoa | 6978487 | 39,327.3 | 8.31 | 14 | Catalytic activity | +1.85 |
| 13 | NADH dehydrogenase (ubiquinone) 1 alpha | Ndufa10 | 46391108 | 31,680.8 | 5.44 | 9 | Energy metabolism | +2.39 |
| 14 | NADH dehydrogenase (ubiquinone) 1 alpha | Ndufa10 | 46391108 | 31,680.8 | 5.44 | 12 | Energy metabolism | −1.83 |
| 15 | Glyceraldehyde-3-phosphate dehydrogenase | Gpd | 56611127 | 35,771.2 | 8.14 | 6 | Energy metabolism | +1.38 |
| 16 | Voltage-dependent anion channel-like protein | — | 540011 | 31,699.6 | 7.44 | 7 | Unknown | −2.1 |
| 17 | Glyceraldehyde-3-phosphate dehydrogenase | Gpd | 8393418 | 35,805.2 | 8.14 | 7 | Energy metabolism | +6.8 |
| 18 | Similar to biliverdin reductase B (flavin reductase) | Blvrd | 34855391 | 22,080.3 | 6.29 | 9 | NADPH diaphorase activity | −2.9 |
| 19 | Glutathione-S transferase Yb-1 subunit (EC 2.5.1.18) | Gstm1 | 204503 | 25,899 | 7.63 | 13 | Detoxification | −5.24 |
| 20 | Glutathione-S transferase Yb-1 subunit (EC 2.5.1.18) | Gstm1 | 204503 | 25,899 | 7.63 | 17 | Detoxification | +2.49 |
| 21 | Quinoid dihydropteridine reductase | Dhpr | 11693160 | 25,535.9 | 7.67 | 9 | NADPH dihydropteridine reductase activity | +1.89 |
| 22 | NF, light polypeptide | Nefl | 13929098 | 61,298 | 4.63 | 17 | Protein binding/structural protein | −1.61 |
Maximum fold change values observed after cross-gel analysis of proteomic expression. Negative or positive values indicate low or high expression level, respectively, of the indicated protein in samples treated with neurotoxic 1,2-DAB. Dual protein expression of certain proteins (e.g., NADH) may possibly be explained by a concomitant expression of isoforms, or protein subunits, or post-translationally modified proteins. Protein no. 16 has been removed from the nrNCBI databases.
FIG. 4.

DeCyder display of relative abundance of gelsolin and PDI. Upper pair: relative abundance of gelsolin (spot no. 5). Gelsolin showed 2.42× higher abundance in vehicle (A) relative to 1,2-DAB (B) treated sample. Lower pair: relative abundance of PDI (spot no. 9). PDI showed 2.86× higher abundance in vehicle—(C) relative to 1,2-DAB—(D) treated sample.
DISCUSSION
This is the first study to report on global proteomic changes associated with proximal giant neurofilamentous axonopathy induced by the γ-diketone–like 1,2-DAB. We have identified 22 proteins that were remarkably altered by treatment with axonopathic 1,2-DAB relative to its nonaxonopathic isomer 1,3-DAB or vehicle. Novel findings include downregulation of PDI, a chaperone molecule that regulates protein folding and of gelsolin, an actin-capping and -severing protein with yet unresolved antiapoptotic properties (Endres et al., 1999; Ferrari and Soling, 1999; Furukawa et al., 1997; Harms et al., 2004; Maattanen et al., 2006). Whether the change in expression levels of PDI and gelsolin are linked is unknown, but it is possible they are coupled upstream and downstream effects of 1,2-DAB, the former triggering protein misfolding and the latter reflecting activation of protein degradation. Whether the change in PDI expression is associated with changes in metabolism and function of the endoplasmic reticulum, the main host-organelle of PDI, is another relevant question (Atkin et al., 2006; Bertoli et al., 2004; Ferrari and Soling, 1999).
PDI could conceivably play a critical role in the pathogenesis of γ-diketone axonopathy for several reasons. First, its abundant lysine content (9.9% of total amino acid) makes PDI highly vulnerable to chemical attack by γ-diketones. Second, four of six γ-diketone–vulnerable cysteine residues (SH-group carriers) are located on the two PDI redox active sites that participate in disulfide bond formation and maintenance of the 3D-conformation of the molecule. Third, modification (adduction) of PDI could result in downstream effects on protein-protein interaction, perhaps resulting in changes in cytoskeletal disorganization of the type seen after local and/or systemic treatment with 2,5-HD or 1,2-DAB (Zagoren et al., 1983; Tshala-Katumbay et al., 2005). Fourth, changes in PDI expression/activity might prevent proper folding of nascent cytoskeleton proteins. For example, NF proteins synthesized in the anterior horn cell and destined for transport to the axon might lose their binding properties and accumulate in proximal axonal spheroids or even fail to exit the cell body. It is noteworthy that changes in PDI expression and its S-nitrosylation have been proposed to play a role in the pathogenesis of neurodegenerative diseases such as the ALS, which has axonal spheroids indistinguishable from those induced by 1,2-DAB (Atkin et al., 2006; Kozarova et al., 2007; Nakamura and Lipton, 2008). We suggest that more studies are conducted to understand the role of PDI and that of redox assistants in protein folding (glutathione and Ero-1) (Atkin et al., 2006; Bertoli et al., 2004; Cuozzo and Kaiser, 1999; Molteni et al., 2004) in the pathogenesis of γ-diketone axonopathy.
Gelsolin is another novel protein actor in 1,2-DAB axonopathy that for several reasons may have mechanistic importance in the genesis of motor neuron/axon pathology. First, it modulates the formation (polymerization-depolymerization) of the actin (microfilament) network. Changes in gelsolin expression and/or activity might disrupt the interactions and/or the structural organization of the cytoskeleton, resulting for example in the separation of NFs and microtubules seen in early stages of γ-diketone axonopathy. Second, gelsolin is implicated in regulating apoptotic cell death, a phenomenon that has been reported in cell cultures treated with γ-diketones but that has still to be demonstrated in vivo. And third, mutation in the gene that codes for gelsolin has been linked to the pathogenesis of the Finnish type familial amyloid polyneuropathy, a neurodegenerative disease with mechanisms that have yet to be understood (Harms et al., 2004; Ikeda et al., 2007; Roch et al., 2007; Spinardi and Witke, 2007; Tanskanen et al., 2007).
Several other proteins were selectively altered in animals treated with 1,2-DAB versus 1,3-DAB or vehicle. NF-L showed a reduced expression that was unexpected in the absence of comparable reductions in NF-H, which has higher lysine content and in mutated form has been implicated in a rare familial form of ALS (Al-Chalabi et al., 1999; Robberecht, 2000). However, NF may be bystanders in the genesis of γ-diketone axonopathy because knocking out the genes that code for NF proteins does not affect susceptibility to 2,5-HD, the aliphatic γ-diketone cousin of 1,2-DAB (Sickles et al., 1994; Stone et al., 2001). Proteins or enzymes associated with energy metabolism have been previously implicated in the genesis of γ-diketone axonopathy (Sabri et al., 1979; Sabri, 1984) and were well represented (ACLY, Hadha, ODP2, Pkm2, Hmgcs1, Ndufa10, and Gpd) among the 22 proteins studied here. ACLY activity was proposed as a biomarker of Wallerian degeneration in peripheral nerves in the late 60′s (Bonavita et al., 1968). Pkm2 and Hadha, a subunit of the mitochondrial trifunctional protein (TFP), may also be of interest because mutations in the genes encoding for TFP subunits and oxidative damage of pkm2 have been respectively implicated in systemic organ failure associated with encephalopathy and mild cognitive impairment in aging subjects (Butterfield et al., 2006; Choi et al., 2007). Changes in the expression level of Gstm1 may be related to an initiation of a detoxification process (Brock et al., 1981; Coles et al., 1984). It is not known whether regulation of Blvrd and Dhpr, which influence nicotinamide adenine dinucleotide phosphate, reduced (NADPH) metabolism, may trigger an oxidative response (Armarego et al., 1984; Florczyk et al., 2008; Sun et al., 2007). Oxidative damage has been recently proposed in the genesis of 1,2-DAB axonopathy in cell cultures (Kim et al., 2007). However, the underlying mechanisms and whether this occurs in vivo have yet to be elucidated. Whether changes in expression levels of endoplasmin, a heat shock protein and/or Gdi2, a protein involved in intracellular trafficking of vesicles (Rivero et al., 2002; Shisheva et al., 1999), are related to 1,2-DAB treatment is also unknown and needs to be investigated.
Although the known functional properties of the 22 proteins are important to consider in relation to the genesis of motor neuron degeneration, there is an abundant need for caution in interpreting the findings. First and foremost, the proteins derive from whole spinal cord tissue, which represents a mixture of neuronal, glial and other cells. Second, the 22 selected proteins undoubtedly represent a fraction of the totality of protein changes and experimentation was done on a limited number of animals. Third, changes in protein expression may arise from the direct effects of 1,2-DAB in adducting proteins, from changes in protein breakdown or in protein synthesis, or in combinations of the foregoing. Changes in protein synthesis are unlikely to be responsible for the segregation of microtubules from NF seen in gamma-diketone axonopathy because this phenomenon is observed immediately following application of 2,5-HD to the sciatic nerve (Zagoren et al., 1983). We have found unaltered mRNA expression levels of PDI, NF-L and gelsolin (data not shown) suggesting that changes in expression of cytoskeleton proteins is unlikely to be caused by a decrease in protein synthesis but may be associated with either protein fragmentation and/or enzymatic degradation. Increase in expression levels of certain proteins may reflect the initiation of homeostatic and/or repair mechanisms following intoxication with 1,2-DAB.
Taken in concert, our global proteomic approach has revealed proteins that are selectively involved in the pathogenesis of 1,2-DAB axonopathy. Changes in PDI are most intriguing because of its established role in the maintenance of the three-dimensional structure of proteins required for normal enzymatic and other functions. Observed changes in the expression of cytoskeletal proteins (gelsolin) could be a downstream effect of alterations in PDI expression. Changes in gelsolin expression could affect cytoskeletal integrity, an early and perhaps key pathological event that precedes development of axonal spheroids.
FUNDING
National Institute of Health (K01NS052183, P42ES10338, U19ES11384); and Oregon Worker Benefit Fund.
Acknowledgments
The technical expertise of Dan Austin (OHSU, Portland OR) and the guidance of Paul Desjardin and Roger Butterworth (Université de Montréal, Canada) are appreciated.
References
- Al-Chalabi A, Andersen PM, Nilsson P, Chioza B, Andersson JL, Russ C, Shaw CE, Powell JF, Leigh PN. Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Hum. Mol. Genet. 1999;8:157–164. doi: 10.1093/hmg/8.2.157. [DOI] [PubMed] [Google Scholar]
- Armarego WL, Randles D, Waring P. Dihydropteridine reductase (DHPR), its cofactors, and its mode of action. Med. Res. Rev. 1984;4:267–321. doi: 10.1002/med.2610040302. [DOI] [PubMed] [Google Scholar]
- Atkin JD, Farg MA, Turner BJ, Tomas D, Lysaght JA, Nunan J, Rembach A, Nagley P, Beart PM, Cheema SS, et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J. Biol. Chem. 2006;281:30152–30165. doi: 10.1074/jbc.M603393200. [DOI] [PubMed] [Google Scholar]
- Bertoli G, Simmen T, Anelli T, Molteni SN, Fesce R, Sitia R. Two conserved cysteine triads in human Ero1alpha cooperate for efficient disulfide bond formation in the endoplasmic reticulum. J. Biol. Chem. 2004;279:30047–30052. doi: 10.1074/jbc.M403192200. [DOI] [PubMed] [Google Scholar]
- Bonavita V, Piccoli F, Serra S. Acetyl-CoA synthetase, NADP malate dehydrogenase and ATP citrate lyase in the degenerating peripheral nerve. Brain Res. 1968;10:245–251. doi: 10.1016/0006-8993(68)90127-3. [DOI] [PubMed] [Google Scholar]
- Brock N, Pohl J, Stekar J. Detoxification of urotoxic oxazaphosphorines by sulfhydryl compounds. J. Cancer Res. Clin. Oncol. 1981;100:311–320. doi: 10.1007/BF00410691. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: Insights into the development of Alzheimer's disease. Neurobiol. Dis. 2006;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Choi JH, Yoon HR, Kim GH, Park SJ, Shin YL, Yoo HW. Identification of novel mutations of the HADHA and HADHB genes in patients with mitochondrial trifunctional protein deficiency. Int. J. Mol. Med. 2007;19:81–87. [PubMed] [Google Scholar]
- Coles B, Ketterer B, Beland FA, Kadlubar FF. Glutathione conjugate formation in the detoxification of ultimate and proximate carcinogens of N-methyl-4-aminoazobenzene. Carcinogenesis. 1984;5:917–920. doi: 10.1093/carcin/5.7.917. [DOI] [PubMed] [Google Scholar]
- Cuozzo JW, Kaiser CA. Competition between glutathione and protein thiols for disulphide-bond formation. Nat. Cell Biol. 1999;1:130–135. doi: 10.1038/11047. [DOI] [PubMed] [Google Scholar]
- DeCaprio AP. Mechanisms of in vitro pyrrole adduct autooxidation in 2,5-hexanedione-treated protein. Mol. Pharmacol. 1986;30:452–458. [PubMed] [Google Scholar]
- DeCaprio AP. n-Hexane neurotoxicity: A mechanism involving pyrrole adduct formation in axonal cytoskeletal protein. Neurotoxicology. 1987;8:199–210. [PubMed] [Google Scholar]
- Endres M, Fink K, Zhu J, Stagliano NE, Bondada V, Geddes JW, Azuma T, Mattson MP, Kwiatkowski DJ, Moskowitz MA. Neuroprotective effects of gelsolin during murine stroke. J. Clin. Invest. 1999;103:347–354. doi: 10.1172/JCI4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari DM, Soling HD. The protein disulphide-isomerase family: Unravelling a string of folds. Biochem. J. 1999;339:1–10. [PMC free article] [PubMed] [Google Scholar]
- Florczyk UM, Jozkowicz A, Dulak J. Biliverdin reductase: New features of an old enzyme and its potential therapeutic significance. Pharmacol. Rep. 2008;60:38–48. [PMC free article] [PubMed] [Google Scholar]
- Furukawa K, Fu W, Li Y, Witke W, Kwiatkowski DJ, Mattson MP. The actin-severing protein gelsolin modulates calcium channel and NMDA receptor activities and vulnerability to excitotoxicity in hippocampal neurons. J. Neurosci. 1997;17:8178–8186. doi: 10.1523/JNEUROSCI.17-21-08178.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms C, Bosel J, Lautenschlager M, Harms U, Braun JS, Hortnagl H, Dirnagl U, Kwiatkowski DJ, Fink K, Endres M. Neuronal gelsolin prevents apoptosis by enhancing actin depolymerization. Mol. Cell Neurosci. 2004;25:69–82. doi: 10.1016/j.mcn.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Mizushima K, Fujita Y, Watanabe M, Sasaki A, Makioka K, Enoki M, Nakamura M, Otani T, Takatama M, Okamoto K. Familial amyloid polyneuropathy (Finnish type) in a Japanese family: Clinical features and immunocytochemical studies. J. Neurol. Sci. 2007;252:4–8. doi: 10.1016/j.jns.2006.09.022. [DOI] [PubMed] [Google Scholar]
- Kato S. Amyotrophic lateral sclerosis models and human neuropathology: Similarities and differences. Acta Neuropathol. 2008;115:97–114. doi: 10.1007/s00401-007-0308-4. [DOI] [PubMed] [Google Scholar]
- Kim MK, Kim KS, Chung JH, Kim JH, Kim JR, Chung HY, Kim MS. Environmental metabolite, 1,2-diacetylbenzene, produces cytotoxicity through ROS generation in HUVEC cells. J. Toxicol. Environ. Health A. 2007;70:1336–1343. doi: 10.1080/15287390701428895. [DOI] [PubMed] [Google Scholar]
- Kim MS, Hashemi SB, Spencer PS, Sabri MI. Amino acid and protein targets of 1,2-diacetylbenzene, a potent aromatic gamma-diketone that induces proximal neurofilamentous axonopathy. Toxicol. Appl. Pharmacol. 2002;183:55–65. doi: 10.1006/taap.2002.9456. [DOI] [PubMed] [Google Scholar]
- Kozarova A, Sliskovic I, Mutus B, Simon ES, Andrews PC, Vacratsis PO. Identification of redox sensitive thiols of protein disulfide isomerase using isotope coded affinity technology and mass spectrometry. J. Am. Soc. Mass. Spectrom. 2007;18:260–269. doi: 10.1016/j.jasms.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Maattanen P, Kozlov G, Gehring K, Thomas DY. ERp57 and PDI: Multifunctional protein disulfide isomerases with similar domain architectures but differing substrate-partner associations. Biochem. Cell. Biol. 2006;84:881–889. doi: 10.1139/o06-186. [DOI] [PubMed] [Google Scholar]
- Molteni SN, Fassio A, Ciriolo MR, Filomeni G, Pasqualetto E, Fagioli C, Sitia R. Glutathione limits Ero1-dependent oxidation in the endoplasmic reticulum. J. Biol. Chem. 2004;279:32667–32673. doi: 10.1074/jbc.M404992200. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Lipton SA. Emerging roles of s-nitrosylation in protein misfolding and neurodegenerative diseases. Antioxid. Redox. Signal. 2008;10:87–102. doi: 10.1089/ars.2007.1858. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. 2007 Available at: http://www.R-project.org. Accessed November 26, 2007. [Google Scholar]
- Rivero F, Illenberger D, Somesh BP, Dislich H, Adam N, Meyer AK. Defects in cytokinesis, actin reorganization and the contractile vacuole in cells deficient in RhoGDI. EMBO J. 2002;21:4539–4549. doi: 10.1093/emboj/cdf449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht W. Genetics of amyotrophic lateral sclerosis. J. Neurol. 2000;247:2–6. [PubMed] [Google Scholar]
- Roch JA, Hermier M, Jouvet A, Nighoghossian N. [Cerebral amyloid angiopathy] Rev. Neurol. (Paris) 2007;163:134–137. doi: 10.1016/s0035-3787(07)90366-5. [DOI] [PubMed] [Google Scholar]
- Sabri MI. In vitro effect of n-hexane and its metabolites on selected enzymes in glycolysis, pentose phosphate pathway and citric acid cycle. Brain Res. 1984;297:145–150. doi: 10.1016/0006-8993(84)90551-1. [DOI] [PubMed] [Google Scholar]
- Sabri MI, Moore CL, Spencer PS. Studies on the biochemical basis of distal axonopathies—I. Inhibition of glycolysis by neurotoxic hexacarbon compounds. J. Neurochem. 1979;32:683–689. doi: 10.1111/j.1471-4159.1979.tb04550.x. [DOI] [PubMed] [Google Scholar]
- Shisheva A, Chinni SR, DeMarco C. General role of GDP dissociation inhibitor 2 in membrane release of Rab proteins: Modulations of its functional interactions by in vitro and in vivo structural modifications. Biochemistry. 1999;38:11711–11721. doi: 10.1021/bi990200r. [DOI] [PubMed] [Google Scholar]
- Sickles DW, Pearson JK, Beall A, Testino A. Toxic axonal degeneration occurs independent of neurofilament accumulation. J. Neurosci. Res. 1994;39:347–354. doi: 10.1002/jnr.490390312. [DOI] [PubMed] [Google Scholar]
- Spencer PS, Kim MS, Sabri MI. Aromatic as well as aliphatic hydrocarbon solvent axonopathy. Int. J. Hyg. Environ. Health. 2002;205:131–136. doi: 10.1078/1438-4639-00138. [DOI] [PubMed] [Google Scholar]
- Spinardi L, Witke W. Gelsolin and diseases. Subcell. Biochem. 2007;45:55–69. doi: 10.1007/978-1-4020-6191-2_3. [DOI] [PubMed] [Google Scholar]
- Stone JD, Peterson AP, Eyer J, Oblak TG, Sickles DW. Neurofilaments are nonessential to the pathogenesis of toxicant-induced axonal degeneration. J. Neurosci. 2001;21:2278–2287. doi: 10.1523/JNEUROSCI.21-07-02278.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun GY, Horrocks LA, Farooqui AA. The roles of NADPH oxidase and phospholipases A2 in oxidative and inflammatory responses in neurodegenerative diseases. J. Neurochem. 2007;103:1–16. doi: 10.1111/j.1471-4159.2007.04670.x. [DOI] [PubMed] [Google Scholar]
- Tanskanen M, Paetau A, Salonen O, Salmi T, Lamminen A, Lindsberg P, Somer H, Kiuru-Enari S. Severe ataxia with neuropathy in hereditary gelsolin amyloidosis: A case report. Amyloid. 2007;14:89–95. doi: 10.1080/13506120601116393. [DOI] [PubMed] [Google Scholar]
- Tshala-Katumbay DD, Palmer VS, Kayton RJ, Sabri MI, Spencer PS. A new murine model of giant proximal axonopathy. Acta Neuropathol. (Berl.) 2005;109:405–410. doi: 10.1007/s00401-005-0982-z. [DOI] [PubMed] [Google Scholar]
- Zagoren JC, Politis MJ, Spencer PS. Rapid reorganization of the axonal cytoskeleton induced by a gamma diketone. Brain Res. 1983;270:162–164. doi: 10.1016/0006-8993(83)90807-7. [DOI] [PubMed] [Google Scholar]