

Abstract
Thioredoxin-2 (Trx2) is a multifunctional, mitochondria-specific protein, which inhibits cell death. The mitochondrial permeability transition (MPT) is a distinct mechanism for cell death activated by oxidants and linked to both necrotic and apoptotic morphologies. We studied mitochondria from Trx2 transgenic mice to determine whether Trx2 protects against oxidant-induced MPT. All experiments were performed in isolated mitochondria. Results showed that Trx2 protected against MPT induced by exogenously added peroxide. Unexpectedly, Trx2 also protected against the MPT induced by Ca2+ in the absence of added peroxide. The results indicate that in addition to protecting against oxidative stress, Trx2 is an endogenous regulator of the MPT.
Keywords: transgenic mice, cell death mechanisms, apoptosis, necrosis, calcium
Thioredoxins are small proteins that perform a variety of functions dependent upon binding interactions and oxidoreductase activity catalyzed by a characteristic dithiol motif (Arner and Holmgren, 2000). Mammalian thioredoxin-2 (Trx2) is a mitochondrial form originally identified in pig heart (Bode stein and Follmann, 1991) and cloned and characterized from a rat heart cDNA library (Spyrou et al., 1997). Although thioredoxin-1 has been studied extensively, less information is available on Trx2.
Overexpression of human Trx2 (hTrx2) showed inhibition of peroxide-induced cell death in 143B osteosarcoma cells (Chen et al., 2002) and interaction with the mitochondrial respiratory chain to increase the mitochondrial membrane potential and increase resistance to etoposide-induced cell death in HEK-293 cells (Damdimopoulos et al., 2002). Mitochondria contain two thioredoxin-dependent peroxidases, peroxiredoxin-3 and -5 (Kang et al., 1998; Seo et al., 2000), and these activities provide a mechanism for Trx2-dependent antioxidant activity. A central role for Trx2 in mitochondria was further demonstrated in Trx2 knockout mice (Nonn et al., 2003b). Trx2 deficiency was embryonic lethal at gestational day 10.5 and embryos showed massive apoptosis. The timing coincided with the maturation of mitochondrial function and the transition in the embryo from anaerobic to aerobic metabolism (Nonn et al., 2003b). A direct interaction of Trx2 with Prx3 was recently demonstrated in a study using a dominant-negative form of Trx2 (Zhang et al., 2007a).
A mechanism for Trx2 control of oxidant-induced apoptosis was revealed by studies showing that reduced Trx2 binds to and inhibits apoptosis signal-regulating kinase-1 (ASK-1) (Zhang et al., 2004). Upon Trx2 oxidation and release, ASK-1 activates caspase-mediated apoptosis. Other studies showed that increased Trx2 blocked tumor necrosis factor-α (TNF-α)–induced apoptosis in HeLa cells, a process mediated by mitochondrial reactive oxygen species (ROS) (Hansen et al., 2006). In these cells, TNF-α treatment oxidized Trx2, and overexpression of Trx2 eliminated the mitochondrial ROS signal and blocked apoptosis (Hansen et al., 2006). Relevance of the antiapoptotic function of Trx2 to disease was recently demonstrated by targeted overexpression of Trx2 in vascular endothelium of mice (Zhang et al., 2007b). Results showed that Trx2 protected against vascular pathology in the apoE2-knockout mouse model for cardiovascular disease (Zhang et al., 2007b). Thus, the accumulated data strongly support a role for Trx2 in protecting against oxidant-induced apoptosis.
Considerable evidence also shows that mitochondria-mediated cell death occurs following activation of the mitochondrial permeability transition (MPT). The MPT is a high amplitude swelling of mitochondria (Hunter et al., 1976) triggered by the opening of a pore which allows rapid influx of ions and associated water (Bernardi, 1999). The MPT gained early interest as a central mechanism of cell death because mitochondrial swelling is a hallmark of necrosis (Imberti et al., 1993). Considerable research has also supports a role for the MPT in apoptosis (Nagahara et al., 2000; Precht et al., 2005; Skulachev, 1996; Zhang et al., 2008). The MPT pore is activated by Ca2+ in the presence of diverse agents, including those that induce oxidative stress (Gunter and Pfeiffer, 1990). Oxidants disrupt cellular Ca2+ regulation, and increased cytoplasmic Ca2+ is an early event in oxidative stress preceding cell death (Orrenius et al., 2003).
In the present study, we developed transgenic (Tg) Trx2 mice to permit isolation of a sufficient quantity of high quality mitochondria to test whether Trx2 protected against activation of the MPT. The results from these studies show that isolated liver mitochondria from Trx2 Tg mice are protected against peroxide-induced MPT compared with wild-type (WT) littermate controls. Unexpectedly, an increased Ca2+ concentration was required to activate the MPT in the absence of added peroxide. The results demonstrate that in addition to protection against H2O2-induced mitochondrial damage and activation of apoptosis by ASK-1, Trx2 can inhibit the MPT, potentially providing a mechanism to protect against this form of mitochondria-mediated cell death.
MATERIALS AND METHODS
Trx2 Tg mouse.
Tg mice were generated and maintained at the Emory University Transgenic Mouse Core Facility under approval of the Emory Institution Animal Care and Use Committee. Expression of the hTrx2 transgene was achieved by using the Cre-LoxP system (Kuhn and Torres, 2002). hTrx2 cDNA (Chen et al., 2002) was first cloned downstream of a chloramphenicol acetyltransferase (CAT) gene and a stop codon under the control of a chicken β-actin gene (CAG) promoter, which consists of a CAG promoter and a cytomegalovirus enhancer. The construct was microinjected into fertilized eggs isolated from C57/BL6 mice and transplanted into CD-1 female pseudopregnant mice. Germ line transmission was confirmed by PCR analyses using DNA isolated from mouse tail sampling and primers: 5′-GCC AAT CAG CTT CTT CAG GAA GGC-3′ and 5′-CAC CAT GGC TCA GCG ACT TCT TCT-3′. These primers did not identify endogenous mouse Trx2. PCR for Cre was performed with primers: 5′-ACC TGA AGA TGT TCG CGA TTA TCT-3′ and 5′-ACC GTC AGT ACG TGA GAT ATC TT-3′. The F1 mice carrying the hTrx2 gene were crossed with Protamine-Cre mice to remove the CAT gene and the stop codon so that the hTrx2 protein can be expressed. To normalize the genetic background for proteomic and MPT assays, the Tg mice were bred successively with 129SVev WT mice (Taconic) and maintained in this genetic background; for each experiment, comparisons were made between littermates. Mice were maintained under standard 12-h dark–light cycle at 20°C. Purina Rodent Chow 5001 diet and water were given ad libitum.
Immunoprecipitation, Western blot analysis, and activity measurements.
Tissues were washed 3× with PBS and homogenized in RIPA lysis buffer containing 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 0.15M NaCl, 10mM sodium phosphate, pH 7.2, and protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). Homogenates were centrifuged and supernatants were stored at −80°C for immunoprecipitation and Western blot analysis. Protein concentration was measured via the Bradford method (BioRad Life Science, Hercules, CA), and equal amounts of protein were used for Western blot analysis following SDS-polyacrylamide gel electrophoresis (PAGE) on 15% gels and blotting to nitrocellulose membrane. Membranes were probed with rabbit anti-Trx2 (Hansen et al., 2006), rabbit anti-cytochrome c (Cell Signaling Technology, Boston, MA), and mouse anti-V5 (Invitrogen, Carlsbad, CA) antibodies. A goat anti-GAPDH antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was used for verification of equal protein loading. An IRDye 800 conjugated affinity purified anti-rabbit or mouse IgG (Rockland Immunochemicals, Gilbersville, PA) or Alexa-Fluor-680-conjugated anti-goat (Invitrogen) was used as secondary antibody. Redox-Western blot analysis of Trx2 redox potential was performed as described (Hansen et al., 2006). Bands were visualized using an Odyssey scanner (Li-Cor Biosciences, Lincoln, NE) and quantified using the Odyssey 1.1 software (Li-Cor Biosciences, Lincoln, NE).
For immunoprecipitation, tissue extracts (400 μg protein) were mixed with 1.5 μl of anti-V5 antibody for 4 h at 4°C and then rotated with 50 μl of protein G magnetic beads (Qiagen, Valencia, CA) for 3 h at 4°C. Protein G beads with protein complex were denatured by loading buffer at 95°C for 5 min and run on 15% SDS-PAGE. Membranes were probed with a rabbit anti-Trx2 antibody.
Trx2 and thioredoxin reductase-2 (TrxR2) were measured in disrupted mitochondria using the insulin reduction assay (Sasada et al., 1999; Yang et al., 2004).
Mitochondria isolation.
Mouse liver mitochondria were isolated following homogenization by differential centrifugation (Savage et al., 1991), and aliquots of the supernatant were kept for Western blot analysis as the cytosolic fraction. The pellets were either suspended in incubation buffer for MPT assay or RIPA buffer for Western blot analysis. For experiments to determine whether Trx2 was present in mitochondria from tissues other than liver, mitochondria were isolated from liver, heart, and skeletal muscle using the mitochondrial isolation kit (Sigma-Aldrich, St. Louis, MO) per the manufacturer's directions.
Submitochondrial localization of Trx2 by digitonin fractionation.
Isolated mitochondria were treated with digitonin (0–4 mg/10 mg protein) for 20 min on ice and centrifuged at 9500 × g at 4°C for 15 min. The pellet was suspended in 150 μl of RIPA buffer, and aliquots of supernatant and pellet were stored at −80°C for subsequent Western blot analysis. Adenylate kinase (AK) was used as a marker for the intermembrane space with the criterion that release of Trx2 in parallel with AK would indicate an intermembrane space location, whereas release of AK at concentrations lower than that required for Trx2 would indicate matrix location (Das, 1981; Kuylenstierna et al., 1970). Heat shock protein-60 (HSP60) was used as a matrix marker and was measured by Western blot (mouse monoclonal, Santa Cruz with Alexa-Fluor-680 donkey-anti-mouse secondary antibody, Invitrogen).
MPT assay.
Freshly isolated liver mitochondria from both WT and Trx2 Tg mice were suspended in incubation medium containing 250mM sucrose, 10mM (3-[N-morpholino]propanesulfonic acid]), 3mM K2PO4, and 5mM sodium succinate, pH 7.25. Mitochondria (300 μg protein) were suspended into incubation medium with varied concentrations of Ca2+ or tert-butylhydroperoxide (tBH) to give a total volume of 1 ml, and MPT was determined spectrophotometrically at 540 nm (Savage et al., 1991).
Ca2+ content of mitochondria.
Mitochondrial Ca2+ was measured by the method of Savage et al. (1991). Aliquots of mitochondria incubated as above in the absence and presence of cyclosporine A (0.5μM) were taken at 0, 2, 4, 6, 8, 10, 20, and 30 min after addition of Ca2+ and were centrifuged for 1 min at 13,000 × g. The supernatant was completely removed and the mitochondrial pellet was treated with 10% perchloric acid and centrifuged to remove protein. Following neutralization with 1M potassium carbonate and removal of the precipitated potassium perchlorate, 20 μl was added to 50μM Fura-2 (Sigma) in 0.1M Tris (pH 8.0). Fluorescence was measured with excitation at 340 and 380 nm and emission at 510 nm; the ratio was used to calculate Ca2+ concentration relative to a standard curve.
Statistics.
Data are expressed as mean ± SEM and analyzed by paired t-test, ANOVA with post hoc analysis by Student-Newman-Keuls test as appropriate.
RESULTS
Characteristics of Trx2 Tg Mice
To facilitate distinction between endogenous Trx2 and the transgene product, we incorporated a V5 epitope at the C-terminus of hTrx2 (Chen et al., 2002). hTrx2 differs from mouse Trx2 in two amino acids (human, mouse: I5V; M80I) (Damdimopoulos et al., 2002; Tanaka et al., 2002) in the mature mitochondrial form, and these differences are not known to affect catalytic or binding activities. Fractionation of liver tissue into cytosolic and mitochondrial fractions showed that Trx2 was present in the mitochondria but not in the cytoplasm (Fig. 1A). Mitochondrial fractions from heart and skeletal muscle also showed specific localization of Trx2 to the mitochondria and not cytoplasm (not shown). Activity measurements in isolated mitochondria with the insulin reduction assay showed that Trx2 activity was increased 63% in Tg mitochondria relative to WT littermate controls, whereas there was no significant change in TrxR2 activity (Fig. 1B). No detectable differences in redox potential of Trx2, as measured by the redox-Western blotting (Hansen et al., 2006), or mitochondrial membrane potential, as estimated by tetramethyl rhodamine methyl ester (Savage et al., 1991), were apparent. Cytochrome c content by Western blot analysis was not significantly different in liver extracts or isolated mitochondria from Tg and WT mice (data not shown).
FIG. 1.

Mitochondrial localization of the hTrx2 and increase in mitochondrial Trx2 activity. (A) To examine distribution of hTrx2-V5, liver homogenates (WCL: whole cell lysate) were separated into mitochondrial (Mito) and cytosolic (Cyt) fractions by differential centrifugation, resolved by SDS-PAGE and analyzed by Western blotting with antibodies to the V5 epitope to detect hTrx2-V5, Trx2 to detect both hTrx2-V5 and endogenous mouse Trx2 (mTrx2), and cytochrome c (Cyt c) as a marker for mitochondria. Data are representative of three separate experiments. Identical results were obtained for heart and skeletal muscle fractionation studies. (B) Mitochondrial Trx2 and TrxR2 activities were measured with the NADPH-insulin reduction assay for liver mitochondria with Tg values being expressed relative to a paired WT littermate. Data are from duplicate assays on three pairs of mitochondrial preparations for Trx2 and for duplicate assays on two pairs of mitochondrial preparations for TrxR2. *Significantly different from WT value, p < 0.05.
The location of Trx2 within the mitochondria was evaluated using digitonin fractionation of isolated liver mitochondria. Results showed that at low concentrations of digitonin, Trx2 was retained in the pellet, but the intermembrane space marker AK was released into the supernatant fraction (Fig. 2). At higher digitonin concentrations, Trx2 was released into the supernatant fraction (outer membrane and intermembrane space proteins) over the same digitonin concentration range which released the matrix protein, HSP60 (Fig. 2). These results show that Trx2 is principally, if not exclusively, present in the matrix space or complexed with inner membrane-associated proteins.
FIG. 2.

Submitochondrial compartmentation of Trx2. Experiments were performed in which increasing concentrations of digitonin were added to isolated mitochondria. Mitochondria were pelleted by centrifugation to allow detection of released proteins in the supernatant. Western blotting was used to determine whether Trx2 was present in the intermembrane space or in the matrix space. Trx2 was released (top and middle panels) only at high concentrations of digitonin, whereas the intermembrane space marker AK (bottom panel) was released at low concentration of digitonin. Data from a single experiment which is representative of data from three separate mitochondrial preparations.
Trx2 Protected against Ca2+-Induced MPT
Experiments were performed to determine whether increased Trx2 altered the MPT in mitochondria isolated from livers of Trx2 Tg mice compared with those from WT littermates. For each experiment, a sibling pair consisting of an hTrx2 Tg and a WT animal was selected, and mitochondria were prepared, diluted to equivalent protein concentrations (0.3 mg/ml) and analyzed in parallel. In the presence of 3mM inorganic phosphate and 80μM Ca2+, mitochondria from both Tg and WT animals showed characteristic MPT responses and both were sensitive to cyclosporine A (CsA; Fig. 3A). The maximal ΔA540 was not significantly different for liver mitochondria from Tg and WT mice when exposed to this Ca2+ overload condition (0.35 ± 0.03 and 0.34 ± 0.02; p = 0.71, n = 4). This result indicated that there was no substantial difference in the volume or extent of swelling of the mitochondria due to the transgene. To determine whether there was a difference in Ca2+ loading capacity, Tg and WT littermate control mitochondria were incubated as in Figure 3A and assayed for mitochondrial Ca2+ content. In the absence of CsA, maximal Ca2+ loading occurred at 4–6 min and was not significantly different between Tg and WT mitochondria (131 ± 7 and 139 ± 15 nmol/mg protein, respectively). In the presence of CsA, the extent of Ca2+ loading was similar to that without CsA and was not significantly different between Tg and WT mitochondria (140 ± 3 and 139 ± 4 nmol/mg protein, respectively). Previous research showed that overexpression of Trx2 in cells increased mitochondrial membrane potential (Damdimopoulos et al., 2002), but we did not find any apparent difference between Tg and WT mitochondria. Although these data do not provide information on rates of Ca2+ uptake, they suggest that there is a similar membrane potential driving force and a similar Ca2+ loading capacity.
FIG. 3.
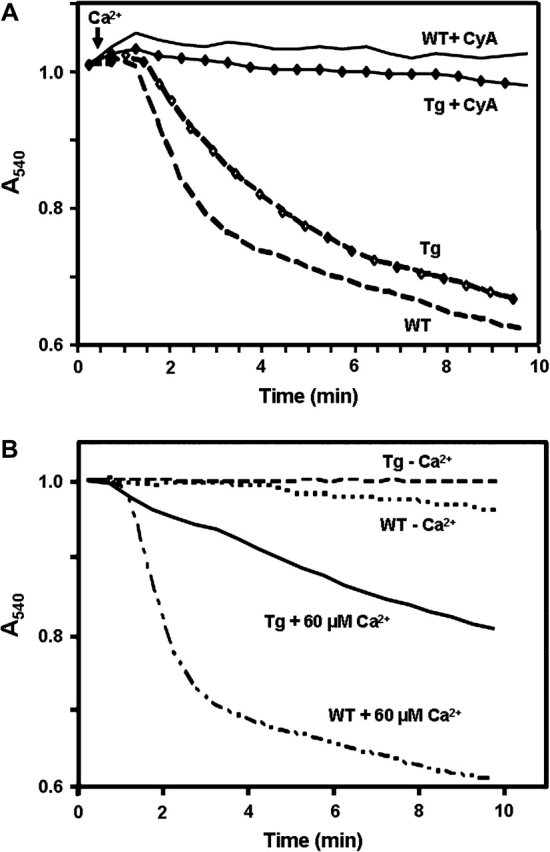
The MPT in liver mitochondria from Trx2 Tg mice. (A) The MPT in Tg mitochondria was similar to that observed in WT littermates and was inhibited by cyclosporine A (CsA). Mitochondria were suspended in media containing 3mM inorganic phosphate, 5mM sodium succinate and 80μM Ca2+ at 20° and activation of the MPT was monitored at 540 nm. Data are representative of results from four pairs of liver mitochondria and two pairs of heart mitochondria. (B) Mitochondria from Trx2 mice were resistant to Ca2+-dependent activation of the MPT. Although the MPT was similar in mitochondria from Tg and WT littermates when 80μM Ca2+ was present, at 60μM and lower concentrations, the MPT was decreased. Conditions for B were identical to A except that Ca2+ was lower, and this resulted in delayed time course for MPT. Data are representative of experiments from four littermate pairs of Tg and WT mice.
The oxidant-induced MPT is dependent upon Ca2+, so initial experiments were designed to obtain an appropriate Ca2+ concentration which would be sufficient to quantify the oxidant sensitivity but did not rapidly induce the MPT alone. Concentration-dependence studies showed that at Ca2+ concentrations of 20μM or lower, the MPT was not observable within 15 min in either Tg or WT mitochondria. However, at higher concentrations, the MPT was activated in both WT and Tg mitochondria, with the Tg mitochondria showing a resistance to Ca2+ (Fig. 3B). To obtain quantitative comparisons, we measured the time required to obtain 20% of the maximal MPT response. With 60μM Ca2+ respective times for WT and Tg mitochondria were 1.8 ± 0.3 min for WT and 4.2 ± 0.3 min for Tg. Comparable results were obtained with mitochondrial preparations from hearts of Tg and WT mice (data not shown). Thus, the results show that the hTrx2 significantly delayed activation of the MPT in response to Ca2+.
Trx2 Protected against Oxidant-Induced MPT
To determine whether Trx2 protected against oxidant-induced MPT, 20μM Ca2+ was selected to allow sensitive detection of the effects of increasing concentrations of tBH on the MPT. Pilot studies were done with a range of Ca2+ concentrations and provided results which qualitatively agreed with the data described below. At Ca2+ concentrations below 20μM, less activation occurred and less stimulation was seen with tBH. At higher concentrations, greater activation occurred and this was stimulated by peroxide. Consequently, there was a relatively narrow range of conditions which were useful.
For each respective Tg and WT pair, the time courses differed with the characteristic that Tg mitochondria had a slower time course of response at all tBH concentrations. For example, with 20μM Ca2+ and 3mM inorganic phosphate, 10% of the maximal MPT absorbance change occurred at 16.5 ± 1.6 min in WT and 18.9 ± 1.1 min in Tg mitochondria. The addition of 12.5μM tBH decreased the time to induce a 10% change to 14.0 ± 1.3 min in WT and 18.1 ± 2.0 min in Tg mitochondria. At 25μM, 10% effect occurred at 12.8 ± 2.3 min in WT and 18.0 ± 2.6 min in Tg mitochondria. At 50μM, 10% effect occurred at 8.5 ± 1.5 min in WT and 11.9 ± 0.6 min in Tg mitochondria.
To graphically illustrate the difference in sensitivity, the percentage of maximal MPT was determined at 20 min (Fig. 4). For this purpose, the maximal MPT was taken as the absorbance change occurring with 80μM Ca2+, a concentration above which no further change is observed Liver mitochondria from Tg and WT mice were assayed in the presence of 20μM Ca2+ and 3mM inorganic phosphate, with additions of 12.5, 25, or 50μM tBH. Results showed that data for respective Tg and WT pairs were significantly different at all tBH concentrations. Consequently, the data show that hTrx2 expression increased the resistance of mitochondria to oxidant-induced MPT. The decreased activation and apparent lack of dose response for the tBH treatment of hTrx2 mitochondria were consistent with two effects, a resistance of the MPT to activation by Ca2+ and a resistance of the Ca2+-dependent MPT to tBH.
FIG. 4.

Trx2 protection against oxidant-induced MPT. To determine whether Trx2 protected against oxidant-induced toxicity, liver mitochondria from Tg and WT mice were assayed for the MPT in the presence of 20μM Ca2+ and 3mM inorganic phosphate, with additions of 0, 12.5, 25, or 50μM tBH. Data are presented as the percentage of the maximal MPT response obtained under the conditions indicated at 20 min. Results are means ± SEM from experiments on four pairs of Tg and WT littermates. Data for respective Tg and WT pairs were significantly different at all tBH concentrations. Data were analyzed by ANOVA with post hoc analysis by Student-Newman-Keuls test. *Significant at p < 0.05.
DISCUSSION
In recent years, a major shift in oxidative stress research has occurred with the recognition that H2O2 and redox-sensitive cysteines of proteins function in cell signaling and control. This has resulted in a redefinition of oxidative stress to include disruption of redox signaling and control along with macromolecular damage as toxicologic consequences (Jones, 2006). The implications of this redefinition for toxicologic mechanisms involving mitochondria are important because of the integration of redox signaling and control with other central mechanisms, including H2O2 signaling, Ca2+ signaling and kinase signaling, as activators of cell death. In this context, the finding that oxidation of Trx2 can result in activation of Ask-1 as an apoptosis mechanism (Zhang et al., 2004) raised the question whether earlier results implicating the MPT were misinterpreted. The current results show explicitly that the content of Trx2 in mitochondria protects against activation of the MPT. Although this does not establish involvement of the MPT in any specific cell toxicity model, it shows that in addition to inhibition of Ask-1, Trx2 also protects against the MPT. Thus, the results show that one cannot simply discount previous findings of oxidative effects on the MPT due to the discovery of the Ask-1 mechanism. Whether the function of Trx2 in blocking two different cell death pathways represents a coordinated control of death mechanisms or alternative mechanisms that are differentially expressed in cells is not known; however, the results highlight the need to discriminate between MPT- and Ask-1–mediated mechanisms in cell toxicity research.
Accumulating data show that Trx2 can protect against toxicity by multiple mechanisms. Reduced Trx2 binds to ASK-1 and inhibits its activity, thereby protecting against apoptosis (Zhang et al., 2004). Trx2 interacts with Prx3 (Zhang et al., 2007a) suggesting that Trx2 supports peroxide reduction in mitochondria. In support of this conclusion, overexpression of Trx2 blocked TNF-α–induced ROS production (Zhang et al., 2007a). Overexpression of Prx5, another thioredoxin-dependent peroxidase found in mitochondria, also protected against peroxide-induced mitochondrial DNA damage (Banmeyer et al., 2004). Thus, the present data add to the accumulating evidence that Trx2 provides central protection against mitochondrial mechanisms of toxicity.
During recent years, it has become increasingly apparent that multiple mechanisms exist for mitochondria-mediated cell death (Dietze et al., 2001; Nagahara et al., 2000; Precht et al., 2005). These include different pathways for release of cytochrome c and activation of caspases, as well as activation of mitochondrial ASK-1 (Dietze et al., 2001; Lim et al., 2008; Zhang et al., 2008). A number of studies implicate the MPT in activation of cell death by apoptosis and necrosis (Lim et al., 2008; Precht et al., 2005; Tsujimoto et al., 2006), and Trx2 has been shown to protect against cell death in cell culture experiments (Chen et al., 2002; Hansen et al., 2006). In vivo, knockout mice are embryonic lethal, showing massive apoptosis at a time of mitochondrial maturation (Nonn et al., 2003a). In addition, conditionally deficient chicken DT40 B-cells were found to undergo apoptosis upon repression of the transgene (Tanaka et al., 2002). Similar studies are needed to determine whether mitochondria in cells from the Tg mice are protected against oxidant-induced MPT and whether mice with conditional repression of Trx2 have increased susceptibility. Available evidence shows that overexpression of Trx2 protects against oxidant-, etoposide-, and TNF-α–induced cell death (Chen et al., 2002; Damdimopoulos et al., 2002; Hansen et al., 2006). Whether any of these mechanisms are specifically linked to prevention of the MPT is not known, although HeLa cells overexpressing Trx2 are protected against loss of the mitochondrial membrane potential dye, JC-1 (J. M. Hansen, unpublished observation).
The in vitro swelling assay is a highly artificial system to study the MPT but it is a standardized protocol for measuring MPT in isolated mitochondria. However, cellular studies on Trx2 protection against oxidant-induced cell death do not show whether Trx2 has an effect on the MPT as opposed to activation of the Ask-1 pathway. Thus, even though the in vitro assay is only a model for the physiologic function, it provides direct evidence that Trx2 affects the MPT without contribution from non-mitochondrial factors. Many factors induce the MPT, including oxidative stress (Bernardi, 1999). The MPT can be facilitated by thiol oxidants such as diamide and phenylarsine oxide and inhibited by antioxidants, such as dithiothreitol (Fagian et al., 1990; Lenartowicz et al., 1991). Due to the reversibility and oxidant/reductant sensitivity, proposed mechanisms include redox-sensitive cysteine residue regulation (Halestrap and Brennerb, 2003). Two distinct mechanisms are implicated in thiol regulation of the MPT (Chernyak and Bernardi, 1996; Costantini et al., 1996, 2000). One is sensitive to oxidants and the other to the redox state of matrix NADP+ (nicotinamide adenine dinucleotide phosphate) on the MPT pore. However, many mitochondrial proteins contain redox-sensitive thiols, and the present data do not provide any clarification concerning which of these could be responsible for the redox sensitivity of the MPT. Redox proteomic methods (Fratelli et al., 2002; Leichert et al., in press) provide important new opportunities to identify such mechanistic links.
Furthermore, whether Trx2 directly interacts with components of the pore or only indirectly regulates the pore remains uncertain. Trx2 overexpression in 143B osteosarcoma cells did not increase the rate of exogenous peroxide elimination (Chen et al., 2002), but these measurements may not reflect the amount of ROS elimination within the mitochondria. Other studies show increased ROS in cells with decreased Trx2 (Tanaka et al., 2002) and decreased ROS in TNF-α-treated cells overexpressing Trx2 (Hansen et al., 2006). Thus, Trx2 could potentially regulate the MPT either directly by interacting with MPT pore components or indirectly by altering the mitochondrial concentrations of oxidants which affect the function of the MPT pore complex.
In summary, Trx2 is present in the matrix of mitochondria and protects mitochondria from calcium- and oxidant-induced MPT. This supports the interpretation that Trx2 can protect against cell death by mechanisms mediated by the MPT. Further study is required to identify the protein targets and outline specifics of this potential mechanism.
FUNDING
National Institutes of Health (ES009047) to D. J., and (ES014668) to J. C.
References
- Arner ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000;267:6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- Banmeyer I, Marchand C, Verhaeghe C, Vucic B, Rees JF, Knoops B. Overexpression of human peroxiredoxin 5 in subcellular compartments of Chinese hamster ovary cells: Effects on cytotoxicity and DNA damage caused by peroxides. Free Radic. Biol. Med. 2004;36:65–77. doi: 10.1016/j.freeradbiomed.2003.10.019. [DOI] [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Bode stein J, Follmann H. Characterization of two thioredoxins in pig heart including a new mitochondrial protein. Z. Naturforsch. C. 1991;46:270–279. doi: 10.1515/znc-1991-3-418. [DOI] [PubMed] [Google Scholar]
- Chen Y, Cai J, Murphy TJ, Jones DP. Overexpressed human mitochondrial thioredoxin confers resistance to oxidant-induced apoptosis in human osteosarcoma cells. J. Biol. Chem. 2002;277:33242–33248. doi: 10.1074/jbc.M202026200. [DOI] [PubMed] [Google Scholar]
- Chernyak BV, Bernardi P. The mitochondrial permeability transition pore is modulated by oxidative agents through both pyridine nucleotides and glutathione at two separate sites. Eur. J. Biochem. 1996;238:623–630. doi: 10.1111/j.1432-1033.1996.0623w.x. [DOI] [PubMed] [Google Scholar]
- Costantini P, Belzacq AS, Vieira HL, Larochette N, de Pablo MA, Zamzami N, Susin SA, Brenner C, Kroemer G. Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl-2-independent permeability transition pore opening and apoptosis. Oncogene. 2000;19:307–314. doi: 10.1038/sj.onc.1203299. [DOI] [PubMed] [Google Scholar]
- Costantini P, Chernyak BV, Petronilli V, Bernardi P. Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J. Biol. Chem. 1996;271:6746–6751. doi: 10.1074/jbc.271.12.6746. [DOI] [PubMed] [Google Scholar]
- Damdimopoulos AE, Miranda-Vizuete A, Pelto-Huikko M, Gustafsson JA, Spyrou G. Human mitochondrial thioredoxin. Involvement in mitochondrial membrane potential and cell death. J. Biol. Chem. 2002;277:33249–33257. doi: 10.1074/jbc.M203036200. [DOI] [PubMed] [Google Scholar]
- Das SK. The submitochondrial localization adenylate kinase: An enzymatic marker for the inner surface of the outer membrane of lung mitochondria in guinea pig. Biochem. Biophys. Res. Commun. 1981;103:1145–1148. doi: 10.1016/0006-291x(81)90242-4. [DOI] [PubMed] [Google Scholar]
- Dietze EC, Caldwell LE, Grupin SL, Mancini M, Seewaldt VL. Tamoxifen but not 4-hydroxytamoxifen initiates apoptosis in p53(-) normal human mammary epithelial cells by inducing mitochondrial depolarization. J. Biol. Chem. 2001;276:5384–5394. doi: 10.1074/jbc.M007915200. [DOI] [PubMed] [Google Scholar]
- Fagian MM, Pereira-da-Silva L, Martins IS, Vercesi AE. Membrane protein thiol cross-linking associated with the permeabilization of the inner mitochondrial membrane by Ca2+ plus prooxidants. J. Biol. Chem. 1990;265:19955–19960. [PubMed] [Google Scholar]
- Fratelli M, Demol H, Puype M, Casagrande S, Eberini I, Salmona M, Mengozzi M, Duffieux F, Miclet E, Bachi A, et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 2002;99:3505–3510. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Brennerb C. The adenine nucleotide translocase: A central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 2003;10:1507–1525. doi: 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- Hansen JM, Zhang H, Jones DP. Mitochondrial thioredoxin-2 has a key role in determining tumor necrosis factor-alpha-induced reactive oxygen species generation, NF-kappaB activation, and apoptosis. Toxicol. Sci. 2006;91:643–650. doi: 10.1093/toxsci/kfj175. [DOI] [PubMed] [Google Scholar]
- Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J. Biol. Chem. 1976;251:5069–5077. [PubMed] [Google Scholar]
- Imberti R, Nieminen AL, Herman B, Lemasters JJ. Mitochondrial and glycolytic dysfunction in lethal injury to hepatocytes by t-butylhydroperoxide: Protection by fructose, cyclosporin A and trifluoperazine. J. Pharmacol. Exp. Ther. 1993;265:392–400. [PubMed] [Google Scholar]
- Jones DP. Redefining oxidative stress. Antioxid. Redox Signal. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- Kang SW, Chae HZ, Seo MS, Kim K, Baines IC, Rhee SG. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J. Biol. Chem. 1998;273:6297–6302. doi: 10.1074/jbc.273.11.6297. [DOI] [PubMed] [Google Scholar]
- Kuhn R, Torres RM. Cre/loxP recombination system and gene targeting. Methods Mol. Biol. 2002;180:175–204. doi: 10.1385/1-59259-178-7:175. [DOI] [PubMed] [Google Scholar]
- Kuylenstierna B, Nicholls DG, Hovmoller S, Ernster L. Effect of trypsin on mitochondrial and microsomal enzymes. Eur. J. Biochem. 1970;12:419–426. doi: 10.1111/j.1432-1033.1970.tb00868.x. [DOI] [PubMed] [Google Scholar]
- Leichert LI, Gehrke F, Gudiseva HV, Blackwell T, Ilbert M, Walker AK, Strahler JR, Andrews PC, Jakob U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. U. S. A. 2008;105(24):8197–8202. doi: 10.1073/pnas.0707723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenartowicz E, Bernardi P, Azzone GF. Phenylarsine oxide induces the cyclosporin A-sensitive membrane permeability transition in rat liver mitochondria. J. Bioenerg. Biomembr. 1991;23:679–688. doi: 10.1007/BF00785817. [DOI] [PubMed] [Google Scholar]
- Lim PL, Liu J, Go ML, Boelsterli UA. The mitochondrial superoxide/thioredoxin-2/Ask1 signaling pathway is critically involved in troglitazone-induced cell injury to human hepatocytes. Toxicol. Sci. 2008;101:341–349. doi: 10.1093/toxsci/kfm273. [DOI] [PubMed] [Google Scholar]
- Nagahara Y, Ikekita M, Shinomiya T. Immunosuppressant FTY720 induces apoptosis by direct induction of permeability transition and release of cytochrome c from mitochondria. J. Immunol. 2000;165:3250–3259. doi: 10.4049/jimmunol.165.6.3250. [DOI] [PubMed] [Google Scholar]
- Nonn L, Berggren M, Powis G. Increased expression of mitochondrial peroxiredoxin-3 (thioredoxin peroxidase-2) protects cancer cells against hypoxia and drug-induced hydrogen peroxide-dependent apoptosis. Mol. Cancer Res. 2003a;1:682–689. [PubMed] [Google Scholar]
- Nonn L, Williams RR, Erickson RP, Powis G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol. Cell. Biol. 2003b;23:916–922. doi: 10.1128/MCB.23.3.916-922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: The calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Precht TA, Phelps RA, Linseman DA, Butts BD, Le SS, Laessig TA, Bouchard RJ, Heidenreich A. The permeability transition pore triggers Bax translocation to mitochondria during neuronal apoptosis. Cell Death Differ. 2005;12:255–265. doi: 10.1038/sj.cdd.4401552. [DOI] [PubMed] [Google Scholar]
- Sasada T, Nakamura H, Ueda S, Sato N, Kitaoka Y, Gon Y, Takabayashi A, Spyrou G, Holmgren A, Yodoi J. Possible involvement of thioredoxin reductase as well as thioredoxin in cellular sensitivity to cis-diamminedichloroplatinum (II) Free Radic. Biol. Med. 1999;27:504–514. doi: 10.1016/s0891-5849(99)00101-x. [DOI] [PubMed] [Google Scholar]
- Savage MK, Jones DP, Reed DJ. Calcium- and phosphate-dependent release and loading of glutathione by liver mitochondria. Arch. Biochem. Biophys. 1991;290:51–56. doi: 10.1016/0003-9861(91)90590-f. [DOI] [PubMed] [Google Scholar]
- Seo MS, Kang SW, Kim K, Baines IC, Lee TH, Rhee SG. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 2000;275:20346–20354. doi: 10.1074/jbc.M001943200. [DOI] [PubMed] [Google Scholar]
- Skulachev VP. Why are mitochondria involved in apoptosis? Permeability transition pores and apoptosis as selective mechanisms to eliminate superoxide-producing mitochondria and cell. FEBS Lett. 1996;397:7–10. doi: 10.1016/0014-5793(96)00989-1. [DOI] [PubMed] [Google Scholar]
- Spyrou G, Enmark E, Miranda-Vizuete A, Gustafsson J. Cloning and expression of a novel mammalian thioredoxin. J. Biol. Chem. 1997;272:2936–2941. doi: 10.1074/jbc.272.5.2936. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Hosoi F, Yamaguchi-Iwai Y, Nakamura H, Masutani H, Ueda S, Nishiyama A, Takeda S, Wada H, Spyrou G, et al. Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J. 2002;21:1695–1703. doi: 10.1093/emboj/21.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y, Nakagawa T, Shimizu S. Mitochondrial membrane permeability transition and cell death. Biochim. Biophys. Acta. 2006;1757:1297–1300. doi: 10.1016/j.bbabio.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Yang X, Wu X, Choi YE, Kern JC, Kehrer JP. Effect of acrolein and glutathione depleting agents on thioredoxin. Toxicology. 2004;204:209–218. doi: 10.1016/j.tox.2004.06.056. [DOI] [PubMed] [Google Scholar]
- Zhang D, Lu C, Whiteman M, Chance B, Armstrong JS. The mitochondrial permeability transition regulates cytochrome c release for apoptosis during endoplasmic reticulum stress by remodeling the cristae junction. J. Biol. Chem. 2008;283:3476–3486. doi: 10.1074/jbc.M707528200. [DOI] [PubMed] [Google Scholar]
- Zhang H, Go YM, Jones DP. Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in parallel with mitochondrial GSH system in protection against oxidative stress. Arch. Biochem. Biophys. 2007a;465:119–126. doi: 10.1016/j.abb.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Zhang H, Luo Y, Zhang W, He Y, Dai S, Zhang R, Huang Y, Bernatchez P, Giordano FJ, Shadel G, et al. Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. Am. J. Pathol. 2007b;170:1108–1120. doi: 10.2353/ajpath.2007.060960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Al-Lamki R, Bai L, Streb JW, Miano JM, Bradley J, Min W. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ. Res. 2004;94:1483–1491. doi: 10.1161/01.RES.0000130525.37646.a7. [DOI] [PubMed] [Google Scholar]