

Abstract
For the past several years, it has been demonstrated that the NAD-dependent protein deacetylase Sirt1 and nicotinamide phosphoribosyltransferase (Nampt)-mediated systemic NAD biosynthesis together play a critical role in the regulation of metabolism and possibly aging in mammals. Based on our recent studies on these two critical components, we have developed a hypothesis of a novel systemic regulatory network, named “NAD World”, for mammalian aging. Conceptually, in the NAD World, systemic NAD biosynthesis mediated by intra- and extracellular Nampt functions as a driver that keeps up the pace of metabolism in multiple tissues/organs, and the NAD-dependent deacetylase Sirt1 serves as a universal mediator that executes metabolic effects in a tissue-dependent manner in response to changes in systemic NAD biosynthesis. This new concept of the NAD World provides important insights into a systemic regulatory mechanism that fundamentally connects metabolism and aging and also conveys the ideas of functional hierarchy and frailty for the regulation of metabolic robustness and aging in mammals.
Index entries/key words: NAD World, metabolism, aging, Sirt1, Nampt, systemic NAD biosynthesis, pancreatic β cells, neurons, robustness, frailty
Introduction
For the past couple of decades, aging research has rapidly gained momentum, and studies using experimental model organisms, such as yeast, worms, and flies, have identified a number of evolutionarily conserved regulators and signaling pathways for the control of aging and longevity. One such pathway is insulin/insulin-like growth factor-I (IGF-I) signaling (IIS) (1-4). The importance of IIS pathways for the regulation of aging and longevity has been established in worms and flies (3, 4). Although the significance of IIS pathways still remains to be determined in mammals, the growth hormone (GH)/IGF-I axis has drawn much attention in the field of aging research mainly because naturally occurring or genetically engineered mutant mice that have defects in the production of GH or in GH/IGF-I signaling exhibit delayed aging and significant life span extension (1, 2). Another critical regulator for aging and longevity is the Sir2 (silent information regulator 2) protein family of NAD-dependent protein deacetylases/ADP-ribosyltransferases, called “sirtuins” (5, 6). In 2000, Leonard Guarente and I demonstrated that yeast and mammalian Sir2 proteins have NAD-dependent deacetylase activity and that this activity is essential for the longevity control in yeast (7). Since this first discovery, it has been demonstrated that Sir2 orthologs play an important role in the regulation of aging and longevity in worms and flies (8, 9), and sirtuin biology has been exploding in many different research areas from bacteria to humans (6, 10-12).
A major focus in the current field of aging research is to examine whether those signaling pathways and regulators, as well as newly identified ones, also regulate mammalian aging and longevity. In mammals, however, multiple organs and tissues communicate through a variety of hormones and metabolites in a more complex manner, and therefore, untangling these complex interplays between tissues/organs and drawing a blueprint of a systemic regulatory network for mammalian aging still remains as a big challenge. To achieve this ultimate goal and understand the regulatory mechanism of mammalian aging at a systemic level, there are several fundamental questions that need to be addressed: 1) In the presumed hierarchical structure of the systemic aging-regulatory network, which tissues/organs play a dominant role in the regulation of aging and longevity in mammals? 2) If there are such “control centers of aging,” it is highly likely that they use certain hormones or humoral factors to communicate with other tissues/organs and govern the pace of aging through the body. If so, what are those “aging-regulatory” hormones or humoral factors? 3) Are there any universal molecular regulators for the production and/or the secretion of the presumed “aging-regulatory” hormones or humoral factors?
There is still a long way to go for clear answers to these fundamental questions. Nevertheless, our recent studies on the roles of the mammalian NAD-dependent deacetylase Sirt1 and mammalian NAD biosynthetic pathways in the regulation of metabolism and aging have provided critical insights to develop a hypothesis of a novel systemic regulatory network, named “NAD World”, for mammalian aging (13). Conceptually, the NAD World is comprised of two critical components: nicotinamide phosphoribosyltransferase (Nampt)-mediated systemic NAD biosynthesis as a driver that keeps up the pace of metabolism, and the NAD-dependent deacetylase Sirt1 as a mediator that executes regulatory effects in various tissues in response to changes in systemic NAD biosynthesis. In this review, I will first summarize the physiological significance of Sirt1 function and Nampt-mediated systemic NAD biosynthesis in the regulation of metabolism and aging and then introduce the concept of the “NAD World.”
Sirt1, a key mediator that coordinates metabolic responses to nutritional availability in various tissues
Sir2 family proteins, called “sirtuins,” have been implicated in the regulation of aging and longevity in experimental model organisms, such as yeast, worms, and flies (5, 6). In those organisms, the dosage or activity of Sir2 proteins determines the length of their life spans, and in certain genetic backgrounds, Sir2 proteins also play an important role in the life span-extending effect of caloric restriction (CR), a dietary regimen that has been demonstrated to retard aging and extend life span in a wide variety of organisms (14). Mammals have seven sirtuin members, named Sirt1 through Sirt7, and Sirt1 is the mammalian ortholog of the founder protein Sir2 in budding yeast (6, 15, 16). Although it is still unclear whether Sirt1 regulates aging and longevity in mammals, increasing lines of evidence have firmly established that Sirt1 plays critical roles in the regulation of metabolism in response to changes in nutritional availability in multiple tissues (Figure 1).
Figure 1.
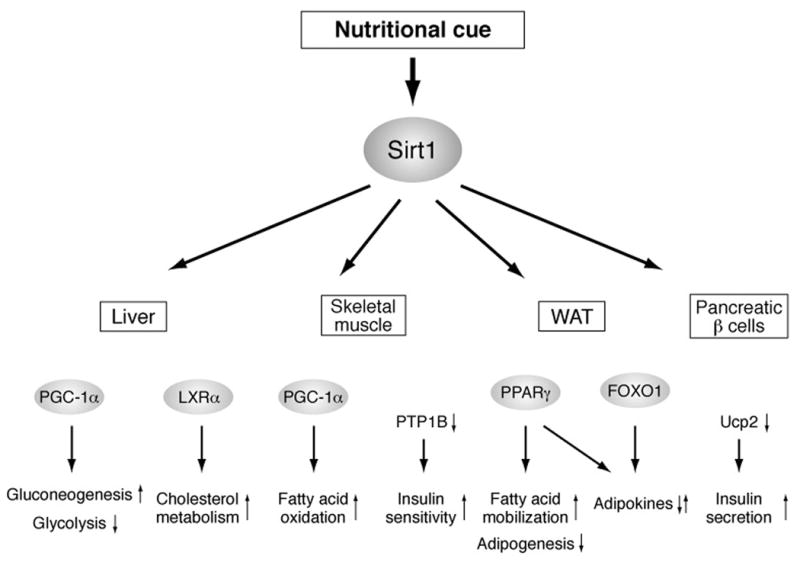
Mammalian Sirt1 as a key mediator that coordinates metabolic responses to nutritional availability in various tissues and organs. Sirt1 regulates the activity of key metabolic transcription factors, such as PGC-1α, LXRα, PPARγ, and FOXO1, and the expression of important effecter genes, such as PTP1B and Ucp2, and mediates diverse metabolic responses in organ/tissue-dependent manners.
In the liver, Sirt1 regulates the gluconeogenic and glycolytic pathways in response to fasting by interacting with and deacetylating PGC-1α, a key transcriptional regulator of glucose production, in an NAD-dependent manner (17, 18). Sirt1 also regulates cholesterol metabolism by deacetylating and activating LXRα, a critical nuclear receptor that controls cholesterol and lipid homeostasis (19). In skeletal muscle, Sirt1-mediated PGC-1α deacetylation is required to induce mitochondrial fatty acid oxidation genes in response to nutrient deprivation (20). Additionally, in skeletal myotube cells, it has been reported that Sirt1 contributes to the improvement of insulin sensitivity through the transcriptional repression of the protein tyrosine phosphatase 1B (PTP1B) gene (21). In white adipose tissue, Sirt1 triggers lipolysis and promotes free fatty acid mobilization in response to fasting by repressing PPARγ, a nuclear receptor that promotes adipogenesis (22). Furthermore, Sirt1 appears to regulate the production and/or the secretion of adipokines, such as adiponectin, through the regulation of FOXO1 and PPARγ (23, 24). In pancreatic β cells, we and others have demonstrated that Sirt1 promotes glucose-stimulated insulin secretion in part by repressing the expression of uncoupling protein 2 (Ucp2), a mitochondrial proton transporter that uncouples respiration from ATP production, and increasing cellular ATP levels (25, 26). Indeed, pancreatic β cell-specific Sirt1-overexpressing (BESTO) transgenic mice show enhanced insulin secretion and improved glucose tolerance in response to glucose challenge (26). These findings clearly suggest that Sirt1 mediates multiple physiological responses to changes in nutritional input in various tissues and integrates them into a coordinated metabolic response at a systemic level.
Most recently, two groups have reported that Sirt1 regulates the circadian clock oscillatory mechanism and affects the expression of circadian clock genes (27, 28). Sirt1 forms a complex with Clock and Bmal1, key regulators of the circadian clock that heterodimerize and activate transcription of target genes, including the Period (Per1, 2, and 3) and Cryptochrome (Cry1 and 2) genes, through E-box cis-regulatory elements (29). Although there are some discrepancies between results from these groups, Sirt1 deacetylates Bmal1 or Per2 proteins and regulates the amplitude and the duration of circadian gene expression. Because the core molecular clock machinery has been demonstrated to be one of the most powerful modifiers of metabolism (30), it is likely that Sirt1 might provide another layer of metabolic regulation through the epigenetic control of the core circadian clock oscillatory mechanism.
The findings that Sirt1 mediates a variety of metabolic effects in response to nutritional cues, particularly to low nutritional input, and coordinates the production and the secretion of key metabolic hormones, such as insulin and adiponectin, and essential metabolites, such as glucose, cholesterol, and fatty acids, place this protein at a central position as the universal molecular regulator that likely plays a critical role in the systemic regulation of aging and longevity in mammals. This notion has been confirmed, at least in part, by the studies on the significance of Sirt1 in the induction of physiological responses to CR. For example, Sirt1-deficient mice do not exhibit the CR-induced increase in physical activity (31). It has also been reported that Sirt1-deficient mice are metabolically inefficient and unable to adapt to CR normally (32). On the other hand, the whole-body Sirt1 transgenic mice display phenotypes that mimic some of the physiological changes in response to CR, including decreased blood insulin and glucose levels, improved glucose tolerance, reduced fat mass and circulating free fatty acid and leptin levels, reduced total blood cholesterol levels, enhanced oxygen consumption, and improved activity in rotarod assays (33). These findings provide strong support for the function of Sirt1 as the universal mediator that may integrate a variety of metabolic responses to the systemic regulation of aging and longevity in mammals.
An interesting question is how Sirt1 as a metabolic regulator could be evolved for the regulation of aging and longevity. One possible explanation is that Sir2 orthologs including Sirt1 might have been selected through evolution to have individuals survive through famine or other nutritionally scarce conditions until the environment changes to a more favorable one for reproduction (34). Because Sir2 orthologs translate the availability of NAD, essentially the currency of energy metabolism, to transcriptional regulation of genes important for metabolic adaptation, Sirt1 might have been placed at a center of this survival-assurance metabolic program through the process of natural selection. Therefore, through the evolution of this particular metabolic program that assures longer survival under nutritionally scarce conditions, Sirt1 might have been evolved to be the universal molecular regulator that is capable of delaying aging and extending life span as the consequence of assuring longer survival in mammals. Comparative studies on Sirt1 in diverse mammalian species might provide important clues to the evolutional aspect of Sirt1 function as the universal molecular regulator that connects metabolism and aging in mammals.
Systemic NAD biosynthesis mediated by intra- and extracellular nicotinamide phosphoribosyltransferase (Nampt)
While Sirt1 executes its metabolic effects by interacting with specific transcriptional regulators in a tissue-dependent manner, NAD biosynthesis appears to provide a more global regulatory mechanism that fine-tunes Sirt1 enzymatic activity and coordinates its effects at a systemic level. Particularly, Nampt plays a central role in the systemic regulation of NAD biosynthesis in mammals. In this section, I will summarize recent findings on this unique enzyme and discuss the importance of Nampt-mediated systemic NAD biosynthesis in mammals.
NAD is synthesized from three major precursors – tryptophan, nicotinic acid, and nicotinamide (35-37). The latter two are different forms of vitamin B3. In lower eukaryotes and invertebrates, such as yeast, worms, and flies, nicotinic acid is used as a major NAD precursor (Figure 2A) (36, 37). In these organisms, nicotinamide, which is produced from the breakdown of NAD, undergoes deamidation to nicotinic acid, a reaction catalyzed by a nicotinamidase encoded by the Pnc1 gene (Figure 2A) (38, 39). While the pathways starting from tryptophan and nicotinic acid are highly conserved in mammals, these animals predominantly use nicotinamide as a precursor for NAD biosynthesis (35-37). Indeed, the administration of radiolabeled nicotinamide and nicotinic acid to mice has clearly shown that nicotinamide is a better precursor of NAD than nicotinic acid and that nicotinic acid is rapidly cleared by being converted to nicotinamide and excreted as nicotinuric acid (40).
Figure 2.

Major NAD biosynthetic pathways in lower eukaryotes and invertebrates vs. mammals and the enzymatic reaction of nicotinamide phosphoribosyltransferase (Nampt). (A) The NAD biosynthetic pathway in the budding yeast Saccharomyces cerevisiae and invertebrates, such as C. elegans and Drosophila. The de novo pathway from tryptophan is not shown in this scheme. Pnc1, nicotinamidase; Npt1, nicotinic acid phosphoribosyltransferase; Nma1 and Nma2, nicotinic acid mononucleotide adenylyltransferase 1 and 2; Qns1, NAD synthetase; Sir2, silencing information regulator 2; Nic, nicotinamide; Na, nicotinic acid; NaMN, nicotinic acid mononucleotide. (B) The NAD biosynthetic pathway from nicotinamide in mammals. The pathways from tryptophan and nicotinic acid are not shown in this scheme. Sirt1 and other mammalian sirtuins are representative enzymes that catalyze NAD for their enzymatic activities. Nampt, nicotinamide phosphoribosyltransferase; Nmnat, nicotinamide mononucleotide adenylyltransferase; NMN, nicotinamide mononucleotide. (C) The reaction catalyzed by Nampt. PRPP, 5-phosphoribosyl 1-pyrophosphate; PPi, inorganic pyrophosphate.
The NAD biosynthetic pathway from nicotinamide is initiated by a unique enzyme, Nampt (Figures 2B and 2C). In mammals, Nampt catalyzes the conversion of nicotinamide to nicotinamide mononucleotide (NMN) (Figure 2C), which is then converted to NAD by nicotinamide/nicotinic acid mononucleotide adenylyltransferase (Nmnat) (Figure 2B). We and other groups have characterized the biochemical and structural features of Nampt and have firmly established that Nampt belongs to a dimeric class of type II phosphoribosyltransferases (41-46). Strangely, despite the fact that Nampt has ancient origins as an NAD biosynthetic enzyme, no organisms between bacteria and vertebrates have obvious homologs of Nampt, except for a few species (43, 44). Nonetheless, the homology of Nampt proteins between bacteria and vertebrates is unusually high (43). Because no obvious homologs of Pnc1 have been found in vertebrates (37, 39), the presence of Nampt, which allows a more direct pathway for NAD biosynthesis from nicotinamide, clearly distinguishes NAD biosynthesis in mammals from that in yeast and invertebrates.
Interestingly, it has been shown that Nampt has both intra- and extracellular forms (iNampt and eNampt, respectively) in mammals (36). While the function of iNampt has been firmly established as an NAD biosynthetic enzyme, the function of eNampt has been a matter of debate (36, 47-49). We have recently demonstrated that eNampt exhibits robust, even higher NAD biosynthetic activity compared to iNampt (50). eNampt has also been documented as a presumptive cytokine named PBEF (51) or a controversial insulin-mimetic hormone named visfatin (52). Although eNampt clearly shows a cytokine-like function that does not require its enzymatic activity (53), careful assessments will be necessary to distinguish these different functions of eNampt in each biological context. Indeed, the function originally reported as PBEF has not been reconfirmed to date, and the original visfatin paper has recently been retracted because of the irreproducibility of the results (54). Additionally, to avoid the confusing nomenclatures of Nampt/PBEF/visfatin, Nampt has recently been approved as the official nomenclature of the protein and the gene by both the HUGO Gene Nomenclature Committee (HGNC) and the Mouse Genome Nomenclature Committee (MGNC). Therefore, Nampt will be used throughout this review.
We have recently demonstrated that NAD biosynthesis mediated by iNampt and eNampt plays a critical role in the regulation of glucose-stimulated insulin secretion (GSIS) in pancreatic β cells (50). Interestingly, Nampt heterozygous (Nampt+/-) female mice show impaired glucose tolerance due to a defect in GSIS, while males do not show these phenotypes. Primary Nampt+/- islets also show defects in NAD biosynthesis and GSIS. These phenotypes in Nampt+/- mice and islets can be completely ameliorated by administration of NMN, suggesting that the observed defects in GSIS are due to a lack of Nampt-mediated NAD biosynthesis. Consistent with these results, FK866, a potent Nampt inhibitor, significantly inhibits NAD biosynthesis and GSIS in isolated wild-type primary islets, and administration of NMN restores normal NAD biosynthesis and GSIS in FK866-treated wild-type primary islets. Surprisingly, only Nampt+/- females, but not males, show reduced plasma levels of eNampt and NMN (50). This disparity in plasma eNampt and NMN levels between Nampt+/- males and females provides a logical explanation to why only Nampt+/- females show defects in glucose metabolism and why NMN administration can restore normal function in those females. These findings strongly suggest that Nampt-mediated NAD biosynthesis plays an important role in the regulation of glucose homeostasis and also that the maintenance of high NMN levels by eNampt in blood circulation is critical for normal β cell function, probably because pancreatic islets have very low levels of iNampt compared to other tissues (50).
This study has provided a novel physiological framework of systemic NAD biosynthesis (Figure 3) (13, 50). While NMN is synthesized from nicotinamide by iNampt in a variety of tissues/organs, a significant amount of NMN may also be synthesized by eNampt in blood circulation and distributed to organs/tissues through blood circulation. We are now able to detect NMN as well as other NAD intermediates in both mouse and human plasma (unpublished observations). Therefore, it will be of great importance to elucidate how exactly NMN is synthesized in mouse and human plasma. Once NMN gets to each tissue, NMN is transported from blood circulation to the inside of cells and converted rapidly to NAD by Nmnat. Based on our studies with pancreatic islets (50), exogenous NMN clearly stimulates intracellular NAD biosynthesis, suggesting that there must be a transporter for the uptake of NMN. Currently, nothing has been known for this putative NMN transporter, and therefore, further studies will be required to identify the NMN transporter.
Figure 3.

Systemic NAD biosynthesis mediated by intra- and extracellular Nampt. See texts for details. NMN, nicotinamide mononucleotide; iNampt and eNampt, intra- and extracellular nicotinamide phosphoribosyltransferase; Nmnat, nicotinamide/nicotinic acid mononucleotide adenylyltransferase.
Inside the cell, NAD biosynthesis might be compartmentalized because there are three different forms of Nmnat, Nmnat1-3, which are localized in the nucleus, cytoplasm, and mitochondria, respectively (35). Indeed, this particular idea has long been discussed (55, 56), and it has also been suggested that the rapid turnover of NAD occurs in a compartment in which the NAD does not equilibrate with the pyridine nucleotide involved in glycolysis, such as NADH (55). Therefore, it will also be of great importance to examine how NAD biosynthesis is regulated in each subcellular compartment as well as in an extracellular compartment, and this could be a separate critical issue from how NAD/NADH ratio is regulated in these compartments. Alterations in NAD levels could affect activities of important enzymes in metabolic pathways, such as glycolysis or fatty acid oxidation, and other NAD-consuming enzymes, such as NAD-dependent sirtuins, poly(ADP-ribose)polymerases (PARPs), and non-sirtuin mono-ADP-ribosyltransferases. In the following section, I will focus on the connection between Sirt1 and Nampt-mediated NAD biosynthesis, partly because we have shown that Nampt-mediated NAD biosynthesis plays an important role in the regulation of Sirt1 activity (43, 57). Nonetheless, it does not exclude a possibility that Nampt-mediated systemic biosynthesis is also important for the regulation of other NAD-dependent enzymes.
The extracellular biosynthesis and the distribution of NMN through blood circulation may be particularly important for organs/tissues that do not have sufficient levels of iNampt to synthesize NAD from nicotinamide. Based on our findings described above, pancreatic β cells are definitely one such tissue. For pancreatic β cells, circulating NMN functions as an essential plasma metabolite that can modulate their insulin-secreting function through NAD biosynthesis (50). It will be of great interest to examine the role of circulating NMN in other tissues that also have low levels of iNampt, such as brain (50). Given that fully differentiated adipocytes are natural produces of eNampt (50), adipose tissue may play an important role in the regulation of metabolic functions of those tissues through secretion of eNampt and extracellular biosynthesis of NMN (see Figure 4). Although further investigation will be necessary, Nampt-mediated systemic NAD biosynthesis might function as a global regulatory mechanism that coordinates the pace of metabolism and possibly aging at a systemic level.
Figure 4.

A conceptual scheme of the NAD World. The NAD World is a novel systemic regulatory network for metabolism and aging that is comprised of two critical components: Nampt-mediated systemic NAD biosynthesis (see Figure 3) as a driver that keeps up the pace of metabolism in tissues/organs and the NAD-dependent deacetylase Sirt1 (see Figure 1) as a universal mediator that executes metabolic effects in various tissues in response to changes in systemic NAD biosynthesis. See texts for details.
NAD World: A new systemic regulatory network for metabolism and aging
As summarized above, Sirt1 and Nampt-mediated systemic NAD biosynthesis together play a critical role in the regulation of metabolism and possibly aging in mammals. Nampt-mediated systemic NAD biosynthesis functions as a driver or a pacemaker that keeps up the pace of metabolism in tissues/organs through intra- and extracellular biosynthesis of NMN, and the NAD-dependent deacetylase Sirt1 serves as a universal mediator that executes metabolic effects in various tissues in response to changes in systemic NAD biosynthesis. These two critical components comprise a novel systemic regulatory network for metabolic regulation in mammals, named “NAD World” (Figure 4). This new concept of the NAD World also provides important insights into a systemic regulatory mechanism that fundamentally connects metabolism and aging in mammals.
The NAD World functions to orchestrate metabolic responses to a variety of nutritional and environmental cues, contributing to the maintenance of the robustness of metabolic regulation at a systemic level (Figure 4). However, every system has a frailty point. In the NAD World, frailty points are tissues/organs that rely on circulating NMN to maintain adequate levels of NAD biosynthesis because of their very low levels of iNampt. Among those tissues/organs, pancreatic β cells and brain (neurons), both of which have very low levels of iNampt (50), are likely the most critical frailty points because of their systemic impacts on many other tissues/organs. If systemic NAD biosynthesis starts declining, these two frailty points would respond first to this change and start having functional problems due to inadequate NAD biosynthesis and thereby reduced Sirt1 activity (Figure 5). We have recently found that plasma NMN levels measured by HPLC significantly decrease over age in both male and female mice (57). This reduction in systemic NAD biosynthesis significantly decreases Sirt1 activity in pancreatic β cells and abolishes all phenotypes of enhanced β cell function in aged BESTO mice. Strikingly, administration of NMN significantly enhances GSIS in both aged wild-type control and BESTO female mice (57). Aged BESTO females show a further increase in GSIS compared to controls so that improved glucose tolerance, which used to be observed in younger BESTO mice (26), is restored in aged BESTO females (57). Similar to the case of the Nampt+/- males (50), the aged BESTO males do not respond to NMN administration, and further investigation will be necessary to clarify this sex-dependent difference. Nonetheless, these findings strongly suggest that Nampt-mediated systemic NAD biosynthesis declines over age, resulting in reduced Sirt1 activity and GSIS in aged β cells.
Figure 5.

A model of mammalian aging as the process of robustness breakdown triggered by a decline in systemic NAD biosynthesis. Pancreatic β cells and neurons are likely the most critical frailty points in the NAD World due to their very low levels of iNampt and systemic impacts on many other tissues/organs. When systemic NAD biosynthesis levels decline and reach frailty thresholds for pancreatic β cells and neurons, these two cell types start having functional problems, which eventually spread to other peripheral tissues/organs through insulin secretion and central metabolic regulation. This cascade of robustness breakdown might be the central process of aging that causes a variety of age-associated diseases, including impaired glucose tolerance (IGT), type 2 diabetes, dementia, and many others. See texts for details.
In pancreatic β cells, a decrease in systemic NAD biosynthesis causes reduced insulin secretion and impaired glucose tolerance, as demonstrated in Nampt+/- mice (50), and possibly type 2 diabetes when the problem persists (Figure 5). Indeed, a progressive age-associated decline in β cell function has been suggested to be one of the major contributing factors to the pathogenesis of type 2 diabetes, one of the major age-associated complications in our modern society (58-60). Therefore, although further investigation will be necessary to examine whether the age-associated decline in Nampt-mediated systemic NAD biosynthesis also causes β cell dysfunction in humans, pancreatic β cells are definitely an important frailty point in the NAD World that is susceptible to changes in Nampt-mediated systemic NAD biosynthesis. Then, how about brain or neurons? Interestingly, it has long been known that one of the triad symptoms in pellagra, the vitamin B3 deficiency, is dementia (61). Therefore, in brain, it is likely that the age-associated decline in systemic NAD biosynthesis causes functional deficits in neurons and results in neurological problems, including dementia (Figure 5). Whereas the role of Sirt1 in the central nervous system has not been fully investigated yet, it is conceivable that Sirt1 might regulate critical molecular processes in brain that connects metabolism and aging in response to changes in systemic NAD biosynthesis (Figure 4). Investigation is currently underway to identify such Sirt1-mediated key molecular processes in brain. Once pancreatic β cells and neurons start having functional problems due to inadequate NAD biosynthesis, other peripheral tissues/organs would also be affected through insulin secretion and central metabolic regulation so that the metabolic robustness would gradually deteriorate over age at a systemic level. This cascade of robustness breakdown triggered by a decrease in systemic NAD biosynthesis might be the central process of aging (Figure 5). In this regard, aging is not a plethora of random events, but the process that occurs according to a functional hierarchy determined by the susceptibility (frailty) to systemic NAD biosynthesis. Pancreatic β cells and neurons, two major cell types that cause serious age-associated complications, are placed at the top of this functional hierarchy due to their frailty in the system of the NAD World.
The concept of the NAD World indicates an important direction to seek the answers to the three questions cast at the beginning of this review. First, which tissues/organs play a dominant role in the regulation of aging and longevity in mammals? In the NAD World, pancreatic β cells and brain (neurons) are the most attractive candidates due to their susceptibility to changes in systemic NAD biosynthesis and their importance in metabolic regulation. Of course, these two tissues have complex interactions with other tissues/organs for metabolic regulation, and therefore this notion does not exclude the importance of other tissues/organs in the regulation of aging and longevity. Nonetheless, their frailty to systemic NAD biosynthesis is key to connect the functional hierarchy in the NAD World to the regulation of aging in mammals. Second, what are “aging-regulatory” hormones or humoral factors? As well as a variety of hormones, novel metabolites in blood circulation, such as NMN, may function as such “aging-regulatory” factors. Given that circulating NMN is produced by eNampt mainly secreted by adipose tissue (50), there might be a feedback regulatory mechanism by which the production and the secretion of Nampt is regulated among pancreatic β cells, adipose tissue, and brain. It will also be of great importance to examine whether NMN administration can prevent or treat age-associated complications and possibly cause delayed aging and the extension of life span. Third, are there any universal molecular regulators for the production and/or the secretion of the presumed “aging-regulatory” hormones or humoral factors? In the NAD World, Sirt1 is at the central position as the major universal molecular regulator that plays a critical role in the systemic regulation of aging and longevity in mammals, as discussed in this review. However, other mammalian sirtuin members might also function as such universal regulators that connect metabolism and aging (15, 16). Further studies will reveal the importance of other mammalian sirtuin members in the NAD World.
The concept of the NAD World also conveys the ideas of functional hierarchy and frailty for the systemic regulation of metabolic robustness and aging. Given that effective trade-offs between robustness and frailty have been suggested in a highly optimized tolerance (HOT) architecture underlying complex biological networks, such as metabolism (62-64), whether improving the frailty by simply enhancing Sirt1 activity always increases the entire robustness of our physiological system still remains unclear. For example, while the system could become more robust against nutritional perturbation by enhancing Sirt1 activity, it could become more fragile to an unexpected perturbation, such as virus infection (63). This possibility might be well illustrated by the physiological effects of CR. As discussed in the previous section, it has been shown that Sirt1 is required for the metabolic adaptation to CR (31, 32) and also that increasing Sirt1 dosage partially mimics the physiological responses to CR (33). Therefore, Sirt1 activity seems to be enhanced in CR, possibly through an increase in Nampt-mediated NAD biosynthesis in response to low nutritional input (43, 65). On the other hand, it has also been reported that calorically restricted animals show reduced natural killer cell function and increased mortality in response to influenza virus infection (66-68). Therefore, detailed analysis of the system structure and its dynamics of the NAD World will be critical before trying to intervene the complex systemic regulatory network for aging and longevity in mammals. Nonetheless, in the next several years, this new NAD World will be further explored, and it may not be so long before we find a way to reverse-engineer this complex system and thereby achieve “productive aging” in our society.
Acknowledgments
I thank all members of the Imai lab for their helpful discussions and comments. I apologize to those whose work is not cited due to the focus of this review and space limitations. This work was supported by grants from the National Institute on Aging (AG024150), Ellison Medical Foundation, and Longer Life Foundation to S. I.
References
- 1.Berryman DE, Christiansen JS, Johannsson G, Thorner MO, Kopchick JJ. Role of the GH/IGF-1 axis in lifespan and healthspan: Lessons from animal models. Growth Horm IGF Res. 2008;18:455–471. doi: 10.1016/j.ghir.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown-Borg HM. Hormonal control of aging in rodents: The somatotropic axis. Mol Cell Endocrinol. 2008 doi: 10.1016/j.mce.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like signals. Science. 2003;299:1346–1351. doi: 10.1126/science.1081447. [DOI] [PubMed] [Google Scholar]
- 5.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 6.Imai S, Guarente L. Sirtuins: A universal link between NAD, metabolism, and aging. In: Guarente L, Partridge L, Wallace S, editors. The Molecular Biology of Aging. New York: Cold Spring Habor Laboratory Press; 2007. pp. 39–72. [Google Scholar]
- 7.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 8.Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci USA. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 10.Guarente L. Sirtuins in aging and disease. Cold Spring Harb Symp Quant Biol. 2007;72:483–488. doi: 10.1101/sqb.2007.72.024. [DOI] [PubMed] [Google Scholar]
- 11.Starai VJ, Takahashi H, Boeke JD, Escalante-Semerena JC. A link between transcription and intermediary metabolism: a role for Sir2 in the control of acetyl-coenzyme A synthetase. Curr Opin Microbiol. 2004;7:115–119. doi: 10.1016/j.mib.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Westphal CH, Dipp MA, Guarente L. A therapeutic role for sirtuins in diseases of aging? Trends Biochem Sci. 2007;32:555–560. doi: 10.1016/j.tibs.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Imai S, Kiess W. Therapeutic potential of SIRT1 and NAMPT-mediated NAD biosynthesis in type 2 diabetes. Front Biosci. 2008 doi: 10.2741/3428. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bishop NA, Guarente L. Genetic links between diet and lifespan: shared mechanisms from yeast to humans. Nat Rev Genet. 2007;8:835–844. doi: 10.1038/nrg2188. [DOI] [PubMed] [Google Scholar]
- 15.Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 16.Schwer B, Verdin E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008;7:104–112. doi: 10.1016/j.cmet.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 18.Rodgers JT, Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci USA. 2007;104:12861–12866. doi: 10.1073/pnas.0702509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell. 2007;28:91–106. doi: 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 20.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun C, Zhang F, Ge X, Yan T, Chen X, Shi X, Zhai Q. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab. 2007;6:307–319. doi: 10.1016/j.cmet.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 22.Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Oliveira RM, Leid M, McBurney MW, Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiao L, Shao J. SIRT1 regulates adiponectin gene expression through Foxo1-C/EBPalpha transcriptional complex. J Biol Chem. 2006;281:39915–39924. doi: 10.1074/jbc.M607215200. [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Qiang L, Farmer SR. Identification of a domain within peroxisome proliferator-activated receptor gamma regulating expression of a group of genes containing fibroblast growth factor 21 that are selectively repressed by SIRT1 in adipocytes. Mol Cell Biol. 2008;28:188–200. doi: 10.1128/MCB.00992-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J, McDonagh T, Lemieux M, McBurney M, Szilvasi A, Easlon EJ, Lin SJ, Guarente L. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006;4:e31. doi: 10.1371/journal.pbio.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C, Permutt MA, Imai S. Increased dosage of mammalian Sir2 in pancreatic β cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2:105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, Schibler U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134:317–328. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 28.Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, Guarente LP, Sassone-Corsi P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008;134:329–340. doi: 10.1016/j.cell.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lowrey PL, Takahashi JS. Genetics of the mammalian circadian system: Photic entrainment, circadian pacemaker mechanisms, and posttranslational regulation. Annu Rev Genet. 2000;34:533–562. doi: 10.1146/annurev.genet.34.1.533. [DOI] [PubMed] [Google Scholar]
- 30.Ramsey KM, Marcheva B, Kohsaka A, Bass J. The clockwork of metabolism. Annu Rev Nutr. 2007;27:219–240. doi: 10.1146/annurev.nutr.27.061406.093546. [DOI] [PubMed] [Google Scholar]
- 31.Chen D, Steele AD, Lindquist S, Guarente L. Increase in activity during calorie restriction requires Sirt1. Science. 2005;310:1641. doi: 10.1126/science.1118357. [DOI] [PubMed] [Google Scholar]
- 32.Boily G, Seifert EL, Bevilacqua L, He XH, Sabourin G, Estey C, Moffat C, Crawford S, Saliba S, Jardine K, Xuan J, Evans M, Harper ME, McBurney MW. SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS ONE. 2008;3:e1759. doi: 10.1371/journal.pone.0001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, Steele AD, Crowe H, Marmor S, Luo J, Gu W, Guarente L. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007;6:759–767. doi: 10.1111/j.1474-9726.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- 34.Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005;6:298–305. doi: 10.1038/nrm1616. [DOI] [PubMed] [Google Scholar]
- 35.Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Enzymology of NAD+ homeostasis in man. Cell Mol Life Sci. 2004;61:19–34. doi: 10.1007/s00018-003-3161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Revollo JR, Grimm AA, Imai S. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol. 2007;23:164–170. doi: 10.1097/MOG.0b013e32801b3c8f. [DOI] [PubMed] [Google Scholar]
- 37.Rongvaux A, Andris F, Van Gool F, Leo O. Reconstructing eukaryotic NAD metabolism. Bioessays. 2003;25:683–690. doi: 10.1002/bies.10297. [DOI] [PubMed] [Google Scholar]
- 38.Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003;423:181–185. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghislain M, Talla E, Francois JM. Identification and functional analysis of the Saccharomyces cerevisiae nicotinamidase gene, PNC1. Yeast. 2002;19:215–324. doi: 10.1002/yea.810. [DOI] [PubMed] [Google Scholar]
- 40.Collins PB, Chaykin S. The management of nicotinamide and nicotinic acid in the mouse. J Biol Chem. 1972;247:778–783. [PubMed] [Google Scholar]
- 41.Khan JA, Tao X, Tong L. Molecular basis for the inhibition of human NMPRTase, a novel target for anticancer agents. Nat Struct Mol Biol. 2006;13:582–588. doi: 10.1038/nsmb1105. [DOI] [PubMed] [Google Scholar]
- 42.Kim MK, Lee JH, Kim H, Park SJ, Kim SH, Kang GB, Lee YS, Kim JB, Kim KK, Suh SW, Eom SH. Crystal Structure of Visfatin/Pre-B Cell Colony-enhancing Factor 1/Nicotinamide Phosphoribosyltransferase, Free and in Complex with the Anti-cancer Agent FK-866. J Mol Biol. 2006;362:66–77. doi: 10.1016/j.jmb.2006.06.082. [DOI] [PubMed] [Google Scholar]
- 43.Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 44.Rongvaux A, Shea RJ, Mulks MH, Gigot D, Urbain J, Leo O, Andris F. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur J Immunol. 2002;32:3225–3234. doi: 10.1002/1521-4141(200211)32:11<3225::AID-IMMU3225>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 45.van der Veer E, Nong Z, O'Neil C, Urquhart B, Freeman D, Pickering JG. Pre-B-cell colony-enhancing factor regulates NAD+-dependent protein deacetylase activity and promotes vascular smooth muscle cell maturation. Circ Res. 2005;97:25–34. doi: 10.1161/01.RES.0000173298.38808.27. [DOI] [PubMed] [Google Scholar]
- 46.Wang T, Zhang X, Bheda P, Revollo JR, Imai S, Wolberger C. Structure of Nampt/PBEF/visfatin, a mammalian NAD(+) biosynthetic enzyme. Nat Struct Mol Biol. 2006;13:661–662. doi: 10.1038/nsmb1114. [DOI] [PubMed] [Google Scholar]
- 47.Arner P. Visfatin—a true or false trail to type 2 diabetes mellitus. J Clin Endocrinol Metab. 2006;91:28–30. doi: 10.1210/jc.2005-2391. [DOI] [PubMed] [Google Scholar]
- 48.Sethi JK. Is PBEF/visfatin/Nampt an authentic adipokine relevant to the metabolic syndrome? Curr Hypertens Rep. 2007;9:33–38. doi: 10.1007/s11906-007-0007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stephens JM, Vidal-Puig AJ. An update on visfatin/pre-B cell colony-enhancing factor, an ubiquitously expressed, illusive cytokine that is regulated in obesity. Curr Opin Lipidol. 2006;17:128–131. doi: 10.1097/01.mol.0000217893.77746.4b. [DOI] [PubMed] [Google Scholar]
- 50.Revollo JR, Körner A, Mills KF, Satoh A, Wang T, Garten A, Dasgupta B, Sasaki Y, Wolberger C, Townsend RR, Milbrandt J, Kiess W, Imai S. Nampt/PBEF/visfatin regulates insulin secretion in β cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6:363–375. doi: 10.1016/j.cmet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994;14:1431–1437. doi: 10.1128/mcb.14.2.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, Watanabe E, Takagi T, Akiyoshi M, Ohtsubo T, Kihara S, Yamashita S, Makishima M, Funahashi T, Yamanaka S, Hiramatsu R, Matsuzawa Y, Shimomura I. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science. 2005;307:426–430. doi: 10.1126/science.1097243. [DOI] [PubMed] [Google Scholar]
- 53.Li Y, Zhang Y, Dorweiler B, Cui D, Wang T, Woo CW, Brunkan CS, Wolberger C, Imai S, Tabas I. Extracellular Nampt promotes macrophages survival via a non-enzymatic interleukin-6/STAT3 signaling mechanism. J Biol Chem. 2008 doi: 10.1074/jbc.M805866200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, Watanabe E, Takagi T, Akiyoshi M, Ohtsubo T, Kihara S, Yamashita S, Makishima M, Funahashi T, Yamanaka S, Hiramatsu R, Matsuzawa Y, Shimomura I. Retraction. Science. 2007;318:565b. doi: 10.1126/science.318.5850.565b. [DOI] [PubMed] [Google Scholar]
- 55.Bernofsky C. Physiology aspects of pyridine nucleotide regulation in mammals. Mol Cell Biochem. 1980;33:135–143. doi: 10.1007/BF00225285. [DOI] [PubMed] [Google Scholar]
- 56.Yang H, Lavu S, Sinclair DA. Nampt/PBEF/Visfatin: a regulator of mammalian health and longevity? Exp Gerontol. 2006;41:718–726. doi: 10.1016/j.exger.2006.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramsey KM, Mills KF, Satoh A, Imai S. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in β cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell. 2008;7:78–88. doi: 10.1111/j.1474-9726.2007.00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Basu R, Breda E, Oberg AL, Powell CC, Dalla Man C, Basu A, Vittone JL, Klee GG, Arora P, Jensen MD, Toffolo G, Cobelli C, Rizza RA. Mechanisms of the age-associated deterioration in glucose tolerance: contribution of alterations in insulin secretion, action, and clearance. Diabetes. 2003;52:1738–1748. doi: 10.2337/diabetes.52.7.1738. [DOI] [PubMed] [Google Scholar]
- 59.Iozzo P, Beck-Nielsen H, Laakso M, Smith U, Yki-Jarvinen H, Ferrannini E. Independent influence of age on basal insulin secretion in nondiabetic humans. European Group for the Study of Insulin Resistance. J Clin Endocrinol Metab. 1999;84:863–868. doi: 10.1210/jcem.84.3.5542. [DOI] [PubMed] [Google Scholar]
- 60.Muzumdar R, Ma X, Atzmon G, Vuguin P, Yang X, Barzilai N. Decrease in glucose-stimulated insulin secretion with aging is independent of insulin action. Diabetes. 2004;53:441–446. doi: 10.2337/diabetes.53.2.441. [DOI] [PubMed] [Google Scholar]
- 61.Roe DA. A PLAGUE OF CORN The Social History of Pellagra. Ithaca and London: Cornell University Press; 1973. [Google Scholar]
- 62.Carlson JM, Doyle J. Highly optimized tolerance: Robustness and design in complex systems. Phys Rev Lett. 2000;84:2529–2532. doi: 10.1103/PhysRevLett.84.2529. [DOI] [PubMed] [Google Scholar]
- 63.Csete M, Doyle J. Bow ties, metabolism and disease. Trends Biotechnol. 2004;22:446–450. doi: 10.1016/j.tibtech.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 64.Zhou T, Carlson JM, Doyle J. Mutation, specialization, and hypersensitivity in highly optimized tolerance. Proc Natl Acad Sci USA. 2002;99:2049–2054. doi: 10.1073/pnas.261714399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, de Cabo R, Sauve AA, Sinclair DA. Nutrient-sensitive mitochondrial NAD(+) levels dictate cell survival. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gardner EM. Caloric restriction decreases survival of aged mice in response to primary influenza infection. J Gerontol A Biol Sci Med Sci. 2005;60:688–694. doi: 10.1093/gerona/60.6.688. [DOI] [PubMed] [Google Scholar]
- 67.Ritz BW, Aktan I, Nogusa S, Gardner EM. Energy restriction impairs natural killer cell function and increases the severity of influenza infection in young adult male C57BL/6 mice. J Nutr. 2008;138:2269–2275. doi: 10.3945/jn.108.093633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roecker EB, Kemnitz JW, Ershler WB, Weindruch R. Reduced immune responses in rhesus monkeys subjected to dietary restriction. J Gerontol A Biol Sci Med Sci. 1996;51:B276–279. doi: 10.1093/gerona/51a.4.b276. [DOI] [PubMed] [Google Scholar]