

Abstract
The inflammatory bowel diseases (IBDs), Crohn's disease and ulcerative colitis, are chronic inflammatory disorders caused by dysregulated immune responses in genetically predisposed individuals. Although the precise etiology of IBD remains unclear, accumulating data, including genome-wide association studies, have advanced our understanding of its immunopathogenesis. This review highlights the role in gut homeostasis and IBD pathogenesis of autophagy, the interleukin (IL)-23/IL-17 axis, and a novel member of the tumor necrosis factor family, TL1A. It focuses on advances in our understanding of IBD from the past year, including advances in genetics and immunobiology.
Introduction
Crohn's disease (CD) and ulcerative colitis (UC) are complex polygenic conditions that are caused by dysregulated activation of effector immunologic pathways in genetically predisposed individuals. Multiple genetic factors contribute to susceptibility to inflammatory bowel disease (IBD). Combined with recent key technologic advances, better understanding of genetic variation within the human genome has lead to the development of genome-wide association studies (GWAS) to identify genetic risk factors for complex polygenic disease in an unbiased manner.
The main genetic associations in IBD can be divided into genes that contribute to innate and adaptive immune responses. In the innate immune arm, the association of CD with polymorphisms in NOD2 (CARD15) and the two autophagy-related genes, ATG16L1 and IRGM, implicate defects in the recognition and handling of intracellular bacteria in the immunopathogenesis of IBD. In the adaptive immune arm, CD has been considered a T-helper-1 (Th1) condition mediated by the interleukin (IL)-12/interferon (IFN)-γ/tumor necrosis factor (TNF) cytokine axis (reviewed in [1]). In contrast, the T-cell response in UC appears to be Th2-dominant (IL-4, IL-13) and mediated by specialized cells such as natural killer (NK) T cells. Tregs, an immune-modulating subset of CD4+ T cells, can suppress the differentiation and function of Th1 and Th2 cells. The cytokine profile in CD and UC is summarized in Table 1. The immunopathologic concept of IBD is evolving in light of recent studies that have unveiled novel effector pathways in IBD, including the involvement of the IL-23/IL-17 axis. Recent data implicate an important role of TL1A in the IL-23/IL-17 axis.
Table 1. Cytokine profile in inflammatory bowel disease.
| Cytokine | Crohn's disease | Ulcerative colitis |
|---|---|---|
| Innate immune response | ||
| IL-1β | I | I |
| IL-6 | I | I |
| IL-8 | I | I |
| IL-12 | I | N |
| IL-18 | I | N |
| IL-23 | I | N |
| IL-27 | I | N |
| TNF-α | I | I |
| TL1A | I | I |
| Adaptive immune response | ||
| IL-5 | N | I |
| IL-13 | N | I |
| IL-17 | I | I |
| IL-21 | I | N |
| IL-22 | I | I |
| IFN-γ | I | I |
| LIGHT | I | I |
| TL1A | I | I |
I—increase; IFN—interferon; IL—interleukin; N—normal; TNF—tumor necrosis factor.
Genome-Wide Association Studies
As a result of recent advances in genetic statistical theory, the availability of cheap, high-throughput genotyping, and the development of the HapMap, researchers are now able to perform genetic association studies on a scale that until recently was thought to be impossible. Within the past few years, numerous GWAS have been published in an increasing number of genetically complex diseases, with CD leading the way. GWAS look at tens of thousands to hundreds of thousands of single nucleotide polymorphisms (SNPs) across the human genome in both individuals with disease and healthy controls. The allele frequencies of these SNPs in the cases and controls are statistically compared to identify any association between a SNP and the disease or condition in question. Independent confirmation of association is needed for findings generated in a GWAS. It is also important to understand that most SNPs included in a GWAS are chosen for methodologic genotyping reasons, not for their perceived function. Therefore, any confirmed SNP associations that are seen with a GWAS are likely to be in linkage disequilibrium with a true disease susceptibility allele rather than being the causal allele itself, unless the researchers have been extremely fortunate. Details of GWAS performed in IBD are listed in Table 2 [2–9,10••].
Table 2. Genome-wide association studies and meta-analysis in inflammatory bowel disease: summary of study designs, populations, and findings.
| Study | Population | SNPs analyzed, n | Cases/controls, n | Phenotype | Genes or loci identified |
|---|---|---|---|---|---|
| Yamazaki et al. [3] | Japanese and British Caucasian | 72,000 | 94/752* | Japanese: CD British: CD and UC | TL1A(TNFSF15) |
| Duerr et al. [4] | North American Caucasian | 304,000 | 946/977 | Ileal CD | IL23R, ATG16L1, PHOX2B, chromosome 10†, FAM92B, NCF4 |
| Hampe et al. [5] | German Caucasian | 7000‡ | 735/368 | CD | ATG16L1 |
| Libioulle et al. [2] | Belgian/French Caucasian | 302,000 | 547/928 | CD | IL23R, chromosome 5p13§ |
| Wellcome Trust Case Control Consortium [6] | British Caucasian | 469,000 | 1748/2938 | CD | IL23R, ATG16L1, chromosome 3†, chromosome 5p13§, IRGM, NKX2-3, PTPN2 |
| Franke et al. [7] | German Caucasian | 92,000 | 393/399 | CD | Chromosome 5p13§, NELL1 |
| Raelson et al. [8] | Quebec Caucasian | 164,000 | 382 trios | CD | IL23R, chromosome 17† |
| Fisher et al. [9] | British Caucasian | 10,886¶ | 905/1465** | UC | ECM1, MST1, BTNL2, HLA-DRB1 |
| Barrett et al. [10••] | North American, British, and Belgian/French populations | NA | 3230/4829 | – | IL23R, ATG16L1, IRGM, MST1, PTGER4, 5q31†, TL1A(TNFSF15), ZNF365, NKX2-3, NOD2, PTPN2, PTPN22, ITLN1, 1q24†, 1q32†, IL12B, CDKAL1, 6q21†, CCR6, 7p12†, 8q24†, 10p11†, C11orf30, LRRK2, MUC19, 13q14†, STAT3, JAK2, 21q21†, ICOSLG, ORMDL3 |
Study performed in 94 cases and 752 controls as stage 1. Findings reproduced in 490 Japanese cases and 345 controls and in 347 British IBD trios, 363 cases, and 372 controls.
A number of the studies identified association with genetic regions in which it was unclear which was the specifi c “causative” gene.
All nonsynonymous (amino acid–changing) polymorphisms.
A number of groups identified association with a variant in a gene desert on chromosome 5p13. Use of an expression database suggests that this region may be involved in regulation of the prostaglandin E receptor 4 gene (PTGER4).
Nonsynonymous and MHC tagging SNPs.
Study performed in 905 patients with UC and 1465 controls. Positive findings were tested in 936 UC cases and 1470 controls. CD—Crohn's disease; MHC—major histocompatibility complex; NA—not applicable; SNP—single nucleotide polymorphism; UC—ulcerative colitis.
More recently, the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), the Wellcome Trust Case Control Consortium (WTCCC), and the Franco-Belgian GWAS group have pooled their data to perform a meta-analysis [10••]. Using this data set with strict statistical criteria and independent replication, the group have identified 30 genetic loci that are associated with CD. These associations are listed in Table 2. Remarkably, these genetic variants account for only 20% of the genetic susceptibility to CD in this population, suggesting that a considerable amount of genetic research remains to be done.
Innate Immunity
Antigen presentation to T cells by professional antigen-presenting cells (APCs) such as dendritic cells (DCs) is critical for the activation of adaptive immune responses. There are three main pathways to transport luminal antigen to lamina propria (LP). The first pathway is mediated by microfold cells through the capture of luminal antigens and their presentation to T cells. The second pathway, performed by LP DCs, involves dendritic cell extension to the intestinal lumen across epithelial cells, facilitating the capture of luminal antigens (Fig. 1). The third pathway involves the neonatal Fc receptor (FcRn), which serves as the vehicle for transporting LP immunoglobulin G (IgG) across the colonic epithelial layer into the lumen, where the IgG can bind enteric bacterial antigens to form immune complexes (IC). The FcRn then recycles the antigens/IgG IC back across the colonic epithelial cells into the LP for processing by APCs (eg, DCs), which can present the antigens to T cells.
Figure 1.
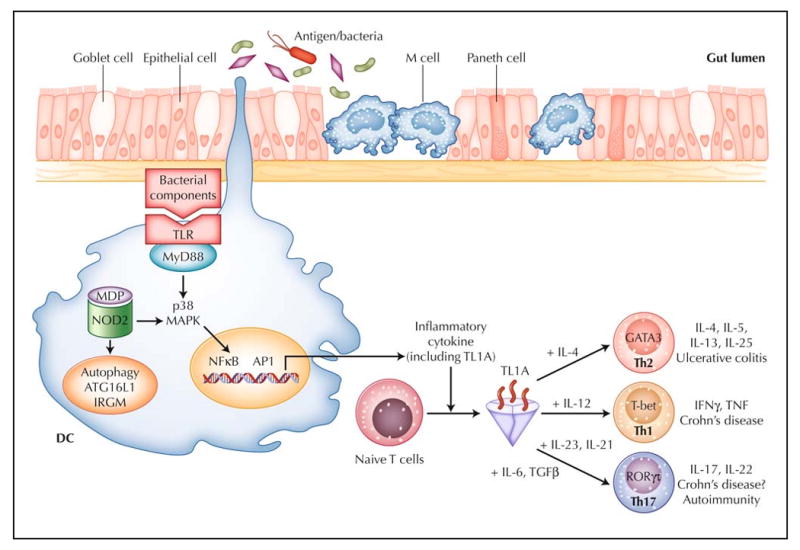
Working hypothesis of inflammatory bowel disease. The intestinal immune system is in close apposition to luminal antigen/bacteria, separated by a single layer of epithelial cells. Goblet cells contribute to the formation of the protective mucus layer, M cells and dendritic cells (DCs) sample intestinal luminal contents. Over-response to antigens, either through the Toll-like receptors (TLR), the intracellular sensor NOD2, or antigen processing via autophagy, results in stimulated DCs that recruit and generate various T-helper-cell subtypes (Th1, Th2, and Th17). TL1A appears to be a critical factor in the generation of Th1, Th2, and Th17 cells. For each T-helper-cell differentiation program, specific transcription factors and cytokine milieu are required. Terminally differentiated T helper cells are characterized by a specific combination of effector cytokines that orchestrate the effector function of the adaptive immune system. Ulcerative colitis appears to be predominately Th2-mediated, whereas Crohn's disease is a predominately Th1- and Th17-mediated process. GATA—GATA binding protein; IFN—interferon; IL—interleukin; IRGM—immunity-related guanosine triphosphatase, M; MAPK—mitogen-activated protein kinase; MDP— muramyl dipeptide; MyD88—myeloid differentiation factor 88; NOD2—nucleotide-binding oligomerization domain containing 2; TGFβ—transforming growth factor β; TNF—tumor necrosis factor; RORγτ—retinoic acid-binding orphan receptor–γτ.
One of the ways that the host distinguishes foreign from self antigen is through a pattern recognition receptor (PRR), which recognizes specific molecular patterns of pathogens. One group of PRRs is the Toll-like receptors (TLRs), which have variable specificities for sensing microbial products. TLR activation can stimulate signaling cascades leading to proinflammatory cytokine production and induction of co-stimulatory signals to initiate effector adaptive immune response (Fig. 1). Nucleotide oligomerization domain (NOD) proteins are cytosolic PRRs involved in the intracellular recognition of bacterial products. The identification of NOD2 as a CD susceptibility gene indicates defective innate immune responses and implicates the role of altered intracellular processing of bacterial components in IBD. Recent genetic associations between autophagy genes, NOD2, and CD further highlight the importance of intracellular processing of bacterial components in mucosal homeostasis.
Defects in autophagy associated with Crohn's disease
Autophagy, from the Greek words auto (oneself) and phagy (to eat), is a biologic process of membrane traffcking in which autophagosomes engulf both organelles and cytosolic macromolecules, followed by fusion of the autophagosome with a lysosome to form an autolysosome in which sequestered material is degraded. The degraded content can then be loaded onto HLA class II molecules or to compartments where recognition by TLRs may occur. For example, a TLR4 signaling pathway was recently found to regulate autophagy associated with innate immunity [11]. Autophagy is important for cellular homeostatic functions, including structural remodeling, generating energy, degrading damaged or long-lived cytoplasmic components, and protecting against invading microorganisms. Its protective role in infectious disease is an important part of the innate immune armamentarium and links to adaptive immunity by delivering foreign antigens necessary for immune recognition. Several recent studies advance our understanding of physiological signals and molecular pathways that regulate and execute autophagy and its role in both health-promoting and disease-associated states. Notably, ATG16L1 and IRGM have been identified as new CD susceptibility genes [12–14].
The ATG16L1 gene product is comprised of an N-terminal ATG16 domain thought to be essential for interaction with other autophagy proteins such as ATG5 and ATG12, a coiled-coil domain postulated to mediate homodimeric interactions, and seven C-terminal WD repeats (tryptophan-aspartate repeats) thought to create stable platforms that can coordinate the formation of the multimeric protein complex that is essential for the formation of the autophagosome [15]. ATG16L1 is broadly expressed by intestinal epithelial cells; APCs; and CD4+, CD8+, and CD19+ primary human T cells [5,12]. Given the important role of autophagy in the traffcking of antigens necessary for immune recognition, mutations affecting important factors involved in autophagy may result in the immune dysregulation seen in IBD. A German CD study using an early GWAS found a nonsynonymous amino acid change—a threonine-to-alanine substitution at position 300 (T300A)—in a screen of about 7000 nonsynonymous coding variants [5]. The association between CD and the same SNP (T300A) in ATG16L1 is confirmed by both the North American GWAS and the WTCCC GWAS [6,12].
IRGM is a member of a family of genes encoding IFN-inducible immunity-related guanosine triphosphatases (IRGs), which are involved in pathogen clearance. IRGM is expressed in several tissues, including colon, small intestine, peripheral blood leukocytes, and monocytes that have been found to have an important role in eliminating the intracellular pathogens Toxoplasma gondii and Listeria monocytogenes [13]. In gene-knockdown experiments, IRGM is shown to induce autophagy to control the intracellular mycobacterial load [16]. Consistent with the idea that defects in autophagy may lead to IBD, multiple IRGM SNPs are found to be associated with CD [6]. In contrast to ATG16L1, however, sequencing of the IRGM coding region did not detect any causal amino acid–changing SNPs [13]. Therefore, the genetic variants conferring susceptibility to CD may lie in regulatory sequences that affect either IRGM expression, transcript splicing, or the rate of protein translation [13].
Nod proteins and autophagy
NOD proteins are a distinct subset of PRRs with important roles in innate immunity as cytoplasmic sensors of microbial components, allowing regulation of infammatory processes and apoptosis. The importance of these PRRs is highlighted by the fact that mutations in the gene encoding NOD2 occur in a subset of patients with CD (reviewed in [1]). NOD2 is a general sensor of most bacteria because it recognizes muramyl dipeptide (MDP), which is a component of both Gram-positive and Gram-negative bacteria. NOD2 stimulation leads to activation of the NFκB and mitogen-activated protein kinase (MAPK) pathway leading to the production of pro-inflammatory mediators (Fig. 1).
There are several contradictory findings on the role of NOD protein in the pathogenesis of IBD. One unanswered question is whether NOD2 mutations confer a gain-of-protein or loss-of-protein function. Perhaps the mutations have different effects in different contexts (ie, in the APC versus epithelial cells or in the mucosa versus the periphery). A recent study showed that NOD2 activation by MDP induces autophagy in DCs that is dependent on both ATG16L1 and components of the NOD2 signaling pathway (Fig. 1) [17]. In addition, DCs from CD patients with NOD2 susceptibility variants are also defective in autophagy induction [17].
Taken together, the associations of SNPs in ATG16L1 and IRGM with CD and the ability of NOD2 to induce autophagy via ATG16L1 indicate that proteins involved in autophagic machinery are an important biologic pathway in chronic inflammatory diseases of the digestive tract. One hypothesis is that alterations in an individual's intracellular processing of bacteria affect how the innate immune system interacts with gut microflora and may contribute to the pathogenesis of CD. Further understanding of the relevant autophagic processes, including elucidation of the molecular mechanisms by which ATG16L1 and IRGM variants contribute to CD susceptibility, will have significant impact on our understanding of IBD.
Adaptive Immunity
Recognition of commensal-derived antigens by the adaptive immune system or its stimulation by the innate immune system plays a key role in the pathogenesis of IBD. Both Th1 and Th2 T cells have been shown to cause chronic gut inflammation, with CD having a predominantly Th1 cytokine profile and UC having a Th2 cytokine profile. In addition to the Th1/Th2 theory, recent studies have unveiled the critical involvement in the pathogenesis of CD of a third subset of effector T helper cells, Th17 cells.
IL-23 and intestinal inflammation
IL-23 is a member of the IL-12 family of heterodimeric cytokines and comprises IL-12p40 (common to IL-12) and the IL-23–specific p19 subunit [18]. In response to microbial antigens or through CD40, activated DCs produce IL-23 and engage the heterodimeric IL-23 receptor, which comprises IL-23R and IL-12RB1 subunits [18]. Janus kinase 2 (JAK2) is then activated, resulting in JAK2 autophosphorylation and tyrosine phosphorylation of IL-23R, which in turn result in the recruitment, phosphorylation, homodimerization, and nuclear translocation of signal transducer and activator of transcription 3 (STAT3). These effects also promote Th17 cell responses and orchestrate additional innate and T cell–mediated inflammatory pathways [18]. IL-23 has been shown to be important in a number of autoimmune and inflammatory diseases, including rheumatoid arthritis, multiple sclerosis, psoriasis, ankylosing spondylitis, and IBD [4,19,20].
Data from immunologic studies suggest that IL-23 plays a significant role in the development of GI mucosal inflammation [21], and the critical role of the IL-23 pathway in IBD pathogenesis was confirmed by the association of several SNPs throughout the IL23R gene with CD and UC [4,22]. This study demonstrates that the rare allele of a nonsynonymous polymorphism, Arg381Gln, confers a threefold increase in protection against CD and is more modestly associated with UC [4]. The Arg381Gln polymorphism is located in a region that is highly conserved across species. Association of IL-23R variants with IBD was also confirmed by the WTCCC [6]. The importance of the IL-23R signaling pathway in IBD is further underscored by the fact that polymorphisms within many genes encoding IL-23R signaling components (including IL-12B, STAT3, and JAK2) are found to be associated with CD in a meta-analysis of three GWAS studies in Caucasians [10••]. Furthermore, variants within IL-23R, IL-12B, and STAT3 have also been genetically associated with UC [7,9].
Numerous studies have demonstrated that the IL-23R signaling pathway contributes to immunopathogenesis of IBD by promoting the proinflammatory state [18]. This effect is further supported by a recent study in which the transfer of bacteria-reactive CD4+ Th17 cells in severe combined immunodeficiency (SCID) mice induced more severe colitis than the transfer of CD4+ Th1 cells [23•]. Administration of anti–IL-23p19 antibodies significantly ameliorates inflammation in the bacteria-reactive Th17 cell–mediated colitis model, highlighting the importance of IL-23 in promoting gut inflammation and indicating a potential new therapeutic approach for this antibody in CD patients [23•].
Emerging data have demonstrated that IL-23 may promote inflammation directly, by inducing inflammatory mediators such as IL-17, and also indirectly, by inhibiting Treg cells, as IL-23 may act directly on T cells to inhibit Foxp3, a transcription factor important for the differentiation of Tregs [24]. That fact that CD103+ DCs induce Foxp3+ Treg cells but CD103− DCs do not appears to be due to high amounts of IL-23p19 mRNA that is expressed in CD103− DCs [25]. Using a T-cell transfer model, the frequency of Foxp3+ cells in the colon that are derived from naive T cells is increased in the absence of IL-23, supporting the hypothesis that IL-23 inhibits Foxp3 and reduces regulatory T-cell induction [26]. In addition, Foxp3-deficient T cells induced colitis when transferred into mice lacking IL-23p19, suggesting that the proinflammatory effect of IL-23 may be mediated mainly by its inhibition of Foxp3 [26]. Together, these results illustrate an important role for IL-23 in reducing Treg cells, thereby permitting the development of gut inflammation.
The IL-23/IL-17/TL1A axis in intestinal inflammation
The effects of IL-23 on CD4+ T cells have been associated with the Th17 cell response. Th17 cells are characterized by the production of IL-17 (IL-17A) and IL-17F but not IFN-γ or IL-4. Differentiation of this Th17 subset is induced through activation of retinoic acid–binding orphan receptor– γτ (RORγτ) signaling (Fig. 1), depending on the pleiotropic cytokine transforming growth factor (TGF)β, which is also linked to the development of Foxp3+ regulatory Treg cells [27–29]. Reciprocal Th17 versus Treg cell differentiation is regulated by proinflammatory cytokines such as IL-6 for differentiation to Th17 and by retinoic acid for differentiation to Treg cells [27,29,30]. The expansion and survival of Th17 cells are subsequently mediated by IL-23 [27,31]. Recently, several studies have revealed that TL1A, a member of the TNF superfamily, is also important for Th17 differentiation and proliferation, making it part of the IL-23/IL-17 axis [32,33••]. These findings are discussed further in the next section.
The latest studies show that IL-21, an IL-2 cytokine family member expressed by activated CD4+ T cells and NK T cells, is a key regulator in the generation of Th17 cells [24,34,35]. This finding is substantiated by a recent study showing that IL-21–deficient mice are protected from experimental colitis, possibly through the failure to generate Th17 response, including induction of IL-17, IL-17F, and RORγτ [36]. Additionally, lack of IL-21, either through neutralizing IL-21 antibodies or by gene knockout of IL-21, impairs the differentiation of naive T cells into Th17 cells [36]. Moreover, IL-21 can substitute for IL-6 in driving IL-17 induction, suggesting that IL-21 either acts downstream of IL-6 or can initiate an alternative pathway that drives Th17 cell polarization [34,36].
The IL-23/IL-17/TL1A axis is important for host defense against many diverse pathogens. In the gastrointestinal tract, this axis may have important immune protective effects against pathogens: IL-23−/− mice exhibited enhanced susceptibility and mortality following infection with Citrobacter rodentium [37]. Interestingly, IL-23−/− mice are still able to generate mucosal Th17 responses, indicating that protective responses to intestinal pathogens mediated by IL-23 need not involve IL-17 [37]. IL-17 and IL-17F induce proinflammatory cytokines, chemokines, and metalloproteinases [27]. However, other cytokines may contribute to Th17 effector function; Th17 cells also produce other pro-inflammatory cytokines, including IL-21 and IL-22 [27]. Treating IL -10−/− mice with neutralizing IL-17 antibodies had little impact on colitis, indicating that IL-17 synergizes with other factors, possibly TL1A, to mediate gut mucosal inflammation [38].
TL1A (TNFSF15)
TL1A (TNFSF15), an IBD-associated gene, is a central immune regulator in IBD. It is a member of the TNF superfamily that binds to death domain receptor 3 (DR3, TNFRSF25) and is expressed on endothelial cells, lymphocytes, plasma cells, monocytes, and DCs [39–41]. Accumulating evidence indicates that the TL1A/DR3 signaling pathway mediates gut mucosal inflammation. TL1A and DR3 expression is increased on T cells and macrophages in the mucosa of patients with IBD [41,42]. Additionally, neutralizing anti–mouse TL1A antibody attenuated inflammation in experimentally induced chronic colitis and a Gαi2−/− T-cell transfer colitis model, suggesting a role for TL1A in the pathology of mucosal inflammation in IBD [33••].
Recent GWAS revealed a significant association of genetic variants of the TL1A (TNFSF15) gene with CD in a large cohort of Japanese patients, in several European cohorts [3,43], in US Jewish patients [44], and in combined data from the NIDDK IBD Genetics Consortium, Belgian-French IBD Consortium, and the WTCCC [10••]. TL1A is the only gene that has been associated with both Asian and Caucasian IBD. Haplotypes A and B are associated with IBD susceptibility in non-Jewish Caucasian CD and UC. However, TL1A haplotype B is a risk haplotype in Jewish CD patients with antibody titers for the Escherichia coli outer membrane porin C (OmpC) [45]. Moreover, monocytes from Jewish OmpC+ patients carrying the risk haplotype B express higher levels of TL1A in response to E. coli stimulation [45]. These results show that TL1A genetic variations contribute to exacerbated induction of TL1A that may lead to an exaggerated immune response and chronic inflammation. Further work is clearly needed to determine the exact disease-associated variants within this gene that will further explain the complicated correlations between genotype and function in these different ethnic groups.
We have previously shown that TL1A is induced by the FcγR signaling pathway [46•] and by enteric microorganisms in antigen APCs [47]. Microbial activated TL1A was in part mediated by TLR1, TLR2, TLR4, THR6, and TLR9 signaling pathways and was dependent on downstream p38 MAPK and NF-κB activation (Fig. 1) [47]. There are also negative regulators of the TL1A signaling pathway; these may maintain gut immune homeostasis. Our group showed that TLR8 signaling can inhibit TL1A production; TLR8 may represent a novel therapeutic target in IBD [48].
TL1A plays an important role in modulating adaptive immune response (Fig. 1). In the Th1 effector arm, TL1A augments IFN-γ production by IL-12/IL-18 stimulated CD4+/CCR9+ T cells, which are specifically enriched in the intestinal immune compartment [49]. Furthermore, in autologous monocyte–T cell cocultures, TL1A production by monocytes potentiates IFN-γ production by CD4+ T cells [46•,47]. In addition to mediating Th1 response, the role of TL1A in Th2-mediated functions and disease pathology was demonstrated in a mouse model of allergic lung inflammation, in which TL1A signaling is required to exert Th2 effector function in Th2-polarized CD4 cells and co-stimulate IL-13 production by activated NK T cells [50]. Additionally, antibody blockade of TL1A inhibits lung inflammation and production of Th2 cytokines such as IL-13. Together, this study implicates TL1A in augmenting Th2 effector function [50]. Using a TL1A-deficient mouse, a recent report show that TL1A−/− DCs exhibited a reduced capacity in supporting Th17 differentiation and proliferation. Consistent with these data, TL1A−/− mice display decreased clinical severity in experimental autoimmune encephalomyelitis, a Th17-mediated autoimmune disease model [32]. Furthermore, IFN-γ production (induced by IL-12) and IL-17 production (induced by IL-23) can be synergistically enhanced by TL1A. Moreover, neutralizing TL1A antibody can attenuate two models of chronic colitis by downregulating Th1 and Th17 activation [33••]. Interestingly, a report using DR3-deficient mice shows that TL1A/DR3 signaling is not essential for polarization of naive CD4+ T cells into Th1, Th2, or Th17 effector cell subtypes [51]. Instead, DR3 expression is required on T cells for immunopathology, local T-cell accumulation, and cytokine production, suggesting that TL1A/DR3 signaling is important to co-stimulate antigen-induced expansion of primed T cells in the target organ of T cell–mediated autoimmune and inflammatory diseases.
Together, TL1A/DR3 signaling appears to have a pleiotropic effect, including amplifying the innate immune response; modulating adaptive immunity by augmenting Th1, Th2, and Th17 effector cell function; and affecting T-cell accumulation and immunopathology of inflamed tissue (Fig. 1). Given the immune modulatory effects of TL1A/DR3 signaling, blocking it is a promising therapeutic strategy in a variety of T cell–dependent autoimmune diseases, including IBD.
Conclusions
The past year has seen considerable progress in elucidating the etiopathogenesis of IBD. The identification of 30 separate loci containing genes from divergent pathways underlies the complexity of these heterogenous conditions. These genetic findings have further implicated both innate and adaptive immune dysregulation as risk factors for developing IBD. The biologic divergence of these immunogenic parameters explains the heterogeneous clinical manifestations of IBD and the lack of a universal therapeutic response to any single agent. Many challenges remain, including the need to better understand the precise molecular mechanisms of IBD immunopathogenesis. A greater understanding of the “causes” of IBD can be achieved by performing integrated, multiparameter assessments of distinct aspects of the disease biology that are already known, by identifying novel immunogenetic parameters through additional genetic studies, and by characterizing the functional consequences of genetic variants of IBD-associated genes. (It is a sobering thought for researchers that nearly 8 years after the identification of the role of NOD2 in CD pathogenesis, the “functional” consequences of the disease-associated variants remain controversial!) The goal of these multidisciplinary approaches must include the ability to stratify these complex conditions into distinct, biologically homogenous subgroups with more defined pathogenic parameters, permitting more rational and individualized approaches to IBD therapy.
Acknowledgments
Dr. Shih is supported by a National Institutes of Health Gastroenterology training grant (T32 DK07180), Proctor & Gamble Investigator Initiated Grant, and the Specialty Training and Advanced Research (STAR) Program at UCLA.
Footnotes
Disclosures: No potential conflicts of interest relevant to this article were reported.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Shih DQ, Targan SR. Immunopathogenesis of inflammatory bowel disease. World J Gastroenterol. 2008;14:390–400. doi: 10.3748/wjg.14.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Libioulle C, Louis E, Hansoul S, et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 2007;3:e58. doi: 10.1371/journal.pgen.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamazaki K, McGovern D, Ragoussis J, et al. Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn's disease. Hum Mol Genet. 2005;14:3499–3506. doi: 10.1093/hmg/ddi379. [DOI] [PubMed] [Google Scholar]
- 4.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 6.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Franke A, Balschun T, Karlsen TH, et al. Replication of signals from recent studies of Crohn's disease identifies previously unknown disease loci for ulcerative colitis. Nat Genet. 2008;40:713–715. doi: 10.1038/ng.148. [DOI] [PubMed] [Google Scholar]
- 8.Raelson JV, Little RD, Ruether A, et al. Genome-wide association study for Crohn's disease in the Quebec Founder Population identifies multiple validated disease loci. Proc Natl Acad Sci U S A. 2007;104:14747–14752. doi: 10.1073/pnas.0706645104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher SA, Tremelling M, Anderson CA, et al. Genetic determinants of ulcerative colitis include the ECM1 locus and five loci implicated in Crohn's disease. Nat Genet. 2008;40:710–712. doi: 10.1038/ng.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10••.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]; To advance gene discovery further, the NIDDK, the WTCCC, and the Franco-Belgian group pooled their data to perform a meta-analysis of their genome-wide association studies. The investigators were able to confirm 11 previously reported IBD loci and provide genome-wide significant evidence for 21 additional loci.
- 11.Xu Y, Jagannath C, Liu XD, et al. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parkes M, Barrett JC, Prescott NJ, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prescott NJ, Fisher SA, Franke A, et al. A nonsynonymous SNP in ATG16L1 predisposes to ileal Crohn's disease and is independent of CARD15 and IBD5. Gastroenterology. 2007;132:1665–1671. doi: 10.1053/j.gastro.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 15.Xavier RJ, Huett A, Rioux JD. Autophagy as an important process in gut homeostasis and Crohn's disease pathogenesis. Gut. 2008;57:717–720. doi: 10.1136/gut.2007.134254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh SB, Davis AS, Taylor GA, et al. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 17.Cooney RM, Baker JS, Simmons A, et al. NOD2 activation by muramyl dipeptide induces autophagy in dendritic cells in an ATG16L1 dependent pathway. Gastroenterology. 2008;134:A514. abstract. [Google Scholar]
- 18.Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol. 2007;25:221–242. doi: 10.1146/annurev.immunol.22.012703.104758. [DOI] [PubMed] [Google Scholar]
- 19.Kim HR, Cho ML, Kim KW, et al. Up-regulation of IL-23p19 expression in rheumatoid arthritis synovial fibroblasts by IL-17 through PI3-kinase-, NF-kappaB- and p38 MAPK-dependent signalling pathways. Rheumatology (Oxford) 2007;46:57–64. doi: 10.1093/rheumatology/kel159. [DOI] [PubMed] [Google Scholar]
- 20.Vaknin-Dembinsky A, Balashov K, Weiner HL. IL-23 is increased in dendritic cells in multiple sclerosis and down-regulation of IL-23 by antisense oligos increases dendritic cell IL-10 production. J Immunol. 2006;176:7768–7774. doi: 10.4049/jimmunol.176.12.7768. [DOI] [PubMed] [Google Scholar]
- 21.McGovern D, Powrie F. The IL23 axis plays a key role in the pathogenesis of IBD. Gut. 2007;56:1333–1336. doi: 10.1136/gut.2006.115402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor KD, Targan SR, Mei L, et al. IL23R haplotypes provide a large population attributable risk for Crohn's disease. Inflamm Bowel Dis. 2008;14:1185–1191. doi: 10.1002/ibd.20478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23•.Elson CO, Cong Y, Weaver CT, et al. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology. 2007;132:2359–2370. doi: 10.1053/j.gastro.2007.03.104. [DOI] [PubMed] [Google Scholar]; This excellent report shows that bacterial-reactive CD4+ Th17 cells are potent effector cells in chronic colitis. Moreover, blocking IL-23p19 was effective in both preventing and treating active colitis, thus providing evidence that IL-23 is an attractive therapeutic target for IBD.
- 24.Zhou L, Ivanov II, Spolski R, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 25.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Izcue A, Hue S, Buonocore S, et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28:559–570. doi: 10.1016/j.immuni.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bettelli E, Korn T, Oukka M, et al. Induction and effector functions of T(H)17 cells. Nature. 2008;453:1051–1057. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 29.Weaver CT, Harrington LE, Mangan PR, et al. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Mucida D, Park Y, Kim G, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 31.Veldhoen M, Hocking RJ, Atkins CJ, et al. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 32.Pappu BP, Borodovsky A, Zheng TS, et al. TL1A-DR3 interaction regulates Th17 cell function and Th17-mediated autoimmune disease. J Exp Med. 2008;205:1049–1062. doi: 10.1084/jem.20071364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33••.Takedatsu H, Michelsen KS, Wei B, et al. TL1A (TNFSF15) regulates the development of chronic colitis by modulating both T-helper 1 and T-helper 17 activation. Gastroenterology. 2008;135:552–567. doi: 10.1053/j.gastro.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]; This outstanding study demonstrates that TL1A is an important modulator in the development of chronic mucosal inflammation by enhancing Th1 and Th17 effector functions. In addition, the investigators show that neutralizing TL1A antibodies can ameliorate two murine models of colitis and provide evidence that TL1A represents an attractive, novel therapeutic target for the treatment of IBD.
- 34.Korn T, Bettelli E, Gao W, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nurieva R, Yang XO, Martinez G, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 36.Fina D, Sarra M, Fantini MC, et al. Regulation of gut inflammation and th17 cell response by interleukin-21. Gastroenterology. 2008;134:1038–1048. doi: 10.1053/j.gastro.2008.01.041. [DOI] [PubMed] [Google Scholar]
- 37.Mangan PR, Harrington LE, O'Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 38.Yen D, Cheung J, Scheerens H, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Migone TS, Zhang J, Luo X, et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity. 2002;16:479–492. doi: 10.1016/s1074-7613(02)00283-2. [DOI] [PubMed] [Google Scholar]
- 40.Papadakis KA, Prehn JL, Landers C, et al. TL1A synergizes with IL-12 and IL-18 to enhance IFN-gamma production in human T cells and NK cells. J Immunol. 2004;172:7002–7007. doi: 10.4049/jimmunol.172.11.7002. [DOI] [PubMed] [Google Scholar]
- 41.Prehn JL, Mehdizadeh S, Landers CJ, et al. Potential role for TL1A, the new TNF-family member and potent costimulator of IFN-gamma, in mucosal inflammation. Clin Immunol. 2004;112:66–77. doi: 10.1016/j.clim.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 42.Bamias G, Martin C, 3rd, Marini M, et al. Expression, localization, and functional activity of TL1A, a novel Th1-polarizing cytokine in inflammatory bowel disease. J Immunol. 2003;171:4868–4874. doi: 10.4049/jimmunol.171.9.4868. [DOI] [PubMed] [Google Scholar]
- 43.Tremelling M, Berzuini C, Massey D, et al. Contribution of TNFSF15 gene variants to Crohn's disease susceptibility confirmed in UK population. Inflamm Bowel Dis. 2008;14:733–737. doi: 10.1002/ibd.20399. [DOI] [PubMed] [Google Scholar]
- 44.Picornell Y, Mei L, Taylor K, et al. TNFSF15 is an ethnic-specific IBD gene. Inflamm Bowel Dis. 2007;13:1333–1338. doi: 10.1002/ibd.20223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Michelsen KS, Thomas LS, Taylor KD, et al. TL1A haplotypes associated with severe Crohn's Disease (CD) determine increased protein expression. Gastroenterology. 2008;134:P-31. abstract. [Google Scholar]
- 46•.Prehn JL, Thomas LS, Landers CJ, et al. The T cell costimulator TL1A is induced by FcgammaR signaling in human monocytes and dendritic cells. J Immunol. 2007;178:4033–4038. doi: 10.4049/jimmunol.178.7.4033. [DOI] [PubMed] [Google Scholar]; This excellent study demonstrates that the Fcγ receptor signaling pathway can induce TL1A on antigen-presenting cells and is the first report that the DR3/TL1A is at the center of immune response initiation.
- 47.Shih DQ, Kwan LY, Chang EY, et al. Mechanism of induction of inflammatory bowel disease associated gene TL1A by microbes in antigen presentation cells. Gastroenterology. 2008;134:P-42. abstract. [Google Scholar]
- 48.Saruta M, Michelsen KS, Yu Q, et al. TLR8 signaling inhibits the expression of the IBD-associated cytokine TL1A in human monocytes. Gastroenterology. 2008;134:A-506. abstract. [Google Scholar]
- 49.Papadakis KA, Zhu D, Prehn JL, et al. Dominant role for TL1A/DR3 pathway in IL-12 plus IL-18-induced IFN-gamma production by peripheral blood and mucosal CCR9+ T lymphocytes. J Immunol. 2005;174:4985–4990. doi: 10.4049/jimmunol.174.8.4985. [DOI] [PubMed] [Google Scholar]
- 50.Fang L, Adkins B, Deyev V, et al. Essential role of TNF receptor superfamily 25 (TNFRSF25) in the development of allergic lung inflammation. J Exp Med. 2008;205:1037–1048. doi: 10.1084/jem.20072528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meylan F, Davidson TS, Kahle E, et al. The TNF-family receptor DR3 is essential for diverse T cell-mediated inflammatory diseases. Immunity. 2008;29:79–89. doi: 10.1016/j.immuni.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]