

Abstract
Both genetic and environmental factors contribute to the pathogenesis of type 2 diabetes, and it is critical to understand the interplay between these factors in the regulation of insulin secretion and insulin sensitivity to develop effective therapeutic interventions for type 2 diabetes. For the past several years, studies on the mammalian NAD-dependent protein deacetylase SIRT1 and systemic NAD biosynthesis mediated by nicotinamide phosphoribosyltransferase (NAMPT) have demonstrated that these two regulatory components together play a critical role in the regulation of glucose homeostasis, particularly in the regulation of glucose-stimulated insulin secretion in pancreatic beta cells. These components also contribute to the age-associated decline in beta cell function, which has been suggested to be one of the major contributing factors to the pathogenesis of type 2 diabetes. In this review article, the roles of SIRT1 and NAMPT-mediated systemic NAD biosynthesis in glucose homeostasis and the pathophysiology of type 2 diabetes will be summarized, and their potential as effective targets for the treatment and prevention of type 2 diabetes will be discussed.
Keywords: SIRT1, NAMPT, NAD Biosynthesis, Insulin Secretion, Insulin Sensitivity, Type 2 Diabetes, Pancreatic β Cells, Liver, Skeletal Muscle, Adipose Tissue, PBEF, visfatin, Review
2. Introduction
In the pathogenesis of type 2 diabetes, a delicate balance between insulin sensitivity and secretion is compromised by both environmental and genetic factors (1-3). While our current life style with nutrient-dense diets and the lack of enough exercise appears to cause an explosive epidemic of obesity and insulin resistance (4), people who manifest insulin resistance do not necessarily develop type 2 diabetes, implicating that the dysfunction of pancreatic beta cells might play a critical role in the development of overt hyperglycemia that clinically defines this disease (2, 3, 5, 6). Although which defect, insulin resistance vs. beta cell dysfunction, primarily contributes to the pathogenesis of type 2 diabetes has long been a matter of debate, the recent success of genome-wide association studies for type 2 diabetes has shed new light on models explaining how this complex disease develops (7-9). Interestingly, most of the identified gene loci associated with type 2 diabetes have been linked to impaired beta cell function: TCF7L2, KCNJ11, TCF2, SLC30A8, HHEX, CDKAL1, IGF2BP2, CDKN2A/B, and WFS1 are included in this category (7, 9). On the other hand, only three gene loci, FTO, MC4R, and PPARG, have been linked or suggested to be linked to obesity risk and insulin resistance (7-9). While these findings do not rule out the importance of insulin resistance, they clearly underscore the genetic contribution of beta cell dysfunction to the pathogenesis of type 2 diabetes.
Aging is one of the greatest risk factors for developing metabolic complications, such as obesity, glucose intolerance, and type 2 diabetes (10, 11). How aging affects metabolic robustness at a systemic level is still poorly understood, although it has been shown that a progressive decline in beta cell function in the elderly is a major contributing factor to the pathophysiology of type 2 diabetes (12, 13). A similar age-associated impairment of beta cell function has also been demonstrated in rodents (14). Therefore, one would speculate that factors that contribute to age-associated changes in beta cell function could also play critical roles in the pathogenesis of type 2 diabetes. As described in detail below, one such factor is the mammalian nicotinamide adenine dinucleotide (NAD)-dependent protein deacetylase SIRT1 (15-17). For the past several years, there has been an increasing interest in SIRT1 as a new therapeutic target for type 2 diabetes in the field of metabolism and aging research (18, 19). Because SIRT1 absolutely requires NAD for its enzymatic activity (20), mammalian NAD biosynthesis has also drawn much attention in these fields (21, 22). Indeed, it has been demonstrated that the NAD biosynthesis mediated by nicotinamide phosphoribosyltransferase (NAMPT), a key NAD biosynthetic enzyme in mammals, plays a critical role in the regulation of glucose-stimulated insulin secretion in pancreatic beta cells (23).
In this review, we will summarize the roles of SIRT1 and NAMPT-mediated NAD biosynthesis in the regulation of glucose metabolism, particularly in the regulation of glucose-stimulated insulin secretion in beta cells. Their potential as therapeutic targets for the treatment and prevention of type 2 diabetes will also be discussed.
3. SIRT1, a key regulator that connects NAD, metabolism, and aging
Since the first discovery that yeast and mammalian Sir2 proteins have NAD-dependent deacetylase activity and that this activity is essential for the longevity control in yeast (20), Sir2 family proteins, called “sirtuins,” have been emerging as evolutionarily conserved, critical regulators for aging and longevity in a wide variety of experimental organisms (16, 24, 25). Increasing the dosage or activity of Sir2 proteins extends the life spans of yeast, worms, and flies, while deletions or mutations of the Sir2 genes shorten their life spans. In certain genetic backgrounds, Sir2 orthologs are also required for the caloric restriction-mediated life span extension in those organisms. In mammals, there are seven Sir2 family members, named SIRT1 through SIRT7 (15-17, 26). SIRT1 is the mammalian ortholog of the founder protein Sir2 in yeast, and the majority of mammalian sirtuin research has focused on the function of SIRT1. Although it is not yet proven whether SIRT1 regulates aging and longevity in mammals, it has already been established that SIRT1 plays a critical role in the regulation of metabolism in response to nutrient availability and cell survival in response to stress.
3.1. The function of SIRT1 in pancreatic beta cells: A possible therapeutic target to restore beta cell adaptation in response to insulin resistance
Pancreatic beta cells have a highly coordinated mechanism to sense glucose and transduce its signal through multiple regulatory components leading to insulin secretion (Figure 1). Briefly, glucose, which enters the beta cells via GLUT2 glucose transporters, is metabolized during glycolysis, resulting in an increase in the ATP/ADP ratio. Subsequent closure of ATP-dependent K+ (KATP) channels causes membrane depolarization, thereby opening voltage-gated Ca2+ channels. The inward Ca2+ flux ultimately triggers exocytosis of insulin-containing granules. This sophisticated glucose-sensing machinery of insulin secretion places beta cells at a central position in the regulation of glucose homeostasis in mammals.
Figure 1.
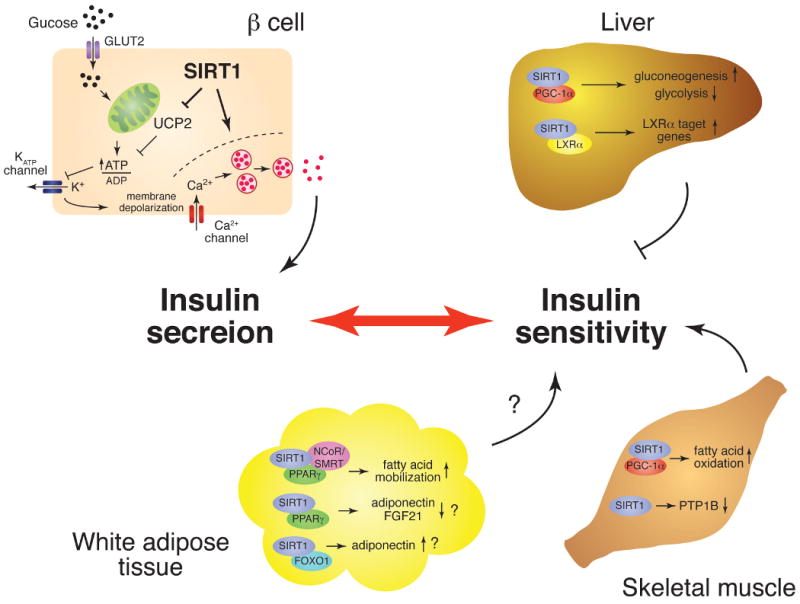
The role of SIRT1 in the regulation of glucose homeostasis. The tissue-specific metabolic functions of SIRT1 in pancreatic beta cells, liver, skeletal muscle, and white adipose tissue are schematically depicted. In pancreatic beta cells, SIRT1 promotes glucose-stimulated insulin secretion and might contribute to beta cell adaptation in response to insulin resistance. In the liver, SIRT1 regulates glucose production through the activation of PGC-1alpha and appears to regulate insulin sensitivity negatively. SIRT1 also regulates LXRalpha function, but the importance of this interaction for insulin sensitivity is unclear. In skeletal muscle, SIRT1 might play an important role in improving insulin sensitivity by increasing fatty acid oxidation through the activation of PGC-1alpha and repressing the expression of PTB1B. In white adipose tissue, SIRT1 promotes fatty acid mobilization by repressing PPARgamma function. SIRT1 also regulates the production of adipokines, such as adiponectin, and FGF21 through FOXO1 and/or PPARgamma. The total effect of SIRT1 in white adipose tissue on insulin sensitivity is still unclear.
When insulin resistance develops in peripheral tissues, beta cells respond to increase insulin secretion and thereby maintain a normal range of blood glucose levels. This ability of beta cells, called adaptation or compensation, is critical to understand the progression of type 2 diabetes (2, 3, 5). Although the underlying mechanism of the beta cell adaptation/compensation is still not fully understood, it is now clear that any failure in this adaptation mechanism causes inadequate insulin secretion in response to insulin resistance, resulting in impaired glucose tolerance and eventually type 2 diabetes (2, 3, 5). The importance of beta cell adaptation in the pathogenesis of type 2 diabetes has been further highlighted by the recent findings that the majority of newly identified genes associated with type 2 diabetes have functions important for beta cell adaptation (7, 9). Therefore, one possible therapeutic approach to prevent and/or treat type 2 diabetes is to restore adequate beta cell adaptation by enhancing the insulin-secreting capability of beta cells (5).
SIRT1 has been demonstrated to positively regulate glucose-stimulated insulin secretion (GSIS) in pancreatic beta cells (Figure 1) (27, 28). Moynihan et al. have demonstrated that an increased dosage of SIRT1 in beta cells significantly enhances GSIS and improves glucose tolerance in beta cell-specific SIRT1-overexpressing (BESTO) transgenic mice (28). In situ-perfused pancreata and isolated primary islets from BESTO mice also show enhanced insulin secretion in response to glucose and KCl, one of the potent non-glucose insulin secretagogues. Interestingly, this SIRT1-mediated enhancement of GSIS is mainly due to an increase in the first-phase insulin secretion. Bordone et al. have also demonstrated that SIRT1-deficient mice and islets show blunted GSIS (27). The SIRT1-mediated enhancement of GSIS is due, at least in part, to the repression of the uncoupling protein 2 (UCP2) gene by SIRT1 (Figure 1) (27, 28). UCP2 is a mitochondrial inner-membrane protein that shunts protons and uncouples respiration from ATP production. The repression of UCP2 by SIRT1 increases the proton gradient across the mitochondrial membrane and thereby enhances ATP production. Indeed, BESTO islets show higher ATP levels in response to glucose, while SIRT1-knockdown beta cells show defects in glucose-stimulated ATP production (27, 28). Glucose-induced ATP production initiates a prompt response of the insulin secretory machinery in beta cells by closing KATP channels. Therefore, the observed increase in ATP levels by SIRT1 is consistent with the increase in the first-phase of GSIS in the BESTO pancreata. Given that the first phase of GSIS is particularly affected when beta cells start failing to compensate (3), enhancing SIRT1 activity in beta cells might be an effective therapeutic approach to restore adequate beta cell adaptation in response to increasing insulin resistance. This idea has been further validated by our study of high-fat diet (HFD)-fed BESTO mice (29). Over the course of the HFD treatment for up to 30 weeks, both male and female BESTO mice were still able to maintain significantly improved glucose tolerance with enhanced GSIS compared to controls, whereas BESTO mice did not differ from controls in HFD-induced weight gain and lipidemia. Therefore, these findings clearly demonstrate that SIRT1 can still function to enhance GSIS and improve glucose tolerance even in the face of diabetogenic dietary conditions, reemphasizing the notion that increasing SIRT1 activity in beta cells provides beneficial effects of enhanced beta cell function on glucose homeostasis (29).
3.2. The role of SIRT1 in the regulation of insulin sensitivity: A promising target to treat insulin resistance?
It is well known that the relationship between insulin sensitivity and insulin secretion can be formulated as a hyperbolic function (2, 5). Because of this relationship, the product of insulin sensitivity and insulin secretion, called the disposition index, is kept constant when individuals stay in a normal range of glucose tolerance. When beta cells start failing to compensate in response to increasing insulin resistance, the hyperbolic curve between insulin sensitivity and insulin secretion starts shifting down, yielding smaller values of the deposition index (2, 5). Therefore, as far as beta cells still maintain their adaptive capability, increasing insulin sensitivity can be an effective approach to restore the normal hyperbolic relationship (5). For example, metformin and thiazolidinediones (TZDs) have been used to increase insulin sensitivity in the treatment of type 2 diabetes. Liver, skeletal muscle, and adipose tissue are classic insulin targets and significantly contribute to systemic insulin sensitivity (30, 31). Therefore, whether and how SIRT1 is involved in the regulation of insulin sensitivity in these organs/tissues is also a critical question to assess a possible connection between SIRT1 and type 2 diabetes.
The liver plays a critical role in maintaining glucose homeostasis through the regulation of glycolysis and gluconeogenesis. During fasting, the liver promotes glycogenolysis and gluconeogenesis and represses glycolysis. In the fasted liver, Rodgers et al. have shown that SIRT1 interacts with and deacetylates PGC-1alpha (peroxisome proliferator-activated receptor-g coactivator 1alpha), a key transcription regulator of glucose production, in an NAD-dependent manner (32). SIRT1 is required for both the induction of gluconeogenic genes and the repression of glycolytic genes by PGC-1alpha in vitro and in vivo (Figure 1) (32, 33). Interestingly, adenovirus-mediated hepatic SIRT1 knockdown results in improved glucose and insulin tolerance in mice, while hepatic SIRT1 overexpression causes moderate glucose intolerance (33). Although further investigation is necessary, these effects of SIRT1 on glucose tolerance and insulin sensitivity in the liver seem to be explained, at least in part, by the stimulatory effect of SIRT1 on PGC-1alpha function. Whatever the molecular mechanism is, it seems conceivable that SIRT1 contributes to the negative regulation of insulin sensitivity in the liver. This notion has been further supported by the recent finding that liver-specific SIRT1 knockout (LKO) mice show improved glucose tolerance and lower levels of blood glucose and insulin in a diabetogenic dietary condition (34). In the liver, it has also been reported that SIRT1 positively regulates LXRalpha function through its deacetylation and controls cholesterol metabolism (Figure 1) (35). Whereas the activation of LXRalpha is suggested to improve glucose tolerance and insulin sensitivity (36, 37), it is unclear whether the activation of LXRalpha by SIRT1 is involved in the regulation of insulin sensitivity.
Skeletal muscle is the major insulin target tissue that accounts for approximately 75% of insulin-stimulated glucose uptake through the body (38). It has also been suggested that insulin resistance is associated particularly with intramyocellular lipid accumulation in both animals and humans, although this association has not always been validated (39). In skeletal muscle cells, Gerhart-Hines et al. have reported that SIRT1 deacetylates PGC-1alpha and induces mitochondrial fatty acid oxidation genes in response to low glucose, resulting in an increase in fatty acid oxidation (Figure 1) (40). Provided that the expression of PGC-1alpha and mitochondrial OXPHOS genes is reduced in skeletal muscle of type 2 diabetes patients (41, 42), the SIRT1-dependent activation of PGC-1alpha function in skeletal muscle might contribute to the improvement of insulin sensitivity. Recently, another study has provided evidence to support the notion that SIRT1 might improve insulin sensitivity in skeletal muscle. In skeletal myotube cells, Sun et al. have shown that SIRT1 improves insulin sensitivity through the transcriptional repression of the protein tyrosine phosphatase 1B (PTP1B) gene (Figure 1) (43). PTP1B is a key insulin receptor phosphatase, and PTP1B-deficient mice have been shown to be more insulin-sensitive and more resistant to diet-induced obesity compared to controls (44). Although these reported effects of SIRT1 should be validated in vivo, the results implicate that SIRT1 might play an important role in improving insulin sensitivity in skeletal muscle.
Compared to liver and skeletal muscle, the contribution of adipose tissue to whole body glucose uptake is relatively small (30). However, this tissue has two unique roles in the regulation of insulin sensitivity. First, adipose tissue regulates lipolysis and mobilization of free fatty acids (FFAs). Circulating FFA levels are elevated in obesity and type 2 diabetes and are associated with insulin resistance in both conditions (2, 31). Picard et al. have reported that SIRT1 regulates the mobilization of free fatty acids in response to fasting (45). Sirt1 is recruited to the promoters of genes, such as aP2, that are controlled by PPAR-gamma (peroxisome proliferator-activated receptor-gamma), a nuclear hormone receptor that promotes adipogenesis and fat storage, in response to fasting (Figure 1). SIRT1 interacts with and represses the activity of PPAR-gamma through interaction with its co-repressors NCoR (nuclear receptor co-repressor) and SMRT (silencing mediator for retinoid and thyroid hormone receptor), resulting in the down-regulation of genes driving adipogenesis and fat storage. Consistent with these findings, fasting-induced fatty acid mobilization is compromised in SIRT1 heterozygous mice (45). Second, adipose tissue also functions as an endocrine tissue that secretes a variety of adipokines that affect insulin sensitivity, including leptin, TNF-alpha, and adiponectin (6, 46). It has been reported that SIRT1 increases adiponectin transcription in adipocytes by activating FOXO1 and promoting the interaction between FOXO1 and C/EBPalpha (CCAAT/enhancer-binding protein alpha) (Figure 1) (47). On the other hand, it has recently been reported that SIRT1 represses a specific subset of PPAR-gamma target genes, including Ero1alpha-Lalpha and FGF21, and negatively regulates the secretion of adiponectin (through Ero1alpha-Lalpha) and possibly FGF21, both of which are insulin sensitizing factors (Figure 1) (48). Based on these findings, SIRT1 might negatively regulate insulin sensitivity in adipose tissue, but further investigation with in vivo models will be necessary to determine the actual effect of SIRT1 in adipose tissue on systemic insulin sensitivity.
3.3. SIRT1 as a therapeutic target for type 2 diabetes
As summarized above, the effect of SIRT1 on glucose homeostasis is so complex and tissue-dependent that it is hard to predict whether we should activate or inhibit SIRT1 for the treatment and prevention of type 2 diabetes. Nonetheless, increasing lines of evidence have so far suggested that the activation of SIRT1 might be a promising effective intervention for the treatment and prevention of type 2 diabetes (18, 19).
Two independent studies of “whole-body” SIRT1 transgenic mice have provided the first set of supportive evidence for the beneficial effects of SIRT1 activation on glucose homeostasis and insulin sensitivity (Figure 2) (49, 50). Bordone et al. have reported the phenotypes of the transgenic mice in which SIRT1 cDNA is knocked into the beta-actin locus (SIRT1-KI mice) (49). The SIRT1-KI mice show moderate SIRT1 overexpression in several tissues including white and brown adipose tissues and brain, but not in liver and muscle, without affecting beta-actin protein levels. They are leaner than controls and show improved glucose tolerance, reduced total blood cholesterol levels, decreased blood insulin and glucose levels, enhanced oxygen consumption, and improved activity in rotarod assays, all of which have been observed in calorically restricted mice (49, 51). Pfluger et al. have recently reported another line of SIRT1 transgenic mice by introducing a large genomic fragment containing the entire SIRT1 gene into mice (50). These SIRT1 transgenic mice show broader overexpression profiles of SIRT1 compared to SIRT1-KI mice. They do not show any overt phenotypes under a standard diet, while they exhibit significant protection from high-fat diet-induced hepatic steatosis, inflammation, and glucose intolerance. These SIRT1 transgenic mice do not show significant improvement of systemic insulin sensitivity. Although the molecular mechanisms that underlie those phenotypes still remain unclear in both studies, the overall effects of SIRT1 activation are the improvement of glucose tolerance and insulin sensitivity and the protection from hepatic steatosis and inflammation, all of which can potentially ameliorate complications in type 2 diabetes.
Figure 2.
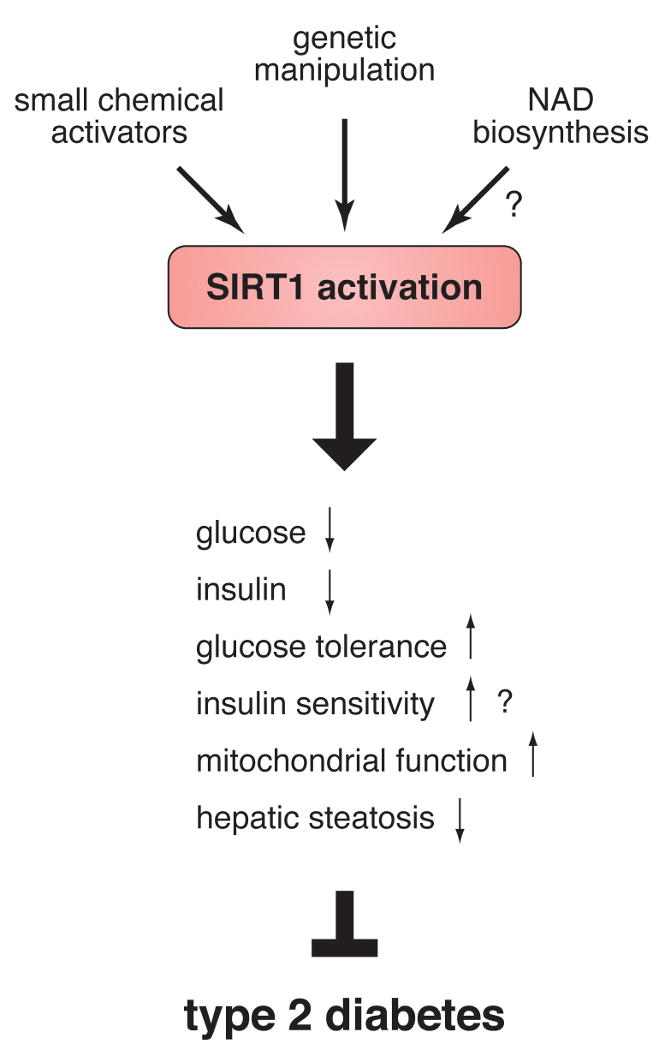
The activation of SIRT1 is a potential therapeutic approach for the treatment and prevention of type 2 diabetes. SIRT1 activation by transgenic manipulation or small chemical activators induces the improvement of glucose tolerance and possibly insulin sensitivity, the enhancement of mitochondrial function, and the protection from hepatic steatosis. Details are described in text.
The second set of supportive evidence has come from studies with small chemical SIRT1 activators (Figure 2) (52-54). Two studies have reported remarkable physiological actions of resveratrol, a putative SIRT1-activating polyphenolic compound, in mice. In one study, Baur et al. have demonstrated that resveratrol (22 mg/kg/day) shifts the physiology of high-fat diet-fed mice toward that of regular diet-fed mice and increases their life expectancy (52). In the other study, Lagouge et al. have also demonstrated that resveratrol (400 mg/kg/day) significantly increases the aerobic capacity and the endurance of high-fat diet-fed or control mice in a running exercise by promoting oxidative phosphorylation and mitochondrial biogenesis in skeletal muscle (53). Most recently, Milne et al. have shown that new small molecule activators of SIRT1, which are structurally unrelated to and 1,000-fold more potent than resveratrol, improve insulin sensitivity, lower plasma glucose, and increase mitochondrial function in diet-induced obese and genetically obese mice (54). Although it is still not definitively proven whether these new compounds specifically target SIRT1 in vivo, these results also support that SIRT1 activation could be an effective therapeutic approach for the treatment of type 2 diabetes.
Given that SIRT1 is a promising pharmaceutical target for the treatment and prevention of type 2 diabetes, another interesting therapeutic approach is to enhance NAD biosynthesis since SIRT1 absolutely requires NAD for its enzymatic activity (Figure 2) (51). Therefore, in the following section, we will assess the physiological importance of mammalian NAD biosynthesis for the regulation of SIRT1 activity and glucose homeostasis.
4. Nicotinamide phosphoribosyltransferase (NAMPT)-mediated systemic NAD biosynthesis
NAD biochemistry has been implicated in a broad range of biological functions, including its classic function as a coenzyme in cellular redox reactions, poly (ADP-ribosyl)ation in DNA repair, mono-ADP-ribosylation in both the immune response and G protein-coupled signaling, and synthesis of cyclic ADP-ribose and nicotinate adenine dinucleotide phosphate (NAADP) in intracellular calcium signaling (22). Recently, it has also been demonstrated that NAD and its derivatives play an important role in transcriptional regulation (55). Indeed, the discovery of the NAD-dependent deacetylase activity of Sir2 proteins has revived an interest in the biology of NAD biosynthesis (16). However, despite the importance of NAD in those biological events, the regulation of NAD biosynthesis in vertebrates remains poorly understood.
4.1. NAMPT, a key NAD biosynthetic enzyme in mammals and its peculiar features
While NAD is synthesized mainly by the de novo pathway via quinolinic acid and by the salvage pathway via nicotinic acid in prokaryotes and lower eukaryotes (Figure 3A), mammals predominantly use nicotinamide rather than nicotinic acid as a precursor for NAD biosynthesis (Figure 3B) (22, 56, 57). In mammals, instead of undergoing deamidation to nicotinic acid, nicotinamide is directly converted to nicotinamide mononucleotide (NMN) by NAMPT (Figure 3C) (22). NMN is then converted to NAD by nicotinamide/nicotinic acid mononucleotide adenylyltransferase (NMNAT). We and other groups have characterized the biochemical nature of the mammalian NAD biosynthesis pathway mediated by NAMPT (58-60). In our study, we have demonstrated that NAMPT is the rate-limiting component in the NAD biosynthetic pathway from nicotinamide and regulates SIRT1 activity in mammalian cells (58). Furthermore, we and other groups have determined crystal structures of the NAMPT apoenzyme, the NAMPT-NMN complex, and the complex of NAMPT and its potent chemical inhibitor FK866, demonstrating that this enzyme is a dimeric type II phosphoribosyltransferase (61-63). These studies have firmly established the biochemical and structural basis of NAMPT as an NAD biosynthetic enzyme.
Figure 3.
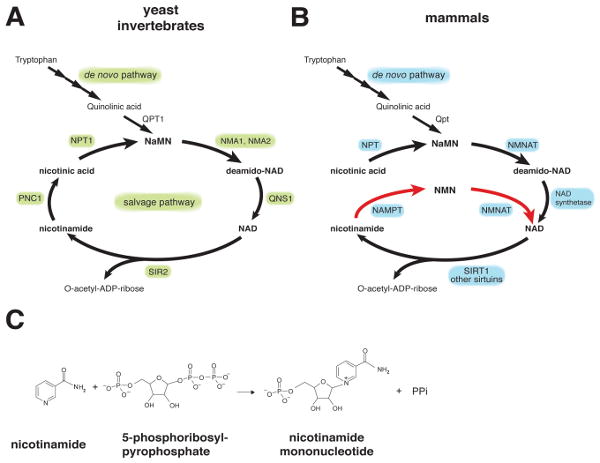
NAD biosynthetic pathways in yeast, invertebrates, and mammals. (A) The NAD biosynthetic pathways in the budding yeast Saccharomyces cerevisiae and invertebrates, such as C. elegans and Drosophila. PNC1, NPT1, NMA1 and NMA2, QNS1, and QPT1 are nicotinamidase, nicotinic acid phosphoribosyltransferase, nicotinic acid mononucleotide adenylyltransferase 1 and 2, NAD synthetase, and quinolinic acid phosphoribosyltransfease, respectively. Only SIR2 is shown as a representative NAD-dependent protein deacetylase. NIC, nicotinamide; NA, nicotinic acid; NaMN, nicotinic acid mononucleotide. B) The NAD biosynthetic pathways in mammals. The de novo pathway and the NAD biosynthetic pathway from nicotinic acid are evolutionarily conserved, while the NAD biosynthetic pathway from nicotinamide (red arrows) is vertebrate-specific and mediated by nicotinamide phosphoribosyltransferase (NAMPT). Nicotinamide is the main precursor for NAD biosynthesis in mammals. While multiple enzymes break NAD into nicotinamide and ADP-ribose, only SIRT1 is shown here. NMN, nicotinamide mononucleotide. C) The reaction catalyzed by NAMPT. PPi, inorganic pyrophosphate.
Interestingly, NAMPT has very ancient origins as an NAD biosynthetic enzyme (58). The entire pyridine nucleotide salvage cycle containing NAMPT, NMNAT, and Sir2 homologues exists even in the vibriophage (64). Despite its ancient origins, NAMPT has a peculiar phylogenetic distribution. No organisms between bacteria and vertebrates have obvious homologs of NAMPT (58, 65). The gene encoding bacterial NAMPT, NadV, was first isolated in Haemophilus ducreyi, and the gene product was found to show significant homology to human pre-B-cell colony-enhancing factor (PBEF) (66), as described below. Indeed, the homology of NAMPT proteins between bacteria and vertebrates is unusually high (58). Interestingly, the organisms that do not have NAMPT homologs, such as yeast, worms, and flies, unanimously have nicotinamidase (PNC1) homologs that convert nicotinamide to nicotinic acid (Figure 3A) (67). It is likely that the organisms that have nicotinamidase use nicotinic acid as a precursor for NAD biosynthesis, while the organisms that have NAMPT use nicotinamide as the main precursor for NAD biosynthesis (22).
Mammalian NAMPT also has a peculiar research history. The gene encoding human NAMPT was originally isolated as a presumptive cytokine named PBEF that reportedly enhances the maturation of B cell precursors in the presence of interleukin-7 (IL-7) and stem cell factor (SCF) (68). Although this particular function of PBEF has not been reconfirmed to date, several groups have since reported cytokine-like functions of NAMPT/PBEF (69-71). Among them, the most controversial function was the one reported by Fukuhara et al. as a “new visceral fat-derived hormone” named visfatin (72). Strikingly, visfatin was reported to exert insulin-mimetic effects in cultured cells and to lower plasma glucose levels in mice by binding to and activating the insulin receptor. Their results immediately drew significant attention to a possible connection between the insulin-mimetic activity of NAMPT/visfatin and metabolic complications, such as obesity and type 2 diabetes. However, subsequent studies have produced conflicting results regarding this possible connection (73-75), partly due to significant differences in immunoassays and sample treatments (76). Furthermore, the original visfatin paper has recently been retracted because of the irreproducibility of the results (77). Therefore, the reported connection between NAMPT/PBEF/visfatin and metabolic complications needs to be carefully re-evaluated by developing a highly accurate, sensitive detection method for this protein. Additionally, to avoid the confusing nomenclatures of NAMPT/PBEF/visfatin, NAMPT has recently been approved as the official nomenclature of the protein and the gene by both the HUGO Gene Nomenclature Committee (HGNC) and the Mouse Genomic Nomenclature Committee (MGNC). Therefore, NAMPT will be used throughout this review.
4.2. The role of NAMPT as a systemic NAD biosynthetic enzyme in the regulation of glucose-stimulated insulin secretion in pancreatic β cells
We have recently demonstrated that NAMPT functions as an intra- and extracellular NAD biosynthetic enzyme (23). Intriguingly, the extracellular form of NAMPT (eNAMPT), also known as PBEF or visfatin, is positively secreted through a non-classical secretory pathway by fully differentiated mouse and human adipocytes, as well as rat and human primary hepatocytes (Garten, A., Petzold, S., Körner, A., Kiess, W., unpublished observation). We have clearly shown that eNAMPT does not exert insulin mimetic effects on adipogenesis, glucose uptake, cellular insulin signaling, and blood glucose levels in mice but rather exhibits robust, even higher, NAD biosynthetic activity compared to its intracellular form (iNAMPT) (23). Currently, the underlying molecular mechanism by which eNAMPT is secreted and activated is unknown. Because the FLAG-tagged NAMPT protein shows different enzymatic activities dependent on the cell type and the compartment, it is very likely that a posttranslational modification is responsible for one or both of the higher enzymatic activity and the secretion of eNAMPT.
Most importantly, we have demonstrated that NAMPT-mediated NAD biosynthesis plays a critical role in the regulation of GSIS in pancreatic beta cells (23). While NAMPT homozygous (NAMPT-/-) mice are lethal around embryonic day 10.5, likely due to the lack of adequate NAD biosynthesis, NAMPT heterozygous (NAMPT+/-) mice are born and grow normally through adulthood. Interestingly, NAMPT+/- females show moderately impaired glucose tolerance and a significant defect in GSIS, while males do not show these phenotypes. Primary islets isolated from NAMPT+/- mice also show defects in NAD biosynthesis and GSIS. Remarkably, the defects in GSIS observed in NAMPT+/- mice and islets are ameliorated by administration of NMN, strongly indicating that these defects are due to a lack of the NAD biosynthetic activity of NAMPT. Furthermore, we have shown that FK866, a potent chemical inhibitor of NAMPT (78), significantly suppresses NAD biosynthesis and GSIS in isolated wild-type primary islets and also that the administration of NMN can restore normal NAD biosynthesis and GSIS in FK866-treated wild-type primary islets. These findings clearly indicate the following important points: First, NAMPT-mediated NAD biosynthesis is critical for the regulation of GSIS in pancreatic beta cells. Second, beta cells can uptake NMN from the outside of cells and use it to maintain normal NAD biosynthesis and GSIS. Strikingly, we have found that a high concentration of NMN circulates systemically in mouse plasma (23). Reduced plasma levels of eNAMPT and NMN have been detected in NAMPT+/- females, but not in males. Although it is currently unknown why and how NAMPT+/- males maintain their plasma eNAMPT and NMN levels, these findings suggest that the maintenance of high NMN levels by eNAMPT in blood circulation is critical for normal beta cell function. Since pancreatic islets have very low levels of iNAMPT, it is very likely that they must rely on circulating NMN to maintain normal NAD biosynthesis.
Based on these findings, we propose that NAMPT-mediated systemic NAD biosynthesis plays a critical role in maintaining glucose homeostasis (Figure 4) (22, 23). Circulating NMN functions as an essential plasma metabolite that can modulate GSIS in pancreatic beta cells. Given that fully differentiated adipocytes are natural producers of eNAMPT, adipose tissue may regulate beta cell function through secretion of eNAMPT and extracellular biosynthesis of NMN. It is also possible that other tissues/organs, such as liver, might contribute to eNAMPT secretion. In this scenario, NMN or NMN-derived compounds might function as a potential signal to stimulate beta cell adaptation in response to increasing insulin resistance. Therefore, genetic predispositions that could cause a failure in adequate production of plasma eNAMPT and NMN might contribute to beta cell dysfunction in the presence of insulin resistance and eventually lead to type 2 diabetes. Indeed, it has been reported that individuals homozygous for either of two single-nucleotide polymorphism variants in the NAMPT gene promoter region have lower fasting plasma insulin levels (79). Further investigation will be necessary to address this interesting hypothesis.
Figure 4.
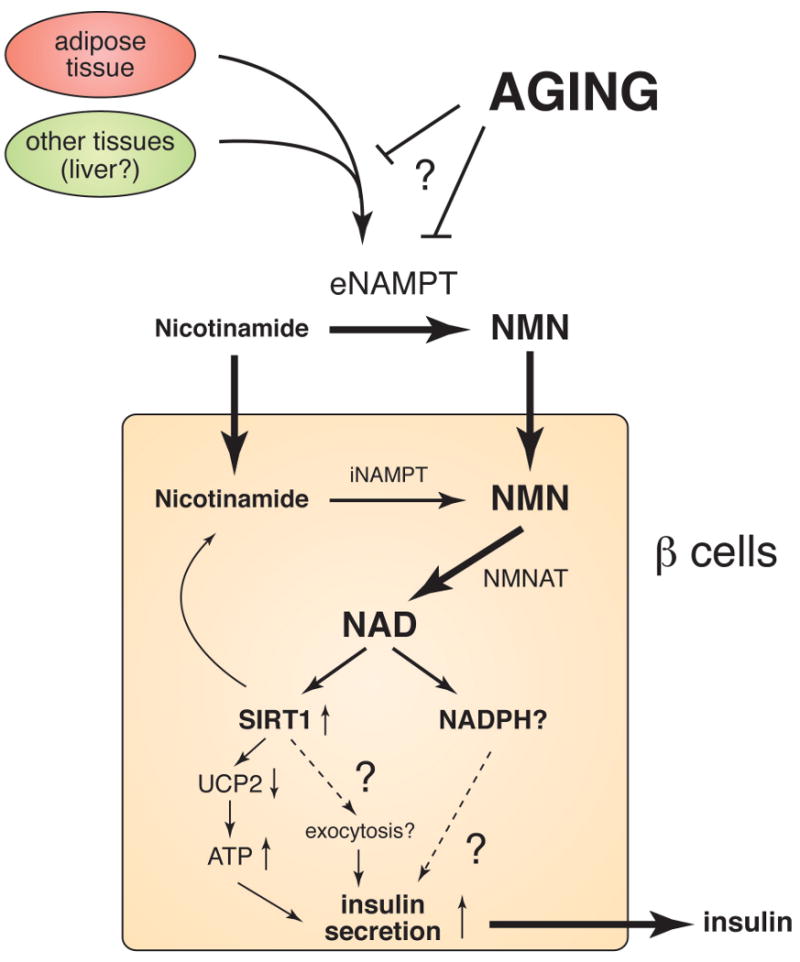
A model for the regulation of insulin secretion by NAMPT-mediated systemic NAD biosynthesis in pancreatic beta cells. Nicotinamide, a form of vitamin B3, is absorbed from the diet and distributed to all organs/tissues through blood circulation. Nicotinamide that enters cells by diffusion and/or transport is converted to nicotinamide mononucleotide (NMN) by intracellular NAMPT (iNAMPT) and then to NAD by NMN adenylyltransferase (NMNAT). A significant fraction of nicotinamide might be converted to NMN by extracellular NAMPT (eNAMPT) in blood circulation. NMN is transported to the inside of cells likely through an unidentified transporter and rapidly converted to NAD by NMNAT. In beta cells, NAD biosynthesis stimulates SIRT1 activity, resulting in enhanced insulin secretion through the repression of UCP2 expression and/or a possible effect on insulin granule exocytosis. NAD biosynthesis might also enhance insulin secretion by increasing other metabolic signals, such as NADPH. Aging affects systemic NAD biosynthesis possibly by affecting the secretion or the activity of eNAMPT, resulting in the reduction in plasma NMN levels and thereby the decrease in SIRT1 activity in pancreatic beta cells with advanced age.
4.3. A connection between SIRT1, NAMPT-mediated systemic NAD biosynthesis, and age-associated decline in beta cell function
As mentioned in the introduction, a progressive age-associated decline in beta cell function has been suggested to be one of the major contributing factors to the pathogenesis of type 2 diabetes (12-14). Based on our studies in BESTO mice, we initially hypothesized that increased SIRT1 dosage or activity in beta cells could provide life-long beneficial effects of enhanced beta cell function on glucose homeostasis and prevent or delay the development of metabolic complications associated with aging. Unexpectedly, we have found that the beneficial phenotypes that younger BESTO mice show are lost completely when the mice reach 18-24 months of age (29). The SIRT1-mediated enhancement of GSIS is blunted in aged BESTO mice and their isolated islets. The repression of UCP2 and the up-regulation of two other genes that are both mediated by SIRT1 are also lost in aged BESTO islets, suggesting that SIRT1 activity decreases with advanced age. Apparently, long-term overexpression of SIRT1 does not confer life-long beneficial effects of improved beta cell function in mice (29).
But why do BESTO mice lose their phenotypes with advanced age? Surprisingly, we have found that while the NAD-biosynthetic capability of aged BESTO islets does not differ from that of young BESTO islets in culture conditions, plasma NMN levels are significantly reduced in aged BESTO mice, indicating that in vivo NAD levels must be reduced in aged BESTO mice (29). Consistent with this finding, NMN administration is able to restore improved glucose tolerance and enhanced GSIS compared to controls in aged BESTO female mice, suggesting that NMN administration restores the activity of overexpressed SIRT1 in aged beta cells and thereby enhances GSIS and improves glucose tolerance in aged BESTO female mice (29). Similar to the case of the NAMPT+/- male mice, the aged BESTO male mice do not respond to NMN administration, and further investigation will be necessary to clarify this sex-dependent difference. Nonetheless, these findings strongly indicate that an age-associated decline in NAMPT-mediated systemic NAD biosynthesis contributes to reduced SIRT1 activity in aged beta cells and thereby the loss of SIRT1-mediated enhancement of GSIS in aged BESTO mice (Figure 4).
Because any failure in NAMPT-mediated systemic NAD biosynthesis presumably causes beta cell dysfunction in response to insulin resistance, the age-associated decline in systemic NAD biosynthesis could be a significant contributing factor to the pathogenesis of type 2 diabetes. If this is the case, NMN administration might be able to restore normal beta cell adaptive function in aged individuals. While the underlying molecular mechanism for the age-associated decline in NAMPT-mediated systemic NAD biosynthesis still needs to be elucidated, our studies provide new insights into this interesting connection between SIRT1, NAMPT-mediated systemic NAD biosynthesis, and the age-associated decline in beta cell function. The activation of SIRT1 in aging beta cells by increasing systemic NAD biosynthesis might be a novel therapeutic approach for the treatment and prevention of age-associated metabolic disorders, such as impaired glucose tolerance and type 2 diabetes (29).
5. Conclusions and perspectives
For the past several years, the fields of sirtuin and NAD biology have been rapidly evolving, and it has been demonstrated that SIRT1 and NAMPT-mediated systemic NAD biosynthesis together play a critical role in the regulation of metabolism and possibly aging in mammals. These two critical components comprise a new systemic regulatory network for metabolic regulation in mammals. In this network, NAMPT-mediated systemic NAD biosynthesis functions as a driver that keeps up the pace of metabolism at a systemic level, and the NAD-dependent deacetylase SIRT1 functions as a mediator that executes regulatory effects in various tissues in response to changes in systemic NAD biosynthesis. This new concept provides important insight into a systemic regulatory mechanism that fundamentally connects metabolism and aging in mammals. Particularly, understanding the physiological role of this regulatory network in glucose homeostasis might provide critical clues to elucidate the molecular mechanism of beta cell adaptation in response to increasing insulin resistance and the pathophysiology of beta cell dysfunction in type 2 diabetes. In this regard, SIRT1 and NAMPT-mediated systemic NAD biosynthesis are two promising therapeutic targets in the same regulatory network for the treatment and prevention of type 2 diabetes. In addition to the use of small SIRT1 chemical activators, manipulating NAD intermediate compounds, such as NMN, might also be an effective approach towards the ultimate conquest of this devastating disease. In the next few years, this new NAD-dependent metabolic network will be further explored, and it may not be so long before new therapeutic interventions based on research on SIRT1 and NAMPT-mediated NAD biosynthesis will be on the horizon.
Acknowledgments
We thank all members of the Imai lab for their helpful discussions and comments. We apologize to those whose work is not cited due to the focus of this review and space limitations. This work was supported by grants from the National Institute on Aging (AG024150), Ellison Medical Foundation, American Diabetes Association, Juvenile Diabetes Research Foundation, the Glenn Award for Research in Biological Mechanisms of Aging, Washington University Clinical Nutrition Research Unit (DK56341), and the National Center for Research Resources (C06RR015502) to S. I. W. K. is supported by a grant from the German Research Council (Deutsche Forschungsgemeinschaft) KFO 152: “Atherobesity”, TP5 and unrestricted grants by Merck Serono, Ipsen and Novo Nordisk.
References
- 1.Bell GI, Polonsky KS. Diabetes mellitus and genetically programmed defects in beta-cell function. Nature. 2001;414:788–791. doi: 10.1038/414788a. [DOI] [PubMed] [Google Scholar]
- 2.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 3.Weir GC, Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes. 2004;53(3):S16–21. doi: 10.2337/diabetes.53.suppl_3.s16. [DOI] [PubMed] [Google Scholar]
- 4.Kiess W, Böttner A, Raile K, Kapellen T, Müller G, Galler A, Paschke R, Wabitsch M. Type 2 diabetes mellitus in children and adolescents: a review from a European perspective. Horm Res. 2003;59(1):77–84. doi: 10.1159/000067829. [DOI] [PubMed] [Google Scholar]
- 5.Ahrén B, Pacini G. Islet adaptation to insulin resistance: mechanisms and implications for intervention. Diabetes Obes Metab. 2005;7:2–8. doi: 10.1111/j.1463-1326.2004.00361.x. [DOI] [PubMed] [Google Scholar]
- 6.Muoio DM, Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- 7.Florez JC. Newly identified loci highlight beta cell dysfunction as a key cause of type 2 diabetes: Where are the insulin resistance genes? Diabetologia. 2008;51:1100–1110. doi: 10.1007/s00125-008-1025-9. [DOI] [PubMed] [Google Scholar]
- 8.Körner A, Kiess W, Stumvoll M, Kovacs P. Poluygenic contribution to obesity: genome-wide strategies reveal new targets. Front Horm Res. 2008;36:12–36. doi: 10.1159/000115335. [DOI] [PubMed] [Google Scholar]
- 9.Perry JR, Frayling TM. New gene variants alter type 2 diabetes risk predominantly through reduced beta-cell function. Curr Opin Clin Nutr Metab Care. 2008;11:371–377. doi: 10.1097/MCO.0b013e32830349a1. [DOI] [PubMed] [Google Scholar]
- 10.Chang AM, Halter JB. Aging and insulin secretion. Am J Physiol Endocrinol Metab. 2003;284:E7–12. doi: 10.1152/ajpendo.00366.2002. [DOI] [PubMed] [Google Scholar]
- 11.Moller N, Gormsen L, Fuglsang J, Gjedsted J. Effects of ageing on insulin secretion and action. Horm Res. 2003;60:102–104. doi: 10.1159/000071233. [DOI] [PubMed] [Google Scholar]
- 12.Basu R, Breda E, Oberg AL, Powell CC, Dalla Man C, Basu A, Vittone JL, Klee GG, Arora P, Jensen MD, Toffolo G, Cobelli C, Rizza RA. Mechanisms of the age-associated deterioration in glucose tolerance: contribution of alterations in insulin secretion, action, and clearance. Diabetes. 2003;52:1738–1748. doi: 10.2337/diabetes.52.7.1738. [DOI] [PubMed] [Google Scholar]
- 13.Iozzo P, Beck-Nielsen H, Laakso M, Smith U, Yki-Jarvinen H, Ferrannini E. Independent influence of age on basal insulin secretion in nondiabetic humans. European Group for the Study of Insulin Resistance. J Clin Endocrinol Metab. 1999;84:863–868. doi: 10.1210/jcem.84.3.5542. [DOI] [PubMed] [Google Scholar]
- 14.Muzumdar R, Ma X, Atzmon G, Vuguin P, Yang X, Barzilai N. Decrease in glucose-stimulated insulin secretion with aging is independent of insulin action. Diabetes. 2004;53:441–446. doi: 10.2337/diabetes.53.2.441. [DOI] [PubMed] [Google Scholar]
- 15.Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 16.Imai S, Guarente L. Sirtuins: A universal link between NAD, metabolism, and aging. In: Guarente L, Partridge L, Wallace D, editors. The Molecular Biology of Aging. Cold Spring Habor Laboratory Press; New York: 2007. [Google Scholar]
- 17.Schwer B, Verdin E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008;7:104–112. doi: 10.1016/j.cmet.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 18.Milne JC, Denu JM. The Sirtuin family: therapeutic targets to treat diseases of aging. Curr Opin Chem Biol. 2008;12:11–17. doi: 10.1016/j.cbpa.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 19.Westphal CH, Dipp MA, Guarente L. A therapeutic role for sirtuins in diseases of aging? Trends Biochem Sci. 2007;32:555–560. doi: 10.1016/j.tibs.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 20.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 21.Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 22.Revollo JR, Grimm AA, Imai S. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol. 2007;23:164–170. doi: 10.1097/MOG.0b013e32801b3c8f. [DOI] [PubMed] [Google Scholar]
- 23.Revollo JR, Körner A, Mills KF, Satoh A, Wang T, Garten A, Dasgupta B, Sasaki Y, Wolberger C, Townsend RR, Milbrandt J, Kiess W, Imai S. Nampt/PBEF/visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6:363–375. doi: 10.1016/j.cmet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 25.Guarente L. Sirtuins in aging and disease. Cold Spring Harb Symp Quant Biol. 2007;72:483–488. doi: 10.1101/sqb.2007.72.024. [DOI] [PubMed] [Google Scholar]
- 26.Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J. Sirtuins: the ‘magnificent seven’, function, metabolism and longevity. Ann Med. 2007;39:335–345. doi: 10.1080/07853890701408194. [DOI] [PubMed] [Google Scholar]
- 27.Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J, McDonagh T, Lemieux M, McBurney M, Szilvasi A, Easlon EJ, Lin SJ, Guarente L. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006;4:e31. doi: 10.1371/journal.pbio.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C, Permutt MA, Imai S. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2:105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 29.Ramsey KM, Mills KF, Satoh A, Imai S. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell. 2008;7:78–88. doi: 10.1111/j.1474-9726.2007.00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kitamura T, Kahn CR, Accili D. Insulin receptor knockout mice. Annu Rev Physiol. 2003;65:313–332. doi: 10.1146/annurev.physiol.65.092101.142540. [DOI] [PubMed] [Google Scholar]
- 31.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 32.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 33.Rodgers JT, Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci USA. 2007;104:12861–12866. doi: 10.1073/pnas.0702509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, Guarente L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22:1753–1757. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell. 2007;28:91–106. doi: 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 36.Laffitte BA, Chao LC, Li J, Walczak R, Hummasti S, Joseph SB, Castrillo A, Wilpitz DC, Mangelsdorf DJ, Collins JL, Saez E, Tontonoz P. Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proc Natl Acad Sci USA. 2003;100:5419–5424. doi: 10.1073/pnas.0830671100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Commerford SR, Vargas L, Dorfman SE, Mitro N, Rocheford EC, Mak PA, Li X, Kennedy P, Mullarkey TL, Saez E. Dissection of the insulin-sensitizing effect of liver X receptor ligands. Mol Endocrinol. 2007;21:3002–3012. doi: 10.1210/me.2007-0156. [DOI] [PubMed] [Google Scholar]
- 38.Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N Engl J Med. 1990;322:223–228. doi: 10.1056/NEJM199001253220403. [DOI] [PubMed] [Google Scholar]
- 39.Corcoran MP, Lamon-Fava S, Fielding RA. Skeletal muscle lipid deposition and insulin resistance: effect of dietary fatty acids and exercise. Am J Clin Nutr. 2007;85:662–677. doi: 10.1093/ajcn/85.3.662. [DOI] [PubMed] [Google Scholar]
- 40.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mootha VK, Handschin C, Arlow D, Xie X, St Pierre J, Sihag S, Yang W, Altshuler D, Puigserver P, Patterson N, Willy PJ, Schulman IG, Heyman RA, Lander ES, Spiegelman BM. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci USA. 2004;101:6570–6575. doi: 10.1073/pnas.0401401101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun C, Zhang F, Ge X, Yan T, Chen X, Shi X, Zhai Q. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab. 2007;6:307–319. doi: 10.1016/j.cmet.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 44.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 45.Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Oliveira RM, Leid M, McBurney MW, Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kiess W, Petzold S, Topfer M, Garten A, Bluher S, Kapellen T, Korner A, Kratzsch J. Adipocytes and adipose tissue. Best Pract Res Clin Endocrinol Metab. 2008;22:135–153. doi: 10.1016/j.beem.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 47.Qiao L, Shao J. SIRT1 regulates adiponectin gene expression through Foxo1-C/EBPalpha transcriptional complex. J Biol Chem. 2006;281:39915–24. doi: 10.1074/jbc.M607215200. [DOI] [PubMed] [Google Scholar]
- 48.Wang H, Qiang L, Farmer SR. Identification of a domain within peroxisome proliferator-activated receptor gamma regulating expression of a group of genes containing fibroblast growth factor 21 that are selectively repressed by SIRT1 in adipocytes. Mol Cell Biol. 2008;28:188–200. doi: 10.1128/MCB.00992-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, Steele AD, Crowe H, Marmor S, Luo J, Gu W, Guarente L. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007;6:759–767. doi: 10.1111/j.1474-9726.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- 50.Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschöp MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci USA. 2008;105:9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Imai S. Is Sirt1 a miracle bullet for longevity? Aging Cell. 2007;6:735–737. doi: 10.1111/j.1474-9726.2007.00344.x. [DOI] [PubMed] [Google Scholar]
- 52.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 54.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin SJ, Guarente L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr Opin Cell Biol. 2003;15:241–246. doi: 10.1016/s0955-0674(03)00006-1. [DOI] [PubMed] [Google Scholar]
- 56.Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Enzymology of NAD+ homeostasis in man. Cell Mol Life Sci. 2004;61:19–34. doi: 10.1007/s00018-003-3161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Magni G, Amici A, Emanuelli M, Raffaelli N, Ruggieri S. Enzymology of NAD+ synthesis. Adv Enzymol Relat Areas Mol Biol. 1999;73:135–182. doi: 10.1002/9780470123195.ch5. [DOI] [PubMed] [Google Scholar]
- 58.Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 59.Rongvaux A, Shea RJ, Mulks MH, Gigot D, Urbain J, Leo O, Andris F. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur J Immunol. 2002;32:3225–3234. doi: 10.1002/1521-4141(200211)32:11<3225::AID-IMMU3225>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 60.van der Veer E, Nong Z, O'Neil C, Urquhart B, Freeman D, Pickering JG. Pre-B-cell colony-enhancing factor regulates NAD+-dependent protein deacetylase activity and promotes vascular smooth muscle cell maturation. Circ Res. 2005;97:25–34. doi: 10.1161/01.RES.0000173298.38808.27. [DOI] [PubMed] [Google Scholar]
- 61.Khan JA, Tao X, Tong L. Molecular basis for the inhibition of human NMPRTase, a novel target for anticancer agents. Nat Struct Mol Biol. 2006;13:582–588. doi: 10.1038/nsmb1105. [DOI] [PubMed] [Google Scholar]
- 62.Kim MK, Lee JH, Kim H, Park SJ, Kim SH, Kang GB, Lee YS, Kim JB, Kim KK, Suh SW, Eom SH. Crystal Structure of Visfatin/Pre-B Cell Colony-enhancing Factor 1/Nicotinamide Phosphoribosyltransferase, Free and in Complex with the Anti-cancer Agent FK-866. J Mol Biol. 2006;362:66–77. doi: 10.1016/j.jmb.2006.06.082. [DOI] [PubMed] [Google Scholar]
- 63.Wang T, Zhang X, Bheda P, Revollo JR, Imai S, Wolberger C. Structure of Nampt/PBEF/visfatin, a mammalian NAD (+) biosynthetic enzyme. Nat Struct Mol Biol. 2006;13:661–662. doi: 10.1038/nsmb1114. [DOI] [PubMed] [Google Scholar]
- 64.Miller ES, Heidelberg JF, Eisen JA, Nelson WC, Durkin AS, Ciecko A, Feldblyum TV, White O, Paulsen IT, Nierman WC, Lee J, Szczypinski B, Fraser CM. Complete genome sequence of the broad-host-range vibriophage KVP40: comparative genomics of a T4-related bacteriophage. J Bacteriol. 2003;185:5220–5233. doi: 10.1128/JB.185.17.5220-5233.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rongvaux A, Andris F, Van Gool F, Leo O. Reconstructing eukaryotic NAD metabolism. Bioessays. 2003;25:683–690. doi: 10.1002/bies.10297. [DOI] [PubMed] [Google Scholar]
- 66.Martin P, Shea R, Mulks M. Identification of a plasmid-encoded gene from Haemophilus ducreyi which confers NAD independence. J Bacteriol. 2001;183:1168–1174. doi: 10.1128/JB.183.4.1168-1174.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ghislain M, Talla E, Francois JM. Identification and functional analysis of the Saccharomyces cerevisiae nicotinamidase gene, PNC1. Yeast. 2002;19:215–324. doi: 10.1002/yea.810. [DOI] [PubMed] [Google Scholar]
- 68.Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994;14:1431–1437. doi: 10.1128/mcb.14.2.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jia SH, Li Y, Parodo J, Kapus A, Fan L, Rotstein OD, Marshall JC. Pre-B cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J Clin Invest. 2004;113:1318–1327. doi: 10.1172/JCI19930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moschen AR, Kaser A, Enrich B, Mosheimer B, Theurl M, Niederegger H, Tilg H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol. 2007;178:1748–58. doi: 10.4049/jimmunol.178.3.1748. [DOI] [PubMed] [Google Scholar]
- 71.Ognjanovic S, Bryant-Greenwood GD. Pre-B-cell colony-enhancing factor, a novel cytokine of human fetal membranes. Am J Obstet Gynecol. 2002;187:1051–1058. doi: 10.1067/mob.2002.126295. [DOI] [PubMed] [Google Scholar]
- 72.Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, Watanabe E, Takagi T, Akiyoshi M, Ohtsubo T, Kihara S, Yamashita S, Makishima M, Funahashi T, Yamanaka S, Hiramatsu R, Matsuzawa Y, Shimomura I. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science. 2005;307:426–430. doi: 10.1126/science.1097243. [DOI] [PubMed] [Google Scholar]
- 73.Arner P. Visfatin--a true or false trail to type 2 diabetes mellitus. J Clin Endocrinol Metab. 2006;91:28–30. doi: 10.1210/jc.2005-2391. [DOI] [PubMed] [Google Scholar]
- 74.Sethi JK. Is PBEF/visfatin/Nampt an authentic adipokine relevant to the metabolic syndrome? Curr Hypertens Rep. 2007;9:33–38. doi: 10.1007/s11906-007-0007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stephens JM, Vidal-Puig AJ. An update on visfatin/pre-B cell colony-enhancing factor, an ubiquitously expressed, illusive cytokine that is regulated in obesity. Curr Opin Lipidol. 2006;17:128–131. doi: 10.1097/01.mol.0000217893.77746.4b. [DOI] [PubMed] [Google Scholar]
- 76.Körner A, Garten A, Bluher M, Tauscher R, Kratzsch J, Kiess W. Molecular characteristics of serum visfatin and differential detection by immunoassays. J Clin Endocrinol Metab. 2007;92:4783–4791. doi: 10.1210/jc.2007-1304. [DOI] [PubMed] [Google Scholar]
- 77.Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, Watanabe E, Takagi T, Akiyoshi M, Ohtsubo T, Kihara S, Yamashita S, Makishima M, Funahashi T, Yamanaka S, Hiramatsu R, Matsuzawa Y, Shimomura I. Retraction. Science. 2007;318:565b. doi: 10.1126/science.318.5850.565b. [DOI] [PubMed] [Google Scholar]
- 78.Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–7442. [PubMed] [Google Scholar]
- 79.Bailey SD, Loredo-Osti JC, Lepage P, Faith J, Fontaine J, Desbiens KM, Hudson TJ, Bouchard C, Gaudet D, Perusse L, Vohl MC, Engert JC. Common Polymorphisms in the Promoter of the Visfatin Gene (PBEF1) Influence Plasma Insulin Levels in a French-Canadian Population. Diabetes. 2006;55:2896–2902. doi: 10.2337/db06-0189. [DOI] [PubMed] [Google Scholar]