

Abstract
Atherosclerosis is an inflammatory disease of the wall of large- and medium-sized arteries that is precipitated by elevated levels of low-density lipoprotein (LDL) cholesterol in the blood. Although dendritic cells (DCs) and lymphocytes are found in the adventitia of normal arteries, their number is greatly expanded and their distribution changed in human and mouse atherosclerotic arteries. Macrophages, DCs, foam cells, lymphocytes, and other inflammatory cells are found in the intimal atherosclerotic lesions. Beneath these lesions, adventitial leukocytes organize in clusters that resemble tertiary lymphoid tissues. Experimental interventions can reduce the number of available blood monocytes, from which macrophages and most DCs and foam cells are derived, and reduce atherosclerotic lesion burden without altering blood lipids. Under proatherogenic conditions, nitric oxide production from endothelial cells is reduced and the burden of reactive oxygen species (ROS) and advanced glycation end products (AGE) is increased. Incapacitating ROS-generating NADPH oxidase or the receptor for AGE (RAGE) has beneficial effects. Targeting inflammatory adhesion molecules also reduces atherosclerosis. Conversely, removing or blocking IL-10 or TGF-β accelerates atherosclerosis. Regulatory T cells and B1 cells secreting natural antibodies are atheroprotective. This review summarizes our current understanding of inflammatory and immune mechanisms in atherosclerosis.
Keywords: inflammation, immune cells, pathophysiology
Introduction
Atherosclerosis is the most common pathological process that leads to cardiovascular diseases (CVD), a disease of large- and medium-sized arteries that is characterized by a formation of atherosclerotic plaques consisting of necrotic cores, calcified regions, accumulated modified lipids, inflamed smooth muscle cells (SMCs), endothelial cells (ECs), leukocytes, and foam cells (1). These features of atherosclerotic plaques illustrate that atherosclerosis is a complex disease, and many components of the vascular, metabolic, and immune systems are involved in this process. Although low-density lipoprotein (LDL) remains the most important risk factor for atherosclerosis, immune and inflammatory mechanisms of atherosclerosis have gained tremendous interest in the past 20 years (1–3). This review focuses on the role of inflammatory cells in atherosclerosis; the molecular mechanisms of their recruitment and retention in atherosclerotic plaque; differentiation, activation, and production of cytokines; as well as other pro- and anti-inflammatory mediators that regulate atherosclerosis and chronic inflammation that accompanies this process. We discuss the association of atherosclerosis with other inflammatory diseases, as well as existing and potential anti-inflammatory treatments for the prevention of atherosclerosis.
In 1829, the term arteriosclerosis was first introduced by Jean Lobstein (4). Within a few years, the associated cellular immune alterations within the arteries were described by two different schools of pathology, resulting in two theories of atherosclerosis. Rudolf Virchow postulated an initial role for aortic cellular conglomerates, emphasizing that cellular pathology is critical in atherosclerosis. In contrast, Carl von Rokitansky suggested that initial injury of the vessel wall owing to mechanical injury and toxins led to endothelial dysfunction and further inflammation (5). Two centuries later, Mayerl et al. (4) analyzed human samples from von Rokitansky's collection and showed T cell accumulation already in early lesions, suggesting that lymphocytes play an essential role in atherosclerosis. In the 1970s, the response-to-injury model was described (1). However, a large number of recent papers have emphasized that the chronic inflammatory response also has an immune component (3). How and why endothelial functions, lipid metabolism, and lipid retention become unbalanced and disturbed is still unclear. Atherosclerosis entails reactivity to self-antigens, but we do not yet know why this occurs so late in life or what role this response might play in atherosclerosis.
Inflammatory Cell Involvement
The presence of leukocytes within atherosclerotic arteries was reported in the early 1980s (6). Initially, investigators thought that only macrophages are predominantly present within atherosclerotic vessels. However, several studies reported the presence of most known leukocytes in both mouse and human aortas (3, 7). The occurrence of inflammatory cells in atherosclerotic lesions (Figure 1) depends on the rate of their recruitment and egress and the balance of proliferation, survival, and apoptosis within the arterial wall. So far, most published studies have investigated short-term leukocyte recruitment, with a small minority studying egress, very few reporting on local proliferation in the vessel wall, and almost none on apoptosis. The reasons for this imbalance are both technical and philosophical. Conducting lymphocyte homing studies is possible, yet finding apoptotic cells in vivo is almost impossible because they are immediately taken up by surrounding phagocytes. Egress studies are technically demanding, and one of the well-established models of egress involves the transplantation of atherosclerotic aortic arches into nonatherosclerotic mice (8). Proliferation can be assessed by dyes such as BrdU (bromodeoxy-uridine) or CFSE (carboxyfluorescein succinimidyl ester), but finding proliferating cells in the vessel wall does not prove that the proliferation occurred locally. Development of more advanced techniques is needed to examine these questions in more detail.
Figure 1.
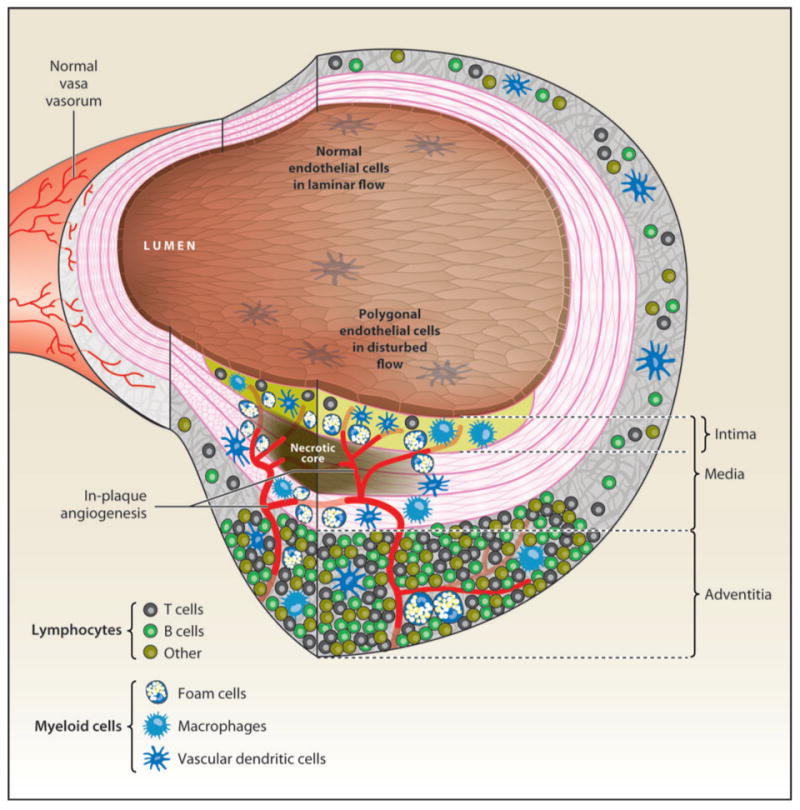
Immune and inflammatory cells in atherosclerosis. Atherosclerotic lesion (foreground, bottom) and relatively unaffected areas. The endothelial cells above the lesion are polygonal in shape (cobblestone), whereas normal endothelial cells are aligned with the direction of flow. The normal intima is so thin as to be invisible at this level of resolution, but it is greatly expanded in the lesion area, where it contains vascular dendritic cells, macrophages, and foam cells (blue) as well as occasional T lymphocytes (gray). The foam cells surround the necrotic core (brown), which is thought to be composed of foam cells that have undergone secondary necrosis. The normal media is populated by smooth muscle cells that are organized by several elastic laminae (magenta lines). These laminae move apart as the smooth muscle cells assume a secretory phenotype and may form foam cells. Myeloid cells (blue) invade the media in the lesion area. The normal adventitia is populated by sparse T cells (gray), B cells (green), and other lymphocytes (brown) as well as vascular DCs (blue). In the lesion area (bottom), the lymphocytes organize into tertiary lymphoid structures, containing high endothelial venules and other vessels. The angiogenic process eventually leads to neovessels invading the intima, a process that is thought to destabilize plaque and precipitate rupture events. The normal adventitia contains some microvessels (vasa vasorum, in background) that do not penetrate the elastic lamina separating the media from the adventitia.
In and Out: Monocyte Recruitment To, Retention In, and Egress from the Artery Wall
DCs and macrophages already reside within the aortas of C57BL/6 mice before atherosclerosis development (9, 10). DCs are observed in the intima of atherosclerosis-predisposed regions of C57BL/6 aorta, and abundant macrophages are found throughout the aortic adventitia (10). Elegant experiments using bone marrow transfer between mice carrying allelic variants of the CD45 common leukocyte antigen demonstrated that monocyte-derived cells in atherosclerotic plaques are of bone marrow origin (11). The mechanisms of monocyte recruitment into noninflamed aortas are not well defined. More is known about monocyte homing to aortas during atherogenesis (12). Monocyte rolling on inflamed endothelium occurs in a P-selectin-dependent manner, and absence of P-selectin results in decreased fatty streaks with concomitant reduction of emigrated macrophages within plaques (13). E-selectin overlaps with P-selectin in supporting rolling. Followed by rolling on inflamed aortic endothelium, monocytes use vascular cell adhesion molecule (VCAM)-1 for slow rolling and tight adhesion (13). Vcam1D4/D4Ldlr−/− mice expressing only 8% of normal VCAM-1 showed decreased early lesion formation (14). There is some evidence that β2 integrins and intercellular adhesion molecule (ICAM)-1 might be involved in the support of monocyte recruitment into aortas, but this was not found in all studies (13).
The chemokine/chemokine receptor network is essential for direction of leukocyte migration in homeostatic and inflammatory conditions (Figure 2). Numerous reports described the important role of the chemokines CXCL1, CCL2, MIF (macrophage migration inhibitory factor), CXCL16, and CX3CL1 and of their receptors CXCR2, CCR2, CXCR2 and CXCR4, CXCR6, and CX3CR1 in the regulation of leukocyte recruitment during atherosclerosis (15).
Figure 2.
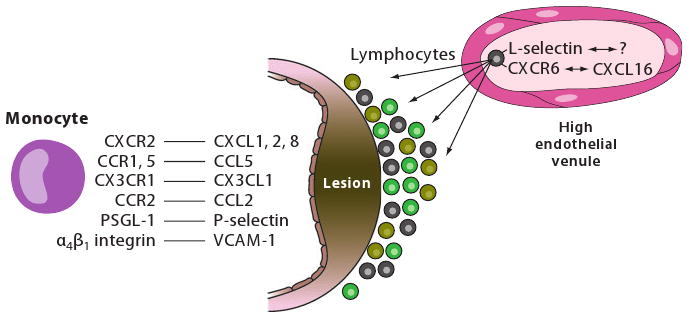
Monocyte recruitment to the atherosclerosis-prone vessel wall. Monocytes use an overlapping network of adhesion molecules and chemokine receptors to enter the artery wall. P-selectin supports rolling and monocyte-platelet interactions. Monocyte α4β1 integrin interacting with endothelial VCAM-1 reduces rolling velocity and leads to firm adhesion. Surface-immobilized chemokines including CXCL1, CXCL2, CXCL4, CCL5, and others can activate monocytes as they roll by, leading to increased adhesiveness of α4β1 integrin through inside-out signaling and receptor clustering. Ly6Chigh monocytes use CCR2, CX3CR1, and CCR5 to migrate to aortas, whereas Ly6Clow monocytes use CCR5. L-selectin, interacting with an unknown ligand, and CXCR6, likely interacting with CXCL16, are partially responsible for lymphocyte recruitment, likely from vasa vasorum and under lesions from high endothelial venules.
Macrophage Polarization
Both M1 and M2 macrophages are found in atherosclerotic lesions. M1 macrophages are the result of classical activation by lipopolysaccharide in the presence of IFN-γ, which leads to production of high levels of IL-12, IL-23, IL-6, IL-1, and TNF-α. Alternatively activated M2 macrophages differentiate in the presence of IL-4, IL-13, IL-1, or vitamin D3 and tend to produce large amounts of IL-10 and express scavenger receptors, mannose receptors, and arginase (20).
Monocyte Subsets
In the circulation of mice, monocytes can be distinguished by differential expression of the Ly6C antigen, CX3CR1, and CCR2 (16). Circulating through lymphoid and nonlymphoid organs under homeostatic conditions, Ly6ChighCCR2+CX3CR1low monocytes are involved in the inflammation caused by microbial infections (17) and have been named inflammatory monocytes. Ly6ClowCCR2lowCX3CR1high resident monocytes may patrol the inside of blood vessels under homeostatic conditions (18) and extravasate during infection with Listeria monocytogenes with differentiation to M2-like, alternatively activated macrophages (see side bar, Macrophage Polarization) (17). Similar subsets of inflammatory and resident monocytes have also been described in human blood (19), but it is not clear whether the mouse and human subsets are truly corresponding.
Recent studies suggest that Ly6Chigh monocytes preferentially migrate into atherosclerosis-prone arteries and predominantly differentiate into aortic macrophages (21), utilizing CX3CR1, CCR2, and CCR5 chemokine receptors (22). These conclusions are based on monocytes loaded with latex beads (22) or cultured in vitro for 24 h (21), both procedures that likely alter the monocyte phenotype (23). The functions of CX3CL1, the only known ligand for CX3CR1, and CCR2 are not completely overlapping because atherosclerosis in Cx3cl1−/−Ccr2−/−Apoe−/− mice is reduced compared with Ccr2−/−Apoe−/− and Cx3cl1−/−Apoe−/− mice (24). Combined inhibition of CCL2, CX3CR1, and CCR5 abrogated monocytosis and almost abolished atherosclerosis (up to 90%) in Apoe−/− mice (25).
LyC6low monocytes require CCR5 for migration to aortas (22), but unknown additional chemokines are likely involved in monocyte recruitment and differentiation in the arterial wall. Monocyte recruitment is also determined by monocyte release from the bone marrow. It is not clear how hyperlipidemia affects monocyte maturation and functions. It is possible and indeed likely that Ly6Chigh and Ly6Clow monocyte-derived macrophages and DCs alter their phenotype under conditions of hypercholesterolemia.
Macrophages
Macrophages were the first inflammatory cells to be associated with atherosclerosis. In a groundbreaking paper, Gerrity and coworkers (6) identified these cells as the main component of the atherosclerotic plaque in porcine specimens. Macrophages produce proinflammatory cytokines, participate in lipid retention and vascular cell remodeling, and express pattern-recognition receptors (PRRs), including scavenger receptors (SRs) and Toll-like receptors (TLRs) that connect the innate and adaptive immune response during atherosclerosis. Macrophages use PRRs to phagocytose different microbes and microbial components. They can also take up modified LDL and form foam cells. There is some controversy about the function of SRs in atherogenesis. Early studies showed that SRs play a strictly proatherogenic role in atherosclerosis, given that Cd36−/−Apoe−/− mice showed protection from atherosclerosis (26). In a separate study, Apoe−/− mice lacking SR-A or CD36 showed increased aortic sinus lesion areas with abundant foam cells, suggesting alternative lipid uptake mechanisms and a possible atheroprotective role for SR-A and CD36 (27). Apoe−/− mice with a combined deficiency of CD36 and SR-A I/II showed no further reduction of atherosclerosis compared with Cd36−/−Apoe−/− mice (28).
Macrophages are extremely plastic cells, taking on many different phenotypes. M1 and M2 macrophages play opposite roles during inflammation, but both are present in atherosclerotic lesions. Using an elegant model of CD11b diphtheria toxin (DT) receptor transgenic mice, Stoneman et al. (29) showed that CD11b+ cells are critical to atherogenesis, but once plaques are established, killing CD11b+ cells does not reduce plaque burden. Because in this model DT administration would eliminate all CD11b+ cells in mice, it is unclear whether the effect from the deletion of CD11b-expressing cells was due to an exclusive role played by macrophages or to a combined role of macrophages, myeloid DCs, and perhaps neutrophils in this model. The ability to form foam cells can also be stimulated by the exposure to lipopolysaccharide (LPS) of Chlamydia pneumoniae through TLR-dependent activation of macrophages resulting in the production of IL-18, IL-12, and IL-15 and promoting a Th1 response with further inflammation within the wall (30). The production of some inflammatory mediators by macrophages is summarized in Figure 3.
Figure 3.

Macrophage functions. Macrophages express scavenger receptors (SRs), TLRs, and other receptors for pathogen-associated molecular patterns (PAMPs). Engagement of these receptors results in release of proinflammatory cytokines IL-1, IL-6, IL-12, IL-15, IL-18, TNF-α, and MIF, as well as anti-inflammatory IL-10 and TGF-β. Vascular endothelial growth factor (VEGF) promotes angiogenesis.
An increase in macrophage apoptosis in early lesions appears to cause the attenuation of atherogenesis, whereas impairment in macrophage apoptosis in the late stage may contribute to secondary necrosis, leading to increased proinflammatory responses and further apoptotic signals for SMCs, ECs, and leukocytes within the plaques (31). Deficiency of phospholipase C β3 resulted in enhanced sensitivity of newly recruited macrophages to 25-OHC- or oxLDL-induced apoptosis in early lesions with concomitant decrease of atherosclerosis (32). Because elimination of phospholipase C β3 leads to no visible effect on the mouse phenotype (32), this may be an attractive target for the modulation of macrophage apoptosis. The adipocyte fatty-acid-binding protein aP2 has an important role in regulating systemic insulin resistance and lipid metabolism and plays a protective role in atherosclerosis (33). Dysregulation in the balance between the influx and efflux of modified LDLs leads to the formation of lipid-laden foam cells. ATP-binding cassette transporter A1 (ABCA1) and G1 (ABCG1) initiate macrophage reverse cholesterol transport in vivo. Combined deficiency of ABCA1 and ABCG1 results in foam cell formation and further acceleration of atherosclerosis (34). Investigators (35) also found that macrophage-specific overexpression of cholesteryl ester hydrolase, which participates in cholesterol efflux, resulted in atherosclerosis reduction in Ldlr−/− mice, demonstrating that enhanced cholesterol efflux and reverse cholesterol transport play important roles in atherosclerosis prevention.
Vascular Dendritic Cells
About a decade after the discovery of vascular macrophages and foam cells, vascular DCs were described in atherosclerotic lesions (36), forming a network of DCs within the intima of arteries but not veins of healthy humans and rabbits. These arterial DCs are CD1a+S-100+lag+CD31−CD83−CD86− and phenotypically similar to Langerhans cells of the skin. Further studies revealed that aortic CD68+CD11c+ DCs have extended long, dendritic-like processes and are mostly located in the intima layers of the lesion-susceptible lesser curvature of the aortic arch (10). Interestingly, the number of intimal DCs, but not the number of CD68+ adventitial macrophages, was significantly reduced in Vcam1D4D/D4D mice, suggesting a role for VCAM-1 in intimal DC recruitment (10).
The function of vascular DCs within healthy and atherosclerosis-prone arteries is poorly understood. DCs are seen in contact with T cells in the atherosclerotic plaques within the zones of neovascularization and near the zones of vasa vasorum with the adventitia. In the immune system, DCs are defined as cells that can present antigen to naive T cells. They constitutively migrate through nonlymphoid organs to secondary lymphoid tissues (17). Whether vascular DCs can present antigen has not been formally demonstrated, but two efforts support this concept: (a) experiments using TCR transgenic mice and antigenic peptide-treated DCs (9) and (b) a study showing that isolated vascular DCs are capable of presenting antigen to transgenic T cells as effectively as bone marrow–derived myeloid DCs (T. Deem & K. Ley, unpublished data) (Figure 4).
Figure 4.

Interactions between DCs and T cells. DCs may present possible atherosclerosis antigens (possibly derived from HSP-60 and oxLDL) to T cells in the context of costimulatory molecules like CD40, OX40L, CD80, and CD86, eliciting T cell differentiation and proliferation. Although a Th1-biased response is documented in atherosclerosis, there is also a significant body of evidence suggesting a possible role for Th2 cells in mature lesions. Whether newly discovered Th17 cells also play a role remains to be investigated. Treg cells have antiatherogenic effects and play a protective role against atherosclerosis, mainly by secreting IL-10 and TGF-β.
In shoulder regions of human unstable plaques, CD83+ DCs are in close proximity to CD40L+ T cells. These DCs produce CCL19 and CCL21 chemokines that might accelerate naive lymphocyte recruitment into atherosclerotic vessels (37). Some DCs within atherosclerosis-prone vessels are IFN-α+ plasmacytoid DCs that responded to pathogenderived motifs (38) and might lead to accelerated apoptosis of CD4+ T cells.
Blood monocytes enter different tissues and differentiate into tissue-resident macrophages or DCs that likely leave nonlymphoid tissue within a few days and migrate back to lymphoid organs through the lymphatic vessels. The number of DCs is significantly elevated in atherosclerosis-prone arteries. Reasons may include accelerated migration into the aorta, reduced emigration out of the vessel wall, increased local proliferation, or decreased apoptosis. In a model of atherosclerosis regression, CCR7-dependent trafficking of monocyte-derived cells out of atherosclerotic plaques was detected during lesion regression (Figure 5), but little emigration was detected from progressive plaques (8, 39).
Figure 5.

Egress of macrophages and DCs from the arterial wall. The factors that control retention of macrophages and DCs in atherosclerotic vessels are not well defined, but sphingosine-1-phosphate (S1P) may have a role in this process. CCR7 expression is necessary for the exit of DCs and macrophages from atherosclerotic plaques.
Mechanisms that might regulate DC responses to modified LDL have not been defined, but some studies suggest that oxLDL plays a role in DC maturation and activation in vivo. Oxidized phospholipids (ox-PLs) alterDC activation; prevent their maturation by blocking TLR3- and TLR4-dependent induction of CD40, CD80, CD83, and CD86; and block IL-12, TNF, and lymphocyte stimulatory capacity (40). OxLDL during the late stage of monocyte differentiation gives rise to phenotypically mature DCs that secrete IL-12 but not IL-10, and it supports both syngeneic and allogeneic T cell stimulation (41).
T Lymphocytes
T lymphocytes mainly reside within the adventitia of normal, noninflamed arteries (7). Short-term adoptive-transfer experiments suggest that this T cell localization is a consequence of constitutive homing of T cells into the aortic wall, which is partially L-selectin dependent (9), and multiphoton microscopy imaging confirms this observation (42). Atherosclerosisprone conditions accelerate T cell recruitment into the aortas in early and advanced atherosclerosis (13). Most of the T cells are TCRαβ+ CD4+ cells with an activated phenotype, and a few express CD8 or TCRγδ (43, 44). Oxidized lipoprotein- and heat shock protein (HSP)-specific T cells are found in atherosclerotic plaques, suggesting local activation and clonal expansion during atherogenesis (45, 46). The set of Vβ and Vα segments are limited within the atherosclerotic lesions with preferential expression of Vβ6 TCR (45). Several studies have also demonstrated CD80-, CD86-, and CD40-CD154-dependent T cell responses to oxLDL (47).
Atherosclerosis is a dynamic process, and the inflammation that accompanies atherosclerosis goes through different stages. There is evidence that early atherosclerosis shows a Th1 response with prevalent production of IFN-γ, IL-6, and IgG2a antibodies against modified LDL (48). T-bet deficiency results in the reduction of lesional SMCs, a switch in the response to HSP-60 toward Th2, an alteration in the T cell–dependent isotype of oxLDL-specific antibodies, an increase in atheroprotective B1-derived antibodies, and reduced atherosclerosis (49). These results suggest that the Th1 response is proatherogenic and affects atherosclerosis not only through the production of proinflammatory cytokines, but also through the regulation of B cell function and antibody production. Severe hypercholesterolemia in Apoe−/− mice induces a switch to a Th2 response and IL-4 production in atherosclerotic lesions, but this Th2 response does not prevent further atherosclerosis development (48).
Adoptive transfer of CD4+ T cells into scid/scid Apoe−/− mice clearly demonstrates the proatherogenic role of CD4+ T cells in these immunodeficient conditions (47). Further studies have demonstrated a more complex role for CD4+ T cells in atherosclerosis. Immunization with MDA-LDL results in CD4+ T cell–independent atheroprotection; however, the atheroprotective immunization with adjuvant injections is CD4+ T cell–dependent (47). It is remarkable and intriguing how CD4+ T cells can play such opposite roles under different conditions. There are at least two sites of action during atherosclerosis: systemic through the secondary lymphoid organs and local within the arteries. Administration of the sphingosine-1-phosphate (S1P) analog FTY720 results in reduced lymphocyte proliferation, IFN-γ production, decreased plasma levels of IL-6, IL-12, and CCL5, and a switch of macrophages to the M2 phenotype, resulting in reduced atherosclerosis in Ldlr−/− (50) and Apoe−/− mice (51). Because FTY720 affects monocyte, T lymphocyte, and B lymphocyte trafficking, it is not clear which population is most responsible for the atheroprotective effect of FTY720.
Regulatory T Cells
The balance between Th1 and Th2 responses is tightly controlled by Tregs, which are critical in maintaining immunological tolerance (52). Atherosclerosis can be considered an autoimmune disease, as significant evidence shows a response to self-antigens such as HSP-60 and LDL during atherosclerosis. Therefore, impaired function or absence of Tregs is likely among the reasons for the local inflammation and proinflammatory response in atherosclerosis. Adoptive transfer of cognate peptide-specific Tregs (Tr1) into Apoe−/− mice diminishes the production of the Th1 cytokine IFN-γ and of IgG2a. Together with elevated IL-10 levels, these phenomena may explain reduced atherosclerosis in recipient mice (53). Adoptive transfer of bone marrow cells deficient in CD4+CD25+ Tregs into irradiated Ldlr−/− donors results in increased lesion size (54). In the same study, transfer of splenocytes deficient in CD4+CD25+ Tregs into Rag2−/−Apoe−/− mice doubled the lesion size compared with mice transferred with wild-type splenocytes. Thus, naturally occurring Tregs that maintain immunological tolerance may play an important role in atherosclerosis prevention (Figure 4).
Which factors might affect the generation and maintenance of Tregs? Recent studies suggest that inducible costimulatory molecule (ICOS) is involved in the Treg response during atherosclerosis. Tregs from Icos−/− mice have an impaired TGF-β-dependent suppressor function compared with wild-type cells (55), suggesting that ICOS is an important molecule controlling Treg functions. Interestingly, the number and activity of Tregs can also be modulated by CD31 (56), but the underlying mechanisms are not known. Unexpectedly, obesity increases Treg numbers and improves Treg function in atherosclerosis-prone mice. Leptin deficiency in Ldlr−/− mice reduces Th1 polarization and improves Treg cell functions, with a significant reduction in lesion development (57). These results identify a critical role for the leptin/leptin receptor pathway in the modulation of the regulatory immune response in atherosclerosis. Another study shows that the oral administration of HSP-60 can induce an increase in CD4+CD25+Foxp3+ Treg cells in HSP-60-treated mice in parallel with decreased atherosclerosis (58). Increased production of IL-10 and TGF-β by lymph node cells in response to HSP-60 was observed after tolerance induction, suggesting key roles for IL-10 and TGF-β in the balancing of the immune response against possible antigens in atherosclerosis.
γδ T Cells
γδ T cells account for 5% of all T cells but are enriched at sites of exposure to antigens such as skin and gastrointestinal mucosa as well as at sites of chronic inflammation. γδ T cells are also observed within the intima of human atherosclerotic vessels at the early stages of atherosclerosis. Interestingly, γδ T cell–deficientApoe−/− mice show no difference in atherosclerosis development throughout the aortic root (59).
Atherosclerosis and Obesity
A growing body of evidence suggests a close association between the immune system, obesity, diabetes, and atherosclerosis (189). Obesity is characterized by an excess of adipose tissue mass and is associated with low-grade inflammation in white adipose tissue, resulting in chronic activation of the immune system. Adipose tissues include adipocytes, preadipocytes, fibroblasts, and macrophages and other types of leukocytes that secrete various cytokine-like hormones, including adiponectin, leptin, resistin, and visfatin, as well as TNF-α, IL-6, IL-1, and CCL2 (190). Adiponectin and leptin are the most abundant adipocyte products and are considered key components in regulating inflammation within the adipose tissues. Serum levels of adiponectin are markedly decreased in obesity, insulin resistance, type 2 diabetes, and atherosclerosis (191). Several reports suggest that adiponectin has antiatherogenic and antithrombotic effects by reducing lipid accumulation in macrophage-derived foam cells (192) and suppressing the production of CXCL10, CXCL11, and CXCL9, leading to reduced T cell homing into atherosclerosisprone aortas (193). Adiponectin can dampen the inflammatory phenotype of ECs and SMCs (190) and curb platelet aggregation (194). In contrast, leptin is considered a proinflammatory and proatherogenic cytokine. Leptin increases the secretion of CCL2 and endothelin-1 by endothelial cells, initiates proliferation and oxidative stress in ECs, and promotes migration and proliferation of SMCs (195). Leptin also facilitates thrombosis by increasing platelet aggregation (196). Treatment with recombinant leptin accelerates atherosclerosis and thrombosis in Apoe−/− mice (197). Paradoxically, leptin resistance is also proatherogenic. Apoe−/− mice lacking the long form of the leptin receptor (db/db) show elevated atherosclerosis compared with control Apoe−/− mice (182). The discovery of adipokines has spawned the concept of an association between atherosclerosis and low-degree inflammation not only within the aortic wall, but also within the surrounding adipose tissues.
Natural Killer Cells
Natural killer (NK) cells were found in early and advanced human atherosclerotic lesions, mainly in the shoulder regions. There is no mouse model that can provide complete NK cell deficiency, but some models, including Il15−/− and Il15ra−/− mice, lack fully functional NK cells (60). Defective cytolysis by NK cells has no effect on atherosclerosis in the model of Ldlr−/− perforin-deficient mice (61). However, Ldrl−/−Lystbeige mutant mice that have defective protein release from cytoplasmic granules show reduced atherosclerosis. Ldrl−/−LystbeigeRag1−/− mutant mice demonstrate increased atherogenesis and lipid levels, which complicate interpretation (61). Because in those models NK cells are still present, other NK cell functions might be involved in atherosclerosis. To further address a possible role of NK cells in atherosclerosis (Figure 6), bone marrow cells from transgenic mice expressing Ly49A under the control of the granzyme A promoter were transferred into lethally irradiated Ldlr−/− mice (62). Absence of fully functional NK cells in this model resulted in 70% reduction of atherosclerotic lesion formation, although it cannot be excluded that some natural killer T cells (NKT cells) and a subset of CD8+ T cells expressing Ly49A might also be affected.
Figure 6.

NK cells. Activated NK cells produce IFN-γ, which promotes a Th1 response, and release perforin and granzymes, which causes apoptosis in target cells.
Natural Killer T Cells
Glycolipid antigens can be presented by CD1 (Figure 7), a major histocompatibility complex (MHC)-like glycoprotein, to CD1-restricted T cells (63). Cellular lipid homeostasis and metabolism play a critical role in atherosclerosis; however, modified lipids may also regulate inflammation through CD1-mediated antigen presentation. The proatherogenic role ofNKT cells has been convincingly demonstrated in different mouse models (60). Cd1d−/−/Apoe−/− mice have a decrease in lesion size up to 25% (64). To show that this effect is dependent on CD1-restricted NKT cell activation, α-galactosylceramide (α-GalCer), a glycolipid that activates NKT cells, was injected into Apoe−/− mice, and it induced a 50% increase in atherosclerosis. In parallel, inflammatory Th1 as well as Th2 cytokines have been observed in Apoe−/− mice that received α-GalCer (64). Interestingly, administration of α-GalCer to Apoe−/− mice with established lesions had no significant effect on size, but it did decrease their collagen content (65). Cd1d−/−Ldlr−/− mice express both IL-12 and IL-10, with unaltered levels of IFN-γ, suggesting that the absence of NKT cells does not significantly alter Th1/Th2 balance (66).
Figure 7.

NKT cells. Dendritic cells present glycolipids on CD1 molecules to NKT cells expressing Vα14 TCR. This results in the production of the Th1 cytokines IFN-γ and TNF-α, the Th2 cytokines IL-4, IL-5, and IL-13, and the anti-inflammatory cytokine IL-10 by the NKT cells and production of IL-12 by the dendritic cells.
Mast Cells
Mast cells are major effector cells in allergy and host defense responses. Upon activation, mast cells release a broad spectrum of proinflammatory cytokines, growth factors, vasoactive substances, and proteolytic enzymes (Figure 8). Although vascular mast cells are rare, they are found within the adventitia and lesions of atherosclerotic plaques, especially in the location of rupture-prone shoulder regions (67). Because mast cells are loaded with proteases such as tryptase and chymase, they might destabilize atherosclerotic plaques. Indeed, mast cells are colocalized within the regions of plaque rupture, and the activation of perivascular mast cells correlates with intraplaque hemorrhage, macrophage and EC apoptosis, vascular leakage, and CXCR2- and very late antigen (VLA)-4-mediated recruitment of leukocytes to the plaque (68). Mast cells alter lipid metabolism by interfering with ApoE- and ApoA-II-dependent cholesterol efflux (69). Mast cell–deficient KitW-sh/W-shLdlr−/− mice show increased collagen content, fibrous cap development, and reduced local inflammation with diminished numbers of T cells and macrophages and reduced atherosclerosis (70). Adoptive transfer of wild-type, but not IL-6- or IFN-γ-deficient, mast cells restores atherosclerosis progression, suggesting that mast cells provide IL-6 and IFN-γ in atherosclerosis.
Figure 8.

Mast cells. Interactions of mast cells with DCs may promote release of proatherogenic TNF-α, INF-γ, and IL-6, a broad spectrum of proteases, the 5-lipoxygenase product leukotriene (LT) B4, and GM-CSF. Mast cells may direct the development of Th1 or Th2 responses.
B Cells
B cells in atherosclerosis were initially discovered within the adventitia, and immunoglobulin-positive cells were detected within atherosclerotic plaques (7). Although investigators did not initially appreciate the impact of B cells on atherosclerosis, recent studies have evaluated the role of B cells in directing the immune response during atherosclerosis (Figure 9). Adoptive transfer of bone marrow from B cell–deficient mice into lethally irradiated Ldlr−/− mice resulted in up to 40% increased lesion size in parallel with decreased production of anti-oxLDL antibodies. This B cell deficiency did not seem to affect Th1 or Treg responses specifically because a simultaneous decrease in IFN-γ, IL-10, and TGF-β was observed (71). Absence of splenocytes upon splenectomy aggravated atherosclerosis in Apoe−/− mice with an associated reduction of anti-oxLDL antibodies. Adoptive transfer of splenic B cells, but not of T cells, from atherosclerosis-prone Apoe−/− mice into young Apoe−/− recipients reduced atherosclerosis (72). These studies indicate that atheroprotective immunity develops during the progression of atherosclerosis and that B cells or their immunoglobulin products may perform protective functions.
Figure 9.

B cells. The B1 subset of B cells is independent of T cell help and produces IgM natural antibodies that appear to have atheroprotective functions. They may be triggered by foreign or self-antigens through their B cell receptor (BCR). Th1-dependent B2 cells produce IgG2a and IgG1. B cells also produce IL-10.
Extensive atherosclerosis in Apoe−/− mice is associated with increased natural antibody titers to oxLDL (73). These IgM autoantibodies to oxLDL recognize ox-PLs containing the phosphorylcholine (PC) head group, and they block binding and degradation of oxLDL by macrophages in vitro (73). The IgM antibodies found in atherosclerosis are structurally and functionally identical to classic natural T15 anti-PC antibodies that are produced by B1 and marginal zone B cells (74, 75). Immunization with malondialdehyde (MDA) leads to the expansion of antigen-specific Th2 cells, elevated production of IL-5, noncognate stimulation of B1 cells, and thus increased production of these antibodies (76). Currently, little information is available about the presence of B1 cells and plasma cells in secondary lymphoid organs and in the aorta under atherosclerosis-prone conditions. In atherosclerotic mice, the spleen is a major source of oxLDL-specific IgM antibodies (73). Much less is known about the local production of natural antibodies within the aorta or their role in preventing modified LDL uptake by macrophages and other cell types.
Neutrophils
Neutrophils are short-lived phagocytic cells with a broad spectrum of biologically active molecules such as myeloperoxidase (MPO) and proteinases (Figure 10). Leukocytosis and especially neutrophilia are independent risk factors for coronary heart disease. CXCR4 and its ligand CXCL12 are involved in the egress of neutrophils from bone marrow and also regulate recruitment of neutrophils to atherosclerotic lesions (77). Chronic blockade of CXCR4 causes neutrophilia and increases neutrophil content in plaques, associated with apoptosis and a proinflammatory phenotype, suggesting a proinflammatory role for neutrophils in atherosclerosis (77). Further studies are necessary to dissect how neutrophils might affect functions of vascular ECs, SMCs, and aortic leukocytes through the production of ROS, enzymes, metalloproteinases, and proinflammatory cytokines. It is unclear whether neutrophils are equally important in human atherosclerosis.
Figure 10.

Neutrophils. Neutrophils, although rare in mature atherosclerotic lesions, interact with the endothelium covering atherosclerotic lesions and may release reactive oxygen species (ROS), myeloperoxidase (MPO), proteases, and the chemokines CXCL1 and CXCL8.
Platelets
Platelets play a major role in the hemostatic process and in thrombus formation upon injury. They can regulate the inflammatory and immune responses through the secretion of inflammatory mediators that modulate leukocyte recruitment into inflamed tissues (78). Activated platelets expressing P-selectin are detected at different stages of atherosclerosis (Figure 11). Bone marrow transfer experiments with P-selectin-deficient mice show that platelet P-selectin contributes to lesion development and assists in calcification of atherosclerotic plaques (79). Activated platelets transiently interact with the endothelium of atherosclerotic carotid arteries of Apoe−/− mice in vivo. This transient interaction results in immobilization of platelet-derived CCL5 and CXCR4 on atherosclerotic endothelium (80, 81). Platelet glycoproteins GPIIb/IIIa, GPIb, and endothelial von Wille-brand factor are at least partially responsible for the platelet-endothelial interactions. Immobilized platelets also interact with leukocytes through P-selectin/P-selectin glycoprotein ligand (PSGL)-1 interactions that activate Mac-1 and VLA-4 integrins and may facilitate firm monocyte adhesion (80). Platelets also initiate rolling of DCs through PSGL-1-dependent interactions between Mac-1 and junctional adhesion molecule (JAM)-C in injured carotid arteries (82). Surprisingly, platelets are also required for CX3CL1-induced leukocyte adhesion at high shear rates. Both soluble and membrane-bound CX3CL1 trigger P-selectin expression on adherent platelets, which facilitates the local accumulation of leukocytes under arterial shear (83). Platelets also help recruit and differentiate progenitor cells into ECs through the interactions of P-selectin and PSGL-1 and by using the β1 and β2 integrins. Platelets bridge inflamed endothelium and circulating blood cells and support recruitment of leukocytes to inflamed atherosclerotic endothelium.
Figure 11.

Platelets. Activated platelets release proinflammatory IL-1β, CD40L, CXCL12, CXCL4, and CCL5 as well as anti-inflammatory TGF-β. Through P-selectin binding to PSGL-1, platelets interact with monocytes.
The Case for a Vascular Immune Response
Anatomical Proximity
Although much evidence supports the involvement of the immune system in a systemic response to hyperlipidemia, a significant body of data also suggests a local immune response within the aortic wall. Atherosclerotic plaques are formed in very specific regions of the aortic tree where flow is disturbed (84). In contrast, very little inflammation or atherosclerosis is found in laminar flow regions (10). Activated by disturbed flow, ECs elevate expression of adhesion molecules and chemokines, which accelerate leukocyte recruitment. Rolling and firm adhesion of monocytes to aortic endothelium was documented in an ex vivo model of the carotid artery and in vivo (13). Eriksson et al. (85) demonstrated a role for L-selectin-dependent secondary capture in leukocyte accumulation in inflammation and atherosclerosis in vivo. However, there is no direct intravital microscopic evidence for lymphocyte recruitment from the arterial lumen into the vessel wall.
Several reports demonstrate T and B lymphocyte accumulation in the aortic adventitia in normal (9) and atherosclerotic vessels (9, 85, 86). Adoptive transfer experiments suggest that lymphocytes accumulate in the adventitia through the migration from the adventitial vasa vasorum rather than from the intimal lumen site (9). Local revascularization correlates with an increase in cellular composition within vulnerable regions of human atherosclerotic plaques (Figure 1). In contrast, the inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis (87). Recently, investigators have shown that vasa vasorum can penetrate the media, enter atherosclerotic plaques, and come close to the arterial lumen (88). This is an important direct demonstration of the existence of a vascular network connecting the adventitia with the plaque tissue. Thus, we now better understand the role of neovascularization in atherosclerosis (87), but further studies are necessary to elucidate the role of small adventitial vessels in the immune response during this disease.
The presence of antigen-presenting cells and T cells within atherosclerosis-prone artery walls is well documented, but there is little information about local antigen-dependent activation of T cells. It remains to be determined whether elevated numbers of lymphocytes, which have been seen in atherosclerotic vessels, are a consequence of the accelerated recruitment of activated cells from draining lymph nodes or of local antigen-induced proliferation that leads to the increased aortic lymphocyte numbers.
One of the possible sites of T cell activation in aorta may be vascular-associated tertiary lymphoid structures (Figure 1). The lymphoid-like structures are formed in a variety of autoimmune-mediated diseases, such as rheumatoid arthritis or Hashimoto's thyroiditis. Conglomerates of leukocytes within the adventitia were reported in the early 1970s; however, only in 1997 did Wick et al. (44) name these conglomerates vascular-associated lymphoid tissues (VALTs). These lymphoid structures are formed within advanced atherosclerosis-prone vessels and contain T and B lymphocytes, plasma cells, CD4+/CD3− inducer (LTi) cells, and some MECA-32+ and HECA-452+ microvessels (9, 86, 89). Follicles located close to the arterial external elastic lamina contain proliferating Ki67+ leukocytes, apoptotic cells, and CD138+ plasma cells, showing local B cell maturation and possible humoral immune response in these structures (86). Whether the VALTs in atherosclerosis are beneficial or proatherogenic is still unclear.
Cytokine Involvement in Atherosclerosis
Cytokines are key players during acute and chronic inflammation. The regulation of cytokine production depends on many factors and is tightly regulated during inflammation. An excellent review by Tedgui et al. (90) analyzed the cytokine biology in atherosclerosis. Many cytokines, such as TNF-α, IL-1, IL-2, IL-3, IL-6, CXCL8, IL-10, IL-12, IL-15, IL-18, IFN-γ, M-CSF, TGF-β1, TGF-β2, and TGF-β3, are detected within atherosclerosis-prone vessels (90). SMCs and ECs produce TNF-α, IL-1, IL-6, CXCL8, and IL-15 and regulate the production of other cytokines in an autocrine and paracrine manner. ECs affect hematopoietic cell proliferation through the production of stem cell factor, IL-3, GM-CSF, G-CSF, and M-CSF (90). Under conditions of hyperlipidemia, macrophages produce TNF-α, IL-1, IL-6, IL-12, IL-15, and IL-18 but also the anti-inflammatory cytokines IL-10 and TGF-β. Proatherogenic mast cells generate IL-6 and IFN-γ that are crucial in the induction of mast cell–dependent acceleration of atherosclerosis. Upon activation, platelets shed IL-1β, CD40L, and CXCL4, which may serve as proatherogenic mediators. Cytokines can affect endothelial permeability, the expression of adhesion molecules, SR, lipid metabolism, and proliferation and migration of SMCs and ECs. Cytokines also influence extracellular matrix composition through the alteration of the expression of matrix metalloproteinase (MMP)-1, -3, -8, -9, and -12 and their inhibitors TIMP-1, -2, and -3 (91).
Neovascularization is a proatherogenic process that greatly depends on the local microenvironment of cytokines and growth factors (87). T cell–derived cytokines are likely responsible for plaque neovascularization because many T cells are found within the regions of microvessels surrounding the plaques. Proatherogenic IL-1β, TNF-α, and leptin, as well as vascular endothelial growth factor (VEGF) and placental growth factor (PIGF), accelerate neovascularization. IL-10, CXCL9, CXCL10, and adiponectin may balance the process of neovascularization and play antiatherogenic and antiangiogenic roles.
TNF-α
Several studies were conducted to analyze the role of TNF-α in atherogenesis, but until recently results were contradictory. TNF-α-deficient Apoe−/− mice showed a reduction in lesion formation, with a concomitant decrease in VCAM-1, ICAM-1, and CCL2 expression (92). In contrast, mice deficient in TNF-α receptor (TNFR) p55 developed larger lesions compared with controls (93). Because of elevated cholesterol levels in p55-deficient mice, it was difficult to interpret whether TNFR deficiency itself resulted in the acceleration of atherosclerosis. Recently, a study with mutant mice clearly identified the roles of transmembrane and soluble forms of TNF in the context of hyperlipidemia. TNF-α affected the development of atherosclerosis at the fatty streak stage, and cleavage of TNF was an important step in activating the proatherogenic properties of TNF-α (94).
IL-1
IL-1 stimulation initiates leukocyte adhesion to ECs and transmigration and serves as a local autocrine and paracrine stimulator of other cytokines (90). Studies with blocking IL-1ra antibodies in Apoe−/− mice and with Ldlr−/− transgenic mice that overexpress IL-1ra or that have a deficiency in IL-1β clearly show that IL-1 is involved in atherogenesis (90).
IL-2
That IL-2 may serve as a proinflammatory, proatherogenic cytokine was shown in experiments with the administration of IL-2 or IL-2-blocking antibodies into Apoe−/− mice (95). However, further experiments are needed to analyze the differential role of IL-2 on Tregs and in lipid metabolism.
IL-6
There are two lines of studies that propose a specific role for IL-6 in atherosclerosis: IL-6 administration into wild-type C57BL/6 mice increases the formation of fatty streaks (96), but Il6−/−Apoe−/− mice show elevated atherosclerosis and decreased leukocyte homing (97, 98). It is important to note that IL-6-dependent regulation of lipid metabolism may have confounded these studies.
IL-12
IL-12 is a key Th1 cytokine that is produced mainly by plaque macrophages and stimulates proliferation and differentiation of NK cells and T cells. IL-12 is detected in the aortas of Apoe−/− mice, and the administration of IL-12 results in enhanced lesion size in Apoe−/− recipients (99). IL-12p40-deficient Il12b−/−Apoe−/− mice have a 52% reduction of plaque area at 30 weeks, but not at 45 weeks of age (100). Some interesting details of IL-12 properties come from a study that analyzed CD4+CD28− T cells in mice. This subset of T cells expresses IL-12 receptor in the absence of antigen stimulation. Upon activation with IL-12, they up-regulate their CCR5 expression, chemotaxis, and transendothelial migration toward CCL5 (101). Together, these results support the notion that IL-12 is proatherogenic and proinflammatory.
IL-18
IL-18 and IL-12 are involved in the generation of Th1 effector cells. Unexpectedly, administration of IL-18 antibodies accelerates lesion development in Apoe−/− mice, but overexpression of IL-18-binding protein abolishes the proinflammatory effects of IL-18, inducing a more stable plaque (90). Il18−/−Apoe−/− mice exhibited reduced expression of I-Ab and IFN-γ, elevated IgG production, and reduced lesions compared with Apoe−/− controls (102). IL-18 seems to enhance atherosclerosis mainly by increasing IFN-γ (103). Interestingly, in Il18−/−Apoe−/− mice, serum cholesterol and triglyceride levels are higher than in Apoe−/− mice, indicating that IL-18 somehow downregulates circulating cholesterol in serum (102).
IFN-γ
IFN-γ administration accelerates atherosclerosis in Apoe−/− mice (104). Conversely, IFN-γ receptor–deficient mice on the Apoe−/− (105) or Ldlr−/− background (106) show decreased atherogenesis. T cell–independent IFN-γ secreted by macrophages, NK cells, and vascular cells seems to be sufficient for disease progression. Moreover, IFN-γ can induce atherosclerosis in scid Apoe−/− mice in the absence of detectable leukocytes by acting on SMCs to prime for growth factor–inducible proliferation (107). IFN-γ is involved not only in early but also in late stages of atherosclerosis. Advanced atherosclerotic lesions can be reduced in size and stabilized in composition by IFN-γ inhibition (108).
IFN-α
IFN-α is a pluripotent inflammatory cytokine typically induced by viral infections. Interestingly, IFN-α produced by aortic plasmacytoid DCs induced a tenfold increase of TNF-related apoptosis-inducing ligand (TRAIL) on CD4+ T cells and enhanced their cytolytic capacity toward SMCs (109). IFN-α also sensitized antigen-presenting cells to pathogen-derived TLR4 ligands by upregulation of TLR4 and intensified TNF-α, IL-12, and MMP-9 production that led to further plaque destabilization (38). Thus, IFN-α provides a possible link between viral infections and immune-mediated complications of atherosclerosis.
CD40/CD40L
T lymphocytes, platelets, ECs, SMCs, macrophages, and DCs express CD40L, whereas CD40 is found on macrophages, ECs, and SMCs from atherosclerosis-prone vessels. The interaction of CD40 with CD40L plays a significant role in thrombosis, but it also contributes to the modulation of the immune response in plaques. Treatment with antibodies against CD40L reduces atherosclerosis in Ldlr−/− mice, with a concomitant decrease of macrophages and T cells and a reduction in VCAM-1 expression (110). Further experiments using Cd40lg−/−Apoe−/− mice have demonstrated a proatherogenic role for CD40L in advanced atherosclerosis by promoting lipid core formation and plaque destabilization. Unfortunately, therapeutic effects directed at CD40L failed clinical trials because of an enhanced thrombosis risk. Cd40−/−Ldlr−/− mice showed no reduction in atherosclerotic lesion formation, suggesting a possible alternative ligand for CD40L (111).
Anti-Inflammatory
IL-4
IL-4 is a Th2 cytokine with an unclear role in atherosclerosis. Neither exogenous administration of IL-4 into Apoe−/− mice nor IL-4 deficiency had any effect on lesion size in mice fed normal or saturated fat diets for 4 weeks (112). However, a long-term diet study in Il4−/−Apoe−/− mice demonstrated a 27% reduction in plaque area at 30 weeks of age and a reduction in aortic arch lesions at 45 weeks of age, compared with Apoe−/− mice (100). Il4−/− mice immunized with HSP-65 showed reduced formation of early atherosclerosis, with concomitant reduction of anti-HSP-65 antibodies and elevated IFN-γ production (113).
IL-5
Immunization with MDA shifts the immune response in atherosclerotic mice toward Th2 and induces significant production of natural antibodies of the EO6/T15 type. IgM antibodies like EO6 (74, 75) found in atherosclerosis are structurally and functionally identical to classic natural T15 anti-PC antibodies that are produced by B1 and marginal zone B cells, with concomitant elevation in IL-5 (73). IL-5 further stimulates B1 cells, leading to increased production of these antibodies. Experiments with bone marrow transferred from Il5−/− or Il5+/+ mice suggest an atheroprotective role for IL-5 (73).
IL-10
IL-10 derived from Th2 cells, B cells, monocytes, and macrophages is an important regulator of the balance between Th1 and Th2 responses. Administration of IL-10 delayed atherosclerosis development. Conversely, IL-10-deficient mice showed increased T cell accumulation and IFN-γ production with diminished collagen content in the atherosclerotic vessels (90). Studies with Il10−/−Apoe−/− confirmed the atheroprotective properties of IL-10 at the early stage of atherosclerosis and showed that IL-10 also promotes the stability of advanced plaques (114).
IL-33
IL-33 is expressed in normal and atherosclerosis-prone arteries (115). The administration of IL-33 significantly reduces atherosclerosis in Apoe−/− mice by mechanisms that involve a switch from a Th1 to a Th2 response, with concomitant increases in IL-4, IL-5, and IL-13, reduction of proinflammatory cytokines, and diminished IFN-γ production (115). IL-33 may also neutralize harmful oxLDL through IL-5-dependent production of antibodies against oxLDL (115).
TGF-β
TGF-β is produced by several cell types, including ECs, SMCs, macrophages, platelets, and Treg cells. Several studies suggest that TGF-β regulates atherosclerosis through the modulation of SMC and EC phenotypes, as well as by regulating Th1 functions. Introducing blocking antibodies against TGF-β or treatment with soluble TGF-β receptor II accelerates atherosclerosis with significant loss of collagen content (90). Apoe−/− mice that express a dominant-negative form of the TGF-β receptor II in T cells, as well as Ldlr−/− irradiated mice that received bone marrow from mice expressing a dominant-negative TGF-β receptor type II under a T cell–specific promoter, both clearly demonstrated a substantial role for TGF-β in controlling the Th1 response in atherosclerosis (116, 117).
Chemokines in Atherosclerosis
Substantial evidence from clinical and experimental research suggests that chemokines and chemokine receptors play critical roles in directing leukocytes into atherosclerosis-prone vessels. Taking into account the chemokine abundance within atherosclerosis-prone arteries and the variety of chemokine receptors on leukocytes, it is clear that a tightly controlled network regulates recruitment, retention, and emigration of leukocytes in the arterial wall (15).
CCL2 was the first chemokine shown to affect atherosclerosis. CCL2 and its receptor CCR2 are most prominently involved in monocyte recruitment from the bone marrow (118) and into the arterial wall (119). Studies with CCL2- and CCR2-deficient mice clearly demonstrate that this pair is mainly involved at the early stages of atherosclerosis (120–123).
The important role of platelets is suggested by their capacity to quickly release the proinflammatory chemokines CCL5 and CXCL4 to endothelium and thus initiate monocyte and T cell recruitment into the vessel wall (80). CXCL4 can induce activation of ECs by inducing expression of E-selectin, NF-κB activation, and enhanced binding of oxLDL to ECs (124). However, Ccr5−/−Apoe−/− mice lacking CCR5, one of the receptors for CCL5, had no significant reduction in early atherosclerosis (125), and Ccr5−/− bone marrow–derived cells affected atherosclerosis only transiently (126). A more recent study found a more than 50% reduction of lesion size in the aortic root and the thoracoabdominal aorta of Apoe−/−Ccr5−/− mice and fewer macrophages and T cells in lesions compared with Apoe−/− mice (127).
CXCL8 induces proliferation and migration of SMCs and ECs and affects neovascularization. In the Apoe−/− mouse model, CXCL1 initiated monocyte arrest through the activation of VLA-4 integrin (128).
Recently, a unique role for chemokines was documented in the shear stress–dependent modulation of atherosclerotic lesion composition (129). Expression of CCL2, CXCL10, and CXCL1 was detected in low shear stress regions, and exclusive expression of CX3CL1 was observed in the low shear stress regions that had thinner fibrous caps and larger necrotic cores (129).
The cytokine MIF is produced by ECs, SMCs, and macrophages in early and advanced atherosclerotic lesions. Binding of MIF to its newly discovered receptor complex of CXCR2 and CD74 resulted in elevated monocyte arrest on atherosclerotic endothelium (130). Studies with Mif−/−Ldlr−/− mice suggest that MIF is involved in atherosclerosis through the regulation of lipid deposition, protease expression, and intimal thickening (131).
CX3CL1 is expressed in atherosclerotic plaques in a transmembrane form and can initiate the arrest of CX3CR1+ NK cells, subsets of T lymphocytes, and monocytes (15). CX3CL1 can also be shed in an ADAM-17-dependent manner. The cleaved form attracts CX3CR1-expressing cells. Several independent studies with Cx3cr1−/−Apoe−/− or Cx3cl1−/−Ldlr−/− mice clearly demonstrate a proatherogenic role for the CX3CL1/CX3CR1 axis in atherosclerosis (15).
The only other known transmembrane chemokine is CXCL16, which has dual functions as SR and soluble chemokine and also is involved in atherogenesis (132). CXCL16 is expressed by SMCs, ECs, and macrophages. Cxcl16−/−Ldlr−/− mice had accelerated atherosclerosis, likely because of the lack of CXCL16 function as an SR (132). In contrast, the absence of CXCR6 in Cxcr6GFP/GFPApoe−/− mice resulted in reduced T cell number within the lesion, dampening the inflammatory response at the lesion site, reducing macrophage infiltration, and diminishing atherosclerosis (133).
CXCL10 is a potent mitogenic and chemotactic factor for SMCs and can modulate the local balance of the effector and regulatory arms of the immune system through the induction of Tregs in aortas (134). Other chemokines, including CCL3, CCL4, CCL11, and CXCL12, are expressed in human and mouse atherosclerotic aortas, but the role of these chemokines in atherosclerosis remains unclear (15).
Inflammation-Regulating Enzymes in Atherosclerosis
5-lipoxygenase
The 5-lipoxygenase (5-LO) pathway is responsible for the production of leukotrienes, inflammatory lipid mediators that have a role in innate immunity but that can also play a proatherogenic role (135). Expression of 5-LO and leukotriene A4 hydrolase in atherosclerotic segments correlates with plaque instability. 5-LO-deficient Ldlr−/− mice show a dramatic decrease in lesions, and bone marrow transplantation experiments suggest that macrophage 5-LO is mainly responsible for atherogenesis (136).
12/15-LO
The proatherogenic effect of 12/15-LO has been established in 12/15-LO-deficient Apoe−/− mice that showed reduced lesions throughout the whole aorta (137). Unexpectedly, these mice also had diminished plasma IgG autoantibodies to oxidized LDL, which suggests that the 12/15-LO pathway affects not only lipid peroxidation but also the adaptive immune response (137). Overexpression of 12/15-LO in C57BL/6 mice leads to the formation of fatty streak lesions, at least partially through the elevated adhesion of monocytes to endothelium (138). Further experiments demonstrated that 12/15-LO expression in bone marrow–derived cells was responsible for the proatherogenic properties of 12/15-LO in vivo (139). 12/15-LO induces the production of IL-6, TNF-α, and CCL2 and therefore connects the metabolic and immune branches of atherosclerosis (140). By contrast, rabbits overexpressing human 15-LO showed reduced lesion size (141). The reason for this difference may be related to the many products of 12/15-LO, some of which are pro- and other anti-inflammatory (142).
Heme Oxygenase -1
Heme oxygenase (HO) catalyzes the rate-limiting step of heme catabolism. The induction of HO-1 reduced monocyte chemotaxis in response to LDL oxidation (143). The absence of HO-1 exacerbated atherosclerosis in HO-1-deficient Apoe−/− mice (144), and macrophages expressing HO-1 are crucial players in this process (145). From a therapeutic point of view, it is important to mention that HO-1 is also involved in antioxidant-dependent protection from atherosclerosis (146).
Paraoxonases
The paraoxonase family consists of three members (PON1, PON2, and PON3) that share structural properties and enzymatic activities, among which is the ability to hydrolyze oxidized lipids in LDL. PON1 prevents oxidation of LDL as well as high-density lipoprotein (HDL), with which PON1 is associated in the serum (147). HDL of Pon1−/−Apoe−/− mice is predisposed to oxidation, and as a consequence lesions in Pon1−/−Apoe−/− mice are larger compared with controls (148). In several other studies using transgenic mice overexpressing PON1, the role of PON1 as inhibitor of lipid oxidation was confirmed (147).
Proinflammatory Mediators
OxLDL
According to the oxidation hypothesis of atherosclerosis, oxLDL plays a pivotal role through the induction of foam cell formation, alteration of nitric oxide signaling, initiation of endothelial activation, and expression of adhesion molecules that accelerate leukocyte homing to the site of atherosclerosis (149). One of the key observations that crystallized the important role of oxLDL in atherosclerosis came from a study that showed heparan sulfate–dependent binding of oxLDL to subendothelial matrix (150). Generation of lectin-like oxLDL receptor-1-deficient Olr1−/−Ldlr−/− mice showed reduced atherosclerosis by luminal obstruction and intima thickness (151). OxLDL can also directly affect the migration of monocytes to the aortic wall by switching from CCR2 to CX3CR1 expression using a peroxisome proliferator-activated receptor γ–dependent pathway (152).
Autoantibodies to oxLDL are found within normal/nondiseased and atherosclerosis-prone lesions. IgG autoantibodies to oxLDL are associated with proatherogenic properties, and IgM autoantibodies to oxLDL, including natural antibodies, have been proposed as atheroprotective (73). The mechanism by which the immune response coordinates the production of these two isotypes in atherosclerosis remains to be identified. Importantly, it is possible to initiate tolerance to oxLDL and MDA-LDL by oral administration of oxLDL or MDA-LDL before the induction of atherogenesis and to promote generation of oxLDL-specific CD4+CD25+Foxp3+ Treg cells (58).
C-Reactive Protein
Elevated plasma C-reactive protein (CRP) is associated with increased risk of atherosclerosis, but the mechanisms have not been fully identified (153). CRP is found within atherosclerotic plaques close to LDL and macrophages. Recently, several reports demonstrated that CRP can modulate endothelial functions and leukocyte activities. CRP also induced the production of IL-1α, IL-1β, IL-6, CXCL1, and CXCL8 by human monocytes in vitro. In contrast to these proinflammatory properties, CRP also displayed anti-inflammatory effects through upregulation of liver X receptor-α (153). CRP binds to minimally modified (mm)LDL and prevents the formation of foam cells from macrophages (154). The functional importance of these observations in vivo and the exact functions of CRP in atherogenesis remain to be investigated.
Advanced Glycation End Products (AGE)
Nonenzymatic modification of proteins by reducing sugars leads to the formation of AGEs in vivo. These reactions take part during aging and substantially accelerate during cancers, diabetes, and atherosclerosis (155). Although the mechanisms are not fully identified, the alterations in glucose and lipid metabolism likely lead to the production of excess aldehydes and formation of AGEs. AGEs act directly or via receptors and participate in the cross-linking proteins of extracellular matrix (155). The receptor for AGE (RAGE) is a member of the immunoglobulin superfamily and is expressed by ECs, SMCs, monocytes, and lymphocytes with enhanced expression in atherosclerotic lesions. Neutralizing AGE by the administration of soluble recombinant RAGE reduced NF-κB induction, VCAM-1 and tissue factor expression, and atherosclerotic lesion burden (156). A critical role for RAGE and its ligands was also demonstrated in RAGE-deficient Apoe−/− mice and in Apoe−/− transgenic mice expressing human dominant-negative RAGE (157). The AGE/RAGE axis has a broad spectrum of effects and elicits oxidative stress, increases endothelial dysfunction, increases production of inflammatory cytokines and tissue factor, elevates expression of adhesion molecules, and, through all these mechanisms, accelerates development of atherosclerosis (155).
Reactive Oxygen Species
Extensive production of ROS has been implicated in atherosclerosis by inducing the chronic activation of the vascular endothelium and components of the immune system. Vascular endothelial ROS released from nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, MPO, xanthine oxidase, lipoxygenases, nitric oxide synthases, and the dysfunctional mitochondrial respiratory chain may play critical roles in ROS generation. In humans, higher expression of NADPH oxidase subunit proteins is associated with increased superoxide (O2−) production and severity of atherosclerosis (158). NADPH oxidase-deficient Apoe−/− mice had significantly less atherosclerosis compared with Apoe−/− mice (159). Further studies clearly demonstrated that superoxide production from both monocytes/macrophages and vascular cells plays a critical role in atherogenesis (160). One of the mechanisms by which superoxide affects atherogenesis is the activation of SMC mitogenic signaling pathways (160). Platelets also produce ROS, and NADPH-induced superoxide production results in enhanced availability of released ADP and amplified platelet recruitment (161). ROS can be neutralized by antioxidants such as superoxide dismutase, catalase, glutathione peroxidase, glutathione reductase, and others, but ROS production can exceed the scavenging capacity of cellular antioxidant systems, and the resulting oxidative stress can damage lipids, membranes, proteins, and DNA. The mechanisms of regulating antioxidant activity are multidirectional, but it is important to mention that laminar shear stress upregulates a mechanosensitive group of antioxidant enzymes, peroxiredoxins in ECs.
Complement System
The complement system plays a central role in innate immunity and also regulates the adaptive response (162). Complement activation is essential to the host's immune defense, but its uncontrolled or inappropriately targeted activation leads to various diseases such as glomerulonephritis, rheumatoid arthritis, psoriasis, and CVDs (162). Emerging evidence suggests that the complement system plays a role in atherosclerosis, although its exact functions and mechanisms of action remain unclear. Modified lipoproteins and apoptotic/necrotic cells have been shown to activate the alternative classical complement pathways. Studies using complement-deficient animals have yielded apparently contradictory conclusions. C6 deficiency protects against diet-induced atherosclerosis in rabbits (163); however, no difference was observed in diet-induced lesion size in C5-deficient Apoe−/− mice (164) or C3-deficient Ldlr−/− mice (165). The classical pathway of complement activation may be protective because it promotes the clearance of apoptotic cells and immune complexes during atherosclerosis. Indeed, C1q-deficient Ldlr−/− mice have more apoptotic bodies within their plaques and larger atherosclerotic lesions (166). The role of the complement system at the advanced stages of atherosclerosis is not known, but examination of human tissues demonstrates activated complement in human vulnerable plaques prone to rupture (167).
Heat Shock Protein 60
There are distinct cellular and humoral reactions against microbial HSPs in humans that participate in host defense. However, because of a high degree of sequence homology between microbial and human HSPs, autoimmune responses may be triggered against human HSPs (168). Anti-HSP antibodies elicit production of proinflammatory cytokines by macrophages and of adhesion molecules by ECs. Autoantibody levels against HSPs are significantly increased in patients with atherosclerosis, and HSP-specific T cells have been observed within atherosclerotic plaques. Most of the known risk factors for atherosclerosis, such as oxLDL, hypertension, infections, and oxidative stress, evoke increased expression of HSPs in ECs, SMCs, and macrophages. Endothelial HSP-60 correlates with site-specific, flow-dependent atherosclerosis development throughout the aortic tree. Altered wall shear stress after ligation of the left common carotid artery induced rapid production of HSP-60 by ECs at this site (169), which may provide conditions for humoral and cellular reactions to endothelial HSPs in the earliest stages of atherosclerosis. Other HSPs such as HSP-90 might also be involved in atherosclerosis (170).
Toll-Like Receptors
There is a significant body of evidence that not only metabolic mediators but also bacterial and viral infections might amplify atherosclerosis and worsen the outcome by promoting a proinflammatory status of the vessel wall. TLRs are the primary receptors of the innate immune system that recognize highly conserved structural motifs of pathogens. TLR2 and TLR4 are also receptors for HSP-60 (171). Under conditions of hyperlipidemia, TLRs likely participate in the regulation of atherosclerosis. Activation of TLRs induces the production of proinflammatory cytokines and nitric oxide in macrophages and the induction of DC maturation, leading to the upregulation of costimulatory molecules such as CD80 and CD86. TLR1, TLR2, TLR4, and TLR5 are expressed in atherosclerotic lesions, and TLR4 can be upregulated by oxLDL. Interestingly, atheroprotective laminar flow downregulates TLR2 expression. Mice lacking MyD88, a signaling molecule downstream of most TLRs, showed reduced atherosclerosis (172). Myd88−/−Apoe−/− and Tlr4−/−Apoe−/− mice exhibited reduced atherosclerosis that was associated with reduction in the circulating levels of the proinflammatory cytokines IL-12 and CCL2, plaque lipid content, numbers of macrophages, and cyclooxygenase 2 immunoreactivity in plaques (173). TLR2 deficiency on non–bone marrow–derived cells resulted in diminished atherosclerosis in Tlr2−/−Ldlr−/− mice, suggesting that an unknown endogenous TLR2 agonist influenced lesion progression by activating TLR2 in cells that were not of bone marrow origin, most likely ECs (174). Thus, at least TLR2 and TLR4 participate in the inflammatory arm of atherosclerosis.
Association with Inflammatory Diseases
Systemic Lupus Erythematosus
Atherosclerosis is associated with many chronic inflammatory diseases. As we start to appreciate possible similarities between atherosclerosis and autoimmune diseases, we must consider the possible impact of chronic inflammation on the acceleration of atherosclerosis. Systemic lupus erythematosus (SLE) is a complex autoimmune disease involving multiple organs that is characterized by autoantibody production (175). In recent years, much attention has been given to the rising incidence of accelerated atherosclerosis and increased risk of CVDs in patients with SLE. Increased production of CCL2, TNF-α, IFN-γ, IL-1, IL-12, and immune complexes, upregulation of adhesion molecules, and increased antibodies to oxLDL may promote atherosclerosis. Gld.Apoe−/− mice that carry an inactivating mutation in the Fas ligand gene (FasL) develop lupus-like autoimmune disorders (176). The gld.Apoe−/− mice displayed enhanced atherosclerosis compared with Apoe−/− mice, accompanied by an increase in lymphocyte proliferation and autoimmunity. The gld.Apoe−/− mice had high levels of apoptotic material both in tissues and in the circulation. This was due, at least in part, to an impaired ability to scavenge apoptotic debris, suggesting that the synergism between atherosclerosis and SLE can be mediated by impaired apoptotic body clearance (176). Fas-deficient Apoe−/− mice also showed increased production of IgG antibodies against dsDNA and cardiolipin, as well as accelerated atherosclerosis (177).
Metabolic Syndrome and Diabetes
Metabolic syndrome is defined as prediabetes, abdominal obesity, elevated LDL cholesterol, and increased blood pressure that significantly correlates with CVD. Despite evidence for a tight correlation between atherosclerosis and metabolic syndrome, mechanisms by which these diseases accelerate each other are not well identified and very little is known about the underlying basis for differential susceptibility to vascular injury in patients with diabetes. Diabetes-accelerated atherosclerosis is observed in type 1 and type 2 diabetic patients, but it is not known whether atherosclerosis is induced through the same mechanisms (178). In type 1 diabetes, hyperglycemia generally occurs in the absence of elevated blood lipid levels, whereas type 2 diabetes is frequently associated with dyslipidemia. Few animal models are available to study diabetes-accelerated atherosclerosis (179). To dissect the role of lipids, glucose, and insulin in atherosclerosis, investigators generated Ldlr−/− mice that express a lymphocytic choriomeningitis virus glycoprotein transgene under control of the insulin promoter (180). Diabetic mice on regular chow diet, in the absence of the lipid abnormalities, developed atherosclerosis with preferential accumulation of macrophages within the aortas. Western diet–fed diabetic mice showed advanced lesions, characterized by extensive intralesional hemorrhage, suggesting that hyperlipidemia induces a formation of more advanced lesions (180). Mice expressing human aldose reductase that may multiply toxic effects of glucose metabolism showed advanced atherosclerosis compared with controls (181). Thus, glucose or products of glucose metabolism are sufficient to induce atherosclerosis in hyperlipidemic conditions.
Type 2 diabetes is much more prevalent than type 1 diabetes and is often preceded by the metabolic syndrome, but it is even more difficult to generate an appropriate mouse model (179). Hyperinsulinemia and hyperglycemia with dyslipidemia induced accelerated atherosclerosis in Apoe−/− mice fed a regular chow diet and lacking the leptin receptor (182) and in leptin-deficient mice on the Apoe−/− or Ldlr−/− background (179). Although these models showed elevated atherosclerosis, the increased lipid levels in experimental groups compared with control made the interpretations of these results difficult.
Adipose tissues release cytokines that regulate not only body weight homeostasis, but also insulin resistance that in turn influences atherosclerosis (183). Several reports suggest that adipose tissues are active regulators of inflammation through the production of adipokines, proinflammatory cytokines, and CRP that can affect immune response, induce endothelial dysfunction, increase oxidative stress, and thus accelerate atherosclerosis (183).
Anti-Inflammatory Drugs and Mediators
Statins
Statins inhibit 3-hydroxyl-3-methylglutaryl coenzyme A reductase, an enzyme crucial to cholesterol synthesis. They reduce total and LDL cholesterol as well as triglycerides and slightly increase HDL cholesterol and reduce the risk for CVD and stroke (184). Although the clinical benefits of statins are mediated in large part through lipid modulation, emerging evidence supports the existence of other mechanisms of action. The beneficial influences may include modification of endothelial function, increased plaque stability, reduced thrombus formation, and, in particular, dampened inflammatory pathways. Statins activate peroxisome proliferative activated receptors (185) and thus, indirectly, control lipid and glucose metabolism, vascular inflammation and thrombosis, and NF-κB-dependent activation of SMCs and monocytes. The level of CRP can be regulated in an LDL-independent manner by statins, but the mechanisms of this influence as well of CRP's role in atherogenesis are not clear (186). Clinical data show decreased levels of IL-6, CXCL8, and CCL2 after treatment with simvastatin. Pretreatment of human monocytes with statins induced downregulation of IL-1, CCL3, and CCL4 and of IL-18, CCR1, and CCR2 (184). Statins also inhibit elevated expression of both ICAM-1 and lymphocyte function–associated antigen 1 (LFA-1) and may reduce leukocyte adhesion and retention within the aortic wall (184).
High-Density Lipoprotein
Efflux of cholesterol from peripheral tissues into plasma, then to the liver and bile, is termed reverse cholesterol transport (187). HDLs mediate most reverse cholesterol transport and thus influence the amount of cellular cholesterols under normal and pathogenic conditions. Several studies with knockout mice for ApoA-1, SR-B1, or ABCA1 dissected the role and importance of reverse cholesterol transport and high-lighted HDL's functions in this process (187). The role of ABCG1 in atherogenesis is less clear because conflicting results have been reported. HDL also has antioxidant, anti-inflammatory, antiapoptotic, and vasodilatory properties. Recently, investigators have also appreciated that HDLs can lose their usual atheroprotective properties through specific chemical and structural alterations and can play a proinflammatory role by alteration of reverse cholesterol transport, enhanced oxidation of LDLs, and increased vascular inflammation (188).
Conclusions
Our understanding of atherosclerosis has progressed remarkably over the past few years. The discovery of subsets of inflammatory and resident monocytes, M1 and M2 macrophages, NKT cells, and Tregs opened new perspectives on the role of inflammation and immune responses in atherosclerosis. New data suggest an important role for chemokine and chemokine receptors in atherosclerosis and highlight a network of cytokines that modulate the immune response and inflammation in the aortic wall. All phases of atherosclerosis are regulated by inflammatory mechanisms that provide overlapping networks of pathways involved in the regulation of immune cell functions, activation of endothelium, and alteration of metabolic parameters. Increasing evidence suggests that components of the immune system may alter lipid metabolism and thus affect atherosclerosis in yet another way. More work is needed to understand the effects of statins on inflammation and the immune system. All these data will help to identify potential therapeutic targets for the prevention and treatment of atherosclerosis and other CVDs.
Summary Points
Atherosclerosis is a complex chronic inflammatory disease that affects large- and medium-size arteries, inducing atherosclerotic plaques and alterations of the phenotypes of vascular cells.
An early step in the development of atherosclerosis is the retention of LDLs in the arterial wall.
Monocyte recruitment into aortas and formation of foam cells are hallmarks of atherosclerosis. Recruitment of immune cells into the aortas is likely modulated by adhesion molecules and chemokines.
There are some candidates for possible autoantigens during atherosclerosis, including HSP-60 and oxLDLs.
The immune response in atherosclerosis-prone conditions is predominantly Th1-biased.
Monocytes, macrophages, DCs, subsets of T cells, NK cells, NKT cells, neutrophils, platelets, and mast cells likely play proatherogenic roles.
Tregs and B cells (through the production of natural antibodies) suppress inflammation during atherosclerosis.
Chronic inflammation produces inflammatory mediators such as modified LDLs, ROS, and AGE that accelerate vascular inflammation and atherosclerosis.
Many autoimmune diseases such as systemic lupus erythematosus and diabetes accelerate atherosclerosis development, probably through the defective clearance of apoptotic cells.
Acknowledgments
This work was supported by NIH grants HL 58108 and 55798 and by the American Heart Association Scientist Development Grant 0730234N. Because of space constraints, it was not possible to cite all of the relevant original papers and many excellent reviews. We apologize to the authors whose important work could not be included in the list of references.
- Atherosclerosis
a chronic inflammatory process characterized by plaque formation within the vessel wall of arteries and extensive necrosis and fibrosis of surrounding tissues
- Atherosclerotic plaques
plaques consisting of foam cells, immune cells, vascular ECs, SMCs, extracellular matrix, and a lipid-rich core
- SMC
smooth muscle cell
- EC
endothelial cell
- Foam cells
cells derived from macrophages or SMCs that took up modified LDL through scavenger receptors
- LDL
low-density lipoproteins
- ApoE
apolipoprotein E
- Apoe−/− mice
these mice develop severe hypercholesterolemia and atherosclerosis plaques that have some similarities to human atherosclerotic plaques
- SR
scavenger receptor
- TLR
Toll-like receptor
- HSP
heat shock protein
- Natural antibodies
these antibodies are found in the absence of exogenous antigens early after birth; they fight bacterial and viral infections, but also recognize self-antigens and carry out a protective function in atherosclerosis
- ROS
reactive oxygen species
- VALT
vascular-associated lymphoid tissue
- LO
lipoxygenase
- HO
heme oxygenase
- AGE
advanced glycation end products
- RAGE
receptors for advanced glycation end products
Footnotes
The authors dedicate this review to the memory of Dr. Ross Gerrity (1945–2008), who discovered monocyte recruitment to atherosclerotic lesions.
Disclosure Statement: The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Literature Cited
- 1.Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8:802–15. doi: 10.1038/nri2415. [DOI] [PubMed] [Google Scholar]
- 3.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–19. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 4.Mayerl C, Lukasser M, Sedivy R, Niederegger H, Seiler R, Wick G. Atherosclerosis research from past to present—on the track of two pathologists with opposing views, Carl von Rokitansky and Rudolf Virchow. Virchows Arch. 2006;449:96–103. doi: 10.1007/s00428-006-0176-7. [DOI] [PubMed] [Google Scholar]
- 5.Methe H, Weis M. Atherogenesis and inflammation—was Virchow right? Nephrol Dial Transplant. 2007;22:1823–27. doi: 10.1093/ndt/gfm112. [DOI] [PubMed] [Google Scholar]
- 6.Gerrity RG, Naito HK, Richardson M, Schwartz CJ. Dietary induced atherogenesis in swine. Morphology of the intima in prelesion stages. Am J Pathol. 1979;95:775–92. [PMC free article] [PubMed] [Google Scholar]; 6. First demonstrated monocyte adhesion to the aortic endothelium and formation of foam cell lesions in areas of enhanced permeability of aortic intima during atherosclerosis development.
- 7.Galkina E, Ley K. Leukocyte influx in atherosclerosis. Curr Drug Targets. 2007;8:1239–48. doi: 10.2174/138945007783220650. [DOI] [PubMed] [Google Scholar]
- 8.Llodra J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci USA. 2004;101:11779–84. doi: 10.1073/pnas.0403259101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galkina E, Kadl A, Sanders J, Varughese D, Sarembock IJ, Ley K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J Exp Med. 2006;203:1273–82. doi: 10.1084/jem.20052205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jongstra-Bilen J, Haidari M, Zhu SN, Chen M, Guha D, Cybulsky MI. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med. 2006;203:2073–83. doi: 10.1084/jem.20060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lessner SM, Prado HL, Waller EK, Galis ZS. Atherosclerotic lesions grow through recruitment and proliferation of circulating monocytes in a murine model. Am J Pathol. 2002;160:2145–55. doi: 10.1016/S0002-9440(10)61163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bobryshev YV. Monocyte recruitment and foam cell formation in atherosclerosis. Micron. 2006;37:208–22. doi: 10.1016/j.micron.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Galkina E, Ley K. Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2292–301. doi: 10.1161/ATVBAHA.107.149179. [DOI] [PubMed] [Google Scholar]
- 14.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, et al. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–62. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]; 14. Established a dominant role of VCAM-1 but not ICAM-1 in the initiation of atherosclerosis.
- 15.Kraaijeveld AO, de Jager SC, van Berkel TJ, Biessen EA, Jukema JW. Chemokines and atherosclerotic plaque progression: towards therapeutic targeting? Curr Pharm Des. 2007;13:1039–52. doi: 10.2174/138161207780487584. [DOI] [PubMed] [Google Scholar]
- 16.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 17.Randolph GJ, Jakubzick C, Qu C. Antigen presentation by monocytes and monocyte-derived cells. Curr Opin Immunol. 2008;20:52–60. doi: 10.1016/j.coi.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–70. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 19.Passlick B, Flieger D, Ziegler-Heitbrock HW. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74:2527–34. [PubMed] [Google Scholar]
- 20.Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–46. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 21.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–94. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho HJ, Shashkin P, Gleissner CA, Dunson D, Jain N, et al. Induction of dendritic cell-like phenotype in macrophages during foam cell formation. Physiol Genomics. 2007;29:149–60. doi: 10.1152/physiolgenomics.00051.2006. [DOI] [PubMed] [Google Scholar]
- 24.Saederup N, Chan L, Lira SA, Charo IF. Fractalkine deficiency markedly reduces macrophage accumulation and atherosclerotic lesion formation in CCR2−/− mice: evidence for independent chemokine functions in atherogenesis. Circulation. 2008;117:1642–48. doi: 10.1161/CIRCULATIONAHA.107.743872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Combadiere C, Potteaux S, Rodero M, Simon T, Pezard A, et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6Chi and Ly6Clo monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–57. doi: 10.1161/CIRCULATIONAHA.107.745091. [DOI] [PubMed] [Google Scholar]
- 26.Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, et al. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. 2000;105:1049–56. doi: 10.1172/JCI9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moore KJ, Kunjathoor VV, Koehn SL, Manning JJ, Tseng AA, et al. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest. 2005;115:2192–201. doi: 10.1172/JCI24061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuchibhotla S, Vanegas D, Kennedy DJ, Guy E, Nimako G, et al. Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc Res. 2008;78:185–96. doi: 10.1093/cvr/cvm093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stoneman V, Braganza D, Figg N, Mercer J, Lang R, et al. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ Res. 2007;100:884–93. doi: 10.1161/01.RES.0000260802.75766.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Byrne GI, Kalayoglu MV. Chlamydia pneumoniae and atherosclerosis: links to the disease process. Am Heart J. 1999;138:S488–S490. doi: 10.1016/s0002-8703(99)70282-6. [DOI] [PubMed] [Google Scholar]
- 31.Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–64. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z, Liu B, Wang P, Dong X, Fernandez-Hernando C, et al. Phospholipase C β3 deficiency leads to macrophage hypersensitivity to apoptotic induction and reduction of atherosclerosis in mice. J Clin Invest. 2008;118:195–204. doi: 10.1172/JCI33139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Makowski L, Boord JB, Maeda K, Babaev VR, Uysal KT, et al. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat Med. 2001;7:699–705. doi: 10.1038/89076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, et al. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest. 2007;117:3900–8. doi: 10.1172/JCI33372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao B, Song J, Chow WN, St Clair RW, Rudel LL, Ghosh S. Macrophage-specific transgenic expression of cholesteryl ester hydrolase significantly reduces atherosclerosis and lesion necrosis in Ldlr−/− mice. J Clin Invest. 2007;117:2983–92. doi: 10.1172/JCI30485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bobryshev YV, Lord RS. Ultrastructural recognition of cells with dendritic cell morphology in human aortic intima. Contacting interactions of vascular dendritic cells in athero-resistant and athero-prone areas of the normal aorta. Arch Histol Cytol. 1995;58:307–22. doi: 10.1679/aohc.58.307. [DOI] [PubMed] [Google Scholar]
- 37.Erbel C, Sato K, Meyer FB, Kopecky SL, Frye RL, et al. Functional profile of activated dendritic cells in unstable atherosclerotic plaque. Basic Res Cardiol. 2007;102:123–32. doi: 10.1007/s00395-006-0636-x. [DOI] [PubMed] [Google Scholar]
- 38.Niessner A, Shin MS, Pryshchep O, Goronzy JJ, Chaikof EL, Weyand CM. Synergistic proinflammatory effects of the antiviral cytokine interferon-α and Toll-like receptor 4 ligands in the atherosclerotic plaque. Circulation. 2007;116:2043–52. doi: 10.1161/CIRCULATIONAHA.107.697789. [DOI] [PubMed] [Google Scholar]
- 39.Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, et al. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci USA. 2006;103:3781–86. doi: 10.1073/pnas.0511043103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bluml S, Kirchberger S, Bochkov VN, Kronke G, Stuhlmeier K, et al. Oxidized phospholipids negatively regulate dendritic cell maturation induced by TLRs and CD40. J Immunol. 2005;175:501–8. doi: 10.4049/jimmunol.175.1.501. [DOI] [PubMed] [Google Scholar]
- 41.Perrin-Cocon L, Coutant F, Agaugue S, Deforges S, Andre P, Lotteau V. Oxidized low-density lipoprotein promotes mature dendritic cell transition from differentiating monocyte. J Immunol. 2001;167:3785–91. doi: 10.4049/jimmunol.167.7.3785. [DOI] [PubMed] [Google Scholar]
- 42.Maffia P, Zinselmeyer BH, Ialenti A, Kennedy S, Baker AH, et al. Images in cardiovascular medicine. Multiphoton microscopy for 3-dimensional imaging of lymphocyte recruitment into apolipoprotein-E-deficient mouse carotid artery. Circulation. 2007;115:e326–28. doi: 10.1161/CIRCULATIONAHA.106.658492. [DOI] [PubMed] [Google Scholar]
- 43.Hansson GK, Holm J, Jonasson L. Detection of activated T lymphocytes in the human atherosclerotic plaque. Am J Pathol. 1989;135:169–75. [PMC free article] [PubMed] [Google Scholar]; 43. Demonstrated presence of activated T cells within atherosclerotic plaques close to HLA-DR + SMCs and macrophages and suggested possible antigen presentation during atherosclerosis.
- 44.Wick G, Romen M, Amberger A, Metzler B, Mayr M, et al. Atherosclerosis, autoimmunity, and vascular-associated lymphoid tissue. FASEB J. 1997;11:1199–207. doi: 10.1096/fasebj.11.13.9367355. [DOI] [PubMed] [Google Scholar]; 44. Proposed the term vascular-associated lymphoid tissues (VALT).
- 45.Paulsson G, Zhou X, Tornquist E, Hansson GK. Oligoclonal T cell expansions in atherosclerotic lesions of apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2000;20:10–17. doi: 10.1161/01.atv.20.1.10. [DOI] [PubMed] [Google Scholar]
- 46.Rossmann A, Henderson B, Heidecker B, Seiler R, Fraedrich G, et al. T-cells from advanced atherosclerotic lesions recognize hHSP60 and have a restricted T-cell receptor repertoire. Exp Gerontol. 2008;43:229–37. doi: 10.1016/j.exger.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 47.Robertson AK, Hansson GK. T cells in atherogenesis: for better or for worse? Arterioscler Thromb Vasc Biol. 2006;26:2421–32. doi: 10.1161/01.ATV.0000245830.29764.84. [DOI] [PubMed] [Google Scholar]
- 48.Zhou X, Paulsson G, Stemme S, Hansson GK. Hypercholesterolemia is associated with a T helper (Th) 1/Th2 switch of the autoimmune response in atherosclerotic apo E-knockout mice. J Clin Invest. 1998;101:1717–25. doi: 10.1172/JCI1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buono C, Binder CJ, Stavrakis G, Witztum JL, Glimcher LH, Lichtman AH. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc Natl Acad Sci USA. 2005;102:1596–601. doi: 10.1073/pnas.0409015102. [DOI] [PMC free article] [PubMed] [Google Scholar]; 49. Clearly demonstrated a proatherogenic role for the Th1 response in atherosclerosis and showed that Th1 cells regulate production of atheroprotective EO6 antibodies.
- 50.Nofer JR, Bot M, Brodde M, Taylor PJ, Salm P, et al. FTY720, a synthetic sphingosine 1 phosphate analogue, inhibits development of atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. 2007;115:501–8. doi: 10.1161/CIRCULATIONAHA.106.641407. [DOI] [PubMed] [Google Scholar]
- 51.Keul P, Tolle M, Lucke S, von Wnuck LK, Heusch G, et al. The sphingosine-1-phosphate analogue FTY720 reduces atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27:607–13. doi: 10.1161/01.ATV.0000254679.42583.88. [DOI] [PubMed] [Google Scholar]
- 52.Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, et al. Foxp3+CD25+CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 53.Mallat Z, Gojova A, Brun V, Esposito B, Fournier N, et al. Induction of a regulatory T cell type 1 response reduces the development of atherosclerosis in apolipoprotein E-knockout mice. Circulation. 2003;108:1232–37. doi: 10.1161/01.CIR.0000089083.61317.A1. [DOI] [PubMed] [Google Scholar]
- 54.it-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12:178–80. doi: 10.1038/nm1343. [DOI] [PubMed] [Google Scholar]; 54. Showed that naturally arising CD4+CD25+ regulatory T cells suppress atherosclerosis development by reducing infiltration of T cells and macrophages into plaques, increasing lesional collagen content and switching to an anti-inflammatory cytokine profile.
- 55.Gotsman I, Grabie N, Gupta R, Dacosta R, MacConmara M, et al. Impaired regulatory T-cell response and enhanced atherosclerosis in the absence of inducible costimulatory molecule. Circulation. 2006;114:2047–55. doi: 10.1161/CIRCULATIONAHA.106.633263. [DOI] [PubMed] [Google Scholar]
- 56.Groyer E, Nicoletti A, it-Oufella H, Khallou-Laschet J, Varthaman A, et al. Atheroprotective effect of CD31 receptor globulin through enrichment of circulating regulatory T-cells. J Am Coll Cardiol. 2007;50:344–50. doi: 10.1016/j.jacc.2007.04.040. [DOI] [PubMed] [Google Scholar]
- 57.Taleb S, Herbin O, it-Oufella H, Verreth W, Gourdy P, et al. Defective leptin/leptin receptor signaling improves regulatory T cell immune response and protects mice from atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2691–98. doi: 10.1161/ATVBAHA.107.149567. [DOI] [PubMed] [Google Scholar]
- 58.van Puijvelde GH, van Es T, van Wanrooij EJ, Habets KL, de Vos P, et al. Induction of oral tolerance to HSP60 or an HSP60-peptide activates T cell regulation and reduces atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2677–83. doi: 10.1161/ATVBAHA.107.151274. [DOI] [PubMed] [Google Scholar]
- 59.Elhage R, Gourdy P, Brouchet L, Jawien J, Fouque MJ, et al. Deleting TCRαβ+ or CD4+ T lymphocytes leads to opposite effects on site-specific atherosclerosis in female apolipoprotein E-deficient mice. Am J Pathol. 2004;165:2013–18. doi: 10.1016/s0002-9440(10)63252-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whitman SC, Ramsamy TA. Participatory role of natural killer and natural killer T cells in atherosclerosis: lessons learned from in vivo mouse studies. Can J Physiol Pharmacol. 2006;84:67–75. doi: 10.1139/y05-159. [DOI] [PubMed] [Google Scholar]
- 61.Schiller NK, Boisvert WA, Curtiss LK. Inflammation in atherosclerosis: lesion formation in LDL receptor-deficient mice with perforin and Lystbeige mutations. Arterioscler Thromb Vasc Biol. 2002;22:1341–46. doi: 10.1161/01.atv.0000024082.46387.38. [DOI] [PubMed] [Google Scholar]
- 62.Whitman SC, Rateri DL, Szilvassy SJ, Yokoyama W, Daugherty A. Depletion of natural killer cell function decreases atherosclerosis in low-density lipoprotein receptor null mice. Arterioscler Thromb Vasc Biol. 2004;24:1049–54. doi: 10.1161/01.ATV.0000124923.95545.2c. [DOI] [PubMed] [Google Scholar]
- 63.Porcelli S, Morita CT, Brenner MB. CD1b restricts the response of human CD4−8− T lymphocytes to a microbial antigen. Nature. 1992;360:593–97. doi: 10.1038/360593a0. [DOI] [PubMed] [Google Scholar]
- 64.Tupin E, Nicoletti A, Elhage R, Rudling M, Ljunggren HG, et al. CD1d-dependent activation of NKT cells aggravates atherosclerosis. J Exp Med. 2004;199:417–22. doi: 10.1084/jem.20030997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakai Y, Iwabuchi K, Fujii S, Ishimori N, Dashtsoodol N, et al. Natural killer T cells accelerate atherogenesis in mice. Blood. 2004;104:2051–59. doi: 10.1182/blood-2003-10-3485. [DOI] [PubMed] [Google Scholar]
- 66.Major AS, Wilson MT, McCaleb JL, Ru SY, Stanic AK, et al. Quantitative and qualitative differences in proatherogenic NKT cells in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2004;24:2351–57. doi: 10.1161/01.ATV.0000147112.84168.87. [DOI] [PubMed] [Google Scholar]
- 67.Lindstedt KA, Mayranpaa MI, Kovanen PT. Mast cells in vulnerable atherosclerotic plaques—a view to a kill. J Cell Mol Med. 2007;11:739–58. doi: 10.1111/j.1582-4934.2007.00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bot I, de Jager SC, Zernecke A, Lindstedt KA, van Berkel TJ, et al. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation. 2007;115:2516–25. doi: 10.1161/CIRCULATIONAHA.106.660472. [DOI] [PubMed] [Google Scholar]
- 69.Lee M, Calabresi L, Chiesa G, Franceschini G, Kovanen PT. Mast cell chymase degrades apoE and apoA-II in apoA-I-knockout mouse plasma and reduces its ability to promote cellular cholesterol efflux. Arterioscler Thromb Vasc Biol. 2002;22:1475–81. doi: 10.1161/01.atv.0000029782.84357.68. [DOI] [PubMed] [Google Scholar]
- 70.Sun J, Sukhova GK, Wolters PJ, Yang M, Kitamoto S, et al. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med. 2007;13:719–24. doi: 10.1038/nm1601. [DOI] [PubMed] [Google Scholar]
- 71.Major AS, Fazio S, Linton MF. B-lymphocyte deficiency increases atherosclerosis in LDL receptor-null mice. Arterioscler Thromb Vasc Biol. 2002;22:1892–98. doi: 10.1161/01.atv.0000039169.47943.ee. [DOI] [PubMed] [Google Scholar]
- 72.Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest. 2002;109:745–53. doi: 10.1172/JCI07272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Binder CJ, Shaw PX, Chang MK, Boullier A, Hartvigsen K, et al. The role of natural antibodies in atherogenesis. J Lipid Res. 2005;46:1353–63. doi: 10.1194/jlr.R500005-JLR200. [DOI] [PubMed] [Google Scholar]
- 74.Shaw PX, Horkko S, Chang MK, Curtiss LK, Palinski W, et al. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J Clin Invest. 2000;105:1731–40. doi: 10.1172/JCI8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Binder CJ, Horkko S, Dewan A, Chang MK, Kieu EP, et al. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med. 2003;9:736–43. doi: 10.1038/nm876. [DOI] [PubMed] [Google Scholar]; 75. Demonstrated that many autoantibodies to oxLDL share genetic and structural similarity with antibodies against common infectious pathogens, suggesting molecular mimicry between epitopes of oxLDL and microbes.
- 76.Binder CJ, Hartvigsen K, Chang MK, Miller M, Broide D, et al. IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J Clin Invest. 2004;114:427–37. doi: 10.1172/JCI20479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zernecke A, Bot I, Djalali-Talab Y, Shagdarsuren E, Bidzhekov K, et al. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ Res. 2008;102:209–17. doi: 10.1161/CIRCRESAHA.107.160697. [DOI] [PubMed] [Google Scholar]
- 78.von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. 2007;100:27–40. doi: 10.1161/01.RES.0000252802.25497.b7. [DOI] [PubMed] [Google Scholar]
- 79.Burger PC, Wagner DD. Platelet P-selectin facilitates atherosclerotic lesion development. Blood. 2003;101:2661–66. doi: 10.1182/blood-2002-07-2209. [DOI] [PubMed] [Google Scholar]
- 80.Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9:61–67. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 81.Schober A, Manka D, von HP, Huo Y, Hanrath P, et al. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation. 2002;106:1523–29. doi: 10.1161/01.cir.0000028590.02477.6f. [DOI] [PubMed] [Google Scholar]
- 82.Langer HF, Daub K, Braun G, Schonberger T, May AE, et al. Platelets recruit human dendritic cells via Mac-1/JAM-C interaction and modulate dendritic cell function in vitro. Arterioscler Thromb Vasc Biol. 2007;27:1463–70. doi: 10.1161/ATVBAHA.107.141515. [DOI] [PubMed] [Google Scholar]
- 83.Schulz C, Schafer A, Stolla M, Kerstan S, Lorenz M, et al. Chemokine fractalkine mediates leukocyte recruitment to inflammatory endothelial cells in flowing whole blood: a critical role for P-selectin expressed on activated platelets. Circulation. 2007;116:764–73. doi: 10.1161/CIRCULATIONAHA.107.695189. [DOI] [PubMed] [Google Scholar]
- 84.Garin G, Berk BC. Flow-mediated signaling modulates endothelial cell phenotype. Endothelium. 2006;13:375–84. doi: 10.1080/10623320601061599. [DOI] [PubMed] [Google Scholar]
- 85.Eriksson EE, Xie X, Werr J, Thoren P, Lindbom L. Importance of primary capture and L-selectin-dependent secondary capture in leukocyte accumulation in inflammation and atherosclerosis in vivo. J Exp Med. 2001;194:205–18. doi: 10.1084/jem.194.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moos MP, John N, Grabner R, Nossmann S, Gunther B, et al. The lamina adventitia is the major site of immune cell accumulation in standard chow-fed apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:2386–91. doi: 10.1161/01.ATV.0000187470.31662.fe. [DOI] [PubMed] [Google Scholar]
- 87.Moreno PR, Purushothaman KR, Sirol M, Levy AP, Fuster V. Neovascularization in human atherosclerosis. Circulation. 2006;113:2245–52. doi: 10.1161/CIRCULATIONAHA.105.578955. [DOI] [PubMed] [Google Scholar]
- 88.Ritman EL, Lerman A. The dynamic vasa vasorum. Cardiovasc Res. 2007;75:649–58. doi: 10.1016/j.cardiores.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Houtkamp MA, de Boer OJ, Van Der Loos CM, Van Der Wal AC, Becker AE. Adventitial infiltrates associated with advanced atherosclerotic plaques: structural organization suggests generation of local humoral immune responses. J Pathol. 2001;193:263–69. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH774>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 90.Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. 2006;86:515–81. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 91.Newby AC. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol Rev. 2005;85:1–31. doi: 10.1152/physrev.00048.2003. [DOI] [PubMed] [Google Scholar]
- 92.Ohta H, Wada H, Niwa T, Kirii H, Iwamoto N, et al. Disruption of tumor necrosis factor-α gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis. 2005;180:11–17. doi: 10.1016/j.atherosclerosis.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 93.Schreyer SA, Peschon JJ, LeBoeuf RC. Accelerated atherosclerosis in mice lacking tumor necrosis factor receptor p 55. J Biol Chem. 1996;271:26174–78. doi: 10.1074/jbc.271.42.26174. [DOI] [PubMed] [Google Scholar]
- 94.Canault M, Peiretti F, Poggi M, Mueller C, Kopp F, et al. Progression of atherosclerosis in ApoE-deficient mice that express distinct molecular forms of TNF-α. J Pathol. 2008;214:574–83. doi: 10.1002/path.2305. [DOI] [PubMed] [Google Scholar]
- 95.Upadhya S, Mooteri S, Peckham N, Pai RG. Atherogenic effect of interleukin-2 and antiatherogenic effect of interleukin-2 antibody in apo-E-deficient mice. Angiology. 2004;55:289–94. doi: 10.1177/000331970405500308. [DOI] [PubMed] [Google Scholar]
- 96.Huber SA, Sakkinen P, Conze D, Hardin N, Tracy R. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 1999;19:2364–67. doi: 10.1161/01.atv.19.10.2364. [DOI] [PubMed] [Google Scholar]
- 97.Schieffer B, Selle T, Hilfiker A, Hilfiker-Kleiner D, Grote K, et al. Impact of interleukin-6 on plaque development and morphology in experimental atherosclerosis. Circulation. 2004;110:3493–500. doi: 10.1161/01.CIR.0000148135.08582.97. [DOI] [PubMed] [Google Scholar]
- 98.Elhage R, Clamens S, Besnard S, Mallat Z, Tedgui A, et al. Involvement of interleukin-6 in atherosclerosis but not in the prevention of fatty streak formation by 17β-estradiol in apolipoprotein E-deficient mice. Atherosclerosis. 2001;156:315–20. doi: 10.1016/s0021-9150(00)00682-1. [DOI] [PubMed] [Google Scholar]
- 99.Lee TS, Yen HC, Pan CC, Chau LY. The role of interleukin 12 in the development of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:734–42. doi: 10.1161/01.atv.19.3.734. [DOI] [PubMed] [Google Scholar]
- 100.Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2003;163:1117–25. doi: 10.1016/S0002-9440(10)63471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang X, Niessner A, Nakajima T, Ma-Krupa W, Kopecky SL, et al. Interleukin 12 induces T-cell recruitment into the atherosclerotic plaque. Circ Res. 2006;98:524–31. doi: 10.1161/01.RES.0000204452.46568.57. [DOI] [PubMed] [Google Scholar]
- 102.Elhage R, Jawien J, Rudling M, Ljunggren HG, Takeda K, et al. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc Res. 2003;59:234–40. doi: 10.1016/s0008-6363(03)00343-2. [DOI] [PubMed] [Google Scholar]
- 103.Whitman SC, Ravisankar P, Daugherty A. Interleukin-18 enhances atherosclerosis in apolipoprotein E−/− mice through release of interferon-γ. Circ Res. 2002;90:E34–38. doi: 10.1161/hh0202.105292. [DOI] [PubMed] [Google Scholar]
- 104.Whitman SC, Ravisankar P, Elam H, Daugherty A. Exogenous interferon-γ enhances atherosclerosis in apolipoprotein E−/− mice. Am J Pathol. 2000;157:1819–24. doi: 10.1016/s0002-9440(10)64820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gupta S, Pablo AM, Jiang X, Wang N, Tall AR, Schindler C. IFN-γ potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest. 1997;99:2752–61. doi: 10.1172/JCI119465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Buono C, Come CE, Stavrakis G, Maguire GF, Connelly PW, Lichtman AH. Influence of interferon-γ on the extent and phenotype of diet-induced atherosclerosis in the LDLR-deficient mouse. Arterioscler Thromb Vasc Biol. 2003;23:454–60. doi: 10.1161/01.ATV.0000059419.11002.6E. [DOI] [PubMed] [Google Scholar]
- 107.Tellides G, Tereb DA, Kirkiles-Smith NC, Kim RW, Wilson JH, et al. Interferon-γ elicits arteriosclerosis in the absence of leukocytes. Nature. 2000;403:207–11. doi: 10.1038/35003221. [DOI] [PubMed] [Google Scholar]
- 108.Koga M, Kai H, Yasukawa H, Yamamoto T, Kawai Y, et al. Inhibition of progression and stabilization of plaques by postnatal interferon-γ function blocking in ApoE-knockout mice. Circ Res. 2007;101:348–56. doi: 10.1161/CIRCRESAHA.106.147256. [DOI] [PubMed] [Google Scholar]
- 109.Niessner A, Sato K, Chaikof EL, Colmegna I, Goronzy JJ, Weyand CM. Pathogen-sensing plasmacytoid dendritic cells stimulate cytotoxic T-cell function in the atherosclerotic plaque through interferon-α. Circulation. 2006;114:2482–89. doi: 10.1161/CIRCULATIONAHA.106.642801. [DOI] [PubMed] [Google Scholar]
- 110.Mach F, Schonbeck U, Sukhova GK, Atkinson E, Libby P. Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature. 1998;394:200–3. doi: 10.1038/28204. [DOI] [PubMed] [Google Scholar]
- 111.Zirlik A, Maier C, Gerdes N, MacFarlane L, Soosairajah J, et al. CD40 ligand mediates inflammation independently of CD40 by interaction with Mac-1. Circulation. 2007;115:1571–80. doi: 10.1161/CIRCULATIONAHA.106.683201. [DOI] [PubMed] [Google Scholar]
- 112.King VL, Cassis LA, Daugherty A. Interleukin-4 does not influence development of hypercholesterolemia or angiotensin II-induced atherosclerotic lesions in mice. Am J Pathol. 2007;171:2040–47. doi: 10.2353/ajpath.2007.060857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.George J, Shoenfeld Y, Gilburd B, Afek A, Shaish A, Harats D. Requisite role for interleukin-4 in the acceleration of fatty streaks induced by heat shock protein 65 or Mycobacterium tuberculosis. Circ Res. 2000;86:1203–10. doi: 10.1161/01.res.86.12.1203. [DOI] [PubMed] [Google Scholar]
- 114.Caligiuri G, Rudling M, Ollivier V, Jacob MP, Michel JB, et al. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol Med. 2003;9:10–17. [PMC free article] [PubMed] [Google Scholar]
- 115.Miller AM, Xu D, Asquith DL, Denby L, Li Y, et al. IL-33 reduces the development of atherosclerosis. J Exp Med. 2008;205:339–46. doi: 10.1084/jem.20071868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Robertson AK, Rudling M, Zhou X, Gorelik L, Flavell RA, Hansson GK. Disruption of TGF-β signaling in T cells accelerates atherosclerosis. J Clin Invest. 2003;112:1342–50. doi: 10.1172/JCI18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gojova A, Brun V, Esposito B, Cottrez F, Gourdy P, et al. Specific abrogation of transforming growth factor-β signaling in T cells alters atherosclerotic lesion size and composition in mice. Blood. 2003;102:4052–58. doi: 10.1182/blood-2003-05-1729. [DOI] [PubMed] [Google Scholar]
- 118.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–17. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 119.Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–9. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Aiello RJ, Bourassa PA, Lindsey S, Weng W, Natoli E, et al. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:1518–25. doi: 10.1161/01.atv.19.6.1518. [DOI] [PubMed] [Google Scholar]
- 121.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–97. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 122.Dawson TC, Kuziel WA, Osahar TA, Maeda N. Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. 1999;143:205–11. doi: 10.1016/s0021-9150(98)00318-9. [DOI] [PubMed] [Google Scholar]
- 123.Gosling J, Slaymaker S, Gu L, Tseng S, Zlot CH, et al. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J Clin Invest. 1999;103:773–78. doi: 10.1172/JCI5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yu G, Rux AH, Ma P, Bdeir K, Sachais BS. Endothelial expression of E-selectin is induced by the platelet-specific chemokine platelet factor 4 through LRP in an NF-κB-dependent manner. Blood. 2005;105:3545–51. doi: 10.1182/blood-2004-07-2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kuziel WA, Dawson TC, Quinones M, Garavito E, Chenaux G, et al. CCR5 deficiency is not protective in the early stages of atherogenesis in apoE knockout mice. Atherosclerosis. 2003;167:25–32. doi: 10.1016/s0021-9150(02)00382-9. [DOI] [PubMed] [Google Scholar]
- 126.Potteaux S, Combadiere C, Esposito B, Lecureuil C, it-Oufella H, et al. Role of bone marrow-derived CC-chemokine receptor 5 in the development of atherosclerosis of low-density lipoprotein receptor knockout mice. Arterioscler Thromb Vasc Biol. 2006;26:1858–63. doi: 10.1161/01.ATV.0000231527.22762.71. [DOI] [PubMed] [Google Scholar]
- 127.Braunersreuther V, Zernecke A, Arnaud C, Liehn EA, Steffens S, et al. Ccr5 but not Ccr1 deficiency reduces development of diet-induced atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2007;27:373–79. doi: 10.1161/01.ATV.0000253886.44609.ae. [DOI] [PubMed] [Google Scholar]
- 128.Huo Y, Weber C, Forlow SB, Sperandio M, Thatte J, et al. The chemokine KC, but not monocyte chemoattractant protein-1, triggers monocyte arrest on early atherosclerotic endothelium. J Clin Invest. 2001;108:1307–14. doi: 10.1172/JCI12877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cheng C, Tempel D, van HR, de Boer HC, Segers D, et al. Shear stress-induced changes in atherosclerotic plaque composition are modulated by chemokines. J Clin Invest. 2007;117:616–26. doi: 10.1172/JCI28180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13:587–96. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 131.Pan JH, Sukhova GK, Yang JT, Wang B, Xie T, et al. Macrophage migration inhibitory factor deficiency impairs atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. 2004;109:3149–53. doi: 10.1161/01.CIR.0000134704.84454.D2. [DOI] [PubMed] [Google Scholar]
- 132.Aslanian AM, Charo IF. Targeted disruption of the scavenger receptor and chemokine CXCL16 accelerates atherosclerosis. Circulation. 2006;114:583–90. doi: 10.1161/CIRCULATIONAHA.105.540583. [DOI] [PubMed] [Google Scholar]
- 133.Galkina E, Harry BL, Ludwig A, Liehn EA, Sanders JM, et al. CXCR6 promotes atherosclerosis by supporting T-cell homing, interferon-γ production, and macrophage accumulation in the aortic wall. Circulation. 2007;116:1801–11. doi: 10.1161/CIRCULATIONAHA.106.678474. [DOI] [PubMed] [Google Scholar]
- 134.Heller EA, Liu E, Tager AM, Yuan Q, Lin AY, et al. Chemokine CXCL10 promotes atherogenesis by modulating the local balance of effector and regulatory T cells. Circulation. 2006;113:2301–12. doi: 10.1161/CIRCULATIONAHA.105.605121. [DOI] [PubMed] [Google Scholar]
- 135.Radmark O, Samuelsson B. 5-lipoxygenase: regulation and possible involvement in atherosclerosis. Prostaglandins Other Lipid Mediat. 2007;83:162–74. doi: 10.1016/j.prostaglandins.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 136.Mehrabian M, Allayee H, Wong J, Shi W, Wang XP, et al. Identification of 5-lipoxygenase as a major gene contributing to atherosclerosis susceptibility in mice. Circ Res. 2002;91:120–26. doi: 10.1161/01.res.0000028008.99774.7f. [DOI] [PubMed] [Google Scholar]
- 137.Cyrus T, Witztum JL, Rader DJ, Tangirala R, Fazio S, et al. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. J Clin Invest. 1999;103:1597–604. doi: 10.1172/JCI5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Reilly KB, Srinivasan S, Hatley ME, Patricia MK, Lannigan J, et al. 12/15-Lipoxygenase activity mediates inflammatory monocyte/endothelial interactions and atherosclerosis in vivo. J Biol Chem. 2004;279:9440–50. doi: 10.1074/jbc.M303857200. [DOI] [PubMed] [Google Scholar]
- 139.Huo Y, Zhao L, Hyman MC, Shashkin P, Harry BL, et al. Critical role of macrophage 12/15-lipoxygenase for atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;110:2024–31. doi: 10.1161/01.CIR.0000143628.37680.F6. [DOI] [PubMed] [Google Scholar]
- 140.Wen Y, Gu J, Vandenhoff GE, Liu X, Nadler JL. Role of 12/15-lipoxygenase in the expression of MCP-1 in mouse macrophages. Am J Physiol Heart Circ Physiol. 2008;294:H1933–38. doi: 10.1152/ajpheart.00260.2007. [DOI] [PubMed] [Google Scholar]
- 141.Serhan CN, Jain A, Marleau S, Clish C, Kantarci A, et al. Reduced inflammation and tissue damage in transgenic rabbits overexpressing 15-lipoxygenase and endogenous anti-inflammatory lipid mediators. J Immunol. 2003;171:6856–65. doi: 10.4049/jimmunol.171.12.6856. [DOI] [PubMed] [Google Scholar]
- 142.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–61. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ishikawa K, Navab M, Leitinger N, Fogelman AM, Lusis AJ. Induction of heme oxygenase-1 inhibits the monocyte transmigration induced by mildly oxidized LDL. J Clin Invest. 1997;100:1209–16. doi: 10.1172/JCI119634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yet SF, Layne MD, Liu X, Chen YH, Ith B, et al. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003;17:1759–61. doi: 10.1096/fj.03-0187fje. [DOI] [PubMed] [Google Scholar]
- 145.Orozco LD, Kapturczak MH, Barajas B, Wang X, Weinstein MM, et al. Heme oxygenase-1 expression in macrophages plays a beneficial role in atherosclerosis. Circ Res. 2007;100:1703–11. doi: 10.1161/CIRCRESAHA.107.151720. [DOI] [PubMed] [Google Scholar]
- 146.Wu BJ, Kathir K, Witting PK, Beck K, Choy K, et al. Antioxidants protect from atherosclerosis by a heme oxygenase-1 pathway that is independent of free radical scavenging. J Exp Med. 2006;203:1117–27. doi: 10.1084/jem.20052321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Marchegiani F, Marra M, Olivieri F, Cardelli M, James RW, et al. Paraoxonase 1: genetics and activities during aging. Rejuvenation Res. 2008;11:113–27. doi: 10.1089/rej.2007.0582. [DOI] [PubMed] [Google Scholar]
- 148.Shih DM, Gu L, Xia YR, Navab M, Li WF, et al. Mice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosis. Nature. 1998;394:284–87. doi: 10.1038/28406. [DOI] [PubMed] [Google Scholar]
- 149.Matsuura E, Kobayashi K, Tabuchi M, Lopez LR. Oxidative modification of low-density lipoprotein and immune regulation of atherosclerosis. Prog Lipid Res. 2006;45:466–86. doi: 10.1016/j.plipres.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 150.Pillarisetti S, Paka L, Obunike JC, Berglund L, Goldberg IJ. Subendothelial retention of lipoprotein (a). Evidence that reduced heparan sulfate promotes lipoprotein binding to subendothelial matrix. J Clin Invest. 1997;100:867–74. doi: 10.1172/JCI119602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mehta JL, Sanada N, Hu CP, Chen J, Dandapat A, et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ Res. 2007;100:1634–42. doi: 10.1161/CIRCRESAHA.107.149724. [DOI] [PubMed] [Google Scholar]
- 152.Barlic J, Zhang Y, Foley JF, Murphy PM. Oxidized lipid-driven chemokine receptor switch, CCR2 to CX3CR1, mediates adhesion of human macrophages to coronary artery smooth muscle cells through a peroxisome proliferator-activated receptor γ-dependent pathway. Circulation. 2006;114:807–19. doi: 10.1161/CIRCULATIONAHA.105.602359. [DOI] [PubMed] [Google Scholar]
- 153.Bisoendial RJ, Kastelein JJ, Stroes ES. C-reactive protein and atherogenesis: from fatty streak to clinical event. Atherosclerosis. 2007;195:e10–18. doi: 10.1016/j.atherosclerosis.2007.04.053. [DOI] [PubMed] [Google Scholar]
- 154.Singh SK, Suresh MV, Prayther DC, Moorman JP, Rusinol AE, Agrawal A. C-reactive protein-bound enzymatically modified low-density lipoprotein does not transform macrophages into foam cells. J Immunol. 2008;180:4316–22. doi: 10.4049/jimmunol.180.6.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Basta G, Schmidt AM, De CR. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res. 2004;63:582–92. doi: 10.1016/j.cardiores.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 156.Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Jr, et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–31. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 157.Harja E, Bu DX, Hudson BI, Chang JS, Shen X, et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J Clin Invest. 2008;118:183–94. doi: 10.1172/JCI32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sorescu D, Weiss D, Lassegue B, Clempus RE, Szocs K, et al. Superoxide production and expression of Nox family proteins in human atherosclerosis. Circulation. 2002;105:1429–35. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- 159.Barry-Lane PA, Patterson C, Van Der MM, Hu Z, Holland SM, et al. p47phox is required for atherosclerotic lesion progression in ApoE−/− mice. J Clin Invest. 2001;108:1513–22. doi: 10.1172/JCI11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vendrov AE, Hakim ZS, Madamanchi NR, Rojas M, Madamanchi C, Runge MS. Atherosclerosis is attenuated by limiting superoxide generation in both macrophages and vessel wall cells. Arterioscler Thromb Vasc Biol. 2007;27:2714–21. doi: 10.1161/ATVBAHA.107.152629. [DOI] [PubMed] [Google Scholar]
- 161.Freedman JE. Oxidative stress and platelets. Arterioscler Thromb Vasc Biol. 2008;28:s11–16. doi: 10.1161/ATVBAHA.107.159178. [DOI] [PubMed] [Google Scholar]
- 162.Gros P, Milder FJ, Janssen BJ. Complement driven by conformational changes. Nat Rev Immunol. 2008;8:48–58. doi: 10.1038/nri2231. [DOI] [PubMed] [Google Scholar]
- 163.Schmiedt W, Kinscherf R, Deigner HP, Kamencic H, Nauen O, et al. Complement C6 deficiency protects against diet-induced atherosclerosis in rabbits. Arterioscler Thromb Vasc Biol. 1998;18:1790–95. doi: 10.1161/01.atv.18.11.1790. [DOI] [PubMed] [Google Scholar]
- 164.Patel S, Thelander EM, Hernandez M, Montenegro J, Hassing H, et al. ApoE−/− mice develop atherosclerosis in the absence of complement component C5. Biochem Biophys Res Commun. 2001;286:164–70. doi: 10.1006/bbrc.2001.5276. [DOI] [PubMed] [Google Scholar]
- 165.Buono C, Come CE, Witztum JL, Maguire GF, Connelly PW, et al. Influence of C3 deficiency on atherosclerosis. Circulation. 2002;105:3025–31. doi: 10.1161/01.cir.0000019584.04929.83. [DOI] [PubMed] [Google Scholar]
- 166.Bhatia VK, Yun S, Leung V, Grimsditch DC, Benson GM, et al. Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am J Pathol. 2007;170:416–26. doi: 10.2353/ajpath.2007.060406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Laine P, Pentikäinen MO, Würzner R, Penttilä A, Paavonen T, et al. Evidence for complement activation in ruptured coronary plaques in acute myocardial infarction. Am J Cardiol. 2002;90:404–8. doi: 10.1016/s0002-9149(02)02498-0. [DOI] [PubMed] [Google Scholar]
- 168.Van EW, Wick G, Albani S, Cohen I. Stress, heat shock proteins, and autoimmunity: how immune responses to heat shock proteins are to be used for the control of chronic inflammatory diseases. Ann NY Acad Sci. 2007;1113:217–37. doi: 10.1196/annals.1391.020. [DOI] [PubMed] [Google Scholar]
- 169.Hochleitner BW, Hochleitner EO, Obrist P, Eberl T, Amberger A, et al. Fluid shear stress induces heat shock protein 60 expression in endothelial cells in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2000;20:617–23. doi: 10.1161/01.atv.20.3.617. [DOI] [PubMed] [Google Scholar]
- 170.Rigano R, Profumo E, Buttari B, Tagliani A, Petrone L, et al. Heat shock proteins and autoimmunity in patients with carotid atherosclerosis. Ann NY Acad Sci. 2007;1107:1–10. doi: 10.1196/annals.1381.001. [DOI] [PubMed] [Google Scholar]
- 171.Frantz S, Ertl G, Bauersachs J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2007;4:444–54. doi: 10.1038/ncpcardio0938. [DOI] [PubMed] [Google Scholar]
- 172.Bjorkbacka H, Kunjathoor VV, Moore KJ, Koehn S, Ordija CM, et al. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat Med. 2004;10:416–21. doi: 10.1038/nm1008. [DOI] [PubMed] [Google Scholar]
- 173.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci USA. 2004;101:10679–84. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest. 2005;115:3149–56. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kotzin BL. Systemic lupus erythematosus. Cell. 1996;85:303–6. doi: 10.1016/s0092-8674(00)81108-3. [DOI] [PubMed] [Google Scholar]
- 176.Aprahamian T, Rifkin I, Bonegio R, Hugel B, Freyssinet JM, et al. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J Exp Med. 2004;199:1121–31. doi: 10.1084/jem.20031557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Feng X, Li H, Rumbin AA, Wang X, La CA, et al. ApoE−/−Fas−/− C57BL/6 mice: a novel murine model simultaneously exhibits lupus nephritis, atherosclerosis, and osteopenia. J Lipid Res. 2007;48:794–805. doi: 10.1194/jlr.M600512-JLR200. [DOI] [PubMed] [Google Scholar]
- 178.Gleissner CA, Galkina E, Nadler JL, Ley K. Mechanisms by which diabetes increases cardiovascular disease. Drug Discov Today Dis Mech. 2007;4:131–40. doi: 10.1016/j.ddmec.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hsueh W, Abel ED, Breslow JL, Maeda N, Davis RC, et al. Recipes for creating animal models of diabetic cardiovascular disease. Circ Res. 2007;100:1415–27. doi: 10.1161/01.RES.0000266449.37396.1f. [DOI] [PubMed] [Google Scholar]
- 180.Renard CB, Kramer F, Johansson F, Lamharzi N, Tannock LR, et al. Diabetes and diabetes-associated lipid abnormalities have distinct effects on initiation and progression of atherosclerotic lesions. J Clin Invest. 2004;114:659–68. doi: 10.1172/JCI17867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Vikramadithyan RK, Hu Y, Noh HL, Liang CP, Hallam K, et al. Human aldose reductase expression accelerates diabetic atherosclerosis in transgenic mice. J Clin Invest. 2005;115:2434–43. doi: 10.1172/JCI24819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wu KK, Wu TJ, Chin J, Mitnaul LJ, Hernandez M, et al. Increased hypercholesterolemia and atherosclerosis in mice lacking both ApoE and leptin receptor. Atherosclerosis. 2005;181:251–59. doi: 10.1016/j.atherosclerosis.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 183.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–80. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 184.Steffens S, Mach F. Drug insight: immunomodulatory effects of statins—potential benefits for renal patients? Nat Clin Pract Nephrol. 2006;2:378–87. doi: 10.1038/ncpneph0217. [DOI] [PubMed] [Google Scholar]
- 185.Martin G, Duez H, Blanquart C, Berezowski V, Poulain P, et al. Statin-induced inhibition of the Rho-signaling pathway activates PPARα and induces HDL apoA-I. J Clin Invest. 2001;107:1423–32. doi: 10.1172/JCI10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–28. doi: 10.1056/NEJMoa042378. [DOI] [PubMed] [Google Scholar]
- 187.Tall AR. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J Intern Med. 2008;263:256–73. doi: 10.1111/j.1365-2796.2007.01898.x. [DOI] [PubMed] [Google Scholar]
- 188.Ansell BJ, Fonarow GC, Fogelman AM. The paradox of dysfunctional high-density lipoprotein. Curr Opin Lipidol. 2007;18:427–34. doi: 10.1097/MOL.0b013e3282364a17. [DOI] [PubMed] [Google Scholar]
- 189.Katagiri H, Yamada T, Oka Y. Adiposity and cardiovascular disorders: disturbance of the regulatory system consisting of humoral and neuronal signals. Circ Res. 2007;101:27–39. doi: 10.1161/CIRCRESAHA.107.151621. [DOI] [PubMed] [Google Scholar]
- 190.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6:772–83. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 191.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 192.Tian L, Luo N, Klein RL, Chung BH, Garvey WT, Fu Y. Adiponectin reduces lipid accumulation in macrophage foam cells. Atherosclerosis. 2008 doi: 10.1016/j.atherosclerosis.2008.04.011. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Okamoto Y, Folco EJ, Minami M, Wara AK, Feinberg MW, et al. Adiponectin inhibits the production of CXC receptor 3 chemokine ligands in macrophages and reduces T-lymphocyte recruitment in atherogenesis. Circ Res. 2008;102:218–25. doi: 10.1161/CIRCRESAHA.107.164988. [DOI] [PubMed] [Google Scholar]
- 194.Kato H, Kashiwagi H, Shiraga M, Tadokoro S, Kamae T, et al. Adiponectin acts as an endogenous antithrombotic factor. Arterioscler Thromb Vasc Biol. 2006;26:224–30. doi: 10.1161/01.ATV.0000194076.84568.81. [DOI] [PubMed] [Google Scholar]
- 195.Yang R, Barouch LA. Leptin signaling and obesity: cardiovascular consequences. Circ Res. 2007;101:545–59. doi: 10.1161/CIRCRESAHA.107.156596. [DOI] [PubMed] [Google Scholar]
- 196.Maruyama I, Nakata M, Yamaji K. Effect of leptin in platelet and endothelial cells Obesity and arterial thrombosis. Ann NY Acad Sci. 2000;902:315–19. doi: 10.1111/j.1749-6632.2000.tb06330.x. [DOI] [PubMed] [Google Scholar]
- 197.Bodary PF, Gu S, Shen Y, Hasty AH, Buckler JM, Eitzman DT. Recombinant leptin promotes atherosclerosis and thrombosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:e119–22. doi: 10.1161/01.ATV.0000173306.47722.ec. [DOI] [PubMed] [Google Scholar]