

Abstract
While most eukaryotic mRNAs are degraded by exonucleases acting on either end of the molecule a subset of mRNAs undergo endonuclease cleavage within the mRNA body. Endonuclease cleavage can be activated by cellular stress, extracellular signals or by ribosome stalling, as might occur at a premature termination codon. Only a few eukaryotic mRNA endonucleases have been identified, and of these polysomal ribonuclease 1 (PMR1) is the best characterized. A notable feature of PMR1-mediated decay is that it acts on specific mRNAs while they are engaged by translating ribosomes. This chapter begins with several procedures used to characterize in vivo endonuclease cleavage of any mRNA by any endonuclease. These include approaches for identifying the 5′ end(s) downstream of an endonuclease cleavage site (S1 nuclease protection and primer extension), and a ligation-mediated RT-PCR approach developed in our lab for identifying the 3′ ends upstream of a cleavage site. We then describe a number of approaches used to characterize PMR1-mediated mRNA decay in cultured cells. PMR1 participates in a number of different complexes and we show several approaches for studying these, and describe techniques for isolating and characterizing PMR1-interacting proteins and its target mRNAs. While the various techniques described here have proven their usefulness in studying PMR1 they can be generalized to studying decay by any other mRNA endonuclease.
Introduction
Most mRNAs are generally thought to be degraded by exonuclease-catalyzed mechanisms in which the mRNA is degraded either in a 3′-5′ manner by the cytoplasmic exosome or in a 5′-3′ manner by enzymes that remove the 5′ cap and degrade the mRNA body with 5′-3′ polarity (reviewed in (Garneau et al., 2007)). In addition to these pathways, a subset of mRNAs undergoes endonuclease cleavage within the mRNA body followed by degradation of the upstream and downstream decay products by exonucleases. While it was generally thought endonuclease-mediated mRNA decay is limited to a few specialized mRNAs, the overall process is functionally analogous to RNA interference, where endonuclease cleavage results in the rapid disappearance of the mRNA body. The mRNA endonucleases that have been identified to date include PMR1, G3BP-1 (Gallouzi et al., 1998), IRE-1 (Hollien and Weissman, 2006), and Dom34 (Lee et al., 2007). While Argonaute 2 can be thought of as an mRNA endonuclease, it is distinguished from this group protein ribonucleases by its requirement for a short antisense RNA. PMR1 was originally identified as an estrogen-induced endonuclease activity that appeared concomitantly with the estrogen-induced destabilization of serum protein mRNAs in Xenopus liver (Pastori et al., 1991b). It differs structurally from most ribonucleases in that it is closely related to the peroxidase gene family, and the active form is processed from a larger precursor (Chernokalskaya et al., 1998). PMR1-mediated mRNA decay also differs fundamentally from other types of mRNA decay in that it forms a specific complex with its translating substrate mRNA, and it is in this context that cleavage initiates mRNA decay. Interestingly, a similar mechanism was recently reported for IRE-1, which is activated by unfold protein to cleave endoplasmic reticulum associated mRNAs (Hollien and Weissman, 2006).
The ability of PMR1 to catalyze mRNA decay depends on its participation in a number of different complexes. PMR1 is inherently unstable, and alterations in its binding to Hsp90 result in rapid degradation by the 26S proteasome (Peng et al., 2007). When examined on sucrose density gradients PMR1 sediments with polysomes and in a lighter mRNP complex at the top of the gradient (Yang and Schoenberg, 2004). Dissociating ribosomes with EDTA releases polysome-bound PMR1 as a complex with its substrate mRNA (termed Complex I) that sediments at ~680 kDa on glycerol gradients (Yang and Schoenberg, 2004). This treatment has no impact on the smaller complex (termed Complex II), which sediments at ~140 kDa on glycerol gradients.
Each of these complexes plays a functional role in PMR1-mediated mRNA decay. PMR1 must be phosphorylated on Y650 in order to join Complex I on polysomes and catalyze mRNA decay (Yang et al., 2004), and this process occurs in Complex II. A key component of the latter complex is the oncogenic tyrosine kinase c-Src. Tyrosine phosphorylation of PMR1 ‘licenses’ PMR1 for binding to polysomes and as such is a requisite step in mRNA decay (Peng and Schoenberg, 2007). PMR1 is also a stress-responsive protein, and in stressed cells the direct interaction of its N-terminal domain with TIA-1 recruits Complex I containing PMR1 and its substrate mRNA to stress granules (Yang et al., 2006), where mRNA decay is stalled. Thus understanding the function of PMR1 in mRNA decay depends on understanding how its participation in a number of macromolecular guides this process.
Our early work used whole animals and primary frog hepatocyte cultures to characterize much of the biology of PMR1. However, the technical difficulty led us to using mammalian cell lines that recapitulate the process of PMR1 targeting to polysomes and the PMR1-mediated mRNA decay. While the work described here focuses on the frog protein expressed in mammalian cells its mammalian ortholog behaves similarly. The latter is somewhat larger, but like frog PMR1 is found on polysomes, is tyrosine phosphorylated, and forms a complex with its substrate mRNA. Whereas most of our work on frog PMR1 uses albumin as a typical substrate mRNA the mammalian ortholog is best characterized for its role in degrading nonsense-containing β-globin mRNA in erythroid cells (Stevens et al., 2002; Bremer et al., 2003).
This chapter is organized roughly in the order of experiments one would follow when first determining if a particular mRNA undergoes endonucleolytic cleavage, followed by experiments used to characterize the behavior and complexes involved in PMR1-mediated mRNA decay. While these approaches are described in the context of this particular enzyme they are generally applicable to studying any other mRNA endonuclease. On a final note, throughout the text we refer to both the frog and mammalian enzyme generically as PMR1. In this context it denotes the fully processed mature protein that participates in the process of endonuclease-mediated mRNA decay. In our published work the frog protein is referred to as PMR60 to distinguish the active, processed 60 kDa form of the protein from the 80 kDa precursor (PMR80) from which it is derived.
I. Identification of endonuclease cleavage sites within mRNA
In general, endonuclease cleavage products including those generated by PMR1 can not be detected because they are rapidly degraded by 5′-3′ or 3′-5′ exonucleases. This makes it difficult to determine how many mRNAs are targeted by endonuclease-mediated decay. If one knows that a particular mRNA is targeted by endonuclease decay it is at least theoretically possible to use techniques like Northern blotting to detect the downstream cleavage product by knocking down Xrn1 or the upstream cleavage product by knocking down one or exosome proteins (Gatfield and Izaurralde, 2004). The protocols described below use S1 nuclease protection and primer extension assays to detect the downstream products of endonuclease cleavage, and a sensitive ligation-mediated PCR assay to detect the upstream products. All three of these have been used successfully in our lab to detect endonuclease-generated decay intermediates in RNA from cultured cells without resorting to knocking down of 5′ or 3′ decay, and products generated by in vitro endonuclease cleavage.
1. Identification of 5′ products generated by endonuclease cleavage in vivo
1a. S1 nuclease protection assay
Overview
S1 nuclease protection assay is a highly sensitive method for the detection, mapping and quantification of specific RNA fragments in total cellular RNAs. The basis of S1 nuclease protection assay is a hybridization of a 5′ end-labeled single-stranded antisense DNA probe to a target RNA, and then digesting unhybridized probe and cellular RNAs with single strand-specific S1 nuclease. Once the reaction is done S1 nuclease is inactivated and the remaining hybridized probe and RNA are precipitated. Finally, the protected probe is electrophoresed on a denaturing polyacrylamide gel and visualized by autoradiography, or more commonly, phosphorimager. With proper controls this can be used to quantify the degree of endonuclease cleavage.
Although double stranded nucleic acids are generally resistant to S1 cleavage can occur under conditions of excess enzyme and high salt (Vogt, 1973). Also, because it does not effectively cleave at single base mismatches S1 nuclease protection can be used to survey the effects of changing a single base across a particular RNA, such as one might encounter when studying the impact of changing the location of a premature termination codon. The susceptibility of mismatches to cleavage by S1 increases with the length of the mismatched sequence but this varies depending on the mismatching sequences (Brookes and Solomon, 1989). Thus if the potential exists for mismatches between the target mRNA and the probe one needs to optimize the reaction conditions to insure that the assay detects genuine cleavage products. Use of dioxane in the S1 nuclease reaction enhances the ability of S1 nuclease to cleave at single base mismatches (Howard et al., 1999). It is imperative that there is some additional sequence at the 3′ end that does not hybridize to the target mRNA so that undigested probe can be differentiated from probe that is protected by hybridization to intact mRNA. We have found it particularly useful to make probes by asymmetric PCR, although one can also use a cDNA clone which is linearized by cleaving within a multiple cloning site to give a 3′ end that does not hybridized to the target mRNA.
Protocol
End-labeling of DNA primer
50 pmol of an antisense oligonucleotide (18–30 nt long) is combined with 10 μl of γ-32P-ATP (3000 Ci/mmol) and 2 μl of polynucleotide kinase (Roche, 10U/μl) in 50 μl of kinase buffer (50 mM Tris-HCl, pH 8.2, 10 mM MgCl2, 5 mM dithiothreitol (DTT), 0.1 mM spermidine, 0.1 mM EDTA).
The mixture is incubated at 37°C for 1 hr, and the reaction is terminated by heating at 65°C for 10 min. If you wish to determine the degree of incorporation at this step dilute 1 μl into 100 μl of PBS and dot 1 μl of this onto a glass fiber filter. This is placed into 10% TCA, rinsed with ethanol, dried and counted.
1 μl of glycogen (Roche, 20 mg/ml), 13 μl of 7.5M ammonium acetate, and 160 μl of ethanol are added to the mixture and labeled oligonucleotides are recovered by precipitation at −20°C.
The precipitated DNA is recovered by centrifuging for 15 min at 12,000 × g in a refrigerated microcentrifuge, washed with 70% ethanol and the pellet is dissolved in 21 μl of water.
Generation of single-stranded probes by asymmetric PCR
A flexible approach is to use asymmetric PCR, since the probe can be prepared from anywhere within a cloned DNA without regard to the location relative to a multiple cloning site. 7 μl of the radiolabeled primer is added to a reaction containing 2.5 μl of 50 mM of MgCl2, 1 μl of 10 mM dNTPs, 0.3 μl of Taq DNA polymerase (Invitrogen), 1.6 pmol of a sense primer that contains additional noncomplementary sequence at 5′ end and 100 ng of template DNA for PCR in 25 μl of PCR buffer (20 mM Tris-HCl, pH 8.4, 50 mM KCl). Note that the ratio of sense to antisense primer in the reaction is 1:10. The DNA template used for probe preparation is a PCR fragment that has been amplified using the same sense primer as above and an antisense primer which is located downstream of the above labeled antisense primer.
The mixture is heated at 94°C for 3 min and PCR amplification is performed for 30 cycles at 94°C for 30 sec, 54°C for 30 sec, and 72°C for 1 min, followed by extension for 5 min at 72°C.
PCR products are separated on 6% polyacrylamide/urea gel and exposed to X-ray film to detect radiolabeled probes.
Radiolabeled probes are eluted from gel slices in 300 μl of probe elution buffer (0.5 M ammonium acetate, 1 mM EDTA, 0.2% SDS) by incubation for 3 ~ 4 hr at 37°C or overnight at 4°C for maximal recovery.
The amount of radioactivity in the recovered probe is determined by scintillation counting.
S1 nuclease protection
5 × 105 cpm of labeled DNA probe is ethanol-precipitated together with 5–50 μg of RNA, or yeast tRNA as a control. The pellet is rinsed with 70% ethanol and dried for 5 min on the bench.
The pellet is dissolved with 30 μl of hybridization buffer (40 mM 1,4-piperazine-diethanesulfonic acid (Pipes), pH 6.4, 1 mM EDTA, 0.4 M NaCl, 80% formamide).
The solution is heated at 95°C for 4 min, transferred immediately to 52°C, and incubated overnight.
S1 nuclease solution is prepared by adding 250 units of S1 nuclease (Promega) to 1 ml of S1 nuclease buffer (0.28 M NaCl, 0.05 M sodium acetate (pH 4.6), 4.5 mM ZnSO4, 20 μg/ml sheared salmon sperm DNA).
300 μl of ice-cold S1 nuclease solution is added to each hybridization mixture.
300 μl of ice-cold S1 nuclease buffer without S1 nuclease is added to yeast RNA control tube for probe integrity.
The reaction mixture is incubated at 28°C for 2 hr followed by addition of 80 μl of stop solution (4M ammonium acetate, 0.1 M EDTA, 50 μg/ml yeast tRNA).
Because the preceding step removes most of the nucleic acid in the reaction 0.5 μl of glycogen carrier (Roche, 20 mg/ml) is added next, followed by 60 μl of 7.5 M ammonium acetate and 400 μl of isopropanol. This then stored at −20°C for at least 1 hr.
The radiolabeled products are recovered by centrifuging at 12,000 ×g for 15 min in a refrigerated microcentrifuge and the pellet is rinsed with 70% ethanol. This is then air-dried for 5 min and dissolved in 5 μl of gel loading buffer (95% formamide, 18 mM EDTA, 0.025% (w/v) SDS, 0.025% (w/v) xylene cyanol, 0.025% (w/v) bromophenol blue).
Samples are heated at 95°C for 3 min, chilled on ice immediately and loaded onto a polyacrylamide/urea gel. Because of the greater amount of radioactivity of the undigested probe only 5% of this is loaded onto the gel.
To pinpoint the exact location of endonuclease cleavage the adjacent lanes should contain a dideoxy sequencing ladder that is prepared using the radiolabeled primer used for generating the S1 probe.
After electrophoresis, the gel is dried and protected fragments are visualized by Phosphorimager. An example of the use of S1 protection to identify an endonuclease cleavage product is shown in Fig. 1. In this experiment S1 protection was used to map a previously unidentified endonuclease cleavage site in nonsense-containing β-globin mRNA expressed in murine erythroleukemia cells.
Figure 1. Identification of a PTC-induced endonuclease cleavage site at position 32 in human β-globin mRNA.

Norm2 are murine erythroleukemia cells stably expressing normal human β-globin mRNA and Thal10 cells are a match line stably expressing the same mRNA with a premature termination codon at codon 60/61 (Stevens et al., 2002). RNA from each of these cell lines was analyzed by S1 nuclease protection using a probe spanning the first 250 nt of β-globin mRNA. A dideoxy sequencing ladder in lanes 1–4 was used to identify a new endonuclease cleavage site at position 32 relative to the 5′ end of the mRNA. Lanes 7 and 8 are darker exposures of lanes 5 and 6.
1b. Primer extension assay
Overview
The primer extension assay is an alternative method for detecting and mapping the 5′ end of endonuclease cleavage products. In this approach a 5′ end-labeled antisense primer is hybridized to the complementary region of a target RNA and extended by reverse transcriptase to generate cDNAs extending to the sites of endonuclease cleavage and/or the 5′ end of the mRNA. The products are analyzed on a denaturing polyacrylamide gel and visualized by Phosphoimager, commonly using a dideoxy sequencing ladder as described above to map precisely the location of the cleavage site. Because secondary structures within an mRNA can result in pausing of reverse transcriptase one must include a control of an in vitro transcript that is run at the same time and electrophoresed in a parallel lane of the gel. Effective hybridization of the primer is the most critical step for success in primer extension analysis, and the amount of template RNA, primer and the hybridization temperature need to be optimized. The particular reverse transcriptase used may also affect the outcome. AMV reverse transcriptase is more robust than MMLV and is less sensitive to the effect of secondary structure. However, MMLV reverse transcriptase generates longer products than AMV reverse transcriptase.
Protocol
1–5 × 105 cpm of a 5′ end-labeled antisense oligonucleotide is ethanol-precipitated with 10–25 μg of total cellular RNA. In a separate tube, 20 ng of in vitro transcript corresponding to the region of your target mRNA and 10 μg of yeast tRNA as a carrier are ethanol-precipitated with the labeled oligonucleotide. This will serve as a control for reverse transcriptase pausing sites. The recovered pellet is washed with 70% of ethanol and dried for 5 min on the bench.
The precipitated RNA and oligonucleotide are dissolved in 10 μl of annealing buffer (50 mM Tris-HCl, pH 8.3, 75 mM KCl), heated at 65°C for 10 min, followed by cooling slowly to 37°C, and then put on ice.
4 μl of RT buffer (125 mM Tris-HCl, pH 8.3, 187.5 mM KCl, 15 mM MgCl2), 1 μl of 10 mM dNTPs, 2 μl of 0.1 M DTT, 1 μl of water and 1 μl of RNaseOUT (Invitrogen, 40U/μl) is added to the annealing mixture and incubated at 42°C for 2 min.
1 μl of M-MLV reverse transcriptase (Invitrogen, 200U/μl) is added and the mixture is incubated for 50 min at 42°C.
The reaction is stopped by adding 20 μl of loading buffer directly or by ethanol precipitation and dissolving the pellet in loading buffer.
The experimental samples and the in vitro transcript control reaction are loaded onto a polyacrylamide/urea gel and visualized by phosphoimager. Any bands seen in both experimental and control samples correspond to reverse transcriptase pausing sites and should be excluded from the list of in vivo cleavage sites.
It is also essential to include a dideoxy sequencing ladder in adjacent lanes to pinpoint the exact site of endonuclease cleavage. Note that the sequencing ladder should use the same radiolabeled primer.
2. Identification of 5′ cleavage products generated by endonuclease in vivo
S1 nuclease protection can be used to detect and map the 3′ end of upstream endonuclease cleavage products; however this requires use of a 3′ end-labeled antisense probe, which is more difficult to prepare than the 5′ end-labeled probes described above. Here, we describe an alternative approach that uses ligation-mediated RT-PCR that we used successfully to identify in vivo decay intermediates of albumin and c-myc mRNA (Hanson and Schoenberg, 2001).
Ligation-Mediated Reverse Transcription-Polymerase Chain Reaction (LM-RT-PCR)
Overview
Protein endonucleases and RISC generate products with a 5′ monophosphate on the downstream fragment and a 3′ hydroxyl on the upstream fragment. 3′ end mapping by LM-RT-PCR takes advantage of the latter. In the first step the total RNA population is ligated to a DNA oligonucleotide bearing a 5′-phosphate and 3′-amino group. The 5′ phosphate allows this to be ligated to the 3′ hydroxyl of the upstream cleavage product and the 3′-amino group blocks concatamerization of the primer during ligation. A primer complementary to the ligated oligonucleotide is used for reverse transcription and PCR is then performed using a 5′-32P-labeled sense primer that is specific to the mRNA of interest and the primer used for reverse transcription. The PCR products are then separated on a denaturing polyacrylamide/urea gel and visualized by Phosphoimager. This application is straightforward and extremely sensitive.
Protocol
Primer Ligation
2 μg of total or cytoplasmic RNA is combined with 1 μg of the ligation primer (5′ P-CCAGGTGGATAGTGCTCAATCTCTAGATCG-NH3) in 15 μl of ligation buffer (50 mM Tris-HCl, pH 8.0, 10 mM MgCl2, 20 mM ATP, 2 mM DTT, 10 μg/ml BSA, 1 mM hexamine cobalt chloride, 25% (w/v) polyethylene glycol 8000, 20 units RNaseOUT).
The mixture is incubated overnight at 4°C using 15 units of T4 RNA ligase (New England BioLabs). The conditions described here are optimized to add the primer onto approximately 50% of the 3′ ends.
The reaction is terminated by extracting twice with an equal volume of phenol: chloroform: isoamyl alcohol (25:24:1) and the products are recovered by adding 1 μl of glycogen, 1/10 volume of 3M sodium acetate (pH 5.4) and 2.5 volumes of cold ethanol. This is kept at −20°C for at least 2 hr to maximize product recovery.
Nucleic acid is recovered by centrifuging at 12,000 ×g for 15 min in a refrigerated microcentrifuge, and the resulting pellet is dissolved in 10 μl of DEPC-treated water.
Labeling of gene specific oligonucleotide primer
500 ng of the desired sense primer is combined with 3 μl of γ-32P-ATP (3000 Ci/mmol) and 1 μl of polynucleotide kinase (Roche, 10U/μl) and 2.5 μl of 10X kinase buffer (500 mM Tris-HCl, pH 8.2, 100 mM MgCl2, 50 mM DTT, 1 mM spermidine, 1 mM EDTA) in final volume of 25 μl.
The mixture is incubated at 37°C for 1 hr, and the reaction is terminated by heating at 65°C for 10 min.
RT-PCR
10 μl of the above reaction mixture is mixed with 1 μl of 10 mM dNTPs and 250 ng of antisense primer (5′-CGATCTAGAGATTGAGCAC-3′) in 1 μl of DEPC-treated water, which is complementary to the last 19 nt of ligation primer.
The solution is heated to 70°C for 10 min and quickly cooled on ice to anneal the primer, followed by addition of 4 μl of 5X RT buffer (250 mM Tris-HCl, pH 8.3, 375 mM KCl, 15 mM MgCl2), 2 μl of 0.1M DTT and 1 μl of RNaseOUT to yield a total volume of 19 μl.
This is heated at 42°C for 2 min followed by the addition of 1 μl of Superscript II reverse transcriptase (Invitrogen, 200U/μl) and incubated for an additional 50 min.
The reaction is terminated by heating at 70°C for 15 min.
3 μl of the above reaction mixture is transferred to a Hot Start tube (Molecular BioProducts) and mixed with 2.5 μl of 10X PCR buffer (200 mM Tris-HCl, pH 8.4, 500 mM KCl), 3 μl of 25 mM of MgCl2, and 3 μl of 10 mM dNTPs.
The wax bead of the Hot Start tube is melted by heating at 75°C for 30 sec followed by cooling quickly on ice.
2 μl of 5′-end-labeled gene specific primer, 0.5 μl of Taq DNA polymerase (Invitrogen) and 11 μl of DEPC-treated water are then added to the mixture in the final volume of 25 μl.
The mixture is heated at 94°C for 2 min and PCR amplification is performed for 25 cycles at 94°C for 1 min, 58°C for 30 sec, and 72°C for 1 min, followed by extension for 3 min at 72°C.
The reaction mixtures are then extracted with phenol: chloroform: isoamyl alcohol and the amplified products are recovered by ethanol precipitation.
The pellet is dissolved in 6 μl of loading buffer, heated at 95°C for 4 min and electrophoresed on a polyacrylamide/urea gel. After electrophoresis, gel is dried and visualized by Phosphorimager.
Two approaches can be used to pinpoint the site of endonuclease cleavage. The easiest is to prepare a sequencing ladder from a cDNA of the target mRNA and subtract from this the size (19 nt) of the ligated primer. If one wishes to obtain precise positional information or look for non-templated nucleotides that might be added after cleavage the gel can be visualized by film autoradiography following which the bands are excised as described above for purifying radiolabeled probes, re-amplified, cloned and sequenced (Hanson and Schoenberg, 2004).
II. Analysis of PMR1-containing complexes
PMR1-mediated mRNA decay depends on its interaction with a number of different complexes, and we generally study these by sedimentation on sucrose and glycerol gradients. Polysome profile analysis using sucrose density gradients is the method of choice for studying the interaction of PMR1 with its translating substrate mRNA. As noted above decay depends on both the binding of PMR1 to polysomes and ongoing translation of its substrate mRNA. Tyrosine phosphorylation at position 650 by c-Src is required for the targeting PMR1 to polysomes, so it is important to know the relative expression and activity of c-Src in cells used for analyzing PMR1-mediated mRNA decay. Dissociating polysomes by adding EDTA to extracts or adding puromycin to cells prior to lysis releases PMR1 in two main complexes; a ~680-kDa complex (termed complex I) that contains PMR1 and its substrate mRNA, and a ~140 kDa complex (complex II), which contains PMR1 and c-Src. As one might expect, complex II is the precursor to complex I and the site where tyrosine phosphorylation occurs. The following approaches describe methods used to study PMR1 in transfected mammalian cells and their application to studying PMR1-mediated mRNA decay.
1. Expression of PMR1 in transfected mammalian cells
Our early work used primary frog hepatocyte cultures to characterize the properties of PMR1. However, these are difficult to culture and transfect, so we investigated mammalian cell lines for their ability to recapitulate the process of PMR1 targeting to polysomes and the PMR1 specific endonuclease activity. As a general rule of thumb the more differentiated epithelial cell lines such as MEL, MCF7 and Jurkat, express the cross-reacting ortholog of frog PMR1, whereas others (eg. Cos-1, U2OS, HeLa, CHO, 3T3) do not. Much of our work has been done using Cos-1 and U2OS osteosarcoma cells since the sedimentation of PMR1 complexes in these cells closely matches that seen in frog liver and hepatocytes cultures. Most work studying the various PMR1-containing complexes uses a catalytically inactive form of PMR1, termed PMR60°, that lacks an endonuclease activity but still participates in all of the relevant complexes. As noted above, this also corresponds to the form of the enzyme that is processed from an 80 kDa precursor. All of our expression plasmids include an N-terminal myc epitope tag for ease of detection. For most experiments 1.5–2 × 106 cells are plated on 100 mm dishes the afternoon prior to transfection. The next morning these are transfected with 6 μg of the plasmid expressing epitope tagged PMR60° using commercial lipid-based transfection reagents (eg. Fugene, Lipofectamine), and cells are harvested 40 hr after transfection.
2. Preparation of postmitochondrial extracts
The protocol described below was developed for use with 100 mm dish and should be adjusted for the amount of sample.
After aspirating the medium cells are washed twice with ice-cold PBS and harvested into 10 ml of PBS using a cell scraper. They are then collected by centrifugation at 500 ×g for 5 min.
The cell pellet is gently resuspended in 0.5 ml of lysis buffer (10 mM Hepes-KOH, pH 7.5, 10 mM KCl, 10 mM MgCl2, 50 mM NaF, 0.5% NP-40, 2 mM DTT, 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 25 μl/ml protease inhibitor cocktail (Sigma-Aldrich), 10 μl/ml phosphatase inhibitor cocktails I and II (Sigma-Aldrich) 400U/ml of RNaseOUT) and the tube is placed on ice for 10 min. Note that the volume of lysis buffer can be adjusted as needed for subsequent steps.
The cells are then homogenized using 15–30 strokes with a chilled Dounce homogenizer (A pestle). Note that this must be done gently and care must be taken to avoid foaming, particularly during the up-stroke. It is a good idea to look at a drop of extract under the microscope after every 5 strokes to monitor the extent of lysis.
The homogenate is centrifuged at 4°C for 10 min at 15,000 ×g to remove cell debris and nuclei and the postmitochondrial supernatant is either used directly (eg. for gradient analysis) or quickly frozen and stored at −80°C. Note that if problems with polysome dissociation are encountered 100 μg/ml of cycloheximide can be added to the PBS used for washing the cells and to the lysis buffer.
3. Sucrose density gradient analysis of PMR1-containing complexes
3a. Linear sucrose density gradients
Place a gradient maker, peristaltic pump and solutions into a cold room or cold box. Each linear 11 ml 10–40% sucrose gradient is prepared in a 14 × 89 mm ultracentrifuge tube (Beckman) using solutions of 10 and 40% sucrose in 10 mM Hepes-KOH, pH 7.5, 10 mM KCl, 10 mM MgCl2, 0.5% NP-40, 0.5 mM PMSF, 25 μl/ml of protease inhibitor cocktail, 100U/ml RNaseOUT, 2 mM DTT, 100 μg/ml cycloheximide. Sucrose gradients are quite stable and can be made the day before harvesting cells.
To insure there is a sufficient amount of sample in each fraction we use postmitochondrial extracts prepared from a 150 mm culture dish containing cells at the density described above.
0.5 ml is layered carefully on top of each gradient, and they are centrifuged at 225,000 ×g for 2–3.5 hr at 4°C in a Sorvall TH641 or Beckman SW41 rotor with slow start and slow stop. The length of time of the centrifugation depends on the desired distribution of polysomes across the gradient, with a broader distribution seen at shorter times.
0.5 ml fractions are collected using a peristaltic pump to drive the solution through a UV monitor into the fraction collector. Alternatively if a monitor is not available one can use a spectrophotometer or Nanodrop instrument to read the absorbance of each fraction at OD260.
Protein samples from each fraction are concentrated by TCA precipitation. Each fraction is diluted with equal volume of water to reduce the concentration of sucrose to <0.5 M. 100% TCA solution is added to sample to final volume of 10–20%, followed by incubation on ice for at least 1 hr. The precipitated protein is recovered by centrifuging for 20 min at 12,000 × g in a refrigerated microcentrifuge, washed with 1 ml of ice-cold acetone and recovered by centrifugation again. The pellet is dried by speed vacuum centrifugation for about 5 min and dissolved in SDS loading buffer.
The distribution of PMR1 is analyzed by western blotting using antibody to the N-terminal myc tag. In addition we commonly use antibody to ribosomal protein S6 (Santa Cruz Biotechnology) to confirm the distribution of polysomes on the gradient. An example of this is shown in Fig. 2. In this particular experiment (from Yang et al., 2004) PMR1-containing fractions were recovered by binding of TAP tagged protein to IgG-Sepharose and material recovered after Tev protease cleavage was analyzed by Western blotting with antibody to the myc tag (upper panel) and S6 (bottom panel). The use of TAP-tagged PMR1 for characterizing complexes is described below. If one co-expresses a PMR1 target, such as albumin mRNA, its distribution can also be analyzed by Northern blot, RPA or semi-quantitative RT-PCR of RNA isolated from gradient fractions.
As an additional control polysomes can be dissociated by adjusting the post-mitochondrial extract to 50 mM EDTA before gradient centrifugation or by adding 200 μg/ml of puromycin to cells 30 min before harvesting.
Figure 2. Sedimentation of PMR1-containing complexes on a linear sucrose density gradient.

Post-mitochondrial extract from Cos-1 cells expressing PMR1-TAP were separated on a linear 10–40% sucrose gradient. In the upper panels odd numbered fractions were recovered on IgG-Sepharose, eluted by Tev protease cleavage and analyzed by Western blotting with antibodies to the myc tag on PMR1 (upper panel) and the phosphotyrosine monoclonal antibody PY20 (middle panel). In the bottom panel even numbered fractions were TCA precipitated and analyzed by Western blotting with antibody to ribosomal protein S6. This figure is adapted from Yang et al. (2004).
3b. Sucrose step gradients
Sucrose step gradients are a simpler method to determine the distribution of PMR1 between polysomes and smaller complexes, such as complex II. In this approach post-mitochondrial extract is centrifuged through a 2 ml gradient using a tabletop ultracentrifuge. The polysomes pellet to the bottom of the gradient and the smaller mRNP complexes accumulate at the interface between two sucrose pads.
1 ml of 35% prepared as described above is placed into the bottom of a 11 × 34 mm ultracentrifuge tube (Beckman) and this is overlaid with 1 ml of 10% sucrose, taking care not to disturb the interface. To facilitate sample recovery the interface and top of gradient should be marked on the tube.
0.2 ml of postmitochondrial extract isolated from cells in 100 mm dish is layered carefully on top and the tube is centrifuged for 1 hr at 147,000 × g in a Sorvall Discovery M120 SE ultracentrifuge at 4°C using an S55S-1123 rotor.
200 μl from top and the interface are removed and saved for analysis, and any remaining solution is removed carefully, taking care not to disturb the polysome pellet on the bottom of the tube. This is then dissolved in 200 μl of lysis buffer.
The distribution of PMR1 is analyzed by Western blotting using antibody to the myc tag, and as above the sedimentation of polysomes to the bottom can be confirmed by Western blotting with S6 antibody. An example of the usefulness of this quick and easy technique is shown in Fig. 3, where step gradients were used to show that native albumin mRNA sediments to the bottom of a step gradient with polysome but albumin mRNA with a strong stem-loop structure that prevents translation initiation sediments with mRNPs (Yang and Schoenberg, 2004).
Figure 3. Separation of polysomes and mRNP complexes on discontinuous sucrose step gradients.
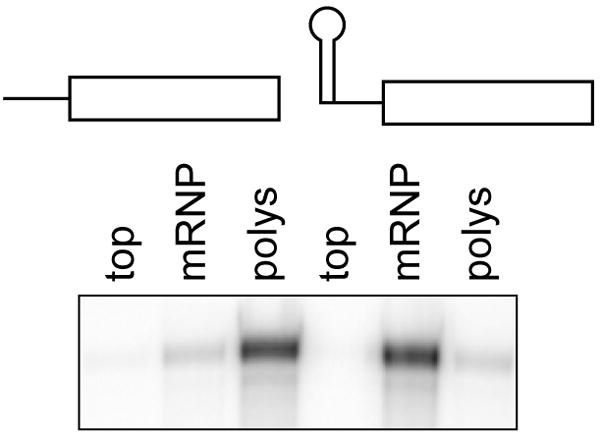
Post-mitochondrial extracts from Cos-1 cells expressing native albumin mRNA or albumin mRNA with a strong stem-loop in the 5′-UTR (SL-albumin) were applied to step gradients consisting of 1 ml each of 10 and 35% sucrose. The top, middle and pellet fractions were collected after centrifugation and adjusted to equal volumes, and RNA recovered from each was analyzed by RPA (adapted from Yang and Schoenberg, 2004).
4. Glycerol gradient analysis of PMR1 containing complexes
As noted above the functional unit of PMR1-mediated mRNA decay is a ~680 kDa complex containing PMR1 and its translating substrate mRNA. This is released from polysomes by treating with EDTA or adding puromycin to cells prior to harvest, and the resulting complexes are analyzed by sedimentation on glycerol gradients, which are more amenable to studying these smaller complexes than sucrose density gradients.
A 11 ml linear density gradient of 10–40% (v/v) glycerol in 10 mM Hepes-KOH, pH 7.5, 10 mM KCl, 10 mM MgCl2, 0.5 % NP-40, 0.5 mM PMSF, 25 μl/ml of protease inhibitor cocktail, 100U/ml RNaseOUT, 2 mM DTT is prepared in a 14 × 89 mm ultracentrifuge tube as described above.
Cells are treated with 200 μg/ml of puromycin for 30 min before harvesting or post-mitochondrial extract is adjusted to 50 mM EDTA to dissociate polysomes. 0.5 ml of the postmitochondrial extract prepared a single 150 mm dish of cells is layered carefully on top of each gradient and centrifuged for 20 hr at 83,000 ×g in a Sorvall TH641 rotor at 4°C. A molecular size marker (GE Healthcare) containing thyroglobulin (MW 669,000), ferritin (MW 440,000), catalase (MW 232,000), lactate dehydrogenase (MW 140,000) and bovine serum albumin (MW, 67,000) is fractionated on a parallel gradient.
0.5 ml of fractions are collected and individual fractions from the gradient containing molecular size markers are assayed using the Bio-Rad (Bradford) protein assay to identify fractions containing each of the markers.
As with sucrose gradient fractions these are diluted with an equal volume of buffer prior to TCA precipitation (See Section II.3a.5).
The sedimentation of PMR1 is analyzed as described above by Western blotting using antibody to the N-terminal myc tag. An example of the usefulness of glycerol gradients for analyzing PMR1-containing complexes is shown in Fig. 4 (modified from Yang et al., 2004). The upper panels show the gradient distribution of native PMR1 or PMR1 containing the Y650F mutation, which prevents phosphorylation by c-Src. Complexes I and II are seen in gradients performed with native PMR1 but not the mutated form, and RT-PCR of RNA recovered with the pooled fractions of each show that target (albumin) mRNA is only recovered with native PMR in Complex I (bottom panels).
Figure 4. Glycerol gradient sedimentation of PMR1-containing complexes.

Cos-1 cells were transfected with plasmids expressing albumin and luciferase mRNA together with TAP-tagged catalytically-inactive PMR1 or the Y650F form of the protein. They were treated with puromycin before harvesting to dissociate polysomes and post-mitochondrial extracts were separated on 10–40% glycerol gradients. The collected fractions were analyzed by Western blot using antibody to myc-tag (upper panel) and the two major complexes are indicated (Complex I - ~680 kDa, Complex II - ~140 kDa). Fractions making up each of these complexes were pooled and in the bottom panels the recovered RNA was analyzed by RT-PCR for albumin and luciferase mRNA (adapted from Yang et al., 2004).
III. Affinity recovery of PMR1-containing complexes
There are numerous approaches available for identifying and characterizing macromolecular complexes. For maximal flexibility we have engineered plasmids expressing PMR1 with an N-terminal myc epitope tag and a C-terminal tandem affinity purification (TAP) (Puig et al., 2001), and used both tags to systematically identify PMR1-interacting proteins and target mRNAs. Both approaches can be used for recovering complexes from transiently transfected cells, but a more powerful approach is to generate stable cell lines expressing tagged PMR1 under a regulated promoter. The following describes approaches for recovering PMR1-containing complexes that take advantage of the gentle recovery afforded by eluting TAP-tagged proteins from IgG-Sepharose by cleaving within the tag with Tev protease, and approaches using immobilized myc monoclonal antibody to immunoprecipitate PMR1 and it associated proteins.
1. IgG-Sepharose selection of PMR1-TAP complexes
The protocol described below was created for use with transient transfected cells in a 100 mm dish or stable cell lines in a 150 mm dish and can be varied depending on the sample size.
Equilibration and Blocking of IgG Sepharose beads
IgG Sepharose 6 Fast Flow (GE Healthcare) is washed twice with at least 5 bed volumes of water by end-over-end rotation at 4°C.
The beads are treated for 5 min with 5 bed volumes of 0.5 M of acetic acid (pH 3.4) and washed four times with at least 5 bed volumes of TST buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% Tween 20).
Step 2 is repeated once.
The beads are washed twice with at least 5 bed volumes of IPP150 buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% NP-40) and the pH of the wash buffer is monitored to insure complete removal of the acetic acid.
To reduce and/or prevent nonspecific binding the beads are incubated for 2 hr at 4cC with 50 μg/ml of BSA or postmitochondrial extract from nontransfected cells.
Unbound protein is removed by washing the beads 5 times with at least 5 bed volumes of IPP150 buffer.
Sample application, washing, and elution with Tev protease
0.5 ml of postmitochondrial extract prepared as described above is either used directly or treated with 50 mM EDTA to dissociate polysomes. 50 μl of extract is kept as an input fraction.
50 μl of pre-blocked IgG Sepharose beads is added to 450 μl of extract and this is incubated for 2 hr at 4°C with end-over-end rotation.
The beads are then washed 4 times with 0.5 ml of IPP150 buffer and twice with Tev protease buffer (50 mM Tris-HCl, pH 8.0, 0.5 mM EDTA, 1mM DTT).
Bound complexes are eluted by adding 100 μl of Tev protease buffer containing 5 μl of Tev protease (Invitrogen, 10U/μl) and incubating at 4°C for 2–5 hr. Alternatively one can perform this step for 1hr at 25°C, but initially both approaches should be compared to insure the increased temperature does not result in dissociation of PMR1-containing complexes.
Complexes recovered in this manner can be subsequently analyzed for PMR1-interacting proteins or RNAs.
The TAP recovery approach is flexible and can used for several downstream applications. One example of this is seen in Fig. 2 where individual sucrose gradient fractions from cells expressing PMR1-TAP were recovered on IgG-Sepharose, released with Tev protease cleavage and analyzed by Western blotting with antibodies to the myc tag on PMR1 and PY20. Another useful application of the TAP tag is for studying the relative strength of protein interactions with PMR1, where IgG-Sepharose-bound complexes can be washed with increasing salt to identify proteins that bind with high affinity versus low-affinity interactions. Because Tev protease cleavage generates PMR1 with a C-terminal calmodulin binding-peptide complexes recovered from IgG-Sepharose can be processed through a second round of selection on calmodulin agarose, followed by elution with EDTA (Puig et al., 2001). This protein is suitable for analysis of post-translational modifications by mass spectrometry. We have also taken advantage of this flexible recovery procedure to look at the interaction of PMR1 with its substrate mRNA. An example of this is seen in the glycerol gradient analysis of Fig. 4, where the fractions corresponding to Complex I and Complex II were pooled, separated on IgG-Sepharose and RNA recovered after Tev protease cleavage was analyzed by RT-PCR. We are currently combining this with microarrays to analyze the scope of PMR1-mediated mRNA decay.
2. Immunoprecipitation of PMR1 using immobilized myc antibody
600 μl of post-mitochondrial extracts from transfected cells or stable cell lines are prepared as described in II.2.
60 μl of extracts is kept as an input fraction and 540 μl is incubated overnight at 4°C with 20 μl of myc monoclonal antibody-coupled beads (Santa Cruz Biotechnology: Cat.No. sc-40AC) using end-over-end rotation.
The beads are recovered by centrifuging at 500 ×g at 4°C for 1 min and the supernatant is kept as the unbound fraction.
The beads are washed at least 5 times with ice-cold IPP150, each time recovering the beads by centrifugation.
50 μl of SDS loading buffer is added to the beads, followed by heating for 5 min at 90°C.
PMR1 and its associated proteins are detected by Western blotting using either a rabbit polyclonal antibody or a mouse monoclonal antibody to the myc tag to detect PMR1. Because PMR1 must be phosphorylated by c-Src to bind to polysomes and catalyze endonuclease cleavage it is important to determine its tyrosine phosphorylation state. Most commonly this is done by probing Western blots of protein recovered as described above with the phosphotyrosine monoclonal antibody PY20 (BD Biosciences). Also, since c-Src binds directly to PMR1 its recovery can be monitored using antibody to this protein (Santa Cruz Biotechnology B-12 Cat.No. sc-8056).
VI. Analysis of PMR1 activity in vivo and in vitro
1. In vivo analysis of PMR1 activity
The in vivo activity of PMR1 is monitored using plasmid vectors expressing a known substrate mRNA together with a known control. Currently the best-characterized substrates are Xenopus serum protein mRNAs (Pastori et al., 1991a) of which we commonly use an albumin minigene. To date the best-characterized mammalian target mRNA for PMR1 is human β-globin (Bremer et al., 2003), and this is degraded by PMR1 in both erythroid and non-erythorid cells. Firefly luciferase mRNA is not degraded by PMR1 and it is commonly used as a co-transfected control for enzymatic activity.
RNase protection assay (RPA) and semi-quantitative RT-PCR with radiolabeled primers are the assays used most commonly to monitor PMR1-induced changes in target mRNA. We most commonly use RPA because it is quite sensitive and enables us to examine substrate and control mRNAs in the same sample. As noted above PMR1 must be phosphorylated by c-Src in order to form a complex with its translating substrate mRNA on polysomes, so the amount and activity of c-Src must be determined any cell line used for analyzing PMR1-mediated mRNA decay.
Protocol
2–3 × 106 cells in a 100 mm dish are transfected with 6 μg of a plasmid expressing myc-tagged catalytically active form of PMR1 or myc-GFP as a control plus 2 μg each of plasmids expressing full-length albumin and luciferase mRNAs. Cells are harvested 40 hr after transfection and total RNA and protein are isolated using Trizol reagent (Invitrogen).
Antisense probes are prepared from cDNA plasmids for each of the target mRNAs using. Each probe should have a specific activity of 3 × 108 cpm/ug.
2–8 × 104 cpm or 150~600 pg of a labeled target RNA probe and a control RNA probe are precipitated together with 5–50 μg of total or cytoplasmic RNA, or yeast RNA as a control by ethanol, the pellet is washed with 70% ethanol and dried for 5 min on the bench. The amount of albumin and luciferase mRNA is then determined by RPA using the Ambion RPA III kit. Products separated on a denaturing polyacrylamide/urea gel are visualized by Phosphorimager to quantify relative differences in albumin/luciferase mRNA, and Western blotting should be performed with antibody to the myc tag on PMR1 or GFP to monitor their expression in each sample, and with antibody to luciferase as a control for relative transfection efficiency.
2. In vitro analysis of PMR1 activity
While the ability to recapitulate PMR1-mediated mRNA decay in cultured cells allows for the analysis of functional domains one must also keep in mind that this is an enzyme, and changes to the protein can have consequences for enzymatic activity. This becomes particularly important when studying mutations that alter the binding of PMR1 to polysomes, since these can modulate decay either by increasing or decreasing polysome targeting, or by interfering with the folding of the active site. To study this we take advantage of the gentle recovery afforded by the TAP tag approach to measure target mRNA degradation in vitro by protein recovered from transfected cells.
PMR1-TAP is recovered from postmitochondrial extracts or complexes recovered by gradient sedimentation by binding onto IgG-Sepharose (see Section III.1). As a specificity control a parallel recovery is done from cells expressing myc-tagged GFP, also containing a TAP tag. Western blotting with antibody to the myc tag is used to assess protein recovery after Tev protease cleavage.
A uniformly labeled transcript is prepared that contains the mapped PMR1 cleavage sites within the 5′ end of albumin mRNA (Chernokalskaya et al., 1997) or another known target mRNA (eg. human β-globin). It is best to use substrates with mapped endonuclease cleavage sites so that one can monitor both loss of input RNA and production of cleavage products. An alternative approach is to prepare a 5′ end-labeled transcript by treating with tobacco acid pyrophosphatase followed by shrimp alkaline phosphatase, and using T4 polynucleotide kinase to label the dephosphorylated RNA with γ-32P-ATP. All radiolabeled RNAs should be gel purified before use in activity assays. Note that luciferase mRNA is not cleaved well in vitro by PMR1 and can be used as a specificity control for PMR1 activity.
Protocol
2–10 × 104 cpm of a labeled RNA transcript is mixed with 5–10 μl of myc-tagged PMR1 or GFP that has been recovered by Tev protease cleavage of protein bound onto IgG-Sepharose in buffer containing 10 mM Tris-HCl, pH 7.5, 50 mM KCl, 2 mM MgCl2, 2 mM DTT.
The mixture is incubated at 25°C for 0, 5, 10, and 30 min
20 μl of formamide gel loading buffer is added and the reaction mixture is heated at 95°C for 5 min to stop the reaction.
5–10 μl of this is loaded onto a denaturing polyacrylamide/urea gel and the dried gel is visualized by phosphorimager.
Generally it is sufficient to use markers of the appropriate size to determine sites of endonuclease cleavage in vitro.
Summary
The protocols described here provide a comprehensive approach to characterizing PMR1-mediated mRNA decay in vivo and in vitro. While these were developed for studying this particular mRNA endonuclease many of these can be used generally to study other endonuclease-catalyzed decay processes.
Acknowledgments
Work in the Schoenberg lab on PMR1 is supported by PHS grant R01 GM38277. Y.O. is supported by a postdoctoral fellowship from Great Rivers Affiliate of the American Heart Association.
References
- Bremer KA, Stevens A, Schoenberg DR. An endonuclease activity similar to Xenopus PMR1 catalyzes the degradation of normal and nonsense-containing human β-globin mRNA in erythroid cells. RNA. 2003;9:1157–1167. doi: 10.1261/rna.5720303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes AJ, Solomon E. Evaluation of the use of S1 nuclease to detect small length variations in genomic DNA. Eur J Biochem. 1989;183:291–296. doi: 10.1111/j.1432-1033.1989.tb14927.x. [DOI] [PubMed] [Google Scholar]
- Chernokalskaya E, Dompenciel RE, Schoenberg DR. Cleavage properties of a polysomal ribonuclease involved in the estrogen-regulated destabilization of albumin mRNA. Nucleic Acids Res. 1997;25:735–742. doi: 10.1093/nar/25.4.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernokalskaya E, Dubell AN, Cunningham KS, Hanson MN, Dompenciel RE, Schoenberg DR. A polysomal ribonuclease involved in the destabilization of albumin mRNA is a novel member of the peroxidase gene family. RNA. 1998;4:1537–1548. doi: 10.1017/s1355838298980451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallouzi IE, Parker F, Chebli K, Maurier F, Labourier E, Barlat I, Capony JP, Tocque B, Tazi J. A novel phosphorylation-dependent RNase activity of GAP-SH3 binding protein: A potential link between signal transduction and RNA stability. Mol Cell Biol. 1998;18:3956–3965. doi: 10.1128/mcb.18.7.3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- Gatfield D, Izaurralde E. Nonsense-mediated messenger RNA decay is initiated by endonucleolytic cleavage in Drosophila. Nature. 2004;429:575–578. doi: 10.1038/nature02559. [DOI] [PubMed] [Google Scholar]
- Hanson MN, Schoenberg DR. Identification of in vivo mRNA decay intermediates corresponding to sites of in vitro cleavage by polysomal ribonuclease 1. J Biol Chem. 2001;276:12331–12337. doi: 10.1074/jbc.M010483200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MN, Schoenberg DR. Application of ligation-mediated reverse transcription polymerase chain reaction to the identification of in vivo endonuclease-generated messenger RNA decay intermediates. Methods Mol Biol. 2004;257:213–222. doi: 10.1385/1-59259-750-5:213. [DOI] [PubMed] [Google Scholar]
- Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- Howard JT, Ward J, Watson JN, Roux KH. Heteroduplex cleavage analysis using S1 nuclease. Biotechniques. 1999;27:18–19. doi: 10.2144/99271bm01. [DOI] [PubMed] [Google Scholar]
- Lee HH, Kim YS, Kim KH, Heo I, Kim SK, Kim O, Kim HK, Yoon JY, Kim HS, Kim Do J, Lee SJ, Yoon HJ, Kim SJ, Lee BG, Song HK, Kim VN, Park CM, Suh SW. Structural and functional insights into Dom34, a key component of no-go mRNA decay. Mol Cell. 2007;27:938–950. doi: 10.1016/j.molcel.2007.07.019. [DOI] [PubMed] [Google Scholar]
- Pastori RL, Moskaitis JE, Buzek SW, Schoenberg DR. Coordinate estrogen-regulated instability of serum protein- coding messenger RNAs in Xenopus laevis. Mol Endocrinol. 1991a;5:461–468. doi: 10.1210/mend-5-4-461. [DOI] [PubMed] [Google Scholar]
- Pastori RL, Moskaitis JE, Schoenberg DR. Estrogen-induced ribonuclease activity in Xenopus liver. Biochemistry. 1991b;30:10490–10498. doi: 10.1021/bi00107a018. [DOI] [PubMed] [Google Scholar]
- Peng Y, Schoenberg DR. c-Src activates endonuclease-mediated mRNA decay. Mol Cell. 2007;25:779–787. doi: 10.1016/j.molcel.2007.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Liu X, Schoenberg DR. Hsp90 stabilizes the PMR1 mRNA endonuclease to degradation by the 26S proteasome. Mol Biol Cell. 2007 doi: 10.1091/mbc.E07-08-0774. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- Stevens A, Wang Y, Bremer K, Zhang J, Hoepfner R, Antoniou M, Schoenberg DR, Maquat LE. Beta-globin mRNA decay in erythroid cells: UG site-preferred endonucleolytic cleavage that is augmented by a premature termination codon. Proc Natl Acad Sci USA. 2002;99:12741–12746. doi: 10.1073/pnas.192442399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt VM. Purification and further properties of single-strand-specific nuclease from Aspergillus oryzae. Eur J Biochem. 1973;33:192–200. doi: 10.1111/j.1432-1033.1973.tb02669.x. [DOI] [PubMed] [Google Scholar]
- Yang F, Peng Y, Murray EL, Otsuka Y, Kedersha N, Schoenberg DR. Polysome-bound endonuclease PMR1 Is targeted to stress granules via stress-specific binding to TIA-1. Mol Cell Biol. 2006;26:8803–8813. doi: 10.1128/MCB.00090-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Schoenberg DR. Endonuclease-mediated mRNA decay involves the selective targeting of PMR1 to polyribosome-bound substrate mRNA. Mol Cell. 2004;14:435–445. doi: 10.1016/j.molcel.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Yang F, Peng Y, Schoenberg DR. Endonuclease-mediated mRNA decay requires tyrosine phosphorylation of polysomal ribonuclease 1 (PMR1) for the targeting and degradation of polyribosome-bound substrate mRNA. J Biol Chem. 2004;279:48993–49002. doi: 10.1074/jbc.M409776200. [DOI] [PMC free article] [PubMed] [Google Scholar]