

Abstract
Apoptotic death of CD8+ T cells can be induced by a population of inhibitory myeloid cells that are double positive for the CD11b and Gr-1 markers. These cells are responsible for the immunosuppression observed in pathologies as dissimilar as tumor growth and overwhelming infections, or after immunization with viruses. The appearance of a CD11b+/Gr-1+ population of inhibitory macrophages (i Macs) could be attributed to high levels of granulocyte-macrophage colony-stimulating factor (GM-CSF) in vivo. Deletion of i Macs in vitro or in vivo reversed the depression of CD8+ T-cell function. We isolated i Macs from the spleens of immunocompromised mice and found that these cells were positive for CD31, ER-MP20 (Ly-6C), and ER-MP58, markers characteristic of granulocyte/monocyte precursors. Importantly, although i Macs retained their inhibitory properties when cultured in vitro in standard medium, suppressive functions could be modulated by cytokine exposure. Whereas culture with the cytokine interleukin 4 (IL-4) increased i Mac inhibitory activity, these cells could be differentiated into a nonadherent population of fully mature and highly activated dendritic cells when cultured in the presence of IL-4 and GM-CSF. A common CD31+/CD11b+/Gr-1+ progenitor can thus give rise to cells capable of either activating or inhibiting the function of CD8+ T lymphocytes, depending on the cytokine milieu that prevails during antigen-presenting cell maturation.
Introduction
To be fully activated, naïve T lymphocytes must come into contact with accessory cells also know as professional antigen-presenting cells (APCs), the most significant of which are activated macrophages and dendritic cells (DCs).1 Besides providing an antigen peptide complexed with a major histocompatibility complex (MHC) molecule, the primary stimulus for T-lymphocyte activation, professional APCs can also supply a “favorable” microenvironment of soluble and surface-bound stimuli composed of cytokines, chemokines, and costimulatory molecules.2,3 T lymphocytes are rapidly activated by exposure to powerful immunogens and, after a peak in reactivity that occurs 4 to 8 days after a primary immunization, their activity subsides before giving way to a memory response. The reasons for this rapid fall in T-cell activity are not completely known. It was proposed that the interleukin 2 (IL-2) decline after antigen clearance might cause “passive” T-cell apoptosis through growth factor deprivation.4 Immune responses are also deactivated by other regulatory circuits that bring about an “active” apoptosis of T lymphocytes; this “propriocidal” form of death, in fact, occurs on T-cell receptor (TCR) engagement in lymphocytes previously exposed to IL-2. This mechanism can control the extent of T-cell activation by eliminating a portion of highly dividing, antigen-reactive lymphocytes, and is mediated by the engagement of the receptors for Fas (Apo-1/CD95), and tumor necrosis factor (TNF).4 Although T cells are made susceptible to death because of their activation by APCs, it is generally thought that lymphocyte death induced by activation is not directly mediated by APCs; the coexpression of receptors and ligands for Fas and TNF causes activated T cells to kill themselves (suicide) and each other (fratricide).
These “self-control” processes of T-lymphocyte expansion, however, do not account for a phenomenon we recently characterized that depends on a nonlymphoid population of accessory cells. We described a profound depression in CD8+ T-cell function that can accompany the tumor-bearing state,5 or immunization with powerful vaccines.6 In these situations as well, the mechanism underlying the decrease in CD8+ T-cell functions involved their apoptotic death. A peculiar set of inhibitory macrophages (iMacs) expressing myeloid surface markers common to both granulocyte (Gr-1/Ly-6G) and macrophage (CD11b) lineages was present in the secondary lymphoid organs of the unresponsive mice and inhibited the generation of effector CD8+ lymphocytes; their removal by specific monoclonal antibodies (mAbs) restored the compromised lymphocyte responses and abrogated the CD8+ lymphocyte apoptosis initiated by antigenic stimulation.5,6 Accessory cells would thus have a central role in the homeostasis of immune responses, as they seem to take part in the initiation and regulation of T-lymphocyte activation.
The powerful regulatory activity of APCs must be tightly monitored, and this is assured, in part, by the cytokines secreted by 2 subsets of CD4+ lymphocytes, Th1 and Th2. Th1 lymphocytes cause delayed-type hypersensitivity, a cell-mediated immune response against intracellular bacteria, whereas Th2 lymphocytes are the most effective activators of humoral responses.7 Macrophages are the final targets and effectors of Th1-mediated responses. Th1-type cytokines, such as interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α), induce enhancement of macrophage antimicrobial pathways, up-regulation of the machinery for antigen presentation, and an increase in costimulatory molecules.8 An equivalent APC target for the Th2-induced responses has not yet been described. However, it was recently advanced that the anti-inflammatory activity of Th2 cytokines, such as IL-4, is due to a direct effect on macrophages.9 IL-4 elicits the expression of a special set of molecules enabling macrophages to participate actively in the resolution of inflammatory processes, as well as in tolerance induction and wound healing.10 Macrophages alternatively activated by IL-4 might thus comprise a distinct set of APCs endowed with the immunosuppressive functions that, over the last decades, have been attributed to classic macrophages.11
The aim of this study was to characterize the phenotype of iMacs and to evaluate the control exerted by Th1- and Th2-type cytokines on their functions. In experiments set out to determine their differentiation state, we found that iMacs isolated from the lymphoid organs of immunosuppressed mice comprised a population of immature myeloid precursors, positive for the markers Gr-1, CD11b, and CD31, that could give rise to distinct cellular populations capable of either activating or suppressing CD8+ T-cell activation. These immature intermediates were normally detectable in the bone marrow, but they could accumulate in the spleen under the influence of the granulocyte-macrophage colony-stimulating factor (GM-CSF) produced during intense lymphocyte stimulation, or by certain growing tumors. When cultured in vitro, CD11b+/Gr-1+/CD31+ cells generated adherent, dominant suppressors of T-lymphocyte function whose suppressive activity was greatly increased by exposure to IL-4. Conversely, Th1 cytokines, such as IL-12 or IFN-γ and TNF-α, induced their maturation into competent, mature APCs. Finally, exposure to GM-CSF and IL-4 led to an intense proliferation, and the generation of nonadherent, myeloid CD11b+/CD11c+/CD8α− DCs. These findings suggest the existence of a pathway for the control of immune responses based on the alternate maturation of bone-marrow—derived APC precursors.
Materials and methods
Cell lines
CT26.WT is a BALB/c (H-2d) carcinogen-induced, undifferentiated colon carcinoma. The mouse mammary adenocarcinoma, TS/A (H-2d), was kindly provided by Dr Guido Forni (University of Turin, Italy). MBL-2 (H-2b) is a Moloney virus-induced lymphoma. B-SC-1 (CCL26, ATCC, Manassas, VA) and Hela S3 (CCL2.2, ATCC) cells were used to prepare all the vaccinia virus (VV) stocks. Cell lines were maintained in Dulbecco modified Eagle medium (DMEM) (Gibco, Paisley, UK), supplemented with 2 mmol/L L-glutamine, 10 mmol/L HEPES, 20 μmol/L 2-mercaptoethanol, 150 U/mL streptomycin, 200 U/mL penicillin, and 10% heat-inactivated fetal bovine serum (FBS) (Gibco).
Recombinant vaccinia viruses
All recombinant (r)VV used in this study were generated by the insertion of the foreign genes into the VV thymidine kinase gene by homologous recombination.12 Virus preparations were propagated from plaque-purified crude virus stocks.13 Viral concentrations were determined by plaque titration on B-SC-1 cells. Construction and preparation of rAd-CMV2, a recombinant adenovirus-expressing β-galactosidase (β-gal-rAd), as well as IL-2-rVV— and IFN-γ-rVV—expressing mouse IL-2 and IFN-γ, respectively, together with β-gal were described previously.14,15
Peptides, antibodies, and cytokines
TPHPARIGL, representing the amino acids 876-884 of β-gal presented in association with H-2 Ld was synthesized by Peptide Technologies (Washington, DC) to a purity of more than 99% as determined by high-performance liquid chromatography (HPLC) and amino acid analysis.
Fluorescein isothiocyanate (FITC)—labeled rat antimouse Ly-6G (Gr-1, Immunokontact, Bioggio, Switzerland); biotinylated rat antimouse CD31 and rat antimouse F4/80 (BMA Biomedicals, Augst, Switzerland); rat antimouse ER-MP20, rat antimouse ER-MP58, and rat antimouse CD31 (a kind gift of M. R. F. de Bruijn, Erasmus University, Rotterdam, Holland); phycoerythrin (PE)—labeled rat antimouse CD11b, FITC-labeled antimouse CD3, FITC- and PE-labeled antimouse CD86, FITC-labeled anti-CD8α, FITC-labeled anti—I-Ad/I-Ed, and FITC-labeled anti-Kd, and isotype-matched controls (Pharmingen, San Diego, CA) were used. Biotinylated rat antimouse CD11c (N418, ATCC) was a gift of U. Grohmann, University of Perugia, Italy. PE antirat F(ab’)2 IgG (Jackson Immunoresearch Lab, West Grove, PA) and TC streptavidin (DakoPatts, Glostrup, Denmark) were used for secondary labeling. The mAb 24G.2 (CD16/CD32, ATCC), which recognizes the extracellular domain of mouse Fcγ-RII and RIII, was purified from mouse ascites and used to block nonspecific mAb binding. Antimouse GM-CSF neutralizing antibody, and normal goat control IgG were purchased from R&D System (Minneapolis, MN).
Recombinant mouse GM-CSF, IL-4, and TNF-α (purchased from PreproTech, Rocky Hill, NJ) and mouse IFN-γ (purchased from R&D System) were resuspended in phosphate-buffered saline (PBS) containing 1% mouse serum (Sigma, St Louis, MO). Mouse IL-12 was a kind gift of U. Grohmann.
Cytofluorometric analysis and cell sorting
Cells were blocked with rat 24G.2 purified mAbs before staining with different amounts of mAb (1-10 μL/106 cells). Isotype-matched mAbs were always included as control. Phenotype analysis and cell sorting were conducted with a Coulter XL Flow Cytometer (Coulter Electronics, Hialeah, FL), equipped with a 488 nm Argon Ion Laser (Coherent, Innova, Santa Clara, CA) running at 15 mW.
Isolation of CD11b+/Gr-1+ splenocytes
A panning technique using 60 mm bacteriologic petri dishes (Falcon 1016, Cockeysville, MD) was used to enrich Gr-1+ splenocytes from tumor-bearing or rVV-immunized mice. Briefly, petri dishes were coated with Gr-1 mAb by adding 3 mL of antibody solution at a concentration of 10 μg/mL in 0.05 mmol/L carbonate/bicarbonate buffer (pH 9.6, Sigma), and then incubated for 3 hours at 37°C. The petri dishes were then washed 3 times with 0.15 mol/L NaCl and incubated overnight at room temperature with 3 mL of 10 mg/mL bovine serum albumin (BSA) in PBS. Spleens were depleted of red cells by incubation in NH4Cl lysis buffer, resuspended in HBSS containing 5% mouse serum, and then kept on ice for 15 minutes; 3 mL of splenocyte preparation (about 2.5 × 107 cells) were dispensed onto petri dishes previously washed with 0.15 mol/L NaCl. After 1 hour at 37°C, petri dishes were washed, and cells still attached to the plastic were provided with complete medium, incubated for 6 hours, and then detached by gentle pipetting with a solution of PBS 2 mmol/L EDTA. In some experiments, CD11b+ splenocytes were isolated with CD11b mAb-coated magnetic beads and magnetic columns (Miltenyi Biotec GmbH, Bergish Gladbach, Germany) according to the manufacturer’s instructions.
Enrichment of spleen-derived dendritic cells
Spleens were collected aseptically, minced, and incubated for 30 minutes at room temperature in HBSS containing collagenase (1 mg/mL, Sigma). Spleens were depleted of red cells by incubation in a NH4Cl lysis buffer and washed. The single cell suspension was resuspended in complete medium (CM) consisting of RPMI 1640 (Euroclone Ltd, Paington-Devon, UK), 10% heat-inactivated FBS (BioWhittaker, Walkersville, MD) supplemented with 2 mmol/L L-glutamine, 1 mmol/L NaPiruvate, 20 μmol/L 2-mercaptoethanol, 150 U/mL streptomycin, 200 U/mL penicillin, and plated in a 150 × 25 mm tissue culture dish (Falcon). Splenocytes were incubated for 2 hours at 37°C, and the nonadherent cells were removed by gentle washing. Adherent cells were further incubated for 18 hours in CM containing 3 ng/mL mouse GM-CSF; the supernatant was then collected, and DCs were finally enriched by centrifugation (1900g) over a 45% Percoll (Sigma) cushion. At this point, more than 80% of the cells were B7-2+, B220−, class II MHC+, and CD11c+.
Immunization protocols
Female BALB/c mice were immunized by intravenous (iv) injection of 0.5 mL PBS containing either 107 plaque-forming unit (PFU) β-gal-rAd or 5 × 106 PFU of the different rVV.
Evaluation of cytotoxic T-lymphocyte responses
At various time intervals after immunization, spleens were collected, separated into single-cell suspensions, and cultured in CM containing 1 μg/mL of peptide. Alternatively, allogeneic mixed lymphocyte cultures (allo-MLCs) were set up by coincubating 3 × 106 BALB/c splenocytes (H-2d) with an equal amount of γ-irradiated C57BL/6n splenocytes (H-2b). The ability of suppressor cells to inhibit cytotoxic T-lymphocyte (CTL) response generation was assessed by adding γ-irradiated cells as the third party to allo-MLC. Briefly, graded doses of suppressor cells derived from cultures of Gr-1+/CD11b+ cells in the presence of different cytokines were added to allo-MLC, consisting of 5 × 106 BALB/c splenocytes and 105 γ-irradiated C57BL/6n DCs. After 5 days, the cultures were tested for their ability to lyse peptide-pulsed or allogeneic targets in a 6-hour 51Cr-release assay using 2 × 104 target cells previously labeled with 3.70 MBq (100 μCi) Na51CrO4 for 90 minutes.15 The amount of 51Cr released was determined by γ-counting, and the percentage specific lysis was calculated from triplicate samples using the formula: [(experimental cpm − spontaneous cpm) / (maximal cpm − spontaneous cpm)] × 100.
Mixed leukocyte reaction
The ability of DCs to stimulate quiescent T cells was assessed by the mixed leukocyte reaction (MLR). Graded doses of γ-irradiated DCs or control cells derived from BALB/c mice were added to microwells containing 2 × 105 allogeneic C57BL/6n splenocytes in a final volume 200 μL per well of CM. After 3.5 days of incubation, cultures were pulsed with 0.037 MBq (1 μCi) per well 3H-thymidine (NEN, Life Science Products, Boston, MA). 3H-thymidine incorporation was measured using a β-scintillation counter (Top-count, Packard Instrument, Meriden, CT).
Results
CD11b+ splenocytes in immunosuppressed mice comprise myelo-monocytic precursors
The suppression of immune response observed in mice bearing a growing tumor or immunized with powerful VV constructs was found to depend on the accumulation of macrophage-related suppressor cells in secondary lymphoid organs.5,6 These iMacs expressed the markers CD11b (Mac-1) and Gr-1 (Ly-6G), and removal of either CD11b+ or Gr-1+ splenocytes restored the deficient T-lymphocyte responses.5,6 Figure 1A shows the morphology of cells selected for the presence or absence of CD11b and Gr-1 from spleens of mice bearing large TS/A tumors. The CD11b/Gr-1 negative population uniformly consisted of mostly small lymphocytes. Conversely, the double-positive fraction was rather polymorphous and included mature myelo-monocytic cells (polymorpho-nuclear cells and monocytes) as well as cells whose morphology recalled immature myeloid cells (arrowheads). To understand whether the difference in the morphology of the CD11b+ cells reflected a phenotypic variance, we used a panel of mAbs selected to recognize markers preferentially expressed by the immature stages of myeloid development.17 In both tumor-bearing and VV-immunized mice, the majority of the CD11b+ splenocytes also expressed the ERMP-20 (Ly-6C) and the ER-MP58 markers (Figure 1B). The coexpression of these antigens with the immature marker CD31 by some CD11b+ cells is consistent with the phenotype of myeloid progenitors that can potentially generate granulocytes, macrophages, and dendritic cells.18 Indeed, enhanced formation of mixed colonies in soft agar was observed in spleens of mice bearing large TS/A nodules and in mice infected with VV, whereas colonies were virtually undetectable in normal spleens (data not shown).
Figure 1. Phenotype and morphology of iMacs in the spleen and bone marrow of immunocompromised mice.

(A) Splenocytes from mice bearing a large (more than 1 cm2) subcutaneous TS/A tumor were stained with FITC—anti-Gr-1 and PE—anti-CD11b antibodies, and were sorted by FACS into double-positive or -negative cells. Cells were spun onto slides and stained with May-Grünwald-Giemsa (MGG). Magnification, × 260. (B) Splenocytes from the same mice bearing TS/A tumor, or immunized 6 days earlier with 5 × 106 PFU of IL-2-rVV (VV) were stained with anti-CD11b, and one of the markers indicated. After gating CD11b+ cells, the cytometric profile for each marker was plotted (histograms to the right of the arrows). Numbers indicate the percentage of positive cells. Isotype-matched mAb were used as control for background staining (shaded areas). The percentage of CD11b+ cells in spleens of normal mice was lower than 4%, with a percentage of CD11b+/Gr-1+ cells around 0.5% to 1% (not shown). (C) CD11b and CD31 expression in the red-cell depleted, unseparated bone marrow cells of normal, tumor-bearing, and VV-immunized mice was assessed by 2-color cytometry. The plots shown in this figure are representative of 3 different experiments.
Staining for CD31 and Gr-1 markers unveiled some heterogeneity between the 2 groups of mice examined: CD11b+ splenocytes of VV-immunized mice showed a lower Gr-1 and a higher CD31 percentage than CD11b+ splenocytes from tumor-bearing mice. This marker distribution indicated that the fraction of immature myeloid cells was larger among the iMacs found in VV-immunized mice. The difference was not confined to the secondary lymphoid organs, such as the spleen, because the percentage of CD11b+/CD31+ cells was also increased in the bone marrow of VV-immunized mice (Figure 1C). In normal mice, CD11b+/CD31+ cells were confined to the bone marrow and were barely detectable in the spleen (less than 1%, data not shown).
To further investigate marker heterogeneity, we isolated splenocytes from TS/A tumor-bearing mice and triple stained them for CD11b, Gr-1, and CD31. Figure 2 shows that the majority of CD11b+/Gr-1+ splenocytes expressed the CD31 marker. After depletion of the Gr-1+ cells by panning with the RB6-8C5 mAbs, the remaining single-positive, CD11b splenocytes were completely negative for CD31 expression. Similarly, Gr-1+, single-positive splenocytes lacked the CD31 antigen (data not shown). These results indicate that the immature marker CD31 is present only on the double-positive splenocytes and not the single-positive cells, which likely are either mature monocytes (CD11b+) or mature granulocytes (Gr-1+).
Figure 2. iMacs isolated from the spleen of tumor-bearing mice are CD11b+/Gr-1+/CD31+.
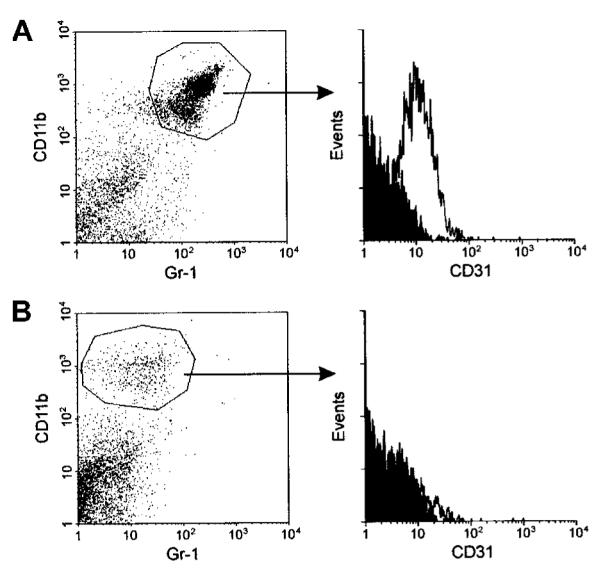
(A) Splenocytes from mice bearing a subcutaneous TS/A tumor were stained with FITC—anti-Gr-1, PE—anti-CD11b, and biotin—anti-CD31 antibodies, followed by Tricolor-streptavidin. After gating CD11b+/Gr-1+ cells (left panel), the cytometric profile for CD31 marker was plotted (histograms to the right of the arrow). Isotype-matched mAbs were used as controls for background staining (shaded areas). (B) Gr-1+ splenocytes from the same mice were depleted by panning with the specific mAbs. The resulting population was stained with the same mAbs used in panel A, and expression of CD31 (right panel) was evaluated among the CD11b+ cells (left panel). Staining with a secondary antirat Ab revealed a number of positive cells <1% confirming that negativity for Gr-1 was not due to Ab competition.
Induction of immunologic unresponsiveness in mice immunized with VV depends on systemic release of GM-CSF
Inoculation of a rVV expressing the mouse IL-2 caused an enhanced activation or expansion of cytotoxic T cells, as assessed by the marked increase in the ex vivo cytotoxic responses to vaccinia determinants and to the heterologous antigen carried by the rVV, the β-gal.15 Although present in the spleen in large number, CD8+ lymphocytes specific for β-gal could not be restimulated in vitro; instead, stimulation with a β-gal peptide triggered their activation-induced cell death. The induction of such immune unresponsiveness was found to depend on iMac activity.6 Several findings associate the appearance of iMacs and immune suppression to the systemic release of the cytokine GM-CSF by mouse tumors.5 The mobilization of iMacs from the bone marrow, or hemopoietic organs like the spleen, could also depend on the release of CSF-like cytokines during an intense immune response, such as that induced by immunization with VV. To test this possibility, BALB/c mice inoculated with different rVVs expressing the antigen β-gal were treated every other day with an antibody that neutralized the biologic effects of the cytokine GM-CSF. Control mice received equal amounts of isotype-matched antibodies. After 6 days, splenocytes were harvested and cultured in the presence of the Ld-restricted β-gal peptide to generate β-gal—specific CTLs. A sustained and specific CTL activity against β-gal—pulsed target cells was elicited after in vitro stimulation of the splenocytes from mice inoculated with an rVV-producing IFN-γ (Figure 3, IFN-γ-rVV) but not from mice that received IL-2-rVV (Figure 3, control serum). By administering an antiserum that neutralized the biologic activity of GM-CSF during immunization with IL-2-rVV, the CTL response was restored to levels almost similar to those induced by IFN-γ-rVV. Thus, secretion of GM-CSF during intense lymphocyte stimulation can account for the immunosuppression of CD8+ lymphocyte responses.
Figure 3. GM-CSF released during immune response to virus cause unresponsiveness to antigen stimulation.
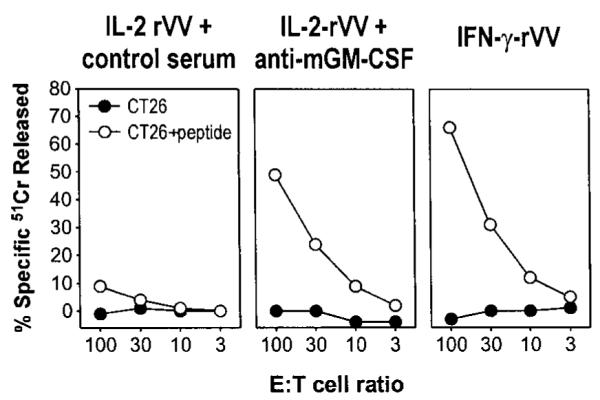
Three BALB/c mice were immunized intravenously with 5 × 106 PFU per mouse of either IL-2-rVV or IFN-γ-rVV recombinant viruses expressing the antigen β-gal, together with the mouse cytokines IL-2 and IFN-γ, respectively. On days 1, 3, and 5, IL-2-rVV immunized mice were inoculated intraperitoneally with 0.2 mL of PBS containing 100 μg of either an antiserum neutralizing mouse GM-CSF activity or the goat control serum. After 6 days, the spleens were removed, pooled, incubated in vitro with 1 μg/mL of the β-gal peptide for 5 days, and then assayed in a 51Cr-release assay against the CT26 cells or with CT26 cells pulsed with the β-gal peptide. E:T cell ratios are indicated; spontaneous release never exceeded 20%.
CD11b+/Gr-1+ cells are the precursors of adherent, macrophage-like suppressor cells
When Gr-1+ cells from tumor-bearing mice were cultured in standard medium, a homogenous population of adherent cells that retained its ability to inhibit T-lymphocyte function was isolated. These cells possessed some markers of APCs, including F4/80 and CD11b, but had lost Gr-1 expression.5 They also differed from “classical” APCs by their lack of uniform expression of costimulatory and MHC class II molecules. We set out to determine whether iMacs expansion under different contexts of immune unresponsiveness could give rise to the same suppressive population.
Spleen cells from BALB/c mice immunized with rVV were enriched with anti—Gr-1 mAb and cultured in vitro in standard medium. After 1 week, most of the cells had died, but a population of adherent, macrophage-like cells survived and could be maintained up to 4 weeks in culture. Phenotypically, they resembled cells obtained from tumor-bearing mice as they expressed F4/80 and CD11cdim, but not B7.1 and class II MHC (I-Ad). B7.2 molecules were scarcely detectable, whereas levels of class I MHC (Kd) molecules were normal (Figure 4A). Finally, they did not express CD8α, which was recently found to mark a subpopulation of DCs with suppressive activity.19 When added as the third party to a culture of splenocytes from immune mice stimulated with the immunodominant, Ld-restricted β-gal peptide, these cells behaved functionally like iMacs as they completely abrogated the generation of β-gal—specific CTL responses (Figure 4B). A similar population of macrophage-like cells with suppressor activity was obtained by enrichment through adherence to plastic of splenocytes from rVV-immunized mice, thus suggesting that the manipulation of membrane molecules through panning or related techniques was not biasing the in vitro maturation of iMacs (data not shown).
Figure 4. iMacs cultured in vitro retain the ability to suppress CTL generation.

Gr-1+ splenocytes from mice immunized with IL-2-rVV were enriched through panning with the specific mAb, and cultured in complete medium. After 6 days, adherent cells were collected in PBS-EDTA by gentle scraping, stained with different mAbs (A), and tested for their suppressive activity (B). (A) The percentage of adherent cells positive for any given marker is reported after subtraction of background staining with isotype-control mAbs. (B) Adherent cells were added at a final concentration of 3% of the total number of cells present in β-gal peptide-stimulated cultures of splenocytes from mice previously immunized with rAd-β-gal (immune). After a 5-day incubation, cultures were assayed in a 51Cr-release assay against either CT26 cells (■) or CT26 cells pulsed with the β-gal peptide (●). E:T cell ratios are indicated.
Cytokine milieu influences iMacs’ differentiation into suppressor or stimulatory antigen-presenting cells
We then tested whether the functional properties of iMacs might be influenced by external factors, like the cytokines produced during the immune response. To this end, we isolated the cells previously shown to be inhibitory, and placed them in culture either alone, or in the presence of Th1 (IFN-γ and TNF-α) or Th2 (IL-4) cytokines. After 6 days of differentiation, the adherent cells were analyzed cytofluorometrically for 3 markers (MHC class II, CD86 and CD11c) of “mature” APCs, and simultaneously added to peptide-stimulated cultures to test their function. Cytokine-treated or -untreated cells constituted 1% of the total cells in a culture of BALB/c splenocytes from mice previously immunized with β-gal and stimulated with the Ld-restricted β-gal peptide (Figure 5).
Figure 5. Differentiation and function of iMacs can be regulated in opposite ways by Th1- and Th2-type cytokines.

To assess the influences of various cytokines on the differentiation of iMacs, we isolated Gr-1+ splenocytes from mice bearing a TS/A tumor. Inhibitory cells were then exposed to either no cytokines, IL-4 (100 ng/mL), IL-12 (20 ng/mL), or a combination of IFN-γ (50 ng/mL) and TNF-α (5 U/mL). After 6 days of culture, the adherent cells were tested for the phenotype (A) and function (B, C). (A) Phenotypic characterization of cells after the 6-day cytokine regimen. Isotype-matched controls are shaded. (B) Cytokine-treated inhibitory cells were added at a final concentration of 6 × 104 cells (1%) to cultures consisting of 6 × 106 BALB/c splenocytes of mice previously immunized with rAd-β-gal and stimulated with the β-gal peptide. Cytolytic activity was assessed by standard 51Cr release assay after an additional 5 days of culture using β-gal peptide pulsed (□) or unpulsed (●) CT26 cells either at E:T ratios starting at 100:1, followed by 3-fold dilutions (100:1, 33:1, 11:1, 3:1, 1:1, 0.3:1). (C) Suppressor cells (iMacs) derived from cultures of Gr-1+ cells in the presence or absence of IL-4 were added to allo-MLC consisting of 5 × 106 BALB/c splenocytes (H-2d) and 105 γ-irradiated C57BL/6n DC (H-2b). The iMacs-to-DC ratio is indicated at the bottom of the panel. The MLC was assessed by standard 51Cr release assay for activity after an additional 5 days of culture using an H-2b target, MBL-2 (●), and a control H-2d target, CT26 (▼), at E:T ratios starting at 100:1, followed by 3-fold dilutions (33:1, 11:1, 3:1).
Cells cultured in plain medium showed the phenotype of the suppressor cells described above, ie, class II MHC−, CD86dim, CD11cdim (Figure 5A). Addition of these cells to the peptide-stimulated cultures caused a reduction in CTL activity against the β-gal peptide-pulsed target cells (Figure 5B). The cells that partially differentiated under the influence of IL-4 showed an up-regulated CD11c expression, but unchanged levels of CD86, as previously reported5; these cells appeared to be more inhibitory. Titration experiments confirmed that cells grown in IL-4 had a greater suppressive activity than the cells maturated in the absence of any cytokine. Even when mixed at a ratio of 1:9 with spleen-derived DCs, they were still able to suppress the allogeneic CTL response, indicating that suppression prevailed over the DC ability to stimulate an allo-MLR (Figure 5C).
When iMacs were cultured with Th1 cytokines, IL-12, or the combination IFN-γ and TNF-α, we observed a significant loss of CD11c expression, and a modest up-regulation of CD86. MHC class II expression was clearly induced only by the latter cytokines (Figure 5A). More importantly, cells differentiated under the influence of Th1 cytokines induced an enhanced anti—β-gal response. These data indicated that the iMacs could differentiate into functional APCs when placed in the appropriate cytokine environment.
CD11b+/Gr-1+ cells can give rise to nonadherent, dendritic cell-like cells in the presence of GM-CSF and IL-4
The ability of iMacs to respond to Th1-type cytokine stimulation by differentiating into functional APCs led us to investigate whether they could also give rise to mature DCs, the most potent APCs.2 Splenocytes from tumor-bearing mice were sorted for CD11b marker expression and cultured in the presence of different cytokines that were shown to affect DC generation.20-22 Cells proliferated in the presence of either IL-4 or GM-CSF after a few days of culture, but the proliferation was maximal on day 10 (Figure 6A,B). Moreover, proliferation was significantly higher in the presence of GM-CSF and IL-4. Only the fraction of CD11b+ cells coexpressing the CD31 marker, likely the most immature, proliferated under the influence of the various cytokines (Figure 6C). On day 5 of culture, about 15% of the nonadherent cells recovered from the culture of CD11b+ splenocytes exposed to GM-CSF and IL-4 presented the classic DC phenotype, ie, contemporaneous expression of CD11c, MHC class II molecules, and CD86 (Figure 7A). These cells were strong stimulators in MLR, surpassing in this property normal splenocytes and adherent cells derived from the same CD11b+ splenocytes (Figure 7B).
Figure 6. Immature CD11b+/CD31+ cells retain the ability to proliferate in the presence of cytokines.

CD11b+ splenocytes from mice bearing a TS/A tumor were enriched with antibody-coated magnetic beads. The cells were plated in triplicate (20 000 cells per well) and then exposed to no cytokines, IL-4 (100 ng/mL), GM-CSF (20 ng/mL), or a combination of GM-CSF and IL-4. After 4 and 9 days of culture (A and B, respectively), cells were pulsed with 3H-TdR, and harvested 18 hours later. Irradiated cells were included as the negative control. (C) 3H-TdR incorporation by CD11b+ cells further sorted into CD31+ and CD31− cells by FACS and cultured for 5 days with the various cytokines. Data are from triplicate wells ± SD.
Figure 7. iMacs differentiate into myeloid DCs under the influence of GM-CSF and IL-4.

To assess the influence of the GM-CSF and IL-4 combination on the iMac differentiation, CD11b+ splenocytes from TS/A tumor-bearing mice were isolated with antibody-coated magnetic beads. Cells were then exposed to a combination of GM-CSF (20 ng/mL) and IL-4 (100 ng/mL). After 5 days of culture, the nonadherent cells (11.5% of the initial number of CD11b+ cells) were collected, and tested for phenotype (A, right panel) and function (B). After removal of nonadherent cells, the remaining cells were further incubated in medium containing GM-CSF and IL-4 for 5 days. The nonadherent cells recovered on day 10 constituted an additional 3.7% of the cells initially seeded. These cells were tested for phenotype (A, left panel) and morphology (C). (A) Cell surface phenotype of nonadherent cells on day 5 or 10 was determined after triple staining with anti-CD86, anti-class II MHC molecules (I-Ad/I-Ed), and anti-CD11c; percentages of I-Ad/I-Ed+/CD86+ also expressing CD11c marker are indicated in parentheses. (B) Cytokine-treated, nonadherent and adherent cells were irradiated and cultured in various numbers with 2 × 105 allogeneic C57BL/6n splenocytes. In control wells, various numbers of γ-irradiated BALB/c splenocytes were used as stimulators. After 3.5 days of culture, cells were pulsed with 3H-TdR, and the results from triplicate wells were corrected for 3H-TdR incorporation by irradiated stimulators alone and allogeneic splenocytes alone. (C) Morphology of cells stained with MGG after the 10-day cytokine regimen. Magnification × 500.
After the complete removal of nonadherent cells form GM-CSF and IL-4 stimulated cultures, the remaining adherent fraction was CD11b+/CD11c+/CD86dim/MHC class IIdim/−.5 These cells were further incubated in the presence of GM-CSF and IL-4 for 5 days. More cells detached from the plastic and were recovered from the culture supernatant that showed an increased percentage of DC markers (Figure 7A); all the cells possessing high levels of class II MHC molecules and CD86 were, in fact, CD11c+, and CD8α− (not shown). May-Grünwald-Giemsa (MGG) staining of the cytospin confirmed that about 50% of the cells in culture had a DC morphology (Figure 7C). These results clearly indicated that under the influence of the appropriate cytokines, a fraction of the adherent cells differentiated into mature DCs. There was no difference in the phenotype and function of DCs obtained from culture with GM-CSF and IL-4 of either CD11b+- or Gr-1+—enriched splenocytes (data not shown), a clear indication that the DC precursors arose from the double-positive iMacs.
Discussion
We previously showed that macrophage-induced immunosuppression (MIS) is the main cause of the profound alteration in CD8+ T-lymphocyte function not only during neoplastic growth, but also during acute, intense stimulation of the immune response by powerful immunogens.5,6 The cells responsible for the MIS exhibited markers typically expressed by monocytes and granulocytes and accumulated in the lymphoid organs of immunocompromised mice as a consequence of systemic GM-CSF release.5,6 On the basis of the present findings, however, the term “inhibitory macrophages” (iMacs) may not be completely appropriate to describe the real nature of the suppressor cells, because they consist of a heterogenous population of mature and immature myeloid cells. This study shows in fact that iMacs exposed to GM-CSF and IL-4 retained the ability of myeloid precursors to proliferate and differentiate into nonadherent myeloid-related CD11c+/CD11b+/CD8α− DCs. This property is peculiar to CD31+/CD11b+/Gr-1+ cells isolated from the spleens of immunocompromised mice; DCs could also be generated from mouse adherent monocytes isolated from normal spleens and cultured for a few days with GM-CSF and IL-4, but monocytes did not proliferate as determined in standard 3H-TdR incorporation assay.22 In the absence of an additional external cytokine influence, however, CD31+/CD11b+/Gr-1+ cells are committed to give rise to adherent, macrophage-like cells that suppress the generation of CTL activity in an MHC-unrestricted fashion. This default pathway is influenced by Th2 and Th1 cytokines that can either enhance the suppressive activity (IL-4) or abrogate it (IL-12 or the combination IFN-γ + TNF-α). Thus, these CD31+/CD11b+/Gr-1+ myeloid cells are the cardinal components of a circuit that can either sustain or down-regulate the T-lymphocyte responses.
The phenotype and function of the iMacs are identical to suppressor cells recently described in mice treated with cyclophosphamide.23 After chemotherapy with this drug, in fact, mice show a transitory impairment in lymphocyte proliferation in response to T-cell mitogens due to colonization of the spleen by a CD11b+/Gr-1+/CD31+ population. The presence of these immature cells correlated with the T-cell proliferation impairment, which depended on their release of nitric oxide in culture.
CD31+/CD11b+ cells were almost undetectable in normal spleens, but could be found in the bone marrow (Figure 1). By means of cell sorting, followed by culture in soft agar, myeloid precursors were found to reside within the CD31hi/ER-MP20−, CD31+/ER-MP20+, and CD31−/ER-MP20hi subsets, that constitute about 15% of nucleated bone marrow cells.18,24 A maturation pathway has been described in which the most immature CD31hi/ER-MP20− cells develop via the CD31+/ER-MP20+ stage into CD31−/ER-MP20hi monocytes. The CD31+/ER-MP20+ cells in the bone marrow contained morphologically recognizable precursors of the granulocytic, erythroid, and lymphoid lineages18; however, 80% expressed the myeloid antigen Gr-1, indicating their commitment to the myeloid lineage.25 Our characterization of double-positive CD11b/Gr-1 cells showed that the majority was also positive for CD31 and ER-MP20 markers, consistent with a more immature phenotype than the monocyte stage (ie, G-CFC, M-CFC).
In contrast to prototype macrophage activation by IFN-γ and LPS, that exerted by IL-4 and glucocorticoids has been referred to as alternative.10 These anti-inflammatory agents induce the expression of molecules that enable macrophages to take active part in the resolution of the inflammatory reaction, tolerance induction, and wound healing.11 The iMacs described in this study share some similarities with the alternatively activated macrophages obtained from IL-4—treated peritoneal macrophages,10,26 but there are some important differences. First, the phenotype of the suppressor cells found in tumor-bearing mice, and in mice undergoing acute stimulation of the immune system was more immature, and second, CD11b+/Gr-1+ cells differentiated in the presence of IL-4 alone showed a modest increase in I-Ad/I-Ed, whereas the same treatment induced class II MHC molecule up-regulation in mature monocytes.26 We favor the hypothesis that an alternative APC differentiation, rather than activation, underlies the generation of the suppressor cells in immunocompromised hosts. Granulocyte-monocyte progenitor cells are unable to mature into functional APCs in the presence of GM-CSF alone. They require additional factors such as IL-12, IFN-γ, TNF-α, or IL-4. The differentiation capacity of CD11b+/Gr-1+/CD31+ myeloid cells in vitro and in vivo could generate a continuum of accessory cell types according to the cytokines and factors present in the microenvironment, which might explain the marker variability found by different groups in mouse myeloid-related suppressor cells: CD11b+; CD11b− and CD11b+; CD31+; Sca-1+/CD11b+; WGA+; WGA+/CD11b+.27-34
Our studies indicate that systemic GM-CSF release suffices to drive the differentiation of precursor cells to iMac, but not mature DCs. It is interesting to compare the effects of different exogenously administered cytokines on the maturation of myeloid cells. Flt3 ligand (Flt3-L) administration in mice was shown to increase 2 DC subpopulations, the lymphoid-related CD11c+/CD11bdim/−/CD8α+, and the myeloid-related CD11c+/CD11bbright/CD8α− DC35 that differentially shaped the Th response after their in vivo administration: Lymphoid-related DCs induced Th lymphocytes to release Th1 cytokines, whereas the myeloid-related DCs triggered a mixed response characterized by release of large amounts of the Th2 cytokines, IL-4 and IL-10, in addition to IFN-γ and IL-2.35 In the same studies, inoculation of polyethylene glycol-modified GM-CSF (pGM-CSF) expanded almost exclusively the myeloid-related DCs. This same group also found that prolonged administration of unmodified GM-CSF did not augment significantly the number of splenic CD11c+ DCs.36 Like our findings, the majority of CD11b recovered in mice treated for 11 days with GM-CSF was, in fact, CD11c negative and Gr-1 positive.35-37 Interestingly, a fraction of these cells was also recovered from the spleens of Flt3-L— and pGM-CSF—treated mice.35-37 An attempt to summarize these findings is shown in Figure 8. GM-CSF is incapable of driving a full maturation of CD31+/CD11b+/Gr-1+ precursors into myeloid DCs, whereas Flt3-L and pGM-CSF supply a thoroughly competent maturation signal. CD31+/CD11b+/Gr-1+ precursors require IL-4, together with GM-CSF, to give rise to mature myeloid DCs (CD11bbright/CD11c+/CD8α−), and at least a part of these cells can arise from an intermediate stage of CD11bbright/CD11cdim/−/CD8α− adherent cells (Figure 7). Why pGM-CSF acts so differently from the natural cytokine is not forthcoming. One explanation could be the short biologic half-life of the natural cytokine compared with pGM-CSF.38 If this were true, one could predict that in vitro culture of the immature precursor in the presence of either low or high GM-CSF concentrations would have different outcomes. This hypothesis is currently being investigated, but recent data seems to confirm a differential effects of high versus low doses of GM-CSF on DC maturation.39
Figure 8. A model for maturation of myeloid suppressors and DC cells in mice.
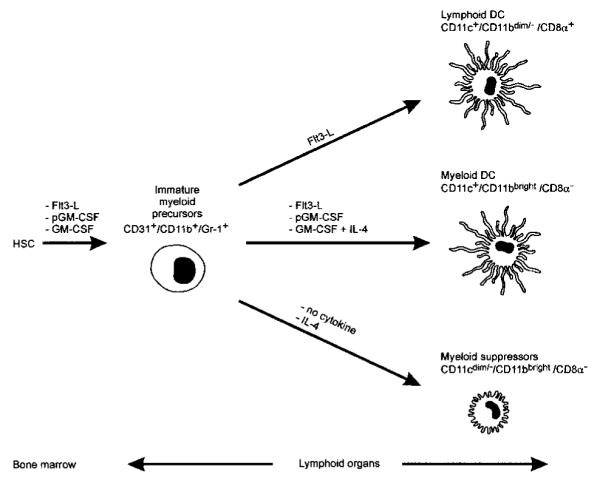
HSC, hemopoietic stem cell; Flt3-L, Flt3 ligand; pGM-CSF, polyethilenglycole-GM-CSF. The diagram originates from the present study and previously published data.35-37
The iMacs are dominant suppressors because their inhibitory activity can overwhelm the powerful antigenic stimulus provided by mature allogeneic DCs when mixed at 1:1 ratio (Figure 5). This ratio can be further lowered in favor of the iMacs derived from cultures with IL-4. We advance that the powerful regulatory activity of iMacs on CD8+ T cells might be part of a feedback circuit that is triggered during the immune response to limit the effects of excessive lymphocyte activation. GM-CSF is released by almost any activated T lymphocyte7; the amount of GM-CSF and duration of its release could be viewed as “gauges” of the immune response intensity. High levels of GM-CSF provoke the appearance of iMacs that induce the apoptosis of activated lymphocytes, and turn off the immune response.6
Human and mouse tumors can override the feedback circuit involving iMacs to escape immune attack by secreting CSF-like factors, and in particular GM-CSF and/or M-CSF.5,27,29,40-43 Our findings have practical implications for the therapy of cancer. Elimination of iMacs in vivo could be a key step to increase the efficiency of antitumor vaccines. In this regard, it was shown that treatment of immunocompetent mice with anti—Gr-1 mAbs reduced the growth of a variant of an ultraviolet light-induced tumor, which had a more aggressive progression than the parental neoplasia44; the effect of in vivo anti—Gr-1 treatment was attributed to the elimination of mature granulocytes, but we advance that the main effect was due to the elimination of the immature precursors. As mature granulocyte destruction by anti—Gr-1 treatment can expose tumor-bearing hosts to lethal bacterial infections, novel protocols that specifically divert the maturation of iMacs toward functional APCs could be combined with powerful immunogens to improve the immunotherapy strategies against cancer.
Acknowledgments
We would like to thank Martha Blalock and Pierantonio Gallo for assistance with graphics; Patricia Segato for editing the manuscript; Vito Barbieri for the technical assistance in mouse studies; and Dina Pozza for the technical help with the slide staining.
Supported by Ministero Italiano della Ricerca Scientifica e Tecnologica (MURST), Italian Association for Cancer Reseach (AIRC), and by the Istituto Superiore Sanitá (ISS), Italy-US cooperation program for the therapy of cancer, Grant 981/A.14. E.A. is supported by a fellowship of the Italian Foundation for Cancer Research (FIRC).
References
- 1.Sprent J, Schaefer M. Antigen-presenting cells for unprimed T cells. Immunol Today. 1989;10:17–23. doi: 10.1016/0167-5699(89)90060-1. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 3.Chambers CA, Allison JP. Costimulatory regulation of T cell function. Curr Opin Cell Biol. 1999;11:203–210. doi: 10.1016/s0955-0674(99)80027-1. [DOI] [PubMed] [Google Scholar]
- 4.Lenardo M, Chan KM, Hornung F, et al. Mature T lymphocyte apoptosis—immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol. 1999;17:221–253. doi: 10.1146/annurev.immunol.17.1.221. [DOI] [PubMed] [Google Scholar]
- 5.Bronte V, Chappel DB, Apolloni E, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999;162:5728–5737. [PMC free article] [PubMed] [Google Scholar]
- 6.Bronte V, Wang M, Overwijk WW, et al. Apoptotic death of CD8+ T lymphocytes after immunization: induction of a suppressive population of Mac-1+/Gr-1+ cells. J Immunol. 1998;161:5313–5320. [PMC free article] [PubMed] [Google Scholar]
- 7.Mosmann TR, Sad S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol Today. 1996;17:138–146. doi: 10.1016/0167-5699(96)80606-2. [DOI] [PubMed] [Google Scholar]
- 8.Paulnock DM. Macrophage activation by T cells. Curr Opin Immunol. 1992;4:344–349. doi: 10.1016/0952-7915(92)90087-u. [DOI] [PubMed] [Google Scholar]
- 9.Goerdt S, Orfanos CE. Other functions, other genes: alternative activation of antigen-presenting cells. Immunity. 1999;10:137–142. doi: 10.1016/s1074-7613(00)80014-x. [DOI] [PubMed] [Google Scholar]
- 10.Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176:287–292. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKnight AJ, Gordon S. Membrane molecules as differentiation antigens of murine macrophages. Adv Immunol. 1998;68:271–314. doi: 10.1016/s0065-2776(08)60562-3. [DOI] [PubMed] [Google Scholar]
- 12.Chakrabarti S, Brechling K, Moss B. Vaccinia virus expression vector: coexpression of beta-galactosidase provides visual screening of recombinant virus plaques. MolCell Biol. 1985;5:3403–3409. doi: 10.1128/mcb.5.12.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moss B, Earl PL. Expression of proteins in mammalian cells using vaccinia viral vectors. In: Ausubel FM, Brent R, Kingston RE, et al., editors. Current Protocols in Molecular Biology. John Wiley; New York, NY: 1998. pp. 16.15.1–16.15.5. [Google Scholar]
- 14.Chen PW, Wang M, Bronte V, Zhai Y, Rosenberg SA, Restifo NP. Therapeutic antitumor response after immunization with a recombinant adenovirus encoding a model tumor-associated antigen. J Immunol. 1996;156:224–231. [PMC free article] [PubMed] [Google Scholar]
- 15.Bronte V, Tsung K, Rao JB, et al. IL-2 enhances the function of recombinant poxvirus-based vaccines in the treatment of established pulmonary metastases. J Immunol. 1995;154:5282–5292. [PMC free article] [PubMed] [Google Scholar]
- 16.Gavin MA, Gilbert MJ, Riddell SR, Greenberg PD, Bevan MJ. Alkali hydrolysis of recombinant proteins allows for the rapid identification of class I MHC-restricted CTL epitopes. J Immunol. 1993;151:3971–3980. [PubMed] [Google Scholar]
- 17.Leenen PJ, de Bruijn MF, Voerman JS, Campbell PA, van Ewijk W. Markers of mouse macrophage development detected by monoclonal antibodies. J Immunol Methods. 1994;174:5–19. doi: 10.1016/0022-1759(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 18.de Bruijn MF, Slieker WA, van der Loo JC, Voerman JS, van Ewijk W, Leenen PJ. Distinct mouse bone marrow macrophage precursors identified by differential expression of ER-MP12 and ER-MP20 antigens. Eur J Immunol. 1994;24:2279–2284. doi: 10.1002/eji.1830241003. [DOI] [PubMed] [Google Scholar]
- 19.Suss G, Shortman K. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand-induced apoptosis. J Exp Med. 1996;183:1789–1796. doi: 10.1084/jem.183.4.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romani N, Gruner S, Brang D, et al. Proliferating dendritic cell progenitors in human blood. J Exp Med. 1994;180:83–93. doi: 10.1084/jem.180.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schreurs MW, Eggert AA, de Boer AJ, Figdor CG, Adema GJ. Generation and functional characterization of mouse monocyte-derived dendritic cells. Eur J Immunol. 1999;29:2835–2841. doi: 10.1002/(SICI)1521-4141(199909)29:09<2835::AID-IMMU2835>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 23.Angulo I, de las Heras FG, Garcia-Bustos JF, Gargallo D, Munoz-Fernandez MA, Fresno M. Nitric oxide-producing CD11b(+)Ly-6G(Gr-1)(+)CD31(ER-MP12)(+) cells in the spleen of cyclophosphamide-treated mice: implications for T-cell responses in immunosuppressed mice. Blood. 2000;95:212–220. [PubMed] [Google Scholar]
- 24.de Bruijn MF, Ploemacher RE, Mayen AE, et al. High-level expression of the ER-MP58 antigen on mouse bone marrow hematopoietic progenitor cells marks commitment to the myeloid lineage. Eur J Immunol. 1996;26:2850–2858. doi: 10.1002/eji.1830261208. [DOI] [PubMed] [Google Scholar]
- 25.de Bruijn MF, van Vianen W, Ploemacher RE, et al. Bone marrow cellular composition in Listeria monocytogenes infected mice detected using ER-MP12 and ER-MP20 antibodies: a flow cytometric alternative to differential counting. J Immunol Methods. 1998;217:27–39. doi: 10.1016/s0022-1759(98)00080-5. [DOI] [PubMed] [Google Scholar]
- 26.Doyle AG, Herbein G, Montaner LJ, et al. Interleukin-13 alters the activation state of murine macrophages in vitro: comparison with interleukin-4 and interferon-gamma. Eur J Immunol. 1994;24:1441–1445. doi: 10.1002/eji.1830240630. [DOI] [PubMed] [Google Scholar]
- 27.Tsuchiya Y, Igarashi M, Suzuki R, Kumagai K. Production of colony-stimulating factor by tumor cells and the factor-mediated induction of suppressor cells. J Immunol. 1988;141:699–708. [PubMed] [Google Scholar]
- 28.Oghiso Y, Yamada Y, Ando K, Ishihara H, Shibata Y. Differential induction of prostaglandin E2-dependent and -independent immune suppressor cells by tumor-derived GM-CSF and M- CSF. J Leukoc Biol. 1993;53:86–92. doi: 10.1002/jlb.53.1.86. [DOI] [PubMed] [Google Scholar]
- 29.Fu YX, Watson GA, Kasahara M, Lopez DM. The role of tumor-derived cytokines on the immune system of mice bearing a mammary adenocarcinoma: I, induction of regulatory macrophages in normal mice by the in vivo administration of rGM-CSF. J Immunol. 1991;146:783–789. [PubMed] [Google Scholar]
- 30.Jaffe ML, Arai H, Nabel GJ. Mechanisms of tumor-induced immunosuppression: evidence for contact-dependent T cell suppression by monocytes. Mol Med. 1996;2:692–701. [PMC free article] [PubMed] [Google Scholar]
- 31.Young MR, Wright MA, Matthews JP, Malik I, Prechel M. Suppression of T cell proliferation by tumor-induced granulocyte-macrophage progenitor cells producing transforming growth factor-beta and nitric oxide. J Immunol. 1996;156:1916–1922. [PubMed] [Google Scholar]
- 32.Johnson BD, Hanke CA, Becker EE, Truitt RL. Sca1(+)/Mac1(+) nitric oxide-producing cells in the spleens of recipients early following bone marrow transplant suppress T cell responses in vitro. Cell Immunol. 1998;189:149–159. doi: 10.1006/cimm.1998.1373. [DOI] [PubMed] [Google Scholar]
- 33.Sugiura K, Inaba M, Ogata H, et al. Wheat germ agglutinin-positive cells in a stem cell-enriched fraction of mouse bone marrow have potent natural suppressor activity. Proc Natl Acad Sci U S A. 1988;85:4824–4826. doi: 10.1073/pnas.85.13.4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brooks-Kaiser JC, Bourque LA, Hoskin DW. Heterogeneity of splenic natural suppressor cells induced in mice by treatment with cyclophosphamide. Immunopharmacology. 1993;25:117–129. doi: 10.1016/0162-3109(93)90015-i. [DOI] [PubMed] [Google Scholar]
- 35.Pulendran B, Smith JL, Caspary G, et al. Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc Natl Acad Sci U S A. 1999;96:1036–1041. doi: 10.1073/pnas.96.3.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maraskovsky E, Pulendran B, Brasel K, et al. Dramatic numerical increase of functionally mature dendritic cells in FLT3 ligand-treated mice. Adv Exp Med Biol. 1997;417:33–40. doi: 10.1007/978-1-4757-9966-8_6. [DOI] [PubMed] [Google Scholar]
- 37.Pulendran B, Lingappa J, Kennedy MK, et al. Developmental pathways of dendritic cells in vivo: distinct function, phenotype, and localization of dendritic cell subsets in FLT3 ligand-treated mice. J Immunol. 1997;159:2222–2231. [PubMed] [Google Scholar]
- 38.Daro E, Pulendran B, Brasel K, et al. Polyethylene glycol-modified GM-CSF expands CD11bhighCD11chigh but not CD11blowCD11chigh murine dendritic cells in vivo: a comparative analysis with Flt3 ligand. J Immunol. 2000;165:49–58. doi: 10.4049/jimmunol.165.1.49. [DOI] [PubMed] [Google Scholar]
- 39.Lutz MB, Suri RM, Niimi M, et al. Immature dendritic cells generated with low doses of GM-CSF in the absence of IL-4 are maturation resistant and prolong allograft survival in vivo. Eur J Immunol. 2000;30:1813–1822. doi: 10.1002/1521-4141(200007)30:7<1813::AID-IMMU1813>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 40.Young MR, Wright MA, Young ME. Antibodies to colony-stimulating factors block Lewis lung carcinoma cell stimulation of immune-suppressive bone marrow cells. Cancer Immunol Immunother. 1991;33:146–152. doi: 10.1007/BF01756134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Young MR, Wright MA, Lozano Y, et al. Increased recurrence and metastasis in patients whose primary head and neck squamous cell carcinomas secreted granulocyte-macrophage colony-stimulating factor and contained CD34+ natural suppressor cells. Int. J Cancer. 1997;74:69–74. doi: 10.1002/(sici)1097-0215(19970220)74:1<69::aid-ijc12>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 42.Rokhlin OW, Griebling TL, Karassina NV, Raines MA, Cohen MB. Human prostate carcinoma cell lines secrete GM-CSF and express GM-CSF-receptor on their cell surface. Anticancer Res. 1996;16:557–563. [PubMed] [Google Scholar]
- 43.Lahm H, Wyniger J, Hertig S, et al. Secretion of bioactive granulocyte-macrophage colony-stimulating factor by human colorectal carcinoma cells. Cancer Res. 1994;54:3700–3702. [PubMed] [Google Scholar]
- 44.Seung LP, Rowley DA, Dubey P, Schreiber H. Synergy between T-cell immunity and inhibition of paracrine stimulation causes tumor rejection. Proc Natl Acad Sci U S A. 1995;92:6254–6258. doi: 10.1073/pnas.92.14.6254. [DOI] [PMC free article] [PubMed] [Google Scholar]