

Abstract
Regulating gene expression at the translational level controls a wide variety of biological events such as development, long-term memory, stress response, transport and storage of certain nutrients, and viral infection. Protein synthesis at steady-state level can be directly measured with western blot or using an easy-to-detect reporter like luciferase. However, these methods do not measure the association of mRNA with ribosomes, which is more meaningful in understanding the mechanism and dynamics of translation. This chapter describes the use of sucrose density gradients for analysis of polysome profiles. RNA or protein samples extracted from gradient fractions are commonly used for further analysis of their association with translating ribosomes. We also describe an in vitro translation system prepared from HeLa S3 cell cytoplasmic extract that shows dependency on the mRNA cap and length of the poly(A) length tail, both features of translation in vivo. This is particularly useful to study the cis- and transacting factors involved in translational control. Lastly we describe a method for transfecting cells with an in vitro prepared RNA to study the impact of poly(A) length on translation. This approach is particularly useful for characterizing cis-acting elements that work in conjunction with poly(A) in regulating translation.
Keywords: translation, sucrose density gradient, polysome, HeLa cytoplasmic extract, in vitro translation, poly(A) tail
1. Introduction
Initiation is the primary regulated step in the translation of most eukaryotic mRNAs (1), and two features common to most mRNAs, the 5′ cap and the 3′ poly(A) tail play principal roles in this (2,3). Much of our current view of the steps involved in initiation derives from the circular polysome model (4), in which eIF4G brings together the 5′ cap bound by eIF4E and the 3′ poly(A) tail bound by poly(A)-binding protein (PABP). Initiation commences with the binding of eIF3 and the met-tRNA-bound 43S ribosome subunit to this complex. Most vertebrate mRNAs exit the nucleus with a poly(A) tail of ~200 residues (5), and this undergoes progressive shortening in the cytoplasm until it reaches a limit or 10–15 residues (6), following which it is no longer associated with eIF4E or PABP (7) and instead becomes associated with Dcp1p, Dcp2p and the cytoplasmic Lsm1p-7p proteins in discrete sites of mRNA degradation termed processing bodies, or P-bodies (7,8). It was known for some time that decreasing poly(A) tail length is associated with reduced translation of maternal mRNAs (ref), and recently a similar process of deadenylation is linked to micro RNA-mediated repression of target mRNAs. Depending on the mRNA poly(A) shortening has been linked to reduced translation or to enhanced degradation (ref). Thus one must evaluate a particular sequence element in the context of differing length poly(A) tails to understand its function in controlling translation.
Physical measurements determined that 12 adenosines constitute the minimum length for binding by PABP (9). On longer tracts PABP binds approximately every 25 residues. The notion that poly(A) is a length-dependent enhancer of translation is supported by experiments performed in vitro using cell extracts (2, 10–12) and in electroporated or RNA transfected cells (12–14). Tethering PABP to mRNA using MS2 fusions can stimulate translation initiation, indicating that the impact of increasing poly(A) length on translation initiation results from the increased availability of PABP bound to the mRNA 3′ end. More recent work with PABP-depleted extracts proved that PABP is an initiation factor that works by enhancing the formation of both the 48S initiation complex and 60S subunit joining (15).
Here we describe a number of approaches used in our lab to analyzing the impact of poly(A) tail length on translation both in vivo and in vitro. Ribosome binding is the hallmark of translation in vivo, and the first half of this chapter describes the use of linear sucrose density gradients to study the efficiency of ribosome loading onto a particular mRNA. This classical approach to separating translating ribosome-bound complexes has proven to be particularly powerful for studying translating mRNP complexes when combined with immunoprecipitation, microarrays and proteomics. While gradient analysis has its place, it is of limited usefulness in evaluating the impact of features such as poly(A) tail length on translational regulation by a particular cis-acting element. For this one must turn to in vitro approaches, and we describe here the preparation of an in vitro translation system derived from cytoplasmic extract of HeLa S3 cells that is dependent on the presence of 5′ cap for initiation and shows increasing translation with increasing length poly(A). We also describe an approach we used successfully to examine the translational efficiency of capped transcripts with varying length poly(A) tails introduced into cells.
2. Materials
2.1. Preparation of sucrose density gradients
Dual chamber gradient maker (Model SG50, Hoefer) fitted with a small spin bar that just fits within the diameter of one chamber.
1 M NaOH, to treat the gradient maker. See Note 1.
DEPC (diethylpyrocarbonate)-treated double-distilled water or water processed through a polishing apparatus such as those sold by Millipore or Barnstead.
High and low concentration sucrose buffers. Start with 15% and 40% sucrose buffers: 15% or 40% w/v sucrose, 20 mM HEPES-KOH (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid) (pH 7.5), 100 mM KCl, 10 mM MgCl2, 100 μg/ml cycloheximide, 1 mM phenylmethylsulfonylfluoride (PMSF), 25 μl/ml protease inhibitor cocktail (Sigma, optional), 10 U/ml RNaseOUT™ (Invitrogen), 1 mM dithiothreitol (DTT). Prepare fresh in DEPC-treated deionized water. See Note 2.
Peristaltic pump, we use an LKB Bromma 2132 Microperpex peristaltic pump.
Centrifuge tubes, we use Beckman centrifuge tubes 9/16 × 3 ½ inches (14 × 89 mm) P.A. part number 331372.
Glass capillary tubes.
Magnetic stirring plate.
2.2. Sample preparation for sucrose density gradient analysis
To insure a sufficient amount of material for application to gradients and to have sufficient material for subsequent analysis of protein and RNA grow enough cells to fill a 150 mm Petri dish. See Note 3.
Cycloheximide, dissolved in 70% ethanol at 100 mg/ml. Store at −20°C for up to 2 weeks.
Phosphate buffered saline (PBS), sterile, ice-cold.
Lysis buffer: 20 mM HEPES-KOH (pH 7.5), 100 mM KCl, 10 mM MgCl2, 0.25% NP-40 (Nonidet P-40), 100 μg/ml cycloheximide, 1 mM PMSF, 25 μl/ml protease inhibitor cocktail (Sigma), 100 U/ml RNaseOUT™ (Invitrogen), 1 mM DTT. Prepare fresh in DEPC-treated double distilled water. See Note 4.
Sterile cell scrapers.
1 ml syringes.
Needles, gauge to vary with size of cells to be lysed.
Ice buckets, filled. Prepare enough so each plate can sit on ice while cells are prepared.
Refrigerated microcentrifuge.
Ultracentrifuge capable of 225,004 ×g. We use a Sorvall UltraPro 80 ultracentrifuge.
Swinging bucket rotor, we use a Sorvall TH-641 swinging bucket rotor. A Beckman SW41 can also be used.
2.3. Sample collection for sucrose density gradient analysis
Glass capillary tubes.
Peristaltic pump.
UV monitor, we use a LKB 2138 Uvicord S UV monitor. This can be substituted with newer models.
Fraction collector, we use a LKB 2112 Redirac fraction collector.
Chart recorder, we use a LKB 2210 two-channel recorder. This can be substituted with newer models.
2.4. Preparation of HeLa cytoplasmic extract for in vitro translation
Complete medium for HeLa S3 cells: Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen) supplemented with 4 mM L-glutamine (Invitrogen) and 10% fetal bovine serum (FBS, Invitrogen).
Phosphate buffered saline (PBS, Amresco).
Hypotonic MC buffer: 10 mM HEPES/KOH, pH 7.4, 10 mM potassium acetate, 0.5 mM magnesium acetate, 5 mM DTT, 1 mM PMSF, 25 μl/ml protease inhibitor cocktail (Sigma). Prepare fresh.
Spinner flasks that are suitable for growing cells in suspension. We use 1 L ProCulture Spinner Flasks (Corning).
Magnetic stirring plate for spinner cell culture.
Dounce homogenizer with a tight-fitting pestle.
Refrigerated microcentrifuge.
2.5. Preparation of m7GpppG-Capped transcripts with varying length poly(A) tails
mMESSAGE mMACHINE™ High Yield Capped RNA Transcription SP6 Kit (Ambion).
Plasmid template that has luciferase coding sequence downstream of a SP6 promoter and varying length poly(A) at the 3′ end. See Note 5.
2.6. Preparation of ApppG-Capped Transcripts
MEGAscript® transcription SP6 kit (Ambion).
ApppG, 20 mM (Ambion).
Plasmid template that has luciferase coding sequence downstream of a SP6 promoter and varying length poly(A) at the 3′ end. See Note 5.
2.7. In vitro translation
HeLa cytoplasmic extract.
In vitro transcripts diluted in DEPC-treated water at the concentration of 15 fmol/μl. See Note 6.
Translation buffer (5×): 30 mM HEPES-KOH, pH 7.4, 330 mM KCl, 5 mM MgCl2, 5 mM ATP, 0.25 mM GTP, 125 μM bovine liver tRNA, 50 μM amino acid mix (Promega), 21 mM β-mercaptoethanol, 50 mM creatine phosphate, 125 ng/μl creatine phosphokinase (optional), 1.2 mM spermidine. Store aliquots for single use at −80°C. These can be used for up to one year. See Note 7.
RNaseOut™ (Invitrogen).
Cycloheximide, 100 mg/ml in 70% ethanol.
Firefly luciferase assay reagent (Promega).
Luminometer for luciferase activity assay. We use the Sirius Luminometer (Berthold Detection Systems).
2.8. Cell transfection with capped and polyadenylated RNA
For LM(tk−) cells used in our lab Complete medium consists of Dulbecco’s Modified Eagle’s Medium (DMEM) (Invitrogen) supplemented with 4 mM L-glutamine (Invitrogen) and 10% fetal bovine serum (FBS, Invitrogen).
OPTI-MEM® medium (Invitrogen).
Lipofectamine 2000™ (Invitrogen).
Dual-Luciferase® Reporter Assay System (Promega).
3. Methods
3.1. Preparation of sucrose density gradients
Treat the gradient maker with 1 M NaOH to remove RNase contamination. Soak at least 20 min then rinse thoroughly with deionized water. While the gradient maker is soaking, prepare sucrose buffers and place on ice.
In cold room, prepare the pump and gradient maker. Disconnect pump from other equipment, attach a capillary tube to one end of the tubing, and place the other end of the tubing over a container to catch waste. Insert the capillary tube into DEPC-treated double distilled water and turn on the pump to run the water through the tubing for a few minutes to clean it. The pump should run at 1 ml/min. Remove tubing from capillary tube and run water out of the tubing.
Next attach the gradient maker to one end of the pump tubing. The gradient maker has two cylindrical chambers connected by a small channel controlled by a stopcock. One chamber connects to an outlet via a second channel; this is also controlled by a stopcock. Connect the gradient maker to the tubing on the pump via this outlet.
Place the gradient maker on the stir plate and place the stir bar in the chamber closest to the outlet. Center the gradient maker so that the stir bar turns smoothly in its chamber.
Close all stopcocks, add some DEPC-treated double distilled water to the chamber furthest from the outlet, turn on the magnetic stirring plate, turn on the pump and open the stopcocks to wash and check the system.
When water has drained from gradient maker and air has entered the tubing, turn off the pump and the magnetic stirring plate and close the stopcocks. Add the higher percentage sucrose buffer to chamber furthest from the tubing connection; add the lower percentage sucrose buffer to the chamber closest to tubing connection. Use an equal volume of each buffer. For example, to pour an 11-ml gradient, put 5.5 ml high-concentration sucrose buffer in the chamber furthest from the outlet, and 5.5 ml low-concentration buffer in the chamber closest to the outlet.
Turn on the magnetic stirring plate to the lowest speed that allows for mixing. Turn on the peristaltic pump and open the stopcock to begin mixing of the sucrose solutions. The lighter sucrose solution should immediately enter the tubing from the gradient maker, and the chamber with the spin bar should show the swirling pattern caused by the mixing of the heavier sucrose solution with the lighter one.
Insert one end of a clean capillary tube into the tubing before the sucrose buffer reaches furthest end of tube and place the glass capillary into the bottom of a centrifuge tube. Mixed sucrose buffers should enter the centrifuge tube at ~1 ml/min. Care should be taken at this step to avoid introducing bubbles. See Note 8.
Gradients are finished when most of each sucrose solution is gone from the gradient maker, before bubbles enter the gradients. Stop the peristaltic pump and carefully remove the glass capillary from gradient. See Note 9.
Gradients are stored at 4°C. This can be done in the rotor bucket, and gradients can be prepared the evening before use. To insure reproducibility between each gradient the gradient maker is rinsed with DEPC-treated double distilled water before the next gradient is prepared. See Note 10.
3.2. Sample preparation for sucrose density gradient analysis
Turn on ultracentrifuge to allow machine to cool to 4°C before use. Pre-cool the rotor in a cold room or refrigerator or place it into the centrifuge to cool before use.
Cycloheximide freezes elongating ribosomes onto mRNA and is useful for stabilizing polysome complexes. If you chose to use this add it to cells at a final concentration of 100 μg/ml within 15 min of cell harvest.
Prepare lysis buffer and keep on ice. Move materials for sample preparation to cold room. Prepare a sufficient number of ice buckets to accommodate all of the plates of cells.
Remove cells from the CO2 incubator and place on ice in a cold room. Wash plates twice with ice-cold PBS and tilt each plate to drain remain PBS.
Add 1 ml ice-cold PBS and remove adherent cells from the surface using a cell scraper. The suspended cells are removed to a 1.5 ml microfuge tube.
Cells are collected by centrifuging at 1000 xg for 2 min, 4°C, after which the PBS is removed leaving the cell pellet.
Add 0.5 ml lysis buffer to each cell pellet and pipet gently to resuspend.
Lyse cells by passage through syringe. See Note 11. The size of the needle and the number of passages is dependent on cell type. See Note 12. Monitor lysis via microscopy. See Note 13.
Pellet nuclei and cell debris by centrifuging at 12,000 ×g for 5 min, 4°C. Carefully load the supernatant (cytoplasmic extract) onto the previously-prepared sucrose gradients.
Use lysis buffer to adjust all of the samples to the same weight.
Add small drop of SpinkoteR grease to screw threads on lids of buckets and screw lids on buckets.
Add all of the buckets to the pre-cooled rotor and carefully place onto the spindle in the ultracentrifuge.
The rotor is centrifuged at 225,000 ×g for 2.5 hours at 4°C. We do not use the brake to slow the rotor, and it takes about 75 minutes for rotor to stop spinning at end of run without the brake.
3.3. Sample collection from sucrose density gradients
The UV monitor, chart recorder and fraction collector are kept in a cold room or refrigerated chromatography cabinet. The UV monitor is turned on when run is over but before rotor stops spinning to allow sufficient time for the lamp to warm up and stabilize (30–60 min prior to use).
The individual gradient fractions are collected into microfuge tubes. These are labeled and placed into the fraction collector to cool. We typically use 11 ml gradients and the added sample volume and void volume within the tubing requires 24 tubes per gradient for collecting 0.5 ml fractions.
When run is finished the rotor is carefully removed from the ultracentrifuge, the buckets are removed from the rotor and placed into their holder and taken to the cold room where forceps are used to remove individual tubes from the buckets.
Prepare the collection apparatus by connecting the output of the UV monitor to a chart recorder and the flow cell within the monitor to the fraction collector. Connect pump tubing to UV monitor inlet tubing, allow outlet from UV monitor tubing to hang over a container to collect waste. Reverse pump direction and rinse tubing once with DEPC-treated double distilled water then introduce bubble into line.
Insert a capillary tube into the end of tubing and place this into 15% sucrose buffer. Turn on pump to and fill the line with sucrose the sucrose solution. At this point the pump is turned off. Adjust electronic zero on the chart recorder before adjusting zero absorbance on the UV monitor. Remove capillary tube from tubing, turn on pump to introduce air bubble into line then stop.
A new capillary tube is placed on the end of the tubing and this is immobilized vertically on a ring stand using a small clamp. The clamp on the ring stand is loosened and the capillary is slowly lowered to the bottom of the tube taking care not to disturb the gradient.
At this point the pump is turned on to and fractions are pumped from the bottom of the tube through the UV monitor and into the fraction collector until all of the solution has been removed an air enters the line, at which time the pump is turned off. A typical profile for absorbance at 254 nm is shown in Fig. 1. In this particular experiment 48 0.25 ml fractions were collected, but 24 0.5 ml fractions provides sufficient separation for most applications. Because these are collected from the bottom of the tube the numbering in this figure starts with the most rapidly-sedimenting fractions. The tubing and flow-cell within the UV monitor are rinsed with DEPC-treated double-distilled water after each gradient and the process is repeated.
When all of the gradients have been collected the apparatus is cleaned by rinsing with DEPC-treated double-distilled water, taking care to clean up any residual sucrose that may have spilled. The individual fractions may be processed immediately or stored at −80°C until ready for further analysis. See Note 14.
Fig. 1.
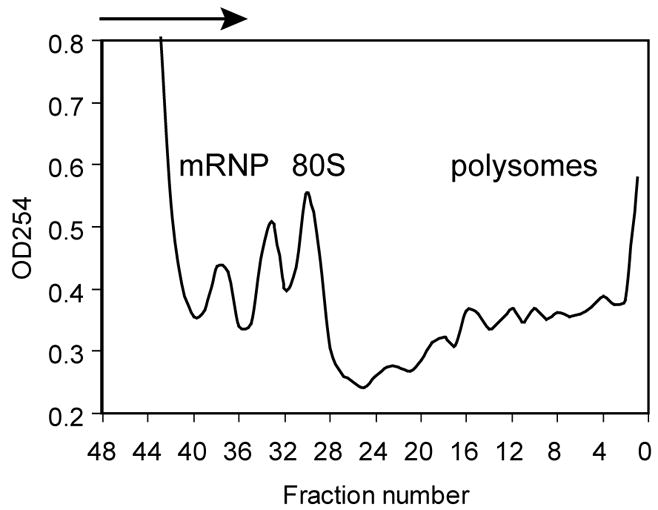
Absorbance profile of RNP and polysome complexes separated on a sucrose density gradient. Cytoplasmic extract of LM(tk−) murine fibroblasts were separated on a 15–40% sucrose density gradient. Shown is the absorbance profile at 254 nm as the indicated fractions pass through a UV monitor.
3.4. Preparation of HeLa cytoplasmic extract for in vitro translation
HeLa S3 cells are maintained in complete medium in plastic cultureware in a humidified 37°C incubator supplemented with 5% CO2. To grow HeLa S3 cells in suspension trypsinized cells are counted and seeded into a 250-ml spinner flask at a concentration of 4–8 × 104 cells/ml. When the cell density reaches 4–6 × 105 cells/ml the entire 250 ml culture is inoculated into a 1-L spinner flask containing 750 ml of medium. The cells are ready for harvest when the cell density reaches 4–6 × 105 cells/ml. See Note 15.
Harvest the cells by centrifugation at 200 ×g for 10 min at 4°C in a Sorvall HS-4 rotor.
Wash the cell pellet three times 10 volumes of ice-cold PBS. See Note 16.
Resuspend the cell pellet with an equal volume of hypotonic MC buffer.
Allow the cells to swell on ice for 5–10 min.
Lyse the cells with 25–30 strokes of a Dounce homogenizer using a tight-fitting (A) pestle. See Note 17.
Remove the cell debris and nuclei by centrifugation at 15,000 ×g for 20 min at 4°C in a refrigerated microcentrifuge. See Note 18.
Collect the supernatant and store in small aliquots at −80°C. See Note 19.
3.5. Preparation of m7GpppG-capped transcripts with varying length poly(A) tails
Set up a 20 μl in vitro transcription reaction on ice following the standard instruction from the mMESSAGE mMACHINE® kit. The reaction mixture include: 1 μg of linear plasmid DNA template, 10 μl 2× NTP mix, 2 μl 10× Reaction Buffer and 2 μl SP6 RNA polymerase See Note 20.
Incubate the reaction mixture at 37°C overnight. See Note 21.
Add 1 μl DNase I and incubate for 15 min at 37°C to remove the plasmid DNA template.
Add 30 μl water and 25 μl Lithium Chloride Precipitation Solution and incubate at 20°C for at least 30 min. See Note 22.
Centrifuge at 15,000 ×g for 15 min at 4°C to pellet the transcript.
Carefully remove the supernatant and wash the pellet once with 1 ml 70% ethanol.
Resuspend the transcript in 30 μl water and measure the concentration with a spectrometer. See Note 23.
3.6. Preparation of ApppG-capped transcripts
Set up a 20 μl in vitro transcription reaction on ice following the standard instruction from the MEGAscript® SP6 kit. The reaction mixture include: 1 μg of linear plasmid DNA template, 2 μl 10× Reaction Buffer and 2 μl each ATP, CTP, and UTP Solution, 0.4 μl GTP solution, 4 μl 20 mM 5′ ApppG and 2 μl SP6 Enzyme Mix. See Note 20.
Incubate the reaction mixture at 37°C overnight. See Note 21.
Add 1 μl DNase I and incubate for 15 min at 37°C to remove the plasmid DNA template.
Add 30 μl water and 25 μl Lithium Chloride Precipitation Solution and incubate at 20°C for at least 30 min. See Note 22.
Centrifuge at 15,000 ×g for 15 min at 4°C to pellet the transcript.
Carefully remove the supernatant and wash the pellet once with 1 ml 70% ethanol.
Resuspend the transcript in 30 μl water and measure the concentration with a spectrometer. See Note 23.
3.7. In vitro translation
Assemble the reaction mixture on ice: 2 μl transcript (30 fmol), 2 μl water, 2 μl 5× translation buffer, 4 μl HeLa cytoplasmic extract and 1 μl (4 units) RNaseOut™. If desired you may include a control 6 fmol of m7GpppG-capped Renilla luciferase transcript with 78 nucleotide poly(A). This is used to normalize results obtained with firefly luciferase transcripts with varying length poly(A) tails (see Fig. 2). If you chose to do this reduce the amount of firefly luciferase RNA in the reaction to 24 fmol.
Incubate the reaction mixture at 37°C for 60–90 min.
Stop the translation reaction by adding 1 μl of 100 μg/μl cycloheximide.
Dilute 2 μl reaction mixture with 8 μl 1× PBS, use 50 μl luciferase assay reagent from the Dual-Luciferase® Reporter Assay kit and measure firefly and Renilla luciferase activity with a luminometer. Results showing the impact of the cap and increasing length poly(A) on translation in vitro (12) are presented in Fig. 2. The first 2 samples consist of luciferase RNA with a non-functional ApppA cap and either no poly(A) or a 98 residue poly(A) tail. The next 5 samples show the translation of capped firefly luciferase mRNA with 0, 14, 20, 54 and 98 residue poly(A) tails. In this figure firefly luciferase translation is normalized to an internal control of Renilla luciferase mRNA (see Northern blot in Fig. 2B), and the fold increase is the amount of activity relative to capped luciferase mRNA with no added poly(A) (cap A0).
Fig. 2.
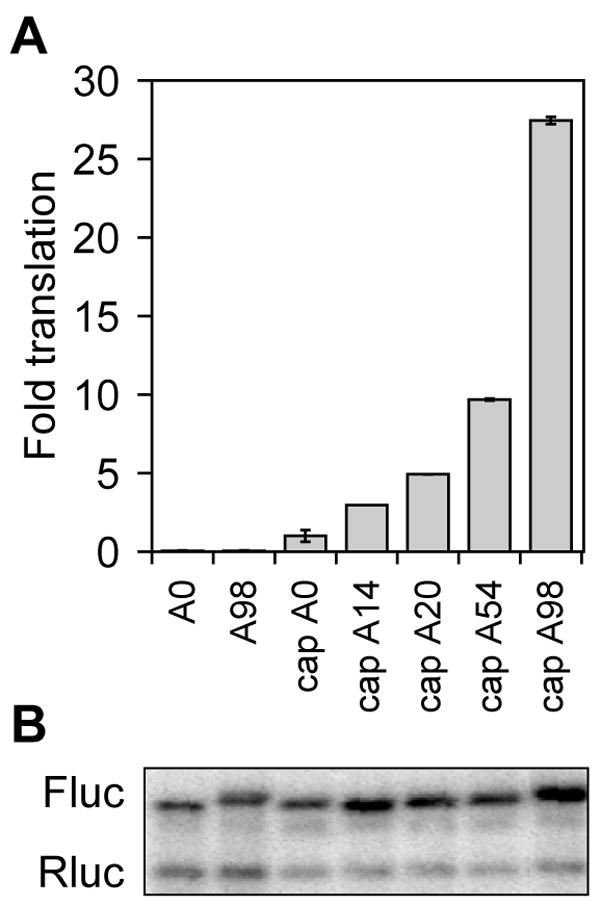
In vitro translation of firefly luciferase mRNA with increasing length poly(A) in HeLa extract. A. Triplicate samples of firefly luciferase mRNA with a non-functional ApppG cap and 0 or 98 nucleotide poly(A) tails (A0, A98), or m7GpppG-capped firefly luciferase mRNA with poly(A) tails of 0, 14, 20, 54 or 98 nucleotides were translated in HeLa cytoplasmic extracts together with a fixed amount of m7GpppG-capped Renilla luciferase mRNA. The relative light units of each luciferase reporter were determined in a luminometer and results with each sample of firefly luciferase were normalized to the internal Renilla luciferase control. The value for m7GpppG-capped firefly luciferase mRNA with no poly(A) (cap A0) was arbitrarily set to one in order to quantify the fold increase in translation observed with increasing length poly(A) on the reporter mRNA. Shown are the mean ± standard deviation for the triplicate determinations. B. Each of the samples in A were pooled and RNA extracted after translation in vitro was analyzed by Northern blot. The blot was hybridized with a mixed probe for firefly (Fluc) and Renilla (Rluc) luciferase mRNA.
3.8. Translation of luciferase mRNA with varying length poly(A) tails in RNA-transfected cells
LM(tk−) cells are grown in complete medium in a humidified 37°C incubator supplemented with 5% CO2. The day before transfection, 1.6 × 104 cells in 100 μl complete medium are seeded into each well of a 96-well culture dish.
Dilute 0.4 μl Lipofectamine 2000® in 10 μl OPTI-MEM® for each well of cells to be transfected. This can be prepared in a master mix. Incubate the mixture for 5–10 min at room temperature.
During the incubation, dilute 160 ng (~240 pmol) firefly luciferase transcript and 40 ng (~120 pmol) m7GpppG-capped Renilla luciferase transcript with 78-nt poly(A) tail in 10 μl OPTI-MEM® for each well of cells to be transfected. See Note 24.
Combine the diluted transfection reagent from step 2 and diluted RNA from step 3 and incubate the mixture for 20 min at room temperature.
Add the transfection mixture to each well of cells, and return the cells to the cell culture incubator.
Harvest the cells 60–90 min after transfection. See Note 25. Wash each well of cells twice with 100 μl PBS, then lyse the cells with 1× Passive Lysis Buffer (diluted in water from the 5× buffer provided by Dual-Luciferase Reporter Assay kit) by incubation at room temperature for 15 min. See Note 26.
Take 10 μl of the cell lysate for firefly and Renilla luciferase activity assay with 50 μl luciferase activity assay reagent from the kit using a luminometer.
An example of the data obtained using this approach is shown in Fig. 3. As seen with in vitro translation using HeLa extract there is little translation of ApppG-capped RNA regardless of poly(A) tail length, and translation of capped luciferase mRNA increases with increasing poly(A) length. A Northern blot of RNA recovered from RNA-transfected cells is an essential control for this experiment to insure that any changes observed in translation are not due to differences in mRNA decay.
Fig. 3.
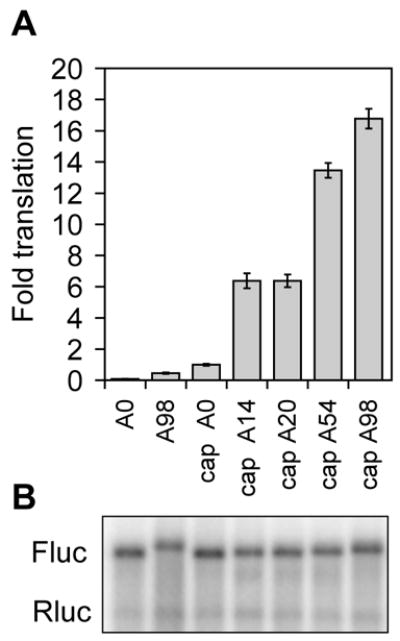
Translation of firefly luciferase mRNA with increasing length poly(A) in RNA-transfected LM(tk−) cells. A. Triplicate cultures of LM(tk−) cells were transfected with the indicated firefly luciferase transcripts with varying length poly(A) tails together with a Renilla luciferase RNA control as in Fig. 2. Firefly luciferase activity was normalized to that of co-transfected Renilla luciferase and the results are plotted as in Fig. 2. B. RNA extracted from the transfected cells was analyzed by Northern blot as in Fig. 2 using a mixed probe for firefly (Fluc) and Renilla (Rluc) luciferase mRNA.
4. Notes
All equipment and buffers should be prepared in a manner that is consistent with preventing RNase contamination and protein degradation. Plastic tubes and tips should be free of DNase and RNase. Solutions should be made with DEPC-treated deionized water, equipment should be soaked in 1 M NaOH. Sample preparation should be performed on ice in the cold room.
The volume of sucrose buffers to prepare depends on the volume of the gradients, the number of gradients, and the volume contained in the tubing connected to the pump. Each gradient is composed of equal volumes of the high and low concentration sucrose buffers. Sufficient amount of the lower concentration sucrose buffer should be prepared to have enough for use in filling the tubing between each gradient and zeroing the UV monitor.
High quality cell cultures are essential to the quality of the resulting polysome profile. Cells should be in log-phase growth at the time of harvest, and while they should not be crowded on the plate it is recommended that they are near confluency to have enough material (protein or RNA) for subsequent studies to be performed with the collected gradient fractions.
Although only 0.5 ml lysis buffer is required per sample, prepare extra for use in balancing samples prior to centrifugation. This should be prepared immediately before use.
The plasmids need to have the gene of interest downstream of a common bacteria phage promoter (T3, T7 or SP6) for transcription in vitro. We use luciferase (either firefly or Renilla) because it is very easy to measure the activity after the in vitro translation reaction. To analyze the relationship between translation efficiency and poly(A) tail length of the mRNA we have prepared a series of plasmids that contain 0, 14, 20, 54 and 98 adenosines downstream of the luciferase coding sequence. It is important to design the templates so that they are linearized by digestion with a restriction enzyme the cuts at the precise end of the poly(A) stretch. The plasmids developed in our lab have an SstII site at this location. In addition we commonly use a capped Renilla luciferase control transcript with a 78 residue poly(A) tail as an internal control in each reaction.
The amount of transcript for the translation reaction needs to be titrated. The amount of protein produced will increase with increasing amounts of input RNA; however, after a certain point the translation reaction becomes less dependent on poly(A) length. For the luciferase RNA used in our lab, 30 fmol of transcript in a 10 μl reaction produces enough protein and also shows good poly(A) length dependency.
The conditions of the translation reaction need to be optimized, especially magnesium and potassium concentrations. While we have provided here concentrations that work reproducibly in our hands subtle differences in sample preparation can affect this.
Both chambers of the gradient maker should drain equally. If not, check that there is no obstruction in the connection between the two chambers. Care should be taken to avoid getting bubbles between the chambers as this is a common source of this problem. Also the entire apparatus should be checked for leaks before used in pouring the gradients.
Bubbles cause mixing of the gradient and care should be taken to prevent this. In the event that bubbles occur in a gradient it is best to pour this out and prepare a new gradient.
Sucrose gradients may be prepared the night before use, but no longer than that. If prepared too far in advance there is enough diffusion that the separation quality is reduced.
Alternately, cells may be lysed with a Dounce homogenizer.
The gauge needle required at this step depends on size of cells to be lysed. A higher gauge needle (ie. smaller diameter) for smaller cells. We use a 28 ½ gauge needle for LM(tk−) cells (mouse fibroblast) and a 25 gauge needle for Cos-1 cells (monkey).
Although some clearing of the cell suspension may be evident after lysis by passing through the syringe it is best to monitor this under the microscope. Lysed cells should appear as nuclei clinging to strips of outer membrane, debris should be obvious in surrounding medium. To prepare wet mount, place about 20 μl of lysed cells on a microscope slide, cover with coverslip and view under phase contrast at 40X magnification. If more than 10% of the cells remain intact the suspension should be passed twice more through the syringe and needle, then examined again under the microscope. Although monitoring the cell lysis is important it is important to minimize the amount of time spent handling these extracts prior to application to the gradient.
RNA and proteins can be recovered from each fraction. We use 2 ml Trizol® (Invitrogen) to extract RNA and 10% trichloroacetic acid (TCA) to precipitate protein from each 0.5 ml fraction. If a protein is particularly abundant gradient fractions can be processed by several spins through a centrifugal microdialyzer (such as a Microcon®) to remove excess sucrose.
The HeLa S3 cells in our lab are HeLa S3 (tet off) from Clontech/BD Bioscience. The doubling time of this cell line is about 24 hr under the conditions described above. If cell density is less than 104/ml, the cells tend to grow more slowly but they are still healthy. It may be necessary to collect cells by centrifugation when replacing the medium every 3–4 days. However, it is critical to harvest the cells at the optimal density for best translation activity.
The cell pellet is about 3.5 ml for every liter of cells at the density of 6 × 105/ml.
It is recommended that cell lysis is monitored as in 13 by phase contrast microscopy. Ideally one will see lysed cell debris and intact nuclei at this step. It is important to minimize nuclear lysis as this nuclear contaminants inhibit translation activity.
If the supernatant is still very cloudy, repeat the centrifugation step. The final extract should be almost clear with a light yellowish color. Try to remove the white fat layer on top of the supernatant, but the extract still works with a little bit fat.
In our experience extracts retain essentially full translational activity through two times of freezing/thawing. We normally store fresh extract in 1 ml aliquots. Prior to use a tube is placed on ice to thaw and any extract that is not used can be re-frozen in 100 μl aliquots and used one more time.
If the gene of interest is downstream of a different promoter (T3 or T7), use the corresponding RNA polymerase instead.
SP6 RNA polymerase is slower than T3 and T7, so the reaction takes longer for maximum production of the transcript. We normally use overnight (~16 hr) incubation. If the T3 or T7 RNA polymerases are used a 2 hr incubation is sufficient.
It is critical to remove the unincorporated m7GpppG cap analog from the transcript, because it will inhibit the translation reaction in later step. LiCl precipitation is a simple and fast way to recover the transcription product while leaving the unincorporated nucleotide including m7GpppG cap analog in the supernatant. Other methods such as spin column chromatography can also be used. However, phenol:chloroform extraction and ethanol (or isopropanol) precipitation is not recommended because the incorporated nucleotides are not removed completely.
It is recommended that each transcript be analyzed by denaturing gel electrophoresis, and this can be used as a purification step to insure the quality of the transcript.
We normally transfect each firefly luciferase transcript in triplicate.
The transcripts transfected in the cells start to degrade 2 hr after transfection. To assess the impact of a particular sequence element on translation it is important to harvest the cells before this. As an additional control it is important to extract RNA and run a Northern blot to insure that RNA decay has not occurred or that any sequence element within the reporter RNA has not preferentially activated decay.
It is optional to remove the cell debris by centrifugation before measuring luciferase activities.
Acknowledgments
This work was supported by PHS grant R01 GM38277 and GM55407 to D.R.S. ELM was supported in part by PHS grant T32 CA09338, and support for core facilities was provided by PHS center grant P30 CA16058 from the National Cancer Institute to The Ohio State University Comprehensive Cancer Center.
References
- 1.Arava Y, Wang Y, Storey JD, Liu CL, Brown PO, Herschlag D. Proc Natl Acad Sci USA. 2003;100:3889–3894. doi: 10.1073/pnas.0635171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raught B, Gingras AC, Sonenberg N. Regulation of ribosomal recruitment in eukaryotes. In: Sonenberg N, Hershey JWB, Mathews MB, Ab G, editors. Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2000. pp. 245–293. [Google Scholar]
- 3.Mangus DA, Evans MC, Jacobson A. Genome Biol. 2003;4:223. doi: 10.1186/gb-2003-4-7-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sachs AB. Physical and functional interactions between the mRNA cap structure and the poly(A) tail. In: Sonenberg N, Hershey JWB, Mathews MB, Ab G, editors. Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2000. pp. 447–465. [Google Scholar]
- 5.Wahle E, Keller W. Trends Biochem Sci. 1996;21:247–250. [PubMed] [Google Scholar]
- 6.Tharun S, Parker R. Mechanisms of mRNA turnover in eukaryotic cells. In: Harford J, Morris DR, editors. mRNA Metabolism and Post-transcriptional Gene Regulation. Wiley; New York: 1997. pp. 181–200. [Google Scholar]
- 7.Tharun S, Parker R. Targeting an mRNA for decapping: displacement of translation factors and association of the Lsm1p-7p complex on deadenylated yeast mRNAs. Mol Cell. 2001;8:1075–1083. doi: 10.1016/s1097-2765(01)00395-1. [DOI] [PubMed] [Google Scholar]
- 8.Sheth U, Parker R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science. 2003;300:805–808. doi: 10.1126/science.1082320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sachs AB. A single domain of yeast poly(A)-binding protein is necessary and sufficient for RNA binding and cell viability. Mol Cell Biol. 1987;7:3268–3276. doi: 10.1128/mcb.7.9.3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Preiss T, Muckenthaler M, Hentze MW. Poly(A)-tail-promoted translation in yeast: implications for translational. RNA. 1998;4:1321–1331. doi: 10.1017/s1355838298980669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bergamini G, Preiss T, Hentze MW. Picornavirus IRESes and the poly(A) tail jointly promote cap-independent translation in a mammalian cell-free system. RNA. 2000;6:1781–1790. doi: 10.1017/s1355838200001679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng J, Schoenberg DR. mRNA with a <20 nt poly(A) tail imparted by the poly(A)-limiting element is translated as efficiently in vivo as long poly(A) mRNA. RNA. 2005;11:1131–1140. doi: 10.1261/rna.2470905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallie DR. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 1991;5:2108–2116. doi: 10.1101/gad.5.11.2108. [DOI] [PubMed] [Google Scholar]
- 14.Grudzien E, Kalek M, Jemielity J, Darzynkiewicz, Rhoads RE. Differential inhibition of mRNA degradation pathways by novel cap analogs. J Biol Chem. 2006;281:1857–1867. doi: 10.1074/jbc.M509121200. [DOI] [PubMed] [Google Scholar]
- 15.Kahvejian A, Svitkin YV, Sukarieh R, M’Boutchou MN, Sonenberg N. Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev. 2005;19:104–113. doi: 10.1101/gad.1262905. [DOI] [PMC free article] [PubMed] [Google Scholar]