

Abstract
Loss-of-function mutations in thyroid hormone transporter monocarboxylate transporter 8 (MCT8) lead to severe X-linked psychomotor retardation and elevated serum T3 levels. Most patients, for example those with mutations V235M, S448X, insI189, or delF230, cannot stand, walk, or speak. Patients with mutations L434W, L568P, and S194F, however, walk independently and/or develop some dysarthric speech. To study the relationship between mutation and phenotype, we transfected JEG3 and COS1 cells with wild-type or mutant MCT8. Expression and function of the transporter were studied by analyzing T3 and T4 uptake, T3 metabolism (by cotransfected type 3 deiodinase), Western blotting, affinity labeling with N-bromoacetyl-T3, immunocytochemistry, and quantitative RT-PCR. Wild-type MCT8 increased T3 uptake and metabolism about 5-fold compared with empty vector controls. Mutants V235M, S448X, insI189, and delF230 did not significantly increase transport. However, S194F, L568P, and L434W showed about 20, 23, and 37% of wild-type activity. RT-PCR did not show significant differences in mRNA expression between wild-type and mutant MCT8. Immunocytochemistry detected the nonfunctional mutants V235M, insI189, and delF230 mostly in the cytoplasm, whereas mutants with residual function were expressed at the plasma membrane. Mutants S194F and L434W showed high protein expression but low affinity for N-bromoacetyl-T3; L568P was detected in low amounts but showed relatively high affinity. Mutations in MCT8 cause loss of function through reduced protein expression, impaired trafficking to the plasma membrane, or reduced substrate affinity. Mutants L434W, L568P, and S194F showed significant residual transport capacity, which may underlie the more advanced psychomotor development observed in patients with these mutations.
MUTATIONS IN THE MCT8 gene (SLC16A2) impressively illustrate the detrimental effects that mutations in X-linked genes can have on psychomotor development. Monocarboxylate transporter 8 (MCT8) is a potent thyroid hormone transporter (1,2), which is expressed in cells of various tissues, including neurons in the central nervous system (CNS). Loss-of-function mutations in MCT8 are associated with severe psychomotor retardation and elevated serum T3 levels in males (3,4,5,6,7,8,9,10,11,12). This indicates that MCT8 plays an essential role in the development of the CNS, most likely by facilitating the supply of thyroid hormone to the neurons.
Reduced thyroid hormone action in the developing brain has long been recognized as an important cause of developmental disorders. Fetal and neonatal hypothyroidism, due to dysgenesis of the thyroid gland or impaired synthesis of thyroid hormone (13,14) or iodine deficiency (15), severely inhibits neurological development (16). Its most severe clinical presentation is referred to as cretinism, a syndrome comprised of growth retardation, impaired cognitive function, spastic diplegia, and deafness (17). However, recently, even mild fetal hypothyroidism due to marginally impaired maternal thyroid function in pregnancy has been associated with adverse neurological outcome (18,19).
Thyroid hormone has to cross numerous membranes before it reaches the nuclear T3 receptor in the neurons, the primary target for thyroid hormone action in the brain. It has become increasingly clear that uptake of thyroid hormone requires transporter proteins and that simple diffusion through membranes is unlikely if not impossible (20,21,22,23). Because T3 is produced locally in the brain, T4 has to be transported over the blood-brain barrier, a process that may include thyroid hormone transporter OATP1C1 (24). T4 is then converted to T3 by type 2 deiodinase (D2) in astrocytes, and transferred to the neurons. As yet it is not known which transporters are involved in the uptake of T4 and the release of T3 from astrocytes. Neurons express MCT8 (25), which is likely to facilitate the uptake of the locally produced T3. Neurons also express type 3 deiodinase (D3); they are considered the main site of thyroid hormone inactivation in the brain (26).
Mutations in MCT8 have been reported in over 25 families around the world (3,4,5,6,7,8,9,10,11,12,27). Affected males show severe psychomotor retardation, hallmarked by hypotonia of the axial muscles, spastic or dystonic quadriplegia, and severe cognitive impairment. Development of speech is usually absent, and the majority of patients are not able to sit, stand, or walk without support. Other symptoms include athetoid movement of hands and arms, paroxysmal dyskinesia, muscle hypoplasia, seizures, nystagmus, and secondary microcephaly. The first identification of this MCT8-related neurological phenotype was published in 1944 (28). Named after the authors, the syndrome is known as Allan-Herndon-Dudley syndrome (OMIM 300523).
In patients with mutations in MCT8, serum thyroid hormone profiles are abnormal. Total and free T4 levels range between low-normal and clearly decreased, whereas T3 levels are strongly increased, and rT3 is decreased. TSH is usually higher than in controls but mostly falls within the normal range (27). The elevated serum T3 level, especially in combination with decreased serum T4, appears to be a specific biochemical marker for mutations in MCT8.
Although psychomotor impairment is severe in all MCT8 patients, some significant differences in development are observed between families. Most notably, the majority of patients with the L568P and L434W mutations reported by Schwartz et al. (6) are able to walk without support, although the gait is ataxic. They also develop limited and dysarthric speech. Elementary speech development is also observed in patients with mutation S194F (6). Independent walking or development of speech was not observed in any of the patients reported by Friesema et al. (3), Dumitrescu et al. (4), Maranduba et al. (7), Holden et al. (9), Kakinuma et al. (10), Herzovich et al. (11), and Jansen et al. (12).
It was recently demonstrated that mutations in MCT8 result in reduced uptake and subsequent metabolism of T3 and T4 in vitro (12). The aim of the present study was to determine whether differences in psychomotor development observed between families correlate with functional characteristics of these mutants in vitro. Possible residual activity of the mutant transporter might underlie the apparent genotype-phenotype relation observed in patients with mutations in MCT8.
Materials and Methods
Plasmids and transfections
The cloning of wild-type human MCT8 (hMCT8) in the expression vector pcDNA3 (pcDNA3-hMCT8) was described previously (2). Point mutations identified in patients (6,9) (Table 1) were introduced in this plasmid using the QuikChange Site-Directed Mutagenesis protocol (Stratagene, Amsterdam, The Netherlands) to produce mutant proteins V235M, L434W, S448X, L568P, S194F, insI189, and delF230. Mutagenesis primers were ordered from Invitrogen (Breda, The Netherlands) without additional purification. Introduction of the mutation was confirmed by sequencing. Construction of pCIneo-hD3 and pcDNA3-rD1 (rat type 1 deiodinase) were described previously (2). A full-length image clone of human μ-crystallin (hCRYM) was obtained from RZPD GmbH (Berlin, Germany) and subcloned into pSG5 (Stratagene) using restriction sites EcoR1 and BamH1. All transfections were performed using 3 μl FuGene-6 transfection reagent (Roche Applied Science, Almere, The Netherlands) per 1000 ng plasmid DNA according to the manufacturer’s protocol. Transfection of empty pcDNA3 vector was used as control in all experiments. Possible differences in transfection efficiency between MCT8 mutants were studied in lysates of MCT8 and hD3 cotransfected JEG3 cells using D3 activity as control as described previously (12). No significant differences between mutants were observed (data not shown). Uptake and metabolism data are presented as mean ± se of four experiments without further corrections.
Table 1.
Mutations and clinical phenotype of MCT8 patients
| Protein | Mutation | No. of patients | Independent walking | Speech | Ref. | |
|---|---|---|---|---|---|---|
| 1 | V235M | 703G→A | 5 | − | − | 6 |
| 2 | L434W | 1301T→G | 9 | Ataxia, awkward gait | Dysarthric, limited | 6 |
| 3 | S448X | 1343C→A | 4 | − | − | 6 |
| 4 | L568P | 1703T→C | 28 | Ataxia, awkward gait | Dysarthric, limited | 6 |
| 5 | S194F | 581C→T | 10 | − | Dysarthric, limited | 6 |
| 6 | insI189 | 565insATC | 1 | − | − | 9 |
| 7 | delF230 | 683delTCT | 6 | − | − | 6 |
−, Absent.
Iodothyronines
Nonradioactive iodothyronines were obtained from Henning (Berlin, Germany) or Sigma Chemical Co. (St. Louis, MO). [3′-125I]T3 and [3′,5′-125I]T4 were obtained from GE Healthcare (Little Chalfont, Buckinghamshire, UK). Radioactive N-bromoacetyl-T3 (BrAc[125I]T3) was synthesized as described previously (29).
Cell culture
COS1 and JEG3 cells were cultured in six-, 12-, or 24-well dishes (Corning, Schiphol, The Netherlands) with DMEM/F12 medium (Invitrogen), containing 9% heat-inactivated fetal bovine serum (Invitrogen) and 100 nm sodium selenite (Sigma-Aldrich).
Iodothyronine transport experiments
COS1 and JEG3 cells were cultured in six-well culture dishes and transfected in duplicate with 500 ng wild-type or mutant pcDNA3-hMCT8 and 500 ng pSG5-hCRYM. hCRYM is a cytosolic thyroid hormone-binding protein. Addition of hCRYM reduces the efflux of T3, greatly increasing the net cellular T3 uptake. After 24 h (COS1) or 48 h (JEG3), cells were washed with assay buffer (Dulbecco’s PBS with Ca2+/Mg2+, 0.1% BSA, 0.1% glucose) and incubated for 30 min at 37 C with 1 nm (2 × 105 cpm) 125I-labeled T3 or T4 in 1.5 ml assay buffer. After incubation, cells were washed with assay buffer, lysed with 0.1 m NaOH, and counted.
Iodothyronine metabolism experiments
JEG3 cells were cultured in 24-well culture dishes (2 cm2) and transfected in duplicate with 100 ng pCIneo-hD3 and 100 ng wild-type or mutant pcDNA3-hMCT8. Two days after transfection, cells were washed with DMEM/F12 plus 0.1% BSA and incubated for 4 or 24 h at 37 C with 1 nm (1 × 106 cpm) [125I]T3 or [125I]T4, respectively, in 0.5 ml DMEM/F12 plus 0.1% BSA. After incubation, the medium was analyzed by HPLC as described previously (2).
Western blotting
Polyclonal antisera were raised in rabbits by Eurogentec SA (Seraing, Belgium) against the keyhole limpet hemocyanin conjugate of the synthetic peptide (C)ELLPGSPNPEEPI (hMCT8 C-terminal amino acid residues 527–539). Antiserum (designated 1306) from the final bleed was IgG purified.
JEG3 cells cultured in six-well plates were transfected with 500 ng wild-type or mutant pcDNA3-hMCT8. After 48 h incubation, the cells were rinsed with PBS, collected in 0.1 m phosphate/2 mm EDTA buffer (pH 7.2), and sonicated on ice. Thirty microliters of homogenate (diluted to 1 μg protein per μl) were mixed with 10 μl 4× loading buffer containing 10 mm dithiothreitol, and denatured at 80 C for 5 min. Samples were separated on 10% SDS-PAGE mini gels, blotted to nitrocellulose membranes, and probed with antiserum 1306 (1:1000) as described previously (30).
Affinity labeling of MCT8 with BrAcT3
COS1 cells in six-well plates were cotransfected with 1000 ng empty pcDNA3, wild-type or mutant pcDNA3-hMCT8, and 1000 ng pcDNA3-rD1. After 24 h, the cells were washed with serum-free DMEM/F12, incubated 4 h at 37 C with 5 × 105 cpm BrAc[125I]T3 and processed as described previously (2). The 75-μg protein samples were analyzed by SDS-PAGE; gels were blotted, and radioactivity on the blots was visualized by phosphor imaging (Typhoon; Amersham Biosciences, Roosendaal, The Netherlands). For statistical analyses, signals of four gels were quantified using ImageQuest software (Amersham).
Immunocytochemistry
JEG3 cells were cultured on 15-mm glass coverslips coated with poly-d-lysine (Sigma) and transfected with 50–100 ng wild-type or mutant pcDNA3-hMCT8. After 48 h, cells were fixed with 4% paraformaldehyde in PBS and permeabilized with 0.2% Triton X-100 in PBS. Samples were blocked for 30 min in PBS containing 2% BSA and stained with rabbit anti-hMCT8 antibody 1306 (1:1000) and monoclonal mouse anti-zona occludens protein 1 (ZO-1; tight junction protein 1) antibody (Invitrogen, 1:250). After secondary staining with goat antirabbit Alexa Fluor 488 and goat antimouse Alexa Fluor 633 (Invitrogen), coverslips were mounted with Prolong Gold containing 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Samples were examined on a Zeiss Axiovert 100 confocal microscope using Zeiss LSM software (Carl Zeiss BV, Sliedrecht, The Netherlands).
mRNA expression in transfected cells
JEG3 cells were cultured in six-well plates and transfected with 500 ng wild-type or mutant pcDNA3-hMCT8. After 48 h, cells were trypsinized, counted, and suspended at 5 × 106/ml. RNA was isolated using the High Pure RNA isolation kit (Roche); cDNA was generated by RT of 500 ng RNA using the TaqMan RT reagents kit (Applied Biosystems, Nieuwekerk a/d IJssel, The Netherlands). Quantitative PCR was performed on the ABI Prism 7700 sequence detection system (Applied Biosystems) using 2 μl cDNA, 12.5 μl TaqMan PCR MasterMix (Applied Biosystems), and a hMCT8-specific primer-probe mix containing forward primer 5′-CCATAACTCTGTCGGGATCCTC-3′ located in exon 1, reverse primer 5′-ACTCACAATGGGAGAACAGAAGAAG-3′ located in exon 2, and probe 5′-FAM-ATACCCATCGGAGGGCTCCGA-TAMRA-3′ located just upstream of the reverse primer. MCT8 mRNA levels were expressed relative to GAPDH mRNA levels, which were obtained using predeveloped human GAPDH TaqMan assay reagents (Applied Biosystems).
Statistical analyses
Comparisons between means were performed using Student’s t tests in the Statistical Package for the Social Sciences (SPSS, Chicago, IL) version 12.
Results
Uptake of iodothyronines in MCT8 and CRYM cotransfected cells
Figure 1A shows the uptake of T3 by JEG3 cells cotransfected with wild-type or mutant pcDNA3-hMCT8 and pSG5-hCRYM after 30 min. Control cells were cotransfected with empty pcDNA3 and pSG5-hCRYM. T3 uptake by control cells was 4.5% after 30 min. Transfection with wild-type hMCT8 increased T3 uptake 5.6-fold (P < 0.001). T3 uptake in cells transfected with the hMCT8 mutants V235M, S448X, insI189 and delF230 did not significantly differ from control. T3 uptake was increased 2.6-fold (P < 0.03) by mutant L434W, 1.8-fold (P = 0.08, NS) by mutant L568P and 1.6-fold (P = 0.12, NS) by mutant S194F. Similar results were obtained for T4 uptake in transfected JEG3 cells as well as for T3 and T4 uptake in transfected COS1 cells (data not shown).
Figure 1.

A, Uptake of T3 by JEG3 cells cotransfected with wild-type (WT) or mutant hMCT8 cDNA and pSG5-hCRYM, shown as percentage of added T3 after 30 min. Empty pcDNA3 plus pSG5-hCRYM served as control. B, Metabolism of T3 after 4 h in JEG3 cells cotransfected with wild-type or mutant hMCT8 and hD3-pCIneo, shown as percentage of T2 and T1 in the medium. Empty pcDNA3 plus hD3-pCIneo served as control. Uptake and metabolism data are presented as mean ± se of four experiments. *, P < 0.05 vs. control.
Metabolism of iodothyronines in MCT8 and D3 cotransfected cells
Figure 1B shows the metabolism of T3 in transfected JEG3 cells. Control cells transfected with hD3 cDNA plus vector without hMCT8 metabolized 13% of T3 after 4 h. Cotransfection of cells with hD3 and wild-type hMCT8 cDNA increased T3 metabolism 4.6-fold to about 60% in 4 h. T3 metabolism in cells cotransfected with hD3 and hMCT8 mutants V235M, S448X, insI189, and delF230 did not significantly differ from control. T3 metabolism was increased 2.6-fold (P < 0.01) by the L434W mutant, 2.2-fold (P < 0.01) by the L568P mutant, and 2.1-fold (P < 0.01) by the S194F mutant. Similar results were obtained after 24 h incubation of JEG3 cells with T4 (data not shown).
Western blotting
Immunoblotting of lysates from JEG3 cells transfected with wild-type or mutant hMCT8 is shown in Fig. 2A. All lanes were loaded with 30 μg protein. As reported before (2), no band of the expected size of hMCT8 (∼60 kDa) could be detected in JEG3 transfected with control plasmid. Transfection with wild-type hMCT8 results in a clear band of the expected size but also a higher band of about 240 kDa. After transfection with the V235M, L568P, insI189, and delF230 mutants, (much) weaker bands of the same sizes were detected, indicating that the expression of the mutant proteins is impaired but not absent. Mutants L434W and S194F are expressed at similar or somewhat higher levels than the wild-type protein. The truncated protein encoded by premature stop mutant S448X cannot be detected because of the loss of the epitope.
Figure 2.
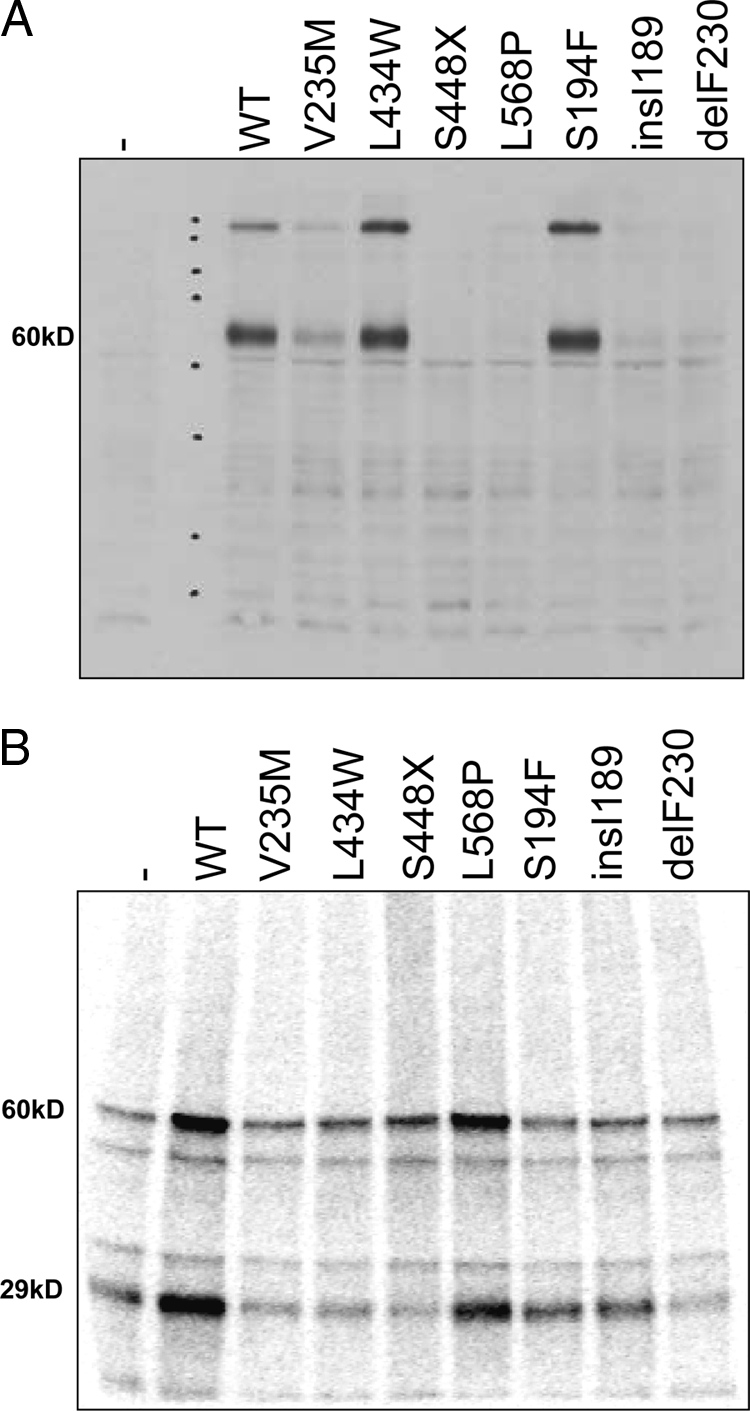
A, Immunoblot of JEG3 cells transfected with wild-type (WT) or mutant hMCT8. All lanes were loaded with 30 μg protein. Transfection with wild-type hMCT8 results in a band of the expected size (60 kDa) but also a higher band of about 240 kDa. Mutants L434W and S194F are expressed at similar or somewhat higher levels than the wild-type protein. V235M is intermediately expressed; faint bands are observed for L568P, insI189, and delF230. The truncated protein encoded by premature stop mutant S448X is not detectable with the antibody used. B, BrAc[125I]T3 affinity labeling of COS1 cells cotransfected with hMCT8 and D1. Transfection of wild-type hMCT8 and mutant L568P increase the labeling of the specific approximately 60-kDa band by 3.5- and 2.7-fold, respectively, vs. cells transfected with empty pcDNA3 (P < 0.05). Labeling of D1 (∼30 kDa) is increased 3.5-fold in cells transfected with wild-type MCT8 and 1.9-fold in L568P-transfected cells and reduced (50–70%) in cells transfected with V235M, L434W, S448X, and delF230 (all P < 0.05).
BrAc[125I]T3 affinity labeling
BrAc[125I]T3 is a specific affinity label for MCT8, which also transports the label, and for D1 (2). Figure 2B shows the autoradiograph of labeled proteins from COS1 cells cotransfected with pcDNA3, wild-type or mutant pcDNA3-hMCT8, and pcDNA3-rD1 after incubation for 4 h at 37 C with BrAc[125I]T3. Incubation of cells transfected with pcDNA3 and rD1 resulted in labeling of a protein of about 60 kDa, consistent with the size of MCT8, and of a protein of about 30 kDa, the size of D1. Two unknown proteins of about 50 and about 37 kDa are also labeled. Transfection with wild-type hMCT8 increases the intensity of the MCT8 and D1 bands 3.5-fold (P < 0.03). Transfection with mutant hMCT8 plasmids does not significantly increase the intensity of the MCT8 band compared with the control, except for mutant L568P. D1 labeling is at control level in cells transfected with S194F and insI189 and significantly reduced in cells transfected with V235M, L434W, S448X, and delF230 (P < 0.01). Mutant L568P shows a 2.7-fold increase of the MCT8 signal and a 1.9-fold increase of the D1 signal (P < 0.01), indicating a relatively high affinity for and transport of BrAcT3.
Immunocytochemistry
After fixation and permeabilization, wild-type or mutant hMCT8 transfected JEG3 cells were stained with hMCT8-specific antibody 1306. ZO-1 antibody was used as plasma membrane marker. For detection, hMCT8 antibody was stained with Alexa Fluor 488 (green) and ZO-1 antibody with Alexa Fluor 633 (red). Cells were mounted using Prolong Gold antifade reagents containing the DNA marker DAPI (blue). Figure 3 shows representative cells for all conditions. In cells transfected with control vector, no specific hMCT8 staining could be observed. In cells transfected with wild-type or mutant MCT8 cDNA, perinuclear MCT8 staining is detected, suggesting protein in the endoplasmic reticulum or Golgi apparatus. Wild-type hMCT8 also colocalizes clearly with the tight-junction marker ZO-1, indicating expression at the plasma membrane of transfected JEG3 cells. For hMCT8 mutants V235M, insI189, and delF230, expression of the protein appears to be predominantly cytoplasmic, whereas mutant S448X is not detected by our MCT8 antibody. Mutants L568P, L434W, and S194F, which are reported here to have residual transport capacity for T3 and T4, are expressed at the plasma membrane.
Figure 3.

Immunocytochemistry of transfected JEG3 cells. hMCT8-specific antibody 1306 is stained green, plasma membrane marker ZO-1 is stained red, and nuclear DNA is stained with DAPI (blue). Wild-type (WT) hMCT8 and mutants L568P, L434W, and S194F colocalize with the plasma membrane marker. Expression of mutants V235M, insI189, and delF230 is mostly limited to the cytoplasm.
Quantitative PCR
To investigate possible effects of the mutations on hMCT8 mRNA stability, we performed quantitative RT-PCR on mRNA isolated from JEG3 cells transfected with wild-type or mutant hMCT8. hMCT8 mRNA expression was corrected for the expression of the housekeeping gene GAPDH (Fig. 4). As reported previously, JEG3 cells do not endogenously express MCT8 mRNA (2). This is reflected in the absence of MCT8 signal in the cells transfected with control vector. No significant differences in mRNA expression were observed between wild-type MCT8 and the various mutants, indicating that loss of function of the mutants investigated here is not associated with reduced mRNA stability. Mutant delF230 mRNA could not be detected with the primer combination used for this RT-PCR, because the reverse primer contains the three deleted nucleotides.
Figure 4.

Quantitative RT-PCR of wild-type (WT) or mutant hMCT8 transfected JEG3 cells. No hMCT8 mRNA is detected in empty vector-transfected controls. No significant differences in expression between wild-type hMCT8 and the various mutants are observed. DelF230 mRNA could not be detected with the primers used in this assay.
Discussion
The first publication on MCT8 (SLC16A2) by Lafrenière et al. in 1994 (31) noted that the gene is located in a region associated with X-linked diseases. The function of MCT8, however, remained elusive until it was demonstrated that it codes for an active and specific thyroid hormone transporter (1). The important physiological role of MCT8 in the development of the CNS became apparent with the identification of patients with mutations in MCT8. These patients, all boys, exhibited elevated serum T3 levels and severe psychomotor retardation, confirming that mutations in MCT8 indeed were a cause of X-linked psychomotor retardation (3,4). In all initially reported patients, motor impairment is such that they are unable to hold up their head and cannot sit, crawl, stand, or walk. None of these patients developed any speech beyond making some sounds.
With the identification of MCT8 mutations in patients diagnosed with the Allan-Herndon-Dudley syndrome (6), it has become clear that mutations are associated with a considerable variety in phenotypical manifestations. Most striking is the ability of most patients with mutations L568P, the original family described by Allan, Herndon, and Dudley, and L434W to walk independently, with an ataxic, awkward gait. Development of walking is delayed, usually after 2 or 3 yr of age, and may be lost later in life. Patients with these mutations usually also show some elementary development of speech, although it is described as largely unintelligible (L568P) (28,32), and dysarthric and difficult to understand (L434W) (33). Patients with the L568P mutation are reported to be able to obey simple commands (28). In one other family, with the mutation S194F, most patients also develop some speech, but this is limited to some words or phrases (6). None of the patients in this family ever developed independent walking.
The apparent correlation between genotype and phenotype raised the question whether mutant proteins differ in functional characteristics regarding the uptake of T3 and/or T4. We recently demonstrated for several MCT8 mutations that transport of T3 and T4 is completely absent in vitro (12). One mutant protein, R271H, showed significant residual transport capacity of 10–20% of wild-type MCT8. The patient with this mutation, however, does not walk or talk or show any other signs indicating more advanced psychomotor development.
In the present study, we demonstrate that mutant proteins L568P, L434W, and S194F significantly increase uptake and subsequent metabolism of T3 and T4 compared with empty vector transfected controls, whereas for mutants V235M, S448X, insI189, and delF230, no significant difference was observed. Residual functionality of the mutants, most clearly demonstrated in the metabolism assays, ranged from 18–27% of wild-type activity for L568P, 35–40% for L434W, and 15–25% for S194F (Table 2).
Table 2.
Functional characteristics, protein expression, and affinity of WT and mutant MCT8
| Protein | T3 uptake (% of WT) | T3 metabolism (% of WT) | Western blot | BrAcT3 affinity labeling | Cellular localization | Ref. |
|---|---|---|---|---|---|---|
| WT MCT8 | 100% | 100% | +++ | +++ | Plasma membrane | 1 |
| V235M | NS | NS | ++ | − | Mostly cytoplasm | 6 |
| L434W | 40% | 35% | +++ | − | Plasma membrane | 6 |
| S448X | NS | NS | ND | − | ND | 6 |
| L568P | 18% | 28% | + | ++ | Plasma membrane | 6 |
| S194F | 15% | 25% | +++ | − | Plasma membrane | 6 |
| insI189 | NS | NS | − | − | Limited expression, mostly cytoplasm | 9 |
| delF230 | NS | NS | − | − | Limited expression, mostly cytoplasm | 6 |
| A224V | NS | NS | +++ | − | Mostly cytoplasm | 3,12 |
| L471P | NS | NS | + | − | Limited expression, mostly cytoplasm | 3,12 |
| R245X | NS | NS | ND | − | ND | 3,12 |
| R271H | 18% | 20% | +++ | − | Plasma membrane | 12 |
| del267-360 | NS | NS | − | − | Limited expression, mostly cytoplasm | 12 |
ND, Not detectable due to lack of epitope; NS, not significant compared with control; WT, wild type. For the Western blot data: −, no expression detected; +, low protein expression; ++, intermediate expression; and +++, high expression. For the BrAcT3 affinity labeling: −, no significant difference compared with cells transfected with empty pcDNA3; ++, intermediate increase of labeling compared with cells transfected with empty pcDNA3; and +++, high increase of labeling compared with cells transfected with empty pcDNA3.
Mutations in MCT8 could lead to a loss of function via several mechanisms, including reduced expression at the RNA or protein level, impaired trafficking to the plasma membrane, and reduced affinity for thyroid hormone. As we demonstrate in Fig. 4, quantitative RT-PCR did not show any significant differences in mRNA expression between wild-type and mutant MCT8. The expression of protein, demonstrated by immunoblotting in Fig. 2A, however, shows clear differences between the mutants. Mutant L434W and S194F proteins are expressed at levels comparable to wild-type MCT8, and V235M shows intermediate expression, whereas mutant insI189, delP230, L568P, and S448X proteins are expressed at much reduced levels or are not detected. Because mRNA expression does not differ between wild-type MCT8 and mutants, low expression of mutant proteins may result from fast degradation. Affinity labeling of MCT8 protein with BrAcT3 (Fig. 2B) is readily observed in COS1 cells transfected with wild-type MCT8 or with mutant L568P but not with other mutants. The significant residual affinity of L568P suggests that this mutant protein is present in higher concentrations than are detected by immunoblotting. Immunocytochemistry (Fig. 3) localizes wild-type MCT8 and mutants L434W, L568P, and S194F at the plasma membrane, whereas the mutants V235M, insI189, and delP230 are mostly located in the cytoplasm.
From these results it becomes apparent that complete loss of thyroid hormone transport function, as observed for mutants V235M, insI189, and delF230, results from decreased protein expression and/or impaired trafficking to the plasma membrane. These mutations are located in highly conserved loci in the first (insI189) and second (delF230 and V235M) transmembrane domain (TMD) and are all likely to affect the helical structures of these domains. For the protein encoded by mutant S448X, expression and localization could not be studied with our antibody. Complete loss of function is, however, expected from the premature stop, truncating the protein in TMD8.
Not surprisingly, all mutants showing significant residual transport capacity are expressed at the plasma membrane, the functional localization of MCT8. From immunoblotting, it appears that the L434W and S194F mutants are expressed at relatively high protein levels. Labeling with BrAcT3, however, did not show a marked increase of MCT8 signal, suggesting that the mutations lead to reduced substrate affinity. This was also observed for mutant R271H (12) (Table 2). The S194F mutation is located in the first extracellular loop, just outside TMD1, whereas mutation L434W is predicted just inside TMD8. R271H is located in the second extracellular loop, between TMDs 3 and 4. Of all mutant proteins studied so far, these three are the only ones showing preserved protein expression but reduced affinity. Although for some members of the MCT family, specific domains have been suggested to be involved in substrate recognition (for example TMDs 8 and 10 in MCT1) (34), little is known about which domains are involved in MCT8. From our findings, it appears that mutations throughout a large part of the protein can affect the affinity for (BrAc)T3 without resulting in increased breakdown of the protein. Whether the extracellular localization of these mutations plays a role in this remains to be elucidated.
Protein expression of mutant L568P, with about 25% residual functionality, appears very limited on immunoblotting. Nonetheless, affinity labeling shows the highest intensity of all mutants tested. The L568P mutation is located in TMD12; possibly, substitution of Leu with the rigid helix-breaking Pro residue influences protein expression, but has little effect on the affinity for (BrAc)T3. However, L568P is the mutation situated most closely to the epitope of antibody 1306, which might impair detection of the protein by immunoblotting.
The observed phenotypical characteristics of patients with mutations in MCT8 appear to be consistent in patients carrying the same mutation. Of 12 patients examined with mutation L568P, 11 walked, as did seven of eight patients carrying the L434W mutation. All four individuals with S194F that were examined developed some speech. It is important to realize, however, that all patients carrying a certain mutation come from one family. Although the residual transport of thyroid hormone we report here possibly contributes to the psychomotor development of these patients, other genetic or environmental factors are likely to contribute to this as well. Comparing patients with the same mutation from different families would provide valuable insight in the effects of residual MCT8 function. Until now, only mutation delF230, which we report here to lack transport capacity, has been reported in two unrelated families (6,12). Patients with this mutation have not been reported to walk independently or develop speech. In general, only patients with mutations showing significant residual transport capacity appear to reach these milestones of advanced psychomotor development.
In conclusion, significant residual uptake and subsequent metabolism of T3 and T4 was demonstrated for mutant MCT8 proteins L434W, L568P, and S194F in vitro. In vivo, these mutations are associated with more advanced psychomotor development than is observed in most MCT8 patients, especially with regard to walking and, to a lesser extent, talking. The functional characteristics of mutant proteins we describe here may underlie the genotype-phenotype relation observed in patients with mutations in MCT8.
Footnotes
J.J. is supported by the Sophia Foundation for Medical Research (Project No. 438). E.C.H.F. and M.H.A.K. are supported by The Netherlands Organization for Scientific Research (NWO Grants 916.36.139 and 916.56.186). Support was provided, in part, by a National Institute of Child Health and Human Development grant (HD26202) to C.E.S. and the South Carolina Department of Disabilities and Special Needs.
Disclosure Statement: All authors have nothing to disclose.
First Published Online January 10, 2008
Abbreviations: BrAcT3, N-Bromoacetyl-T3; CNS, central nervous system; D2, type 2 deiodinase; DAPI, 4′,6-diamidino-2-phenylindole; hMCT8, human MCT8; MCT8, monocarboxylate transporter 8; TMD, transmembrane domain; ZO-1, zona occludens protein 1.
References
- Friesema EC, Ganguly S, Abdalla A, Manning Fox JE, Halestrap AP, Visser TJ 2003 Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem 278:40128–40135 [DOI] [PubMed] [Google Scholar]
- Friesema EC, Kuiper GG, Jansen J, Visser TJ, Kester MH 2006 Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol Endocrinol 20:2761–2772 [DOI] [PubMed] [Google Scholar]
- Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, Barrett TG, Mancilla EE, Svensson J, Kester MH, Kuiper GG, Balkassmi S, Uitterlinden AG, Koehrle J, Rodien P, Halestrap AP, Visser TJ 2004 Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 364:1435–1437 [DOI] [PubMed] [Google Scholar]
- Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S 2004 A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet 74:168–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann K, Dumitrescu AM, Best TT, Hanefeld F, Refetoff S 2005 X-linked paroxysmal dyskinesia and severe global retardation caused by defective MCT8 gene. J Neurol 252:663–666 [DOI] [PubMed] [Google Scholar]
- Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, Ward J, Sanabria J, Marsa S, Lewis JA, Echeverri R, Lubs HA, Voeller K, Simensen RJ, Stevenson RE 2005 Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet 77:41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maranduba CM, Friesema EC, Kok F, Kester MH, Jansen J, Sertie AL, Passos Bueno MR, Visser TJ 2005 Decreased cellular T3 uptake and metabolism in Allan-Herndon-Dudley syndrome (AHDS) due to a novel mutation in the MCT8 thyroid hormone transporter. J Med Genet 43:457–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biebermann H, Ambrugger P, Tarnow P, von Moers A, Schweizer U, Grueters A 2005 Extended clinical phenotype, endocrine investigations and functional studies of a loss-of-function mutation A150V in the thyroid hormone specific transporter MCT8. Eur J Endocrinol 153:359–366 [DOI] [PubMed] [Google Scholar]
- Holden KR, Zuniga OF, May MM, Su H, Molinero MR, Rogers RC, Schwartz CE 2005 X-linked MCT8 gene mutations: characterization of the pediatric neurologic phenotype. J Child Neurol 20:852–857 [DOI] [PubMed] [Google Scholar]
- Kakinuma H, Itoh M, Takahashi H 2005 A novel mutation in the monocarboxylate transporter 8 gene in a boy with putamen lesions and low free T4 levels in cerebrospinal fluid. J Pediatr 147:552–554 [DOI] [PubMed] [Google Scholar]
- Herzovich V, Vaiani E, Marino R, Dratler G, Lazzati JM, Tilitzky S, Ramirez P, Iorcansky S, Rivarola MA, Belgorosky A 2007 Unexpected peripheral markers of thyroid function in a patient with a novel mutation of the MCT8 thyroid hormone transporter gene. Horm Res 67:1–6 [DOI] [PubMed] [Google Scholar]
- Jansen J, Friesema EC, Kester MH, Milici C, Reeser M, Gruters A, Barrett TG, Mancilla EE, Svensson J, Wemeau JL, Busi da Silva Canalli MH, Lundgren J, McEntagart ME, Hopper N, Arts WF, Visser TJ 2007 Functional analysis of monocarboxylate transporter 8 mutations identified in patients with X-linked psychomotor retardation and elevated serum triiodothyronine. J Clin Endocrinol Metab 92:2378–2381 [DOI] [PubMed] [Google Scholar]
- Gruters A, Krude H, Biebermann H 2004 Molecular genetic defects in congenital hypothyroidism. Eur J Endocrinol 151(Suppl 3):U39–U44 [DOI] [PubMed] [Google Scholar]
- Park SM, Chatterjee VK 2005 Genetics of congenital hypothyroidism. J Med Genet 42:379–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JT, Delange F 2001 Damaged reproduction: the most important consequence of iodine deficiency. J Clin Endocrinol Metab 86:2360–2363 [DOI] [PubMed] [Google Scholar]
- Bernal J 2005 Thyroid hormones and brain development. Vitam Horm 71:95–122 [DOI] [PubMed] [Google Scholar]
- Boyages SC, Halpern JP 1993 Endemic cretinism: toward a unifying hypothesis. Thyroid 3:59–69 [DOI] [PubMed] [Google Scholar]
- Pop VJ, Kuijpens JL, van Baar AL, Verkerk G, van Son MM, de Vijlder JJ, Vulsma T, Wiersinga WM, Drexhage HA, Vader HL 1999 Low maternal free thyroxine concentrations during early pregnancy are associated with impaired psychomotor development in infancy. Clin Endocrinol (Oxf) 50:149–155 [DOI] [PubMed] [Google Scholar]
- Haddow JE, Palomaki GE, Allan WC, Williams JR, Knight GJ, Gagnon J, O’Heir CE, Mitchell ML, Hermos RJ, Waisbren SE, Faix JD, Klein RZ 1999 Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl J Med 341:549–555 [DOI] [PubMed] [Google Scholar]
- Hennemann G, Docter R, Friesema EC, de Jong M, Krenning EP, Visser TJ 2001 Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr Rev 22:451–476 [DOI] [PubMed] [Google Scholar]
- Hagenbuch B 2007 Cellular entry of thyroid hormones by organic anion transporting polypeptides. Best Pract Res Clin Endocrinol Metab 21:209–221 [DOI] [PubMed] [Google Scholar]
- Visser WE, Friesema EC, Jansen J, Visser TJ 2007 Thyroid hormone transport by monocarboxylate transporters. Best Pract Res Clin Endocrinol Metab 21:223–236 [DOI] [PubMed] [Google Scholar]
- Taylor PM, Ritchie JW 2007 Tissue uptake of thyroid hormone by amino acid transporters. Best Pract Res Clin Endocrinol Metab 21:237–251 [DOI] [PubMed] [Google Scholar]
- Pizzagalli F, Hagenbuch B, Stieger B, Klenk U, Folkers G, Meier PJ 2002 Identification of a novel human organic anion transporting polypeptide as a high affinity thyroxine transporter. Mol Endocrinol 16:2283–2296 [DOI] [PubMed] [Google Scholar]
- Heuer H, Maier MK, Iden S, Mittag J, Friesema EC, Visser TJ, Bauer K 2005 The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology 146:1701–1706 [DOI] [PubMed] [Google Scholar]
- Courtin F, Zrouri H, Lamirand A, Li WW, Mercier G, Schumacher M, Goascogne CL, Pierre M 2005 Thyroid hormone deiodinases in the central and peripheral nervous system. Thyroid 15:931–942 [DOI] [PubMed] [Google Scholar]
- Friesema EC, Jansen J, Heuer H, Trajkovic M, Bauer K, Visser TJ 2006 Mechanisms of disease: psychomotor retardation and high T3 levels caused by mutations in monocarboxylate transporter 8. Nat Clin Pract Endocrinol Metab 2:512–523 [DOI] [PubMed] [Google Scholar]
- Allan W, Herndon C, Dudley F 1944 Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly. Am J Ment Defic 48:325–334 [Google Scholar]
- Mol JA, Docter R, Kaptein E, Jansen G, Hennemann G, Visser TJ 1984 Inactivation and affinity-labeling of rat liver iodothyronine deiodinase with N-bromoacetyl-3,3′,5-triiodothyronine. Biochem Biophys Res Commun 124:475–483 [DOI] [PubMed] [Google Scholar]
- Kuiper GG, Klootwijk W, Visser TJ 2003 Substitution of cysteine for selenocysteine in the catalytic center of type III iodothyronine deiodinase reduces catalytic efficiency and alters substrate preference. Endocrinology 144:2505–2513 [DOI] [PubMed] [Google Scholar]
- Lafreniere RG, Carrel L, Willard HF 1994 A novel transmembrane transporter encoded by the XPCT gene in Xq13.2. Hum Mol Genet 3:1133–1139 [DOI] [PubMed] [Google Scholar]
- Stevenson RE, Goodman HO, Schwartz CE, Simensen RJ, McLean Jr WT, Herndon CN 1990 Allan-Herndon syndrome. I. Clinical studies. Am J Hum Genet 47:446–453 [PMC free article] [PubMed] [Google Scholar]
- Bialer MG, Lawrence L, Stevenson RE, Silverberg G, Williams MK, Arena JF, Lubs HA, Schwartz CE 1992 Allan-Herndon-Dudley syndrome: clinical and linkage studies on a second family. Am J Med Genet 43:491–497 [DOI] [PubMed] [Google Scholar]
- Rahman B, Schneider HP, Broer A, Deitmer JW, Broer S 1999 Helix 8 and helix 10 are involved in substrate recognition in the rat monocarboxylate transporter MCT1. Biochemistry 38:11577–11584 [DOI] [PubMed] [Google Scholar]