

Abstract
In humans, insulin sensitivity is relatively impaired by diets that are low in oleic acid (OA), a cis monounsaturated fatty acid (MUFA), or rich in trans MUFA or palmitic acid (PA), a saturated fatty acid (FA). Emerging evidence exists that PA, in contrast to OA, causes insulin resistance via stimulation of inflammatory signaling and production of cytosolic lipid compounds (diacylglycerol and ceramide), leading one to presume that dietary or pharmacologic maneuvers that facilitate transport of FA into the mitochondria would be beneficial. However, in some models, insulin resistance is caused by excessive FA transport into the mitochondria, coupled with deficient electron transport and possibly increased reactive oxygen species formation; PA may impair electron transport via effects on gene expression. A research challenge is to determine whether feeding humans diets with markedly different contents of PA and OA would alter insulin sensitivity and/or critical biochemical mechanisms impacting muscle insulin signaling.
Introduction
Obesity and very high intakes of fat have been associated with a markedly increased risk of insulin resistance and the metabolic syndrome [1]. Accumulation of fat in nonadipocytes, such as myocytes, hepatocytes, cardiomyocytes, and pancreatic β cells, and the metabolism of fatty acids (FAs) have been linked to insulin resistance and the metabolic syndrome [1]. Thus, understanding how specific dietary FAs differentially alter insulin sensitivity in skeletal muscle and liver is important to our understanding of how to modify, via dietary interventions, the risk or severity of insulin resistance. Palmitic acid (16:0) (PA) is the principal saturated FA (SFA) in the diet and adipose tissue stores. Oleic acid (cis-9 18:1) (OA) is a monounsaturated FA (MUFA) and a major dietary and storage FA. A healthy diet must provide two “essential” polyunsaturated FAs (PUFAs)—linoleic acid (18:2 n-6 or omega 6) and α-linolenic acid (ALA) (18:3 n-3 or omega 3)—which are found in vegetable oils but cannot be synthesized by humans. Linoleic acid is the precursor of arachidonic acid (20:4 n-6), a biologically important FA, which is the precursor of several important prostaglandins. ALA is a precursor of the very long chain PUFAs contained in marine oils: eicosapentaenoic acid (22:5 n-3) and docosahexaenoic acid (22:6 n-3). Dietary trans MUFAs are produced via partial hydrogenation of PUFAs, in the rumen or via industrial processes. Trans MUFAs are derived from two sources: ruminant fat (including dairy products, lamb, and beef) (vaccenic acid, 18:1 trans-11) and partially hydrogenated oil (elaidic acid, 18:1 trans-9) [2]. Conjugated linoleic acid (CLA) is another form of trans FA [3].
Perhaps because of their ubiquity in human and animal diets, OA and especially PA have been extensively studied in experimental systems designed to study insulin resistance’s etiology. This article emphasizes recent human metabolic studies and studies of rodents and isolated cells germane to the issue of whether dietary PA and OA differentially alter the risk of insulin resistance.
Metabolic Pathways Governing FA Metabolism
Incubating muscle cells with PA decreases glucose uptake, whereas incubating these cells with unsaturated FA, such as palmitoleic acid, OA, linoleic acid, or ALA, increases glucose uptake [4,5]. Therefore, it is important to consider mechanisms for how FAs alter muscle glucose uptake. OA is synthesized from acetyl-coenzyme A (CoA), via formation of PA and stearic acid (18:0), thus assuring a certain degree of unsaturation of FA in cell membranes, which is required to maintain certain physical properties of the membrane (“plasticity”). The penultimate enzyme in this pathway, elongation of long-chain FAs family member 6 (Elovl6), catalyzes the elongation of PA to stearic acid. Genetic knockout of Elovl6 abrogated the development of diet-induced hepatic insulin resistance, otherwise observed in wild-type mice fed a high-fat/high-carbohydrate diet [6••]. The final step in this pathway is catalyzed by stearoyl-CoA desaturase (SCD1) (Fig. 1). Mice lacking normal SCD1 activity are protected from obesity [7], and obese people manifest abnormally high activities of this enzyme in skeletal muscle [8]. Absent or deficient SCD1 activity causes downregulation of acetyl-CoA carboxylase activity, resulting in lower production of malonyl-CoA. This is thought to relieve the inhibitory effects of this compound on carnitine palmitoyltransferase I (CPT-I), a rate-limiting enzyme for catalyzing the inward transport of FA across the inner mitochondrial membrane (permitting β oxidation) [1]. Thus, diminished endogenous formation of stearic acid and OA may prevent, respectively, insulin resistance in liver and skeletal muscle. In rats, markedly increasing the OA intake caused a 80% lower hepatic mRNA expression of SCD1 after 3 weeks [9], suggesting that chronically elevated OA intakes may downregulate its de novo synthesis.
Figure 1.
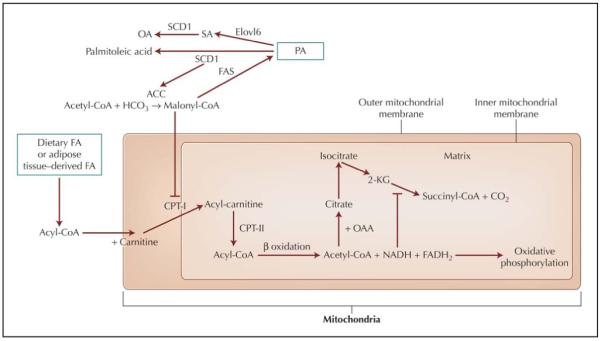
Mechanisms for fatty acid (FA) synthesis and oxidation. ACC—acetyl-coenzyme A carboxylase; CO2—carbon dioxide; CoA—coenzyme A; CPT—carnitine palmitoyltransferase; Elovl6—elongation of long-chain fatty acids family member 6; FADH2—flavin adenine dinucleotide [reduced form]; FAS—fatty acid synthase; 2-HCO3—bicarbonate; 2-KG—2-ketoglutaric acid; OA—oleic acid; OAA—oxaloacetic acid; PA—palmitic acid; SA—stearic acid; SCD1—stearoyl-CoA desaturase.
Acyl-CoA are regenerated within the mitochondrial matrix by CPT-II, where they enter the β oxidation sequence of chain shortening or are converted back to acylcarnitines and then exported from the mitochondria [1]. The “original” acyl-CoA cannot be reformed in the cytosol. β oxidation proceeds via a succession of FA chainshortening steps, which sequentially produce acetyl-CoA and generate reduced nucleotides (NADH, fl avin adenine dinucleotide [reduced form; [FADH2]).
Oxidative phosphorylation (electron transport activity) is required to oxidize NADH to NAD and FADH2 to FAD, and deficiency in this process leads to inhibition of the tricarboxylic acid cycle and, secondarily, β oxidation of FA. Excessive FA oxidation will lead to increased production of NADH and could challenge the oxidative phosphorylation capacity, leading to excessive generation of oxidants [10].
Normal Insulin Signaling
Insulin binding to the skeletal muscle cell induces autophosphorylation of the insulin receptor, a tyrosine kinase, on its tyrosine residues; this leads to tyrosine phosphorylation of insulin receptor substrate-1 (IRS-1) mainly in skeletal muscle (and IRS-2 in the liver) (Fig. 2). Tyrosine-phosphorylated IRS-1 then activates subsequent proteins, eventually leading to activation (by serine phosphorylation) of Akt/protein kinase B (Fig. 2). When activated by serine phosphorylation, Akt causes phosphorylation (inhibition) of AS160 (the Akt substrate of 160 kDa), a RabGAP protein, which has a GTPase (“GAP”) domain toward members of the Rab protein family. In their GTP-bound form, Rabs move vesicles containing the glucose receptor (GLUT4) to the membrane, allowing functional glucose transport. Thus, Akt, by inhibiting GTPase activity on Rabs, stimulates glucose transport. Using incubated muscle, isolated from obese humans but not lean humans, Thrush et al. [11] showed that PA inhibited serine phosphorylation of Akt and phosphorylation of AS160, but the authors presented no comparative OA data.
Figure 2.
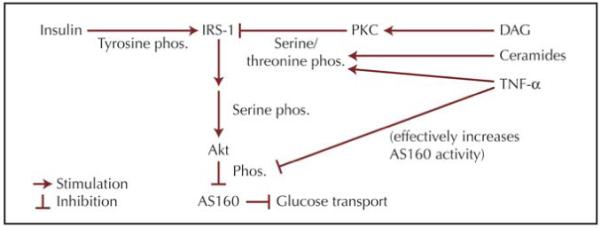
Insulin signaling in the skeletal muscle. DAG—diacylglycerol; IRS-1—insulin receptor substrate-1; phos.—phosphorylation; PKC-protein kinase C; TNF-α—tumor necrosis factor-α.
Relationship of Intramyocellular Lipid Content to Insulin Resistance
In humans, increased lipid accumulation within myocytes correlates with insulin resistance [12]. However, hepatic overexpression of malonyl-CoA decarboxylase in rats caused amelioration of insulin resistance produced by a high-fat diet without lowering muscle triacylglycerol (TAG) concentration [1]. Thus, there are reasons for considering if intramyocellular accumulation of specific FAs within complex lipids and the intramyocellular accumulation of specific lipids, other than TAG (eg, diacylglycerol [DAG] and ceramide), may account for the inverse correlation of intramyocellular lipid and insulin sensitivity [12].
Effects of Dietary FA Composition and FA Composition of Membrane Phospholipid on Skeletal Muscle Insulin Signaling and Sensitivity
One theory for how dietary FAs alter the risk of insulin resistance and type 2 diabetes posits that greater unsaturation of FAs in cell membrane contributes to certain physical properties, such as “plasticity,” which facilitate movement of the glucose receptor to the cell’s surface [13]. In humans, insulin sensitivity is correlated with the amount of docosahexaenoic acid in skeletal muscle phospholipid [14].
Diacylglycerol
DAG is an intermediate in the synthesis of TAG. Skeletal muscle insulin resistance caused by obesity or dietary FAs also might be related to defective mitochondrial oxidation of FAs, leading to the accumulation of DAG or ceramides (synthesized from PA), which in turn inhibit insulin signaling [12]. In contrast to IRS-1 activation via tyrosine phosphorylation, phosphorylation of the serine/threonine residues of IRS-1 impairs the IRS-1 activity [12]. Protein kinase C-τ (PKC-τ) is a serine/threonine kinase, which is activated by DAG or ceramides [15]. In humans, intravenous lipid infusion increased DAG and PKC-τ activity [16]. In cultured myocytes, a higher PA concentration (or lower OA concentration) enhances DAG accumulation and PKC-τ activation [17]. Rats fed a high SFA diet manifest higher muscle concentration of DAG compared with those fed a fat source primarily containing linoleic acid [5].
Ceramide
Palmitoyl-CoA is a precursor for ceramide formation. Ceramide, like DAG, antagonizes insulin signaling in muscle, and it also accumulates in the muscle of obese people [18]. Postulated mechanisms for how ceramides might alter insulin sensitivity have included effects on serine phosphorylation of IRS-1, inflammatory signaling, and apoptosis, but the most consistent effect on insulin signaling seems to be Akt inhibition [18]. Powell et al. [19] showed that PA, not palmitoleic acid, inhibited activation of Akt by a mechanism requiring ceramide synthesis and involving activation of PKC-ζ. Turpin et al. [20] showed that exposure of myocytes to PA increased ceramide content and apoptosis; insulin-stimulated glucose uptake was inhibited by PA.
Cardiolipin
Cardiolipin (diphosphatidylglycerol), a phospholipid containing four FAs, anchors cytochrome c it to the inner mitochondrial membrane [21]. Cytochrome c is dissociated from cardiolipin in the initiation of apoptosis [21]. Reactive oxygen species cause peroxidation of cardiolipin, enabling cytochrome c release from the inner mitochondrial membrane. PA caused a decrease in cardiolipin content of breast cancer cells and cardiomyocytes, apparently by increasing cardiolipin’s degradation rate [21]. In contrast, OA increased cardiolipin content and when coadministered with PA, prevented the decrease in cardiolipin and increased apoptosis caused by PA [21].
FA Effects on Gene Expression
The peroxisome proliferator activated receptors (PPARs) are transcription factors that regulate FA oxidation, adipogenesis, and inflammation, among other cellular processes [1]. Several PPAR family members exist, including PPAR-α, PPAR-γ1 and PPAR-γ2, and PPAR-γ [1]. PPAR-α is expressed predominately in the liver and in brown adipose tissue, the heart, and the kidney. PPAR-γ mRNA is expressed mostly in adipose tissue but also in skeletal muscle. PPAR-γ is expressed to a high degree and is the most abundant PPAR in skeletal muscle, but it is also expressed in the small intestine, colon, heart, vascular smooth muscle cells, and adipose tissue [1].
The binding of the PPAR to its response element depends on activation by ligands such as FA [1]. Activation of PPAR-α or PPAR-γ increases FA oxidation and alters skeletal muscle gene expression in favor of increased FA oxidation (increased malonyl-CoA decarboxylase, CPT-I, and medium-chain acyl-CoA dehydrogenase) (Fig. 1) [1]. PPAR-γ coactivator-1α (PGC-1α) may play a major role in the regulating mitochondrial FA oxidation and energy generation [1]. PGC-1α has been shown to bind and enhance the activity of PPAR-γ in muscle cells, causing increased expression of genes targeted by PPAR-γ [1]. In skeletal muscle, PGC-1α also stimulates mitochondrial biogenesis by coactivating the nuclear respiratory factor [1]. Moreover, abnormalities in the mRNA expression of PGC-1α in skeletal muscle have been implicated in the etiology of type 2 diabetes [1].
High SFA diets or PA downregulate mRNA expression of PGC-1α, possibly via activation of nuclear factor-κB (NF-κB), a transcription factor that regulates the mRNA expression of many cytokines [1,22,23•].
Inflammatory Signaling
Host organisms have evolved in the presence of immunostimulatory microbial products and recognize these molecules through toll-like receptors (TLRs) to modulate immunity against the intact microorganism [24]. Recently, interest has also focused on how TLRs mediate FA effects, such as PA, on skeletal muscle insulin signaling and inflammatory signaling through the NF-κB pathway [24,25].
NF-κB is a transcription factor that is important to the mediation of inflammatory signals. The NF-κB pathway also inhibits insulin signaling [16]. In the cytosol, NF-κB is complexed to the “inhibitor of κB” (IκB) [16]. Stimulation of TLRs and recruitment of MyD88, an adaptor molecule, phosphorylates cytoplasmic IκBα, causing it to be degraded. This leads to NF-κB activation [26]. More than 150 genes are regulated by NF-κB, including cytokines (eg, tumor necrosis factor-α, interleukin-6 [27-29]). Palmitate induces expression of tumor necrosis factor-α [28] and interleukin-6 [29] in muscle cells. Sinha et al. [30] showed that in myotubes PA or linoleic acid stimulated the degradation of IκB and the nuclear translocation of NF-κB (OA was not studied). Evidence now exists in macrophages that intact TLR4 expression is necessary for this PA effect [24]. Unsaturated FAs, such as OA, appear to inhibit the increased NF-κB signaling induced by SFA [25]. NF-κB may downregulate FA oxidation via binding and inhibition of the activity of PPAR-γ [31].
Skeletal muscle also is a target of inflammation induced in other tissues by PA [32]. Adipose tissue is composed of adipocytes and stromal tissue; the latter contains macrophages [32]. Both cells produce cytokines, but not all macrophages present in adipose tissue are inflammatory, with some macrophages exhibiting a so-called alternative, less inflammatory component, and the absence of alternatively activated macrophages in adipose tissue leads to mitochondrial dysfunction and insulin resistance in some genetically altered (knockout) mice [33]. OA may potentiate alternative macrophage activation [33]. Thus, it is conceivable that PA and OA have opposite effects in terms of inducing inflammatory pathways and thus insulin resistance.
FA Oxidation: Differential Effects of Dietary FAs
One might hypothesize that FAs such as PA may play an especially important role in the etiology of insulin resistance if the flux of FAs is great (eg, obesity, high intake of specific FA) and if the FAs are not “cleared” from cytosolic pools by oxidation. The literature suggests that OA and other unsaturated FAs may be more readily oxidized than SFAs [34]. A few studies in humans have addressed differential effects of dietary FA on total FA oxidation. Jones and Schoeller [35] showed that an increased PUFA intake for 7 days caused a 40% increase in fat oxidation in the fed state and a decreased (25%) basal fat oxidation. We reported that compared with a low PA/high OA diet, a high PA diet caused a decrease in the rate of fat oxidation, whereas this rate increased in those fed the high OA diet [34]. We found that relative to baseline, the rate of FA oxidation (percent of resting energy expenditure) increased in females on the high OA diet while decreasing on the high PA diet in the fed and fasted states, but changes in males were not statistically significant [36]. The gender difference seemed to relate mainly to an increased rate of FA oxidation during the high OA diet in females compared with a decrease in men; the high PA diet did not seem to affect the genders differentially [36].
Increased Rate of FA Entry Into the Mitochondria May Decrease Insulin Sensitivity in Muscle
Literature evidence from some models showed that increased (not decreased) FA entry into the mitochondria also may cause insulin resistance [1,37]. Insulin resistance in skeletal muscle is not consistently explained by intramyocellular lipid accumulation (eg, acyl-CoA or DAG) [1], by defective inward mitochondrial transport of FA (low CPT-I activity), or by defective mitochondrial β oxidation of the first two to four FA carbons (initial chain shortening) [1,38]. Koves et al. [37] found that depriving cultured myocytes of carnitine prevented FA from reducing Akt activation. In vivo, inhibiting CPT-I via overproduction of malonyl-CoA protected mice from the adverse effects of a high-fat diet on insulin sensitivity [37]. Although accumulation of tricarboxylic acid cycle intermediates and chain-shortened acylcarnitines reflect defective mitochondrial function, it is not clear how these compounds inhibit insulin signaling [1], but very high rates of FA oxidation also could stress the respiratory chain cycle via excessive NADH production, if electron transport activity is not sufficient. This in turn could lead to increased generation of reactive oxygen species, which might have adverse effects on insulin-signaling proteins [10,39]. In line with this theory, carnitine administration improves insulin sensitivity in healthy volunteers and type 2 diabetic patients, presumably by exporting acyl-CoAs from the mitochondria as acyl-carnitines and by preventing excessive generation of reduced nucleotides via β oxidation [1].
Defective mitochondrial function would seem to be a root cause of insulin resistance, independent of whether there is defective FA entry into the mitochondria (defective oxidation or initial chain shortening) or excessive entry into the mitochondria with accompanying incomplete FA oxidation. However, Turner et al. [38] showed that feeding mice a high-fat, lard-based diet (high OA/high PA) impaired glucose tolerance, increased degradation of the first two PA carbons (with no evidence of incomplete PA oxidation), increased activity of β oxidation enzymes, and increased protein expression of PGC-1α and various components of the respiratory chain. Also, feeding rats a high-fat diet for 2 to 5 weeks increased PA oxidation, increased expression of mitochondrial proteins (including PGC-1α), and mitochondrial DNA copy number [40••]. These studies of rodents and isolated cells present a somewhat confusing and even contradictory picture of how mitochondrial function is affected by FAs. However, as pointed out by Hancock et al. [40••], exercise may play a role in the expression of proteins affecting mitochondrial function (eg, PGC-1α [1]), and perhaps these extraordinarily high-fat diets that are used in rodents to induce obesity and insulin resistance may not reflect completely what happens in obese humans.
Dietary FA Type May Alter the Risk for the Metabolic Syndrome and Type 2 Diabetes
Epidemiologic studies suggest that high SFA intakes increase the risk for type 2 diabetes. However, because OA intake is derived mostly from animal products and not olive oil in populations living outside the Mediterranean areas, it is difficult to separate the effects of SFA (PA) from MUFA (OA) [13,41]. Men with diabetes manifest higher intakes of total fat, SFA, and MUFA than those with normal glucose tolerance and also higher intakes of PUFA than those nondiabetic men with impaired glucose tolerance [41]. A protective effect of MUFA intake on the development of insulin resistance was observed in one study from Italy, in which the MUFA intake was mainly derived from olive oil, not animal products [42]. Salmeron et al. [13] found that total fat, SFA, and MUFA intake were not associated with the risk of diabetes, but PUFA intake reduced and trans fat intake increased the risk of diabetes. They concluded that the risk of type 2 diabetes could be reduced by 40% if the intake of trans FA could be minimized.
Vessby [43] and Riserus [44] have recently reviewed human data on the effects of dietary FA composition on insulin sensitivity. Some small trials involving comparisons of different diet FA compositions on insulin sensitivity did not show any significant effects [43,44]. However, Vessby et al. [45] randomized 162 healthy, nondiabetic men and women, 30 to 65 years of age, body mass index 22 to 32 kg/m2, to a high SFA diet (SFA: 17% kcal; MUFA: 14% kcal; PUFA: 6% kcal) or a high MUFA diet (8%, 23%, and 6%). The diets were prescribed and not strictly controlled. Insulin sensitivity, measured using an intravenous glucose tolerance test, decreased during the high SFA diet but did not change on the high MUFA diet (compared with a baseline test on the patients’ habitual diets). Median fat intake was reported as equal to that prescribed by the protocol: 37% kcal for all patients. In those ingesting less than the median fat intake, the high MUFA diet increased insulin sensitivity and the high SFA diet lowered it [45]. Interestingly, in those ingesting a higher fat intake (mean, 40.2% kcal), the 11.1% difference between the diets was not statistically significant, and insulin sensitivity decreased in both. To what extent those ingesting more than the prescribed fat intake also deviated from the prescribed dietary FA composition cannot be determined from this study. Perez-Jimenez et al. [46] studied 59 young, lean men and women. The patients were evaluated after a control, high SFA diet for 4 weeks and then again after two 4-week crossover limbs involving a high MUFA diet or a low-fat/high-carbohydrate diet. Relative to the baseline (a high SFA diet), the latter two diets seemed to improve insulin sensitivity based on a glucose suppression test [46].
Although this article primarily deals with the effects of dietary FA on insulin sensitivity and the metabolic syndrome, high OA diets may have efficacy with respect to glucose tolerance in patients with type 2 diabetes. Based on a meta-analysis of studies investigating the effects of high MUFA diets in patients with diabetes, MUFA-enriched diets tend to lower blood glucose and 24-hour insulin concentration and improve insulin sensitivity, compared with high-carbohydrate diets [47].
A few studies have addressed specifically the effects of trans MUFA in the diet on insulin sensitivity. Based on studies in rats and myocytes, Tardy et al. [2] concluded that elaidic acid or vaccenic acid, when substituted at a dietary level of 4% kcal for OA, did not alter insulin sensitivity in vivo or insulin signaling (Akt phosphorylation) in vitro. Christiansen et al. [48] compared a high cis MUFA diet with a high SFA diet and with a high trans MUFA diet in obese, diabetic men and women in a crossover design. Compared with the cis MUFA diet, the SFA and trans MUFA diets caused a higher postprandial serum insulin and C-peptide response, indicative of decreased peripheral insulin sensitivity and/or increased insulin secretion [48].
Conjugated linoleic acid
Dietary CLA also is derived from ingested ruminant meats and dairy products. CLA consists of various isomers, including CLA cis-9, trans-11, CLA t10, c12, and CLA c10 t12; the cis-9, trans-11 isomer is the most prevalent isomer of CLA in the human diet [3]. The effects of various CLA isomers on body composition or insulin sensitivity are unclear and seem to be dependent on the species studied, the adiposity and/or physical activity of the humans, and the isomer of CLA used in a particular study, but CLA may diminish insulin sensitivity in humans [3].
Hepatic Insulin Sensitivity and Steatosis
Observational studies suggest that high SFA intake is associated with nonalcoholic fatty liver disease [49]. Musso et al. [49] evaluated dietary habits using a diet record and compared individuals with nonalcoholic steatohepatitis and healthy controls. The patients with nonalcoholic steatohepatitis reported higher SFA intakes (13.7% kcal) and lower PUFA intakes (10.0% of FA) compared with the controls (10.0% and 14.5%, respectively) [49]. Clore et al. [50] showed that an enteral infusion of palm oil (high in PA and OA) induced relative hepatic insulin resistance compared with a safflower oil infusion containing less PA and OA and more linoleic acid.
Conclusions
The two most common FAs in the diet and in tissue stores are PA (an SFA) and OA (a MUFA). Perhaps because in western diets SFA and MUFA often derive from the same foods, some epidemiologic studies have not detected a differential effect of these two dietary FA groups on the risk of insulin resistance and type 2 diabetes, but some studies suggest that decreasing PA intake or increasing OA intake may be beneficial [42,45]. However, lowering trans MUFA intake also is beneficial [13]. A burgeoning basic science literature exists demonstrating potential detrimental PA effects on biochemical mechanisms impacting insulin signaling in skeletal muscle and suggesting that OA may have opposite effects. Less information exists about PUFA, but there are both negative and positive effects of n-6 or n-3 PUFA on biochemical mechanisms regulating insulin sensitivity in skeletal muscle. The attractive feature of studies in isolated cells is the ability to precisely fix the conditions of the experiment, including the concentration of individual FA, but how reliably these studies mimic the human condition is unclear. Thus, in vivo studies are needed to determine whether dietary FA-induced changes in intramyocellular FA composition, inflammatory pathways, and FA metabolism alter biochemical mechanisms, which in aggregate and over a period of time (perhaps months or years) may alter insulin sensitivity.
Acknowledgments
Disclosures
Efforts on this paper were supported, in part, by National Institutes of Health (NIH) Grant No. NIDDK RO1 073284, and research carried out by the author and described in this paper was supported, in part, by NIH Grant R01 DK55384, NIH grants to the General Clinical Research Centers at the University of Texas Medical Branch at Galveston, the Ohio State University, and the University of Vermont (respectively, M01 RR 00073, M01 RR 00034, and M01 RR00109), by the Shriners Hospital for Children, Galveston, TX (SHC Grant No. #8760), and by Ross Products Division of Abbott Laboratories, which provided experimental formula used in these studies.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Muoio DM, Newgard CB. Obesity-related derangements in metabolic regulation. Annu Rev Biochem. 2006;75:367–401. doi: 10.1146/annurev.biochem.75.103004.142512. [DOI] [PubMed] [Google Scholar]
- 2.Tardy AL, Giraudet C, Rousset P, et al. Effects of trans MUFA from dairy and industrial sources on muscle mitochondrial function and insulin sensitivity. J Lipid Res. 2008;49:1445–1455. doi: 10.1194/jlr.M700561-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Riserus U, Arner P, Brismar K, Vessby B. Treatment with dietary trans10cis12 conjugated linoleic acid causes isomerspecific insulin resistance in obese men with the metabolic syndrome. Diabetes Care. 2002;25:1516–1521. doi: 10.2337/diacare.25.9.1516. [DOI] [PubMed] [Google Scholar]
- 4.Dimopoulos N, Watson M, Sakamoto K, Hundal HS. Differential effects of palmitate and palmitoleate on insulin action and glucose utilization in rat L6 skeletal muscle cells. Biochem J. 2006;399:473–481. doi: 10.1042/BJ20060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee JS, Pinnamaneni SK, Eo SJ, et al. Saturated, but not n-6 polyunsaturated, fatty acids induce insulin resistance: role of intramuscular accumulation of lipid metabolites. J Appl Physiol. 2006;100:1467–1474. doi: 10.1152/japplphysiol.01438.2005. [DOI] [PubMed] [Google Scholar]
- 6••.Matsuzaka T, Shimano H, Yahagi N, et al. Crucial role of a long-chain fatty acid elongase, Elovl6, in obesity-induced insulin resistance. Nat Med. 2007;13:1193–1202. doi: 10.1038/nm1662.This paper addresses how obesity and dietary FAs may cause hepatic insulin resistance and suggests that the elongation of PA to stearic acid may play a crucial role in this process.
- 7.Cohen P, Friedman JM. Leptin and the control of metabolism: role for stearoyl-CoA desaturase-1 (SCD-1) J Nutr. 2004;134:2455s–2463s. doi: 10.1093/jn/134.9.2455S. [DOI] [PubMed] [Google Scholar]
- 8.Hulver MW, Berggren JR, Carper MJ, et al. Elevated stearoyl-CoA desaturase-1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metab. 2005;2:251–261. doi: 10.1016/j.cmet.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kakuma T, Lee Y, Unger RH. Effects of leptin, troglitazone, and dietary fat on stearoyl CoA desaturase. Biochem Biophys Res Commun. 2002;297:1259–1263. doi: 10.1016/s0006-291x(02)02375-6. [DOI] [PubMed] [Google Scholar]
- 10.Anderson EJ, Yamazaki H, Neufer PD. Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid-supported respiration. J Biol Chem. 2007;282:31257–31266. doi: 10.1074/jbc.M706129200. [DOI] [PubMed] [Google Scholar]
- 11.Thrush AB, Heigenhauser GJ, Mullen KL, et al. Palmitate acutely induces insulin resistance in isolated muscle from obese but not lean humans. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1205–R1212. doi: 10.1152/ajpregu.00909.2007. [DOI] [PubMed] [Google Scholar]
- 12.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salmeron J, Hu FB, Manson JE, et al. Dietary fat intake and risk of type 2 diabetes in women. Am J Clin Nutr. 2001;73:1019–1026. doi: 10.1093/ajcn/73.6.1019. [DOI] [PubMed] [Google Scholar]
- 14.Baur LA, O’Connor J, Pan DA, et al. The fatty acid composition of skeletal muscle membrane phospholipid: its relationship with the type of feeding and plasma glucose levels in young children. Metabolism. 1998;47:106–112. doi: 10.1016/s0026-0495(98)90202-5. [DOI] [PubMed] [Google Scholar]
- 15.Bollag GE, Roth RA, Beaudoin J, et al. Protein kinase C directly phosphorylates the insulin receptor in vitro and reduces its protein-tyrosine kinase activity. Proc Natl Acad Sci U S A. 1986;83:5822–5824. doi: 10.1073/pnas.83.16.5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 17.Chavez JA, Summers SA. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch Biochem Biophys. 2003;419:101–109. doi: 10.1016/j.abb.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 18.Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45:42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 19.Powell DJ, Turban S, Gray A, et al. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem J. 2004;382:619–629. doi: 10.1042/BJ20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turpin SM, Lancaster GI, Darby I, et al. Apoptosis in skeletal muscle myotubes is induced by ceramides and is positively related to insulin resistance. Am J Physiol Endocrinol Metab. 2006;291:E1341–E1350. doi: 10.1152/ajpendo.00095.2006. [DOI] [PubMed] [Google Scholar]
- 21.Hardy S, El Assaad W, Przybytkowski E, et al. Saturated fatty acid-induced apoptosis in MDA-MB-231 breast cancer cells. A role for cardiolipin. J Biol Chem. 2003;278:31861–31870. doi: 10.1074/jbc.M300190200. [DOI] [PubMed] [Google Scholar]
- 22.Koves TR, Li P, An J, et al. PPARgamma coactivator-1alpha -mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J Biol Chem. 2005;280:33588–33598. doi: 10.1074/jbc.M507621200. [DOI] [PubMed] [Google Scholar]
- 23•.Coll T, Jove M, Rodriguez-Calvo R, et al. Palmitate-mediated downregulation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha in skeletal muscle cells involves MEK1/2 and nuclear factor-kappaB activation. Diabetes. 2006;55:2779–2787. doi: 10.2337/db05-1494.This paper links the palmitate-mediated downregulation of PGC-1α to activation of NF-κB (inflammatory) signaling.
- 24.Shi H, Kokoeva MV, Inouye K, et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276:16683–16689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 27.Ajuwon KM, Spurlock ME. Palmitate activates the NF-kappaB transcription factor and induces IL-6 and TNFalpha expression in 3T3-L1 adipocytes. J Nutr. 2005;135:1841–1846. doi: 10.1093/jn/135.8.1841. [DOI] [PubMed] [Google Scholar]
- 28.Jove M, Planavila A, Sanchez RM, et al. Palmitate induces tumor necrosis factor-alpha expression in C2C12 skeletal muscle cells by a mechanism involving protein kinase C and nuclear factor-kappaB activation. Endocrinology. 2006;147:552–561. doi: 10.1210/en.2005-0440. [DOI] [PubMed] [Google Scholar]
- 29.Jove M, Planavila A, Laguna JC, Vazquez-Carrera M. Palmitate-induced interleukin 6 production is mediated by protein kinase C and nuclear-factor kappaB activation and leads to glucose transporter 4 down-regulation in skeletal muscle cells. Endocrinology. 2005;146:3087–3095. doi: 10.1210/en.2004-1560. [DOI] [PubMed] [Google Scholar]
- 30.Sinha S, Perdomo G, Brown NF, O’Doherty RM. Fatty acid-induced insulin resistance in L6 myotubes is prevented by inhibition of activation and nuclear localization of nuclear factor kappa B. J Biol Chem. 2004;279:41294–41301. doi: 10.1074/jbc.M406514200. [DOI] [PubMed] [Google Scholar]
- 31.Jove M, Laguna JC, Vazquez-Carrera M. Agonist-induced activation releases peroxisome proliferator-activated receptor beta/delta from its inhibition by palmitate-induced nuclear factor-kappaB in skeletal muscle cells. Biochim Biophys Acta. 2005;1734:52–61. doi: 10.1016/j.bbalip.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 32.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Odegaard JI, Ricardo-Gonzalez RR, Red EA, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7:496–507. doi: 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kien CL, Bunn JY, Ugrasbul F. Increasing dietary palmitic acid decreases fat oxidation and daily energy expenditure. Am J Clin Nutr. 2005;82:320–326. doi: 10.1093/ajcn.82.2.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones PJH, Schoeller DA. Polyunsaturated:saturated ratio of diet fat influences energy substrate utilization in the human. Metabolism. 1988;37:145–151. doi: 10.1016/s0026-0495(98)90009-9. [DOI] [PubMed] [Google Scholar]
- 36.Kien CL, Bunn JY. Gender alters the effects of palmitate and oleate on fat oxidation and energy expenditure. Obesity. 2008;16:29–33. doi: 10.1038/oby.2007.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koves TR, Ussher JR, Noland RC, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 38.Turner N, Bruce CR, Beale SM, et al. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes. 2007;56:2085–2092. doi: 10.2337/db07-0093. [DOI] [PubMed] [Google Scholar]
- 39.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 40••.Hancock CR, Han DH, Chen M, et al. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci U S A. 2008;105:7815–7820. doi: 10.1073/pnas.0802057105.A considerable body of literature (Muoio and Newgard [1], and Shulman [12]) suggests that defective mitochondrial function links FAs to insulin resistance, but this paper suggests that feeding rats a high-fat diet for 4 weeks increases mitochondrial protein while also causing decreased insulin sensitivity. Interestingly, the diet caused an increase in PGC-1α protein but not PGC-1α mRNA expression (which actually decreased transiently after 1 week).
- 41.Feskens EJ, Virtanen SM, Rasanen L, et al. Dietary factors determining diabetes and impaired glucose tolerance. A 20-year follow-up of the Finnish and Dutch cohorts of the Seven Countries Study. Diabetes Care. 1995;18:1104–1112. doi: 10.2337/diacare.18.8.1104. [DOI] [PubMed] [Google Scholar]
- 42.Hu FB, van Dam RM, Liu S. Diet and risk of type 2 diabetes: the role of types of fat and carbohydrate. Diabetologia. 2001;44:805–817. doi: 10.1007/s001250100547. [DOI] [PubMed] [Google Scholar]
- 43.Vessby B. Dietary fat and insulin action in humans. Br J Nutr. 2000;83(Suppl 1):S91–S96. doi: 10.1017/s000711450000101x. [DOI] [PubMed] [Google Scholar]
- 44.Riserus U. Fatty acids and insulin sensitivity. Curr Opin Clin Nutr Metab Care. 2008;11:100–105. doi: 10.1097/MCO.0b013e3282f52708. [DOI] [PubMed] [Google Scholar]
- 45.Vessby B, Unsitupa M, Hermansen K, et al. Substituting dietary saturated for monounsaturated fat impairs insulin sensitivity in healthy men and women: the KANWU study. Diabetologia. 2001;44:312–319. doi: 10.1007/s001250051620. [DOI] [PubMed] [Google Scholar]
- 46.Perez-Jimenez F, Lopez-Miranda J, Pinillos MD, et al. A Mediterranean and a high-carbohydrate diet improve glucose metabolism in healthy young persons. Diabetologia. 2001;44:2038–2043. doi: 10.1007/s001250100009. [DOI] [PubMed] [Google Scholar]
- 47.Garg A. High-monounsaturated-fat diets for patients with diabetes mellitus: a meta-analysis. Am J Clin Nutr. 1998;67(3 Suppl):577S–582S. doi: 10.1093/ajcn/67.3.577S. [DOI] [PubMed] [Google Scholar]
- 48.Christiansen E, Schnider S, Palmvig B, et al. Intake of a diet high in trans monounsaturated fatty acids or saturated fatty acids. Effects on postprandial insulinemia and glycemia in obese patients with NIDDM. Diabetes Care. 1997;20:881–887. doi: 10.2337/diacare.20.5.881. [DOI] [PubMed] [Google Scholar]
- 49.Musso G, Gambino R, De Michieli F, et al. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology. 2003;37:909–916. doi: 10.1053/jhep.2003.50132. [DOI] [PubMed] [Google Scholar]
- 50.Clore JN, Stillman JS, Li J, et al. Differential effect of saturated and polyunsaturated fatty acids on hepatic glucose metabolism in humans. Am J Physiol Endocrinol Metab. 2004;287:E358–E365. doi: 10.1152/ajpendo.00360.2003. [DOI] [PubMed] [Google Scholar]