

Abstract
Glutamate racemase (RacE) is a bacterial enzyme that converts L-glutamate to D-glutamate, an essential precursor for peptidoglycan synthesis. In prior work, we have shown that both isoforms co-crystallize with D-glutamate as dimers, and the enzyme is in a closed conformation with limited access to the active site [May et al., (2007) J. Mol. Biol. 371, 1219–1237]. The active site of RacE2 is especially restricted. We now utilize several computational and experimental approaches to understand the overall conformational dynamics involved during catalysis when the ligand enters and product exits the active site. Our steered molecular dynamics simulations and normal mode analysis results indicate that the monomeric form of the enzyme is more flexible than the native dimeric form. These results suggest that the monomeric enzyme might be more active than the dimeric form. We thus generated site-specific mutations that disrupt dimerization, and find that they exhibit significantly higher catalytic rates in the D-Glu to L-Glu reaction direction than the native enzyme. Low resolution models restored from solution X-ray scattering studies correlate well with the first six normal modes of the dimeric form of the enzyme, obtained from NMA. Thus, along with the local active site residues, global domain motions appear to be implicated in the catalytically relevant structural dynamics of this enzyme, and suggest that increased flexibility may accelerate catalysis. This is a novel observation that residues distant from the catalytic site restrain catalytic activity through formation of the dimer structure.
Keywords: Bacillus anthracis, glutamate racemase, disruption of dimerization, normal modes, x-ray solution scattering, global domain motions, catalytically relevant enzyme structural dynamics
Glutamate racemase (RacE) is a bacterial enzyme that converts L-glutamate (L-Glu) to D-glutamate (D-Glu). Maintenance of appropriate L/D-Glu levels is vital for the normal functioning of bacterial cells. L-Glu is the most abundant intracellular amino acid (1) and is found in concentrations of up to 20mM in bacterial cells (2). L-Glu is utilized by many different cellular pathways, while D-Glu is a critical component of the peptidoglycan layer. Glutamate racemase appears to be the primary source of D-Glu for bacterial cell wall biosynthesis and has been found to be an essential enzyme for the survival of many pathogenic species of bacteria (3). It is potentially an attractive target for the development of new antibacterial agents because in addition to being an essential enzyme, it has no known human homolog.
Bacillus anthracis expresses two glutamate racemase isoforms, RacE1 and RacE2. We have characterized both isoforms, and have reported the crystal structures of these enzymes in complex with D-Glu (4). RacE1 is monomeric in the absence of glutamate but dimerizes in its presence (4). RacE2, on the other hand, is a dimer irrespective of the presence of glutamate (4). RacE1 and RacE2 have similar crystal structures when bound to D-Glu; both are dimers and have their active sites on the face opposite to the dimer interface. Each monomer of this enzyme consists of two domains. The active site is located at the interface between the two domains with each domain contributing residues involved in catalysis (Figure 1A). The α-amine group and α-carboxylate group of D-Glu are stabilized within the catalytic site by hydrogen-bonding with various hydrophilic residues surrounding the active site cysteines. The γ-carboxylate is stabilized by hydrogen bonding with residues located on a highly conserved loop near the entrance to the active site while the two methylene groups in the side chain of D-Glu pack against the hydrophobic residues that are present in the tunnel to the active site (Figure 1B). As the substrate for this enzyme is a single amino acid (glutamate) the active site is rather small (as seen in the crystal structure) and needs to open for the glutamate to enter or exit the active site. To date there has been no success in obtaining crystals of the apo enzyme, hence it is not clear how the substrate and product respectively enter and exit the active site. Thus, the conformational changes and conformational dynamics taking place during catalysis are unknown.
Figure 1.

(A) Crystal structure of RacE2. The two domains in each monomer in the dimer are shaded in pink and beige. Domain I (pink) consists of residues 1–95, and 208–270, which form five α-helices encircling a six-stranded parallel β-sheet. Domain II (beige) includes residues 96–207 and is comprised of six α-helices surrounding a four stranded parallel β-sheet. The active site is on the face opposite the dimer interface and is indicated by the presence of D-Glu (red). This figure was generated by PyMOL. (B) Orientation of D-Glu in the active site of RacE2 as seen in the crystal structure, with the α-groups centered in the active site, and the side chain pointing away from the catalytic residues.
To better understand the overall dynamics and functional properties of RacE2 we used a combination of steered molecular dynamics (SMD) simulations to follow the release of D-Glu from the active site of the enzyme and normal mode analysis (NMA) to determine large amplitude highly correlated movements. SMD simulation method has been widely used to explore the binding and unbinding properties of biomolecules and their responses to external mechanical manipulations at the atomic level. It has been successfully applied to identify several ligand binding pathways (5–7). In SMD simulation studies, time-dependent external forces are applied to the ligand to facilitate its unbinding from the active site. SMD simulation can reveal information about the enzyme’s flexibility and its response to the dissociation of the ligand. We used SMD simulations to compare the force required to remove D-Glu from RacE2 as a dimer (its physiological form) and as a monomer. We found significant differences in the forces required to release the ligand from the active sites of the monomeric and dimeric forms with less force required to remove the ligand from the monomer than the dimer. We also used NMA to compare the flexibility of the monomeric and dimeric forms of the enzyme, and found that when the first six normal modes were compared, the active site was more accessible in the monomer than in the dimer.
To better understand the role of dimerization in the overall dynamics and function of glutamate racemase, we generated site specific mutants at the dimer interface, rendering the enzyme monomeric, and characterized the kinetics of these mutants. Surprisingly, disruption of the dimer interface increased enzyme catalytic activity for all mutants studied. The mutants exhibited a ~10 fold and a ~3 fold higher turnover number in the directions catalyzing the conversion of D-Glu to L-Glu and L-Glu to D-Glu, respectively. NMR measurements and solution X-ray scattering studies were also consistent with models of overall collective motions. Thus, we propose that global domain motions play an important role in the catalytically relevant structural dynamics of this enzyme.
EXPERIMENTAL PROCEDURES
Steered Molecular Dynamics
The SMD simulations were performed on the system when it reached an equilibrium state after 1.5ns conventional MD simulation (with explicit solvent model). 10 different snapshots of the structure were then extracted at 100ps intervals from the conventional MD simulation of the ligand bound RacE2 complex. These snapshots were used as starting points for the SMD simulation runs. The SMD procedure involved in NAMD2 (8) was adopted. The bound ligand D-Glu was pulled in the same direction in both the monomeric RacE2 model and the dimeric RacE2 model. To find an appropriate pulling velocity several SMD simulations were carried out at different pulling velocities. A pulling velocity of 0.125 Å/ps was selected as it produced reasonable results without any distortion of protein structure. The force required toattain the speed of 0.125 Å/ps along with the time taken by the ligand to exit the active site was monitored. To ensure that the pulling velocity was not excessive, a second series of three runs each for the monomer and dimeric forms was also performed with a pulling velocity of 0.0125 Å/ps with otherwise equivalent conditions.
Normal Mode Analysis (NMA)
NMA was carried out on two web based servers, NOMAD-Ref web server at http://lorentz.immstr.pasteur.fr/ and the elNemo web server at http://www.igs.cnrs-mrs.fr/elnemo/. Identical results were obtained from both servers.
Plasmid and Mutagenesis
The genes encoding RacE1 and RacE2 were PCR-amplified from genomic DNA isolated from B. anthracis Sterne 34F2, inserted into the pET-15b vector (Novagen), and transformed into E. coli BL21(DE3) as reported previously (4). Site directed mutagenesis was accomplished using the Stratagene Quikchange XL-Site-Directed Mutagenesis kit. The mutations were confirmed by DNA sequencing at the University of Illinois’ Research Resources Center. Protein Purification. The wild type and mutant proteins were expressed and purified following previously reported methods (4). The RacE2 expression protocol was successfully employed to overexpress all the mutant proteins.
Size-Exclusion Chromatography
The oligomeric states of RacE2 and its mutants were determined using a Superdex 200 HR 10/30 (Amersham Biosciences) high-performance, gel-filtration column. The column was equilibrated with equilibration buffer, 50 mM Tris (pH 8.0), 250 mM NaCl, and 2 mM DTT, and was calibrated by running a set of protein standards (with molecular weights and elution volumes as follows: ribonuclease A, 13,700 Da (16.26 mL); chymotrypsinogen, 25,000 Da (15.69 mL); ovalbumin, 43,000 Da (13.76 mL); albumin, 67,000 Da (12.46 mL); catalase, 232,000 Da (10.93 mL); ferritin, 440,000 Da (9.06 mL)). A 10 mg/mL sample of blue dextrin was used to determine the void volume. Blue dextrin and all molecular weight standards were prepared by reconstituting lyophilized samples in the equilibration buffer, and centrifuging at 13,000 g for 5 min to remove any particulates.
To determine the quaternary structure of the apoenzymes, a 200 μL sample of purified, recombinant mutant RacE2 at a concentration of 5 mg/mL was applied to the column, and the elution volume of the enzyme peak was determined using the AKTA FPLC software (Amersham Biosciences). To determine the quaternary structure of the enzymes in the presence of D-Glu, a 200 μL sample of each enzyme was exchanged into the aforementioned equilibration buffer, supplemented with 10 mM D-Glu. The column was equilibrated with the supplemented equilibration buffer, and the enzymes were run over the column as described for the apoenzyme. The apparent molecular weights of RacE2 and its mutants in the absence and presence of 10 mM D-Glu were estimated by comparing their relative mobility (Kav) to those of the standard enzymes via a plot of the log of the molecular weights of the standards versus the Kav. The value of Kav is equal to (VR – VO)/(VC – VO), where VR is the retention volume, VO is the void volume, and VC is the bed volume of the column.
Glutamate Racemase Activity Assays
The enzymatic activity in the L-Glu to D-Glu direction was measured by a continuous, circular dichroism (CD) assay based on methods described previously (4). The enzymatic activity of RacE1 and RacE2 in the reverse direction with D-Glu as the substrate was measured using a continuous, coupled-enzyme assay (4). The initial velocity data were then plotted against substrate concentration and fit to the Michaelis-Menten equation as described below.
Steady-State Kinetic Parameters
The steady-state kinetic parameters for the forward direction catalyzed by RacE2 and its mutants, using L-Glu as substrate, were determined in an assay buffer consisting of 10 mM potassium phosphate, pH 8.2, and 0.2 mM DTT. The concentration of L-Glu was varied between 0.1 and 30.0 mM with a final enzyme concentration of 0.5 μM enzyme. The assay volumes for both enzymes were 3.00 mL, and the path length of the cuvettes was 1.00 cm. Initial velocity data at each substrate concentration were measured in duplicate and averaged. Initial velocity data were plotted against substrate concentration and the data were fit to the Michaelis-Menten equation. The resulting kinetic parameters kcat and Km are reported with their standard error values.
The steady state kinetics parameters in the reverse direction (D-Glu to L-Glu) were determined using the continuous coupled enzyme assay (4), with modification to fit the 96-well plate format. Reactions were carried out at 25°C in the assay mixture that contained 5mM NAD+ (Sigma-Aldrich), 2.5mM ADP (Sigma-Aldrich), 50mM CHES buffer (pH9.2) (Sigma-Aldrich), 0.65mM iodonitrotetrazolium chloride (INT) (Sigma-Aldrich), 37.5 units/mL L-glutamate dehydrogenase (Sigma-Aldrich), 2 units/mL diaphorase (Sigma-Aldrich), and 50 μM D-glutamate (Sigma-Aldrich). The reaction was initiated by the addition of the enzyme to a final concentration of 3μM in a total assay mixture volume of 200μL. Rates were determined by measuring the absorbance at 500nm in 96-well clear microplates (Fisher Scientific).
Protein NMR
The RacE2 mutant R25A was labeled with 2H and 15N using standard protocols (9). Briefly, a 5mL LB culture was inoculated and allowed to grow for 6 hours. This culture (2.5mL) was then transferred to two 50 mL minimal media which were grown overnight. The next day the overnight culture was added to two 500 mL minimal media (1L contains 3g KH2PO4, 12.8g Na2HPO4.7H2O, 1.5g NaCl, 4g D-glucose, 5mL basal vitamin eagle media, 1mL 1M MgSO4, 1mL of 100μg/mL ampicillin and 1g 15NH4Cl in 75% 2H2O). The culture was induced with IPTG (1mM final concentration) when the optical density reached 0.6. The cultures were grown for an additional 4 hrs and then harvested. The protein was purified following standard procedures described above. The protein was finally dialyzed in 50mMTris, 250mM NaCl, and 1mM TCEP at pH 8.0. Two samples, of the enzyme without substrate (at a concentration of 400μM), and of enzyme with substrate (20mM D-Glu was added to 400μM of enzyme and incubated at room temperature for 2 hrs) were prepared. These samples were centrifuged at 16,000g for 30 min and the top 50% of the solution was used for the experiment to minimize the presence of aggregates. The TROSY HSQC was recorded at room temperature on a 900 MHz NMR spectrometer. Data was processed using NMRPipe and analyzed by NMRView (10).
X-ray solution scattering
RacE2 was dialyzed overnight in 50mM Tris, 250mM NaCl, and 1mM DTT at pH8.0 to ensure a precise buffer match for background subtraction of the scattering arising from the buffer from that of the protein sample. The enzyme was studied in the absence and presence of saturating amounts of D-Glu (2mM). The samples for SAXS were at a concentration between 1–2mg/ml, and for WAXS the concentrations were between 6–8 mg/ml. Measurements were carried out at room temperature (23 °C) using the SAXS instrument at the BioCAT beamline of the Advanced Photon Source, Argonne National Laboratory. Data were collected using the 8 cm × 16 cm Aviex CCD area detector at a sample-to-detector distance of 190.7 cm for SAXS and 17.8cm for WAXS. Samples were centrifuged at 16,000g for 30 min and the top 50% of the solution was used for data measurement to eliminate all possible aggregates. Samples were measured in a thermostated quartz capillary flow cell with a 1.5 mm diameter under a constant flow rate of 10 μL/s to prevent potential radiation damage and 20 successive frames of 1s exposure were recorded for each sample and the data averaged for better signal/noise statistics. Each sample measurement was preceded by the measurement of its matching buffer solution. In addition to being used for background subtraction, the buffer measurements also served as a check on beam properties and the cleanliness of the sample cell between switching samples. Samples were checked before and after the SAXS measurements for concentration, specific activity, and oligomerization state (by gel filtration). The samples were still active after exposure to X-rays, with minimal radiation damage. Scattering profiles (intensity I vs. scattering vector Q) were reduced and the SAXS and WAXS data merged by using the program IGOR Pro (WaveMetrics, Inc.) with macros written by the BioCAT staff. Structural parameters and the distance distribution function, P(r), were calculated with the program GNOM using data up to a Q of 0.45Å−1. These values were compared to the theoretical Rg values of the crystal structures calculated using the program CRYSOL.
Ab Initio Modeling
Low resolution molecular shape reconstructions from the experimental scattering data were performed with the program GASBOR (11). The scattering profiles were used up to a Q of 0.45Å−1 for the reconstruction. GASBOR searches a chain compatible spatial distribution of an exact number of dummy residues, corresponding to the Cα atoms of protein amino acids. The Cα chain is folded to minimize the discrepancy between the scattering curve calculated from the folded model and the experimental scattering curve. Over 80 GASBOR calculations were performed, and 20 calculations with the smallest standard deviation relative to the experimental data were selected for averaging by DAMAVER (12) to generate the final model which represents the most probable conformation reconstructed for the protein.
RESULTS
Steered Molecular Dynamics Simulation Studies
We used SMD simulation to investigate the unbinding of D-Glu from the active site of RacE2 in the monomeric and dimeric forms. The computational procedure consisted of applying external forces to pull the ligand out of the active site. A constant pulling velocity of 0.125 Å/ps and 0.0125 Å/ps was applied along a predetermined direction and the force required to achieve this speed was monitored, as a function of time. The simulation results are shown in Figure 2. The overall global shapes of the force profiles under the same pulling velocity for the unbinding simulation differ significantly between the monomeric and dimeric forms of RacE2. Less force is required to pull D-Glu out of the active site of the monomeric form than of the dimeric form. The SMD results in Figure 2A and 2B were generated by averaging 10 different SMD simulations that were done using 10 different starting structures of the monomer and dimer. Molecular dynamics simulations were carried out on both the monomer and dimer separately and once the systems had reached equilibrium 10 different snapshots were selected at a time interval of 100 ps to use as the starting structures for the SMD simulation run. In Figure 2C and 2D we used a slower pulling speed of 0.0125 Å/ps and the SMD results were generated by averaging three different SMD simulations that were done using 3 different starting structures (200 ps apart) from the equilibrium stage of the molecular dynamics simulations run on both systems. In these figures we observe two peaks in the profile of the dimeric form, suggesting large scale conformational rearrangement that temporarily relaxes resistance to D-Glu exit. This appears to occur at the point where the charged D-Glu α-carboxylate and α-amine groups pass through the hydrophobic region in the tunnel leading into the active site (Figure 1B). As a result of the repulsive forces, force on the ligand decreases. However, once the D-Glu α-carboxylate is near the entrance of the active site, it can interact with the residues comprising the γ-carboxylate binding sub-site, thereby increasing the force to remove D-Glu completely out of the active site. We do not see the two peaks in the force profile of the monomeric form, and the maximum force observed during the trajectory is lower for the monomer than the dimer. This could be due to the greater flexibility and hence less resistance to the D-Glu exit.
Figure 2.

SMD simulation results using two different pulling speeds, 0.125 A/ps (A and B) and 0.0125 A/ps (C, and D) for the monomeric (A, and C) and the dimeric (B, and D) forms of RacE2, respectively. A and B were generated by averaging 10 different SMD runs using 10 different starting structures that were selected at a time interval of 100ps from a molecular dynamics simulation run after the system had reached equilibrium. C and D were generated by averaging 3 different SMD runs using 3 different starting structures selected at a time interval of 200 ps from the equilibrium phase of the molecular dynamics simulation. Figs 1A, B are plotted on a 10-fold expanded time scale compared with C and D to compensate for the 10-fold difference in pulling rates.
Normal Mode Analysis (NMA)
We used computational normal mode analysis to explore the conformational dynamics of RacE2. NMA is a method to characterize the collective motions of a group of atoms. It is based on the harmonic approximation of the potential energy function around a minimum energy conformation (13). Each set of normal modes provides information on the probable conformations that the molecule can experience. High frequency normal modes are highly localized motions of side chains, while the lowest frequency modes are collective motions of large groups of atoms, usually whole structural domains. It has been shown that there is usually a single low frequency mode whose direction compares well with conformational change (14). This single mode need not be the lowest frequency mode but is often one of the three lowest frequency ones (14).
Figure 3 shows the lowest normal mode (mode 7) for both the monomeric and dimeric forms of RacE2 respectively (the first six normal modes are the rotation and translation movement of the entire molecule). The collective motions in the monomer are between the two domains which either move within planes that are parallel to each other (as in mode 7) (Figure 3A) or towards and away from each other (as in mode 8). The residues that are the most mobile in the monomer are those that are at the outer ends of the two domains. The collective motions in the dimer differ from those of the monomer in that the three lowest modes in the monomer are primarily oscillatory or twisting motions of the two domains with respect to each other with a relatively stationary “hinge” region between the two domains, while in the dimer, the three lowest modes are primarily oscillatory or twisting motions of the two monomers with respect to each other, while maintaining a relatively stationary dimer interface. Modes 10–12 of the dimer more closely correlate with modes 7–9 of the monomer, but appear to include a mix of motions with respect to the domain “hinge” and the dimer interface. This is partially illustrated in Figure 3 where in modes 7 (Figure 3B) and 9 of the dimer, the two monomers bend towards and away from each other. In mode 8, the two monomers move in a twisting motion originating at the dimer interface. The residues at the dimer interface are relatively immobile when compared to the rest of the protein.
Figure 3.

Normal mode analysis results showing the lowest mode for the monomeric RacE2 (A) and the dimeric RacE2 (B). The two domains in the monomeric unit are shaded in pink and beige and the location of the active site is indicated by the presence of the catalytic cysteines shown in yellow. In each mode two frames representing the two extreme conformations are shown. One frame is in solid color and the other is in transparent grey. The large arrows aid in the visualization of the overall collective motions in the different modes. Dominant motions are hinge like motions about the catalytic site resulting in the opening and closing of the active site in the monomer (A). In the dimer (B) dominant motions are between the two monomers at the dimer interface. This figure was generated by PyMOL
In a plot of the relative displacement of each residue vs. residue number (Figure 4), catalytic residues occupy local minima in both the monomeric and dimeric forms of RacE2. We expect that residues at key mechanical sites at the dimer interface will occupy the local minima in the dimeric form (Figure 4B) and will no longer be at the local minima in the monomeric form (Figure 4A). One such key residue is R25. This residue could possibly be at a key mechanical site that possibly controls global motions (15) in the dimer. This key mechanical site is locked down by hydrogen bonding in the dimer, and hence is relatively immobile, whereas in the monomer it is relatively more mobile. Internal motions between the two domains of the monomer in the dimeric form of the enzyme begin to appear in modes 10 and above. However these motions are still restricted by the locked dimer interface. Thus, the overall global dynamics differ significantly between the monomeric and dimeric forms of RacE2 (Figure 4).
Figure 4.

Plot of the relative displacement of each residue vs. the residue number in modes 7 (red), 8 (green), and 9 (blue) in the RacE2 monomer (A) and RacE2 dimer (B). Catalytic residues (C74 and C185) occupy the minima in both the monomer and dimer. Residues that disrupted dimerization (R25 and K106+R214) are less flexible in the dimer than in the monomer. The overall global dynamics between the monomeric and dimeric forms are qualitatively significantly different.
The collective motions between the two domains of the monomer are more prominent in the monomeric form than in the dimeric form, leading to a greater accessibility of the active site that is located between the two domains. This is clearly seen when the open active site is compared between that of the monomer and dimer. In Figure 5A the active site observed in the crystal structure is closed with it not being possible for the ligand to enter or exit the active site as such without there being some kind of opening event. It is clear from Figures 5B and 5C that there is a greater accessibility of the active site in the monomer when compared to that of the dimer.
Figure 5.

(A) Close-up view of the active site of RacE2 as seen in the crystal structure. Residues lining the active site are colored yellow. D-Glu (red) is present inside the active site and cannot exit the active site without the active site opening. (B) Open active site of RacE2 as seen in one of the frames in mode 7 of the monomer (NMA results). (C) Open active site of RacE2 as seen in one of the frames in mode 10 of the dimer (NMA results). In the dimer, movement between the two domains of the monomer were observed in modes 10 and above, however the accessibility of the active site is less than that seen in the case of the monomer. This figure was generated by PyMOL.
Selection of residues at the dimer interface for mutagenesis
Our computational studies suggest that the monomeric and dimeric forms of RacE2 will exhibit differing conformational dynamics that should be reflected in their kinetic profiles. We proceeded to confirm this speculation through experimental approaches.
The two monomers of RacE2 are held together by hydrogen bonds and van der Waals interactions. The overall surface area at the dimeric interface is ~1545 Å2, which constitutes ~13% of the total solvent accessible surface area of the isolated monomers. The interface is predominantly polar. Residue pairs that formed H-bonds were selected for mutagenesis. All residues were mutated to an alanine. The residues selected for mutagenesis are listed in Table 1. We generated eight mutants, with 7 having single mutations and one having a double mutation. Mutation sites were chosen such that they span the outer region (R25, K29 and Q86), the inner region (K106 and Y221), and the innermost central region (R214 and P99) of the dimer interface. R25 (located at the end of a helix), K29 (located on a large loop), and K106 (located at the end of a helix) are all situated on either side of the dimer interface (Figure 6). Q86 and Y221 are both located at the end of helices but situated on one face of the dimer interface. R214 from one monomer interacts with the side chains of E215, T103, and the backbone of P99 in the other monomer. These residues are situated in the central region of the dimer interface and mutating R214 was expected to disrupt six hydrogen bonds at the dimer interface.
Table 1.
Comparison of kinetic parameters of B. anthracis glutamate racemase, RacE2 (RacE2_WT), and its mutants. Kinetics was determined using two different assay methods as described in materials and methods. Freshly purified enzyme was used for each assay. The kinetic measurements were repeated at least twice (and in some cases three times) with different batches of protein preparations and were found to vary by about two fold with different batches. Representative measurements from one batch (done in triplicate) are shown here.
The quaternary structure (determined by gel filtration) for each mutant is listed as either a monomer (M), dimer (D) or as a monomer-dimer equilibrium (M ↔ D).
| LGlu → DGlu | DGlu → LGlu | Quaternary structure +/−DGlu | |||||
|---|---|---|---|---|---|---|---|
| Km (mM) | kcat (min−1) | kcat/Km (M−1s−1)×103 | Km (mM) | kcat (min−1) | kcat/Km (M−1s−1)×103 | ||
| RacE2_WT | 6.1 ± 0.8 | 1944 ± 84 | 5.3 ± 0.7 | 0.07 ± 0.01 | 13 ± 0 | 3.1 ± 0.4 | D |
| R25A | 5.8 ± 0.4 | 4900 ± 112 | 14.1 ± 1.0 | 0.14 ± 0.03 | 126 ± 7 | 15 ± 3.3 | M |
| R214A/K106A | 11.0 ± 0.9 | 4640 ± 140 | 7.0 ± 0.6 | 0.16 ± 0.03 | 133 ±12 | 13.9 ± 2.9 | M |
| K29A | 7.2 ± 1.0 | 5100 ± 244 | 11.8 ± 1.8 | 0.25 ± 0.03 | 124 ± 19 | 8.3 ± 1.6 | M ↔ D |
| P99A | 4.1 ± 0.3 | 2740 ± 60 | 11.1 ± 3.5 | 0.17 ± 0.02 | 128 ± 10 | 12.5 ± 1.8 | M ↔ D |
| K106A | 10.8 ± 1.4 | 3660 ± 200 | 5.6 ± 0.8 | 0.42 ± 0.03 | 87 ± 0 | 3.5 ± 0.2 | M ↔ D |
| R214A | 10.1 ± 0.9 | 5920 ± 200 | 9.8 ± 0.9 | 0.30 ± 0.03 | 111 ± 14 | 6.2 ± 1.0 | M ↔ D |
| Q86A | 8.8 ± 1.8 | 2400 ± 180 | 4.5 ± 1.0 | 0.1 ± 0.01 | 36 ± 2 | 6.0 ± 0.7 | D |
| Y221A | 9.9 ± 1.2 | 4200 ± 200 | 7.1 ± 0.9 | 0.13 ± 0.01 | 46 ± 2 | 5.9 ± 0.5 | D |
Figure 6.

(A) Crystal structure of RacE2. The two monomers are shown in different colors (yellow and cyan). The residues at the dimer interface that were mutated to Ala are indicated. R25 (red) interacts with the backbone of K106 (blue). K29 (magenta) is located on a large loop on either side of the dimer interface and interacts with the side chain of Asn 136 and the backbones of Lys132 and Ser133 (not shown). R214 (green) is located in the center of the dimer interface and interacts with the side chains of T103, R215 and the backbone of P99 (not shown). (B) Q86 (red) and Y221 (magenta) are both located on one face of the dimer interface with Q86 interacting with the side chain of Y221 from the other monomer. This figure was generated by PyMOL.
Enzyme kinetics and oligomeric state of the dimer interface mutants
Kinetics analysis of these RacE2 mutant enzymes in the D-Glu → L-Glu direction showed a surprising result – the catalytic rates of the mutants were higher than those of the wild type with some of the mutants exhibiting as much as a 10 fold increase in their turnover number (Table 1). The overall catalytic efficiency (kcat/Km) was also about 4 to 5 fold higher in the case of the mutants R25A, R214A/K106A, and P99A in the D-Glu → L-Glu direction. The kinetics in the L-Glu → D-Glu direction were not as significantly affected with at most a 2–3 fold increase in the overall catalytic efficiency. None of the mutants were less active than the wild type enzyme in either direction. Analysis of the oligomeric nature of the mutants by gel filtration revealed that two of the mutants, R25A and K106A/R214A, were completely monomeric at the concentration studied (5mg/mL). Four mutants were in equilibrium between the monomeric and dimeric forms, and two mutants, Q86A and Y221A, were still dimeric irrespective of the presence of substrate.
Determination of substrate specificity and thermal stability of the R25A mutant
Disrupting dimerization increases RacE2 catalytic efficiency. Circular dichroism studies were carried out to determine if disrupting dimerization causes the enzyme to lose specificity for its substrate. D and L-amino acids have differential absorption of left- and right-handed circularly polarized light with the D-enantiomer having a similar signal compared to the L-enantiomer but with opposite sign. Thus, if the enzyme is active we expect to see a change in molar ellipticity, with time, as the L-amino acid gets converted to the D-enantiomer. We monitored the change in molar ellipticity at high concentrations (20mM) of the amino acids, L-glutamine and L-aspartate, upon addition of about 5μM RacE2-R25A. No change in ellipticity was observed, indicating that the enzyme does not lose specificity for its substrate upon monomerization under these conditions. The thermal stability of the enzyme as reflected by the Tm value (midpoint of thermal unfolding) was also determined by differential scanning calorimetry (DSC). The Tm for the monomeric mutant R25A (51°C) was not found to be significantly different from that of the wild type (49°C) (data not shown).
Protein NMR
We used NMR spectroscopy to experimentally gain insight into the conformational dynamics of the monomer in both in the free form and the bound forms. HSQC-NMR spectra for the 2H and 15N-isotopically labeled RacE2 mutant, R25A, were recorded in the absence and presence of saturating concentrations of D-Glu. Two dimensional transverse relaxation-optimized spectroscopy (TROSY) revealed significant shift in several resonances, along with the appearance of several new resonances, when D-Glu was added (Figure 7). Similar observations were made in the case of RacE from H. pylori, where the spectra between the bound and free forms differed significantly (3). This was attributed to the protein being quite flexible and assuming multiple conformations in the free form (3). The monomeric nature of the R25A mutant both in the absence and presence of saturating amounts of D-Glu, at the concentrations used for NMR (~13mg/ml or 400μM), was confirmed by gel filtration studies. This was done to rule out the possibility that the monomeric mutant was dimerizing at higher concentrations, in the presence of D-Glu, resulting in the significant shift in several resonance peaks.
Figure 7.
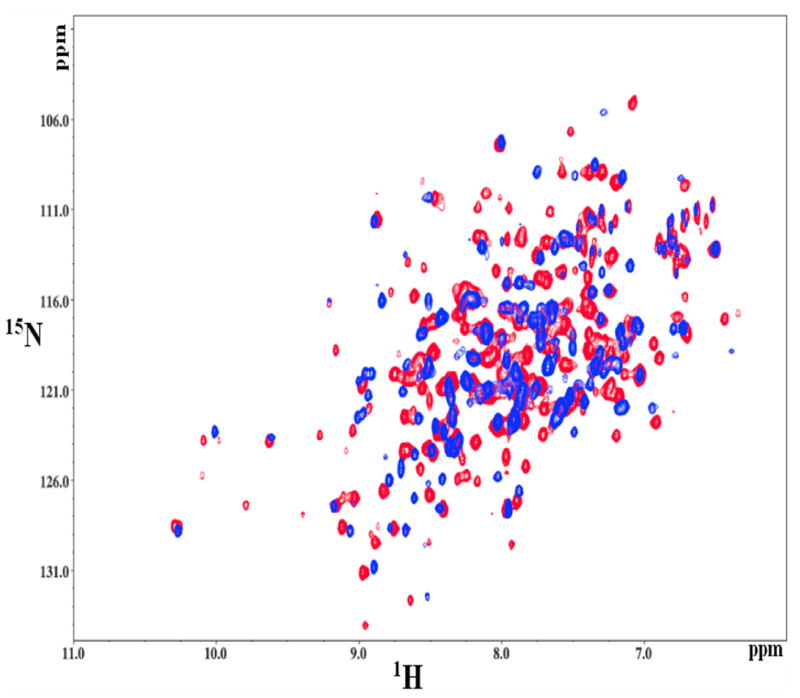
Comparison of the substrate bound (red) and free (blue) RacE2_R25A HSQC. The spectra show significant perturbations in the chemical shifts of many resonances, along with the appearance of multiple additional resonances when the enzyme binds the substrate. Thus, there is a significant conformational change upon substrate binding.
It is possible that the enzyme either undergoes conformational changes upon binding D-Glu, or that substrate binding suppresses conformational flexibility. However, we cannot confirm this due to the lack of resonance assignments. We can conclude that the enzyme is more conformationally stable or rigid upon binding its substrate, as indicated by the reduced line broadening which led to the appearance of new peaks that are completely absent in the apo-enzyme.
X-ray solution scattering studies
We used X-ray solution scattering methods to further evaluate the collective motions predicted by normal mode analysis for the dimeric form of RacE2. Solution scattering experiments measure an ensemble average conformation of all species in the solution. Therefore, the scattering curve corresponds to the average of different protein conformations if the structure exhibits some flexibility. Solution scattering data provides a unique and effective tool for observing gross domain movements in a protein molecule. We measured the solution scattering profiles of RacE2 to reconstruct the structural envelope of RacE2 in solution to compare with the collective motions that were predicted by computational normal mode analysis. The experimental scattering profile is shown in Figure 8. Low angle scattering intensity calibration with cytochrome C (Sigma c7752) indicated that RacE2 exists as a dimer in solution. Analysis of the scattering data by GNOM led to a radius of gyration of 26.4 ± 0.1 Å without D-Glu and 26.8 ± 0.1 Å with D-Glu (Guinier analysis gives similar results of 27.5 ± 0.2 and 27.4 ± 0.2, respectively). The distance distribution function P(r) has a bell shaped appearance for RacE2 both with and without D-Glu which is typical of a globular molecule. We compared the experimental scattering profile with the calculated profile of the crystal structure (with D-Glu) and found that they differed slightly, but the theoretical radius of gyration was only ~2Å lower (with the theoretical Rg being 24.8 Å). Low resolution models of the structure were generated using data up to a Q of 0.45 Å −1 with the program GASBOR (11, 16, 17). These models were averaged and filtered using the program DAMAVER (12). The final two models (of RacE2 with and without D-Glu) were found to be similar to each other. These low resolution models or molecular envelopes reflect the conformational space sampled by the RacE2 dimer. We overlaid the envelope of RacE2 with D-Glu onto the crystal structure (which has D-Glu in its active site) using the SUPCOMB (18) software that is part of the DAMAVER program suite. We expect the crystal structure to be well accommodated in this molecular envelope which is indeed the case (Figure 9).
Figure 8.
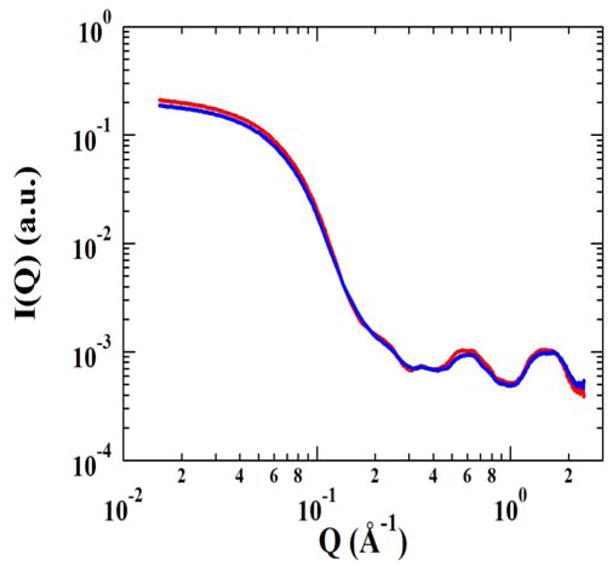
X-ray solution scattering data for RacE2. X-ray scattering data was collected at room temperature in the absence of D-Glu (red) and in the presence of saturating amounts of D-Glu (blue).
Figure 9.

About 20 independent models derived from GASBOR were averaged and filtered (DAMAVER and DAMFILT) to generate the most likely solution structure which was then superimposed (SUPCOMB) onto the crystal structure of RacE2. Solution structure (or molecular envelope) is depicted as a mesh and overlapped onto the crystal structure (ribbon structure). The crystal structure was found to be relatively well accommodated in the molecular envelope. (A), (B), and (C) represent different views with (A) being the side view, (B) is the view where (A) is rotated by 90° through a vertical axis, and (C) is looking down at (A) from above. This figure was generated by PyMOL.
Normal mode analysis identified large amplitude and highly correlated movements between the two monomers in the dimeric form of the enzyme. By comparing the low frequency modes with the molecular envelope reconstructed from solution scattering data one can determine if these movements are consistent with the solution structure. In each of the 6 lowest frequency normal modes (modes 7–12) for the dimer, 2 frames were chosen representing the two extreme conformations (e.g. if the collective motions were between two monomers in the dimer, then the two frames chosen would be when the two monomers are farthest apart from each other and closest to each other). Each of these conformations was superimposed onto the solution structure and found to be relatively well accommodated (Figure 10) with the exception of frames in modes 8 (frame 23) and 9 (frame 25). Not all sterically allowed conformations are equally populated in solution and this could account for these misfits. We expect that if the two extreme conformations are well accommodated in the molecular envelope then the intermediate frames (i.e., the overall predicted movements) should also be well accommodated. Hence, the range of large amplitude collective motions predicted by NMA for the dimeric form of RacE2 appear to be consistent with the low resolution structure determined by X-ray solution scattering analyses just as the crystal structure is consistent with the low resolution structure. However, this does not indicate that the normal modes agree better with the solution structure than the crystal structure.
Figure 10.

Solution structure of RacE2 (depicted as a mesh) overlapped onto the different conformers arising from modes 7, 8 and 9. The open conformations of the active site from mode 7 (A), 8 (B) and 9 (C) are overlapped onto the solution structure. The structures are relatively well accommodated in the molecular envelope. This figure was generated by PyMOL.
Discussion
Glutamate racemase, RacE2, is a fascinating enzyme system. Our NMR results on the monomeric form of RacE2, together with prior NMR work done on glutamate racemase from H. pylori (19) indicate that this enzyme is quite flexible and either adopts multiple conformations in solution or exhibits significant motional averaging. The active site of this enzyme is rather small, and the enzyme must exhibit some conformational flexibility for the substrate and product to enter and exit the active site. To understand this conformational flexibility we used steered molecular dynamics simulations and normal mode analysis. Our computational approaches revealed that the enzyme exhibits different conformational dynamics depending on whether it is a monomer or a dimer. Steered molecular dynamics simulation results indicate that the forces required to pull the bound D-Glu out of the active site differ between the monomeric and the dimeric forms of RacE2. Normal mode calculations provide an alternative to MD simulations for studying collective motions in macromolecules. We focused on the low-frequency motions (global motions as opposed to local motions subject to higher frequency modes) to see if or how the global dynamics and enzymatic function are correlated. The dominant role of slow modes in effecting the functional motions has been suggested in early normal mode analyses (20), and a number of studies have shown that the domain movements are dominated by one or a few normal modes (14, 15, 21). By analyzing the first six relevant low frequency modes in the monomer and dimer, we found that the overall collective motions differ significantly. In the monomeric form, there is significant motion between the two domains of RacE2, which leads to the opening and closing of the active site. In the dimeric form, the lowest frequency modes reflect collective motions about the dimer interface between the two monomers. Through these studies we conclude that there is an increase in overall flexibility of the enzyme in the monomeric form. This leads to the possibility of the monomer being more active than the dimer as there is greater movement between the two domains about the active site. This intriguing possibility was confirmed through experimental methods. We found critical residues at the dimer interface that when mutated to Ala rendered the enzyme monomeric. The kinetic parameters of the two monomeric mutants (R25A and the double mutant R214A/K106A) were such that the turnover number or kcat was 10 fold higher than that of the wild type with only a 2 fold increase in Km value. Changes in the Km values reflect a change in the stability of the ES complex or a change in rate of formation of the ES complex (since Km is the ratio between the rate of dissociation of the ES complex to E and S or P and rate of formation of the ES complex). Since we do not see a significant change in the Km values in the mutants it appears that our mutations are not affecting any of the steps relating to the formation or dissociation of the ES complex. Changes in kcat on the other hand, reflect perturbations of the chemical steps subsequent to initial substrate binding (22), and in our case we see a significant difference in these values in the mutants. The ratio of kcat/Km (also known as catalytic efficiency) can be used to compare the efficiency with which an enzyme catalyzes a particular reaction. We find that the catalytic efficiency in the case of the monomeric mutant is about 5-fold greater than the dimeric wild type enzyme. This increase in catalytic efficiency could be due to an increase in the overall flexibility of the enzyme, specifically to the motion related to the catalytic step. This is an unusual case where the native dimeric form is less active than the non-native monomeric enzyme. As the mutations are not in the vicinity of the active site (the dimer interface is on the face opposite the active site), they are not directly involved in substrate recognition or binding. Thus, we attribute the change in enzyme activity to a change in protein flexibility and/or collective motions of the enzyme.
In other systems, where dimer dissociation has been accompanied by an increase in catalytic rate, the dissociation of a dimer was a mechanism for enzyme activation (23, 24). Mutagenesis of dimeric phosphoribosylanthranilate isomerase from Thermotoga maritima to generate monomeric variants revealed that the monomers were just as active as subunits in the dimer but were thermally less stable (25). In malate dehydrogenase, monomeric mutants were generally catalytically less active although one mutation at the dimer interface increased activity by increasing the stability of the dimer (26). Among others systems, proteins undergo dimerization as a means to increase or decrease catalytic activity because the dimeric form was either more or less active than the monomeric form. However, in all these cases the physiologically active form was the catalytically more efficient form (23, 24, 27–31). In the case of RacE2, monomerization increased catalytic activity but does not significantly affect either its thermal stability or substrate specificity.
Proteins are dynamically flexible molecules in solution. Flexibility is a key determinant of protein function. Knowledge of protein flexibility is thus crucial for understanding the overall protein function and evolution. Flexibility can be brought about by linkers connecting two rigid domains or between two domains that can move relative to each other through a pivot point. Our NMR data on the monomeric RacE2 indicates significant perturbations in chemical shifts of several resonances along with the appearance of new resonances when D-Glu is present (Figure 7). This indicates that the monomeric RacE2 enzyme is a highly dynamic enzyme that may become more conformationally stable or fixed upon binding its substrate. Normal mode analysis predicts large collective motions between the two domains in the monomer with the outer edges of the two domains being more mobile than the rest of the protein.
The enhanced catalytic activity may be due to the differences in the overall collective dynamics of the monomer and dimer. The mutant K29A residue is located on a large loop on either side of the outer region of the dimer interface (Figure 6). Mutating this residue did not render the enzyme completely monomeric because interactions in the inner region of the dimer interface still hold the two monomers together. However, we speculate that this mutation releases the outer lock down between the two domains, as a result of which, they may be able to move more as they would in the monomer. The low mobility of the catalytic cysteines (Figure 4) is probably due to the fact that they are near the crossover region between the two domains that exhibit oppositely correlated motions. The catalytic cysteines are therefore localized near key mechanical sites that control the overall global motions as was illustrated by Yang and Bahar (15). The residues at the dimer interface are more mobile in the monomer than in the dimer consequent to their being locked down in the dimer. This significantly restricts the movements between the two monomeric domains within the dimeric unit, thus regulating and/or limiting the extent of the opening and closing of the active site. We observed this to be the case in modes 10 and above. We thus suggest that there is a functional coupling between catalysis and global conformational dynamics, as there is a communication pathway between the active site and the sites at the dimer interface that control the overall conformational dynamics.
A small number of modes are useful to probe structural flexibility and understand the differences in collective motions between proteins. However, we are aware that a much larger number of modes are required for properly investigating correlated motions in proteins.
The first six low frequency computed normal modes (modes 7–12) were found to compare well with the average solution structure determined by the low resolution structure reconstructed from the solution scattering data. The average molecular envelope reflects the conformational space sampled by the RacE2 dimer. With the exception of a few misfits (discussed in the results section), the overall movements are well accommodated in the experimentally determined low resolution solution structure. It should be noted that not all sterically allowed conformations are equally populated in solution. Thus, the predicted normal modes for the dimeric form of RacE2 are consistent with the solution scattering data. The predicted normal modes suggest that the conformational dynamics between monomeric and dimeric forms of RacE2 differ significantly. This difference in conformational dynamics appears to be strongly correlated with the observed increase in catalytic efficiency of the monomeric form.
Conclusions
Understanding the relationship between protein structure, function and dynamics is of utmost importance in the drug discovery field for the design of effective inhibitors. However, relatively few studies have investigated the connection between enzyme function and overall conformational dynamics. We found that the conformational dynamics of the RacE2 isoform differ depending on whether the enzyme was a monomer or a dimer. We also found single point mutations as well as a double mutation that affect the structure and catalytic activity of RacE2 by disrupting its dimerization. The change in conformational dynamics upon monomerization appears to enhance the catalytic efficiency. Thus, dimerization appears to have a significant impact on the dynamics of RacE2. No information is available regarding the oligomeric nature of glutamate racemase in vivo. We speculate that the in vivo concentration-dependent monomer-dimer equilibrium may be a regulatory mechanism for controlling the rate of D-Glu production as needed for cell wall or spore capsule synthesis.
Our data does not directly demonstrate that catalysis is controlled by the conformational dynamics of the enzyme. However, we have a large number of correlations consistent with catalysis being controlled by the internal dynamics of the enzyme and consequently provide a potentially testable hypothesis for future work.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (AI056575).
Funded by: • NIH; Grant Number 5U19AI056575
We thank Drs. Anjali Mahajan, Benjamin Ramirez and Hyun Lee for assistance in obtaining the NMR spectra shown in Figure 7 as well as for helpful discussions. Construction of the Center for Structural Biology was supported by NCRR NIH grant 1C06RR016560. Funding from the NIH grant 5P41GM068944 enabled the acquisition and support of the 900 MHz NMR instrument in the Center’s NMR Facility.
Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract no. W-31-109-ENG-38. BioCAT is a National Institutes of Health-supported Research Center RR-08630.
The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Center for Research Resources or the National Institutes of Health.
Abbreviations
- L-Glu
L-glutamate
- D-Glu
D-glutamate
- RacE1
glutamate racemase isozyme 1 from B. anthracis
- RacE2
glutamate racemase isozyme 2 from B. anthracis
- NMA
normal mode analysis
- ΔANR
(pX01-, pX02-) B. anthracis
- DTT
dithiothreitol
- TB
Terrific Broth
- CD
circular dichroism
- Q
scattering vector
- Rg
radius of gyration
References
- 1.Newsholme P, Procopio J, Lima MM, Pithon-Curi TC, Curi R. Glutamine and glutamate--their central role in cell metabolism and function. Cell Biochem Funct. 2003;21:1–9. doi: 10.1002/cbf.1003. [DOI] [PubMed] [Google Scholar]
- 2.Yan D. Protection of the glutamate pool concentration in enteric bacteria. Proc Natl Acad Sci U S A. 2007;104:9475–9480. doi: 10.1073/pnas.0703360104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lundqvist T, Fisher SL, Kern G, Folmer RH, Xue Y, Newton DT, Keating TA, Alm RA, de Jonge BL. Exploitation of structural and regulatory diversity in glutamate racemases. Nature. 2007;447:817–822. doi: 10.1038/nature05689. [DOI] [PubMed] [Google Scholar]
- 4.May M, Mehboob S, Mulhearn DC, Wang Z, Yu H, Thatcher GR, Santarsiero BD, Johnson ME, Mesecar AD. Structural and functional analysis of two glutamate racemase isozymes from Bacillus anthracis and implications for inhibitor design. J Mol Biol. 2007;371:1219–1237. doi: 10.1016/j.jmb.2007.05.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isralewitz B, Izrailev S, Schulten K. Binding pathway of retinal to bacterio-opsin: a prediction by molecular dynamics simulations. Biophys J. 1997;73:2972–2979. doi: 10.1016/S0006-3495(97)78326-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Izrailev S, Stepaniants S, Balsera M, Oono Y, Schulten K. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys J. 1997;72:1568–1581. doi: 10.1016/S0006-3495(97)78804-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grubmuller H, Heymann B, Tavan P. Ligand binding: molecular mechanics calculation of the streptavidin-biotin rupture force. Science. 1996;271:997–999. doi: 10.1126/science.271.5251.997. [DOI] [PubMed] [Google Scholar]
- 8.Kale L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, Schulten K. NAMD2: Greater Scalability for Parallel Molecular Dynamics. Journal of Computational Physics. 1999;151:283–312. [Google Scholar]
- 9.Marley J, Lu M, Bracken C. A method for efficient isotopic labeling of recombinant proteins. J Biomol NMR. 2001;20:71–75. doi: 10.1023/a:1011254402785. [DOI] [PubMed] [Google Scholar]
- 10.Johnson BAB, Richard A. NMRView: a computer program for the visualization and analysis of NMR data. Journal of Biomolecular NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 11.Svergun DI, Petoukhov MV, Koch MH. Determination of domain structure of proteins from X-ray solution scattering. Biophys. 2001;J80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Volkov V, Svergun D. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Cryst. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suhre K, Sanejouand YH. ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32:W610–614. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tama F, Sanejouand YH. Conformational change of proteins arising from normal mode calculations. Protein Eng. 2001;14:1–6. doi: 10.1093/protein/14.1.1. [DOI] [PubMed] [Google Scholar]
- 15.Yang LW, Bahar I. Coupling between catalytic site and collective dynamics: a requirement for mechanochemical activity of enzymes. Structure. 2005;13:893–904. doi: 10.1016/j.str.2005.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toratani T, Kezuka Y, Nonaka T, Hiragi Y, Watanabe T. Structure of full-length bacterial chitinase containing two fibronectin type III domains revealed by small angle X-ray scattering. Biochem Biophys Res Commun. 2006;348:814–818. doi: 10.1016/j.bbrc.2006.07.096. [DOI] [PubMed] [Google Scholar]
- 17.Kozak M. Solution scattering studies of conformation stability of xylanase XYNII from Trichoderma longibrachiatum. Biopolymers. 2006;83:95–102. doi: 10.1002/bip.20531. [DOI] [PubMed] [Google Scholar]
- 18.Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. Journal of Applied Crystallography. 2001;34:33–41. [Google Scholar]
- 19.Fisher SL, Kern G, Newton T, Lundqvist T, Folmer R, Xue Y. 45th Annual ICAAC, American Society for Microbiology.2005. [Google Scholar]
- 20.Karplus M, McCammon JA. Dynamics of proteins: elements and function. Annu Rev Biochem. 1983;52:263–300. doi: 10.1146/annurev.bi.52.070183.001403. [DOI] [PubMed] [Google Scholar]
- 21.Tama F, Gadea FX, Marques O, Sanejouand YH. Building-block approach for determining low-frequency normal modes of macromolecules. Proteins. 2000;41:1–7. doi: 10.1002/1097-0134(20001001)41:1<1::aid-prot10>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 22.Copeland RA. Enzymes: A Practical Introduction to Structure, Mechanism and Data Analysis. 2. John Wiley & Sons, Inc; [Google Scholar]
- 23.Giraldo R, Fernandez-Tornero C, Evans PR, Diaz-Orejas R, Romero A. A conformational switch between transcriptional repression and replication initiation in the RepA dimerization domain. Nat Struct Biol. 2003;10:565–571. doi: 10.1038/nsb937. [DOI] [PubMed] [Google Scholar]
- 24.Marianayagam NJ, Sunde M, Matthews JM. The power of two: protein dimerization in biology. Trends Biochem Sci. 2004;29:618–625. doi: 10.1016/j.tibs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Thoma R, Hennig M, Sterner R, Kirschner K. Structure and function of mutationally generated monomers of dimeric phosphoribosylanthranilate isomerase from Thermotoga maritima. Structure. 2000;8:265–276. doi: 10.1016/s0969-2126(00)00106-4. [DOI] [PubMed] [Google Scholar]
- 26.Breiter DR, Resnik E, Banaszak LJ. Engineering the quaternary structure of an enzyme: construction and analysis of a monomeric form of malate dehydrogenase from Escherichia coli. Protein Sci. 1994;3:2023–2032. doi: 10.1002/pro.5560031115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Batra R, Khayat R, Tong L. Molecular mechanism for dimerization to regulate the catalytic activity of human cytomegalovirus protease. Nat Struct Biol. 2001;8:810–817. doi: 10.1038/nsb0901-810. [DOI] [PubMed] [Google Scholar]
- 28.Remmele RL, Jr, Callahan WJ, Krishnan S, Zhou L, Bondarenko PV, Nichols AC, Kleemann GR, Pipes GD, Park S, Fodor S, Kras E, Brems DN. Active dimer of Epratuzumab provides insight into the complex nature of an antibody aggregate. J Pharm Sci. 2006;95:126–145. doi: 10.1002/jps.20515. [DOI] [PubMed] [Google Scholar]
- 29.Hearne JL, Colman RF. Catalytically active monomer of class mu glutathione transferase from rat. Biochemistry. 2006;45:5974–5784. doi: 10.1021/bi060249e. [DOI] [PubMed] [Google Scholar]
- 30.Vanathi P, Mishra AK, Bhargava P. Regulation of activity of the yeast TATA-binding protein through intra-molecular interactions. J Biosci. 2003;28:413–421. doi: 10.1007/BF02705116. [DOI] [PubMed] [Google Scholar]
- 31.Depaux A, Regnier-Ricard F, Germani A, Varin-Blank N. Dimerization of hSiah proteins regulates their stability. Biochem Biophys Res Commun. 2006;348:857–863. doi: 10.1016/j.bbrc.2006.07.092. [DOI] [PubMed] [Google Scholar]