

Abstract
Background
The retinoblastoma tumor suppressor (RB) is a key regulator of cell cycle progression and is functionally inactivated in the majority of human non-small cell lung cancers (NSCLC). The specific influence of RB on therapeutic response in NSCLC remains elusive.
Materials and Methods
We investigated the consequence of re-introduction of RB on checkpoint response and chemosensitivity in NSCLC cell lines. RB introduction into RB-proficient (NCI-H1299) and -deficient (H1734, H2172) NSCLC cells was achieved by adenoviral infection. RB/E2F target gene expression was determined by immunoblot analysis. Cell cycle response and viability after chemotherapeutic exposure were assessed by flow cytometry and MTT viability assay.
Results
RB reconstitution in RB-deficient lines restored regulation of topoIIα, TS, and cyclin A. Similarly, RB overexpression in RB-proficient cells caused further regulation of some RB/E2F target genes including TS and topoIIα. In addition, RB overexpression resulted in restoration of the G1 arrest mechanism. Exposure of RB-proficient cells to cisplatin, etoposide, or 5-FU elicited arrest in various phases of the cell cycle while lines deficient for RB exhibited different checkpoint responses. However, introduction of RB restored ability to arrest following chemotherapeutic exposure. Chemotherapeutic challenge resulted in varying effects on cellular viability independent of RB status, yet restoration of RB activity conferred partial chemoresistance.
Conclusions
These results demonstrate that Rb reconstitution into RB-deficient NSCLC lines establishes regulation of certain RB/E2F target genes and restores G1 arrest mechanisms. Furthermore, introduction of RB enhances the G1 checkpoint response to chemotherapeutics and decreases chemosensitivity. Knowledge of RB-dependent chemosensitivity may ultimately contribute to individualized therapy based on molecular characterization of tumors.
Introduction
Lung cancer is the greatest cancer killer in the world, causing over one million deaths annually worldwide [1]. More than 170,000 new cases are diagnosed every year in the United States alone [2]. RB inactivation is among the most common abnormalities in lung cancer [3]. Non-small cell lung cancer (NSCLC), the histologic subtype which accounts for over 80% of lung cancers, exhibits RB inactivation via a diversity of mechanisms including mutation [4, 5], deregulated phosphorylation through abnormal CDK4/Cyclin D expression, and loss of p16INK4A activity by aberrant promoter methylation [6] or homozygous deletions or point mutations [7, 8].
RB functions by regulating transcription of numerous target genes, thereby controlling progression through the cell cycle. RB impacts cell cycle progression through transcriptional co-repression, acting on specific promoters [9–11]. For example, RB associates with the E2F family of transcriptional regulators that induce cell cycle progression [12]. When bound by active RB, the function of E2F as a transcriptional activator is antagonized. Upon disruption of this repressor complex, the antiproliferative activity of RB is negated [11, 13, 14].
The activity of RB is modulated by its degree of phosphorylation. In early G1, RB is in an active hypophosphorylated state, during which it inhibits cell cycle progression. When exposed to mitogenic signals, RB is phosphorylated by the action of CDK4 and D cyclins [15, 16]. Subsequent phosphorylation by CDK2/Cyclin E results in hyperphosphorylated RB [17]. These combined events functionally inactivate RB and facilitate progression into S phase [17]. Appropriate coordination of the cell cycle maintains genomic integrity by ensuring faithful replication and partitioning of the genome [18, 19]. Inactivation of cell cycle control mechanisms predisposes cells to the development of genomic instability and cancer [20, 21].
Loss of RB function leads to deregulation of cell cycle control such that cells respond inappropriately to challenge with chemotherapy [22, 23]. The basis for this is not completely understood, but discrete targets of RB-mediated transcriptional repression include genes involved in cell cycle control, and DNA repair. Using microarray analysis of RNA from cells harboring an inducible constitutively-active RB, our has group identified over 200 targets of RB-mediated repression, the majority of which are involved in cell cycle control, DNA repair, and transcription/chromatin structure [24]. Among the targets of RB-mediated repression are certain molecular targets of chemotherapeutic agents. For example, thymidylate synthase (TS) is the target of 5-fluorouracil (5-FU) and topoisomerase IIα (topoIIα) is the target of etoposide (VP-16). This finding, as well as the prior observation that absence of RB causes elevation in TS levels resulting in resistance to 5-fluorodeoxyuridine (which is metabolized to 5-FU) [25], suggested that deregulation of specific RB targets could contribute to altered chemosensitivity. Our group subsequently exploited the Cre/LoxP system for targeted disruption of Rb, thereby showing that RB mediates chemotherapy-induced cell cycle inhibition in adult fibroblasts [26, 27].
Together, these data support the proposition that functional deregulation of RB disrupts cell cycle checkpoint control in human cancers, thereby altering chemosensitivity and survival. In the majority of NSCLC, RB is functionally inactivated through a diversity of mechanisms. Chemotherapy is the primary treatment for advanced NSCLC; it also plays an important adjuvant role after surgical resection of early disease. NSCLC is thus an ideal system to evaluate the effect of discrete mechanisms of RB inactivation on therapeutic response. We therefore hypothesized that RB replacement into RB-deficient NSCLC lines would delineate a role for RB in chemotherapeutic response.
Materials and Methods
Cell Culture
NSCLC lines NCI-H1299, H1734, and H2172, and human embryonic kidney cells transformed with adenovirus type 5 (HEK 293 cells) were purchased from American Type Culture Collection (Manassas, Virginia). The lung cancer cells were grown at 37°C with a 10% CO2 atmosphere in RPMI 1640 (Mediatech, Inc., Herndon, VA) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, penicillin (100 units/mL, Mediatech), and streptomycin (100 μg/mL, Mediatech). HEK 293 cells were grown in DMEM (Mediatech) supplemented as above.
Construction of Recombinant Adenoviral Vector Containing Rb
The retinoblastoma gene (Rb) was excised from pCMV-wtRb [28] using BamHI. The shuttle vector pShuttleCMV-wtRb was constructed by ligating the full length Rb cDNA obtained above into the BglII site of pShuttleCMV (Qbiogene, Inc., Irvine, CA) using T4 DNA ligase (New England Biolabs, Inc., Beverly, MA). The cloned shuttle vector was transformed into DH5α E. coli for amplification. Plasmid DNA was isolated from E. coli using QIAGEN Mini Kits (QIAGEN, Inc., Valencia, CA) and insertion was confirmed by digestion with PacI. Digestion products were resolved in a 1% agarose gel, stained with ethidium bromide, and visualized under UV light. Orientation of the insert was confirmed by PCR analysis, using the primers CMV Forward 5′GGCGTGTACGGTGGGAGG3′ and Rb Reverse 5′TAACCAAGCTCTCTCTCTGA3′ (Integrated DNA Technologies, Inc., Coralville, IA), and were resolved on agarose gels as above. 10 μg of pShuttleCMV-Rb or pShuttleCMV-Empty (control virus) were linearized with PmeI and resolved on a 1% agarose gel. Linearized plasmids were purified using QIAEX II gel extraction kit (QIAGEN). BJ5183 E. coli were transformed with the AdEasy vector (Qbiogene) grown in low salt media and prepared to be electrocompetent. Purified plasmid DNA (1.5 μg) was electroporated (Bio-Rad Gene Pulser II, Bio-Rad Laboratories, Hercules, CA) at 200 Ohms, 25 μF, and 2.25 kV, into the transformed BJ5183 bacteria containing pAd5ΔE1/ΔE3 (AdEasy vector) and plated on LB agar plates with 30μg/mL kanamycin (Sigma, St. Louis, MO). Colonies were expanded and plasmid DNA was isolated as described above. Recombination was confirmed by digestion with PacI and resolved in agarose as above. Recombinant adenoviral plasmids pAd-Rb and pAd-Empty were electroporated as above into DH5α E. coli for large scale amplification. Plasmid DNA was isolated using QIAGEN Midi Kit (QIAGEN), linearized with PacI, and transfected into HEK 293 cells using FuGene 5 Transfection Reagent (Roche Diagnostics Corp., Indianapolis, IN) for propagation.
Purification and Infection of Recombinant Adenovirus
Ad-Rb and Ad-Empty were purified by double cesium chloride gradient and viral titers were determined by the tissue culture infectious dose 50 (TCID50) method as described (Qbiogene). NSCLC lines in logarithmic growth phase were infected at multiplicities of infection (moi) of 100 pfu/cell.
Immunoblot Analysis
Untreated cells were in logarithmic growth phase when harvested. Infections were performed one day after seeding and were harvested 72 hours post-infection. All cell lines were harvested by trypsinization and washed once with PBS after harvesting. Lysis was performed in RIPA buffer containing 150 mM sodium chloride (Fisher Scientific Co., Pittsburgh, PA), 1% NP40 (Amresco, Solon, OH), 0.5% deoxycholate (Sigma), 0.1% SDS (Fisher) and 50 mM tris chloride pH 8.0 (Fisher), with protease inhibitors: PMSF (1 mM, Sigma), NaF (5 mM, Sigma), β-glycerophosphate (13 mg/mL, Sigma), sodium orthovanadate (120 μg/mL, Sigma), benzamide-HCl (10 μM, Sigma), 1,10 phenanthroline HCl (10 μg/mL, Sigma), aprotinin (10 μg/mL, Sigma), leupeptin (10 μg/mL, Roche), and pepstatin (10 μg/mL, Sigma). Samples were then sonicated to complete lysis and stored at −80°C. Protein concentrations were determined by protein assay kit (Bio-Rad), and equal amounts of protein were resolved by SDS-PAGE, and transferred onto nitrocellulose membranes. Specific protein detection was performed with human RB antibody (851 polyclonal antisera [23]), β-Tubulin D-10 mouse monoclonal antibody (Santa Cruz Biotechnologies Inc., Santa Cruz, CA), topoisomerase IIα H-231 polyclonal antibody (Santa Cruz), thymidylate synthase (TS) polyclonal antibody (Taiho Pharmaceutical Co., Ltd., Kawauchi, Tokushima, Japan), and cyclin A H-432 polyclonal antibody (Santa Cruz) followed by horseradish peroxidase-conjugated secondary antibodies (Pierce Biotechnologies Inc., Rockford, IL) and electrochemiluminescence (ECL) reagents (Perkins Elmer Life Sciences Inc., Boston, MA).
Cellular Viability Assays and Chemotherapeutic Treatment
On day 0, NSCLC lines were seeded on 60 mm plates. On day 1, cell lines were infected and PBS (250 μL) was added to uninfected cell lines. On day 2, cells were seeded into 96-well plates at 5,000 cells per well in 90 μL RPMI 1640 medium supplemented as above. Cells were plated in the presence of 16 μM cisplatin (cis-diaminedichloroplatinum II (CDDP)) (Ben Venue Laboratories, Inc., Bedford, OH), 12 μM etoposide (VP-16) (Sigma), 150 μM 5-fluorouracil (5-FU) (Sigma), or PBS (10% v/v final concentration for untreated and positive controls). On day 4, viability was determined by adding 3-(4,5-dimethylthiazol-2-yl)-2,5-dibenzyltetrozolium bromide (MTT) reagent (50 μg in 10 μL PBS) to each well. Cells were incubated for 4 hours then solubilized with 110 μL of 10% Triton X-100 (AMRESCO), 0.1 N HCl (Fisher) in anhydrous isopropanol (Fisher). Colorimetric analysis was performed at 570 nm, and values for control cells were considered as 100% viable. Change in viability was calculated as Ad-Rb infected cells compared to Ad-Empty controls.
Flow Cytometry
On day 0, cells were plated on 100 mm plates and allowed to adhere overnight. On day 1, they were infected or treated with PBS (uninfected control) as above. On day 2, infected and uninfected cell lines were treated with PBS, CDDP, VP-16, or 5-FU as above. On day 4, cells were harvested by trypsinization, washed with PBS, and fixed in ice-cold 70% ethanol. Cells were then washed again with PBS and resuspended in 200 μL PBS, then stained with propidium iodide (1 μg/mL in PBS, Sigma) and RNaseA (40 μg/mL, QIAGEN) 30 minutes before DNA content was analyzed by flow cytometry (FACS Coulter Epics XL, Beckman Coulter, Miami, FL). To verify cell cycle distribution, cells were labeled with BrdU for 4 hours prior to fixation and PI/BrdU bivariate flow cytometric analyses were performed as previously described [29]. The fraction of G1, S, and G2/M populations was determined by analysis using ModFitLT 2.0 (Verity Software House, Topsham, ME) and the sum was normalized to 100%. The increase in the G1 population was determined by comparing Ad-Rb infected cells to Ad-Empty control cells.
Results
Absence of functional RB is not associated with E2F regulated gene products in NSCLC lines
Loss of Rb results in deregulated expression of a host of target genes in cells containing an inducible (tetracycline-regulated) system [24], as well as with targeted disruption of Rb using the Cre-LoxP system [27]. In order to determine if RB level alone determines the expression of corresponding genes, such as cyclin A, TS, and topoIIα, in human lung cancer cells, we utilized a panel of NSCLC lines. RB-deficient lines (NCI-H1734, and H2172) were compared to an RB-proficient line (H1299) [30]. Immunoblot analysis verified the expression of RB in the H1299 cells and its absence in the H1734 and H2172 cells (Fig. 1). RB-deficient cells exhibited elevated levels of topoIIα compared to the RB-proficient line. However, TS and cyclin A levels were comparable in RB-proficient and -deficient lines. Thus, the genetic targets of RB/E2F appear to be variably influenced by RB in the setting of human lung cancer.
FIG. 1.
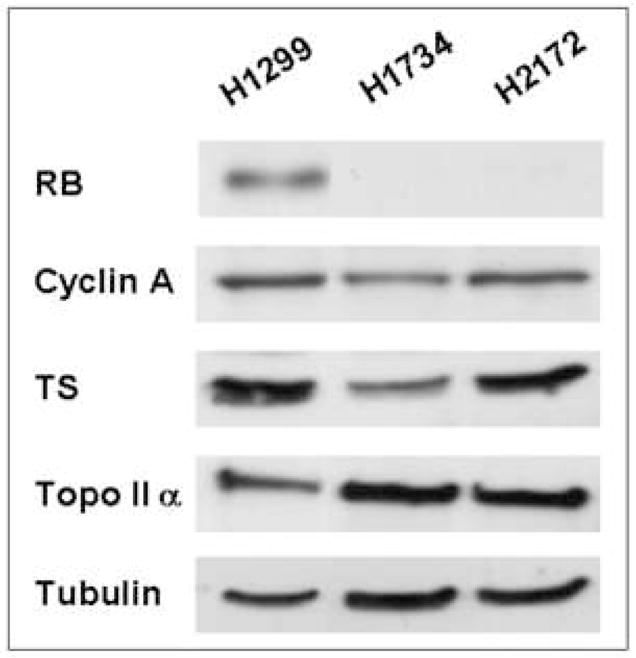
Absence of functional RB is not directly associated with E2F regulated gene products in NSCLC lines. RB-deficient cells (H1734 and H2172) exhibit deregulated levels topoIIα, but not of cyclin A and TS, compared to RB-proficient cells (H1299). Tubulin serves as a loading control. TS = thymidylate synthase; topoIIα = topoisomerase IIα.
Chemotherapeutic exposure of NSCLC cells invokes checkpoint responses unrelated to RB expression
In order to evaluate whether RB status influences the checkpoint responses of NSCLC lines, cells were treated with three distinct chemotherapeutic agents: cisplatin (CDDP) etoposide (VP-16) and 5-fluorouracil (5-FU). Cisplatin is used therapeutically for lung cancer and the in vitro checkpoint response to this agent has been shown to be RB-dependent [22, 23]. Etoposide was evaluated because it has been used in the clinical management of lung cancer and because the molecular target of the drug, topoIIα, is regulated by RB. TS, the target of 5-FU, is also regulated by RB and this agent was therefore tested.
Cell cycle distributions of the three cell lines were assessed by flow cytometric analysis of propidium iodide-labeled cells after 48 hours of chemotherapeutic exposure (16 μM CDDP, 12 μM VP-16, or 150 μM 5-FU), and G1, S, and G2/M-phase percentages were verified using bivariate (PI/BrdU) flow cytometric analysis. There was no absolute relationship between S-phase entry (i.e. proliferative capacity) and RB when comparing the three different cell lines (Fig. 2A). For example, the arrest mechanism induced by cisplatin exposure yielded predominantly S- and G2/M-phase accumulation in the RB-proficient cells compared to the two RB-deficient lines in which the S-phase arrest was slightly less pronounced. Similarly, when compared to untreated cells, 5-FU exposure of the RB-proficient line yielded a cell cycle block in S phase, while the two RB-deficient lines had minimal change in cell cycle distribution. Etoposide induced a G2/M arrest in the RB-proficient H1299 cells, as well as the RB-deficient H2172 cells, but resulted in an S-phase block in the RB-deficient H1734 cells. Cell cycle distributions are summarized, in Figure 2A where G1, S, and G2/M-phase percentages are reported. Thus, the G1 arrest mechanism which is classically controlled by RB activity was not clearly demonstrable in the setting of the assorted checkpoints that were induced by the chemotherapeutic agents, particularly when comparing cell lines containing an otherwise diverse genetic background. While the checkpoints caused by certain chemotherapeutic treatments appear to depend on RB status, this is not consistently demonstrated by comparing different cell lines.
FIG. 2.
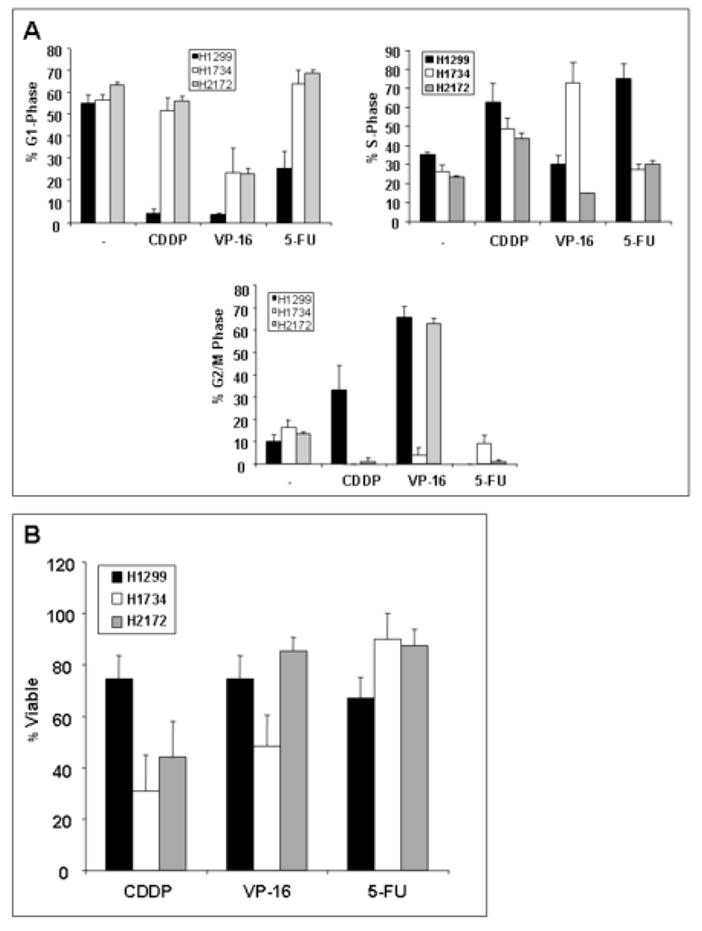
Chemotherapeutic exposure in NSCLC cells invokes variable checkpoint responses and altered cellular viability unrelated to RB expression. (A) Calculated G1, S, and G2/M-phase percentages of propidium iodide-stained cells after chemotherapeutic treatment are shown. (B) MTT assays were utilized to calculate cellular viability. RB-deficient cells (H1734 and H2172) are more sensitive to CDDP treatment then RB-proficient cells (H1299). VP-16 and 5-FU lead to diminished viability independent of RB status when separate cell lines are compared. All values represent average percentages of at least three independent experiments. Error bars indicate one standard deviation. CDDP = cisplatin; VP-16 = etoposide; 5-FU = 5-fluorouracil.
The viability of NSCLC cell lines was assessed after 48 hours of treatment with chemotherapeutic agents as described above. In vitro cellular viability after chemotherapeutic treatment was measured using the MTT assay. Cisplatin caused significantly decreased cellular viability in the RB-deficient setting (H1734 and H2172) (Fig. 2B). However, etoposide resulted in no significant change in viability between the RB-proficient and -deficient lines. Furthermore, 5-FU exposure did not cause altered viability depending on RB status, despite titrating the concentration to levels much higher than required for diminished viability in other cells lines [27]. Therefore, RB status appears to determine sensitivity to cisplatin. But RB alone does not account for differences in chemosensitivity to etoposide and 5-FU when comparing separate lung cancer cell lines.
RB reconstitution restores control of RB/E2F target genes and results in G1 accumulation
In order to more specifically study the role of RB in NSCLC, we manipulated RB expression to evaluate its effects in a consistent genetic background. Recombinant adenovirus was used to reconstitute RB activity in RB-deficient lung cancer cells (Fig. 3A). Adenoviral delivery of RB (Ad-RB) (lanes 4 and 6) downregulated TS, cyclin A, and topoIIα in RB-deficient NSCLC cells compared to controls (lanes 3 and 5). Even in an RB-proficient background (H1299), RB overexpression (lane 2) resulted in further downregulation of RB target genes. Thus, re-introduction of RB activity into RB-deficient lung cancer cells is sufficient to restore regulation of certain RB/E2F target genes. The cell cycle distribution after RB reconstitution was then determined by flow cytometric analysis. Adenoviral delivery of Rb increased the G1 population, particularly in the RB-deficient lung cancer cells (Fig. 3B). Calculated cell cycle percentages are given as G1-S-G2 in Figure 3B. Interestingly, infection with the control virus (Ad-Empty) resulted in a slight alteration in cell cycle distribution, manifested as a trend toward an increase in S-phase. We therefore used the Ad-Empty infected cells as the control for all experiments involving RB reconstitution via adenoviral delivery. These results demonstrate that adenoviral delivery of Rb effectively restores RB activity in NSCLC lines.
FIG. 3.
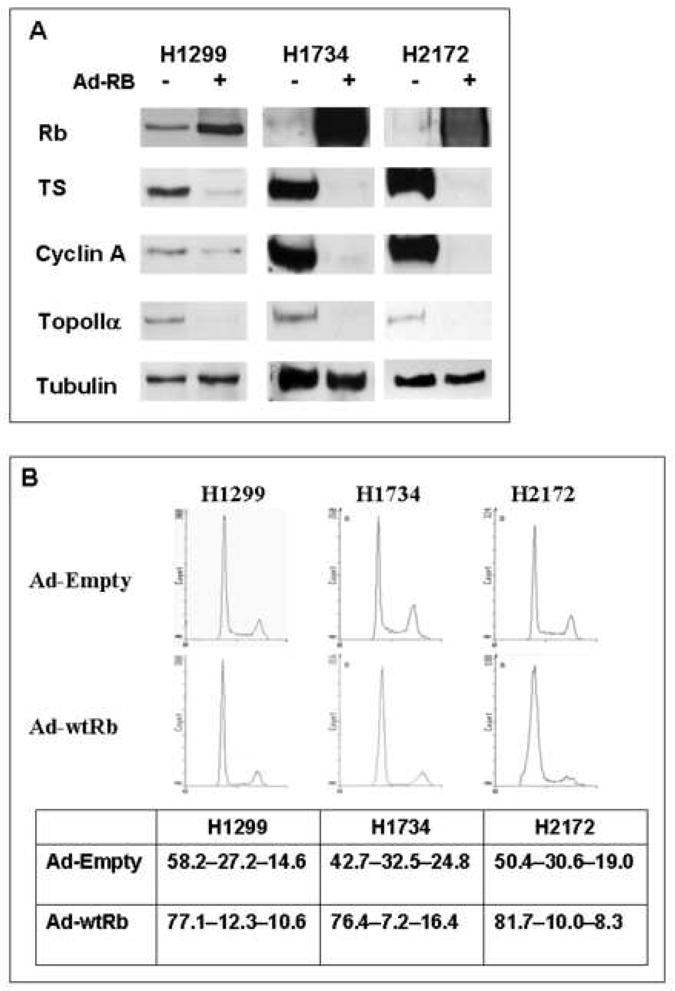
RB reconstitution restores control of RB/E2F target genes and results in G1 accumulation. (A) Adenoviral delivery of RB restores regulation of TS, cyclin A, and topoIIα expression levels in RB-deficient NSCLC cells (H1734 and H2172) compared to control infection. Even in an RB-proficient background (H1299), RB overexpression results in further downregulation of RB/E2F target genes. Tubulin is used as a loading control. Lanes 1, 3, 5 = Ad-Empty (−); lanes 2, 4, 6 = Ad-RB (+); TS = thymidylate synthase; TopoIIα = topoisomerase II alpha. (B) Adenoviral delivery of RB increases the G1 population, particularly in RB-deficient cells (H1734 and H2172). Histograms represent 10,000 cells. Calculated cell cycle distributions are represented as percentages (G1-S-G2/M). Values in the table represent the average of three independent experiments.
RB reconstitution potentiates the G1 arrest in response to chemotherapeutic exposure and decreases chemosensitivity
Cell cycle checkpoint responses resulting from chemotherapeutics were analyzed after adenoviral delivery of Rb or the empty control. In the two RB-deficient cell lines, RB overexpression increased the G1 population after exposure to cisplatin, etoposide, and, to a lesser extent, 5-FU (Fig. 4A). Therefore, reconstitution of RB activity restores a chemotherapeutic-induced G1 checkpoint mechanism in lung cancer cells.
FIG. 4.
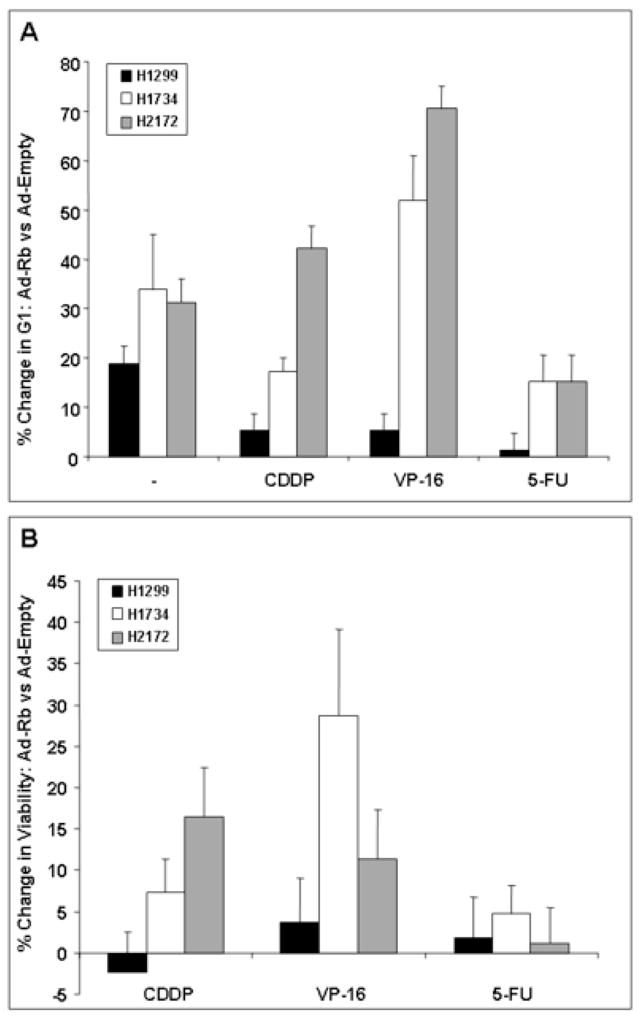
RB reconstitution leads to increased efficiency of the G1 arrest mechanism and increased cellular viability in response to chemotherapeutic exposure. (A) Graph represents the increase in G1 population in Ad-RB infected cells compared to Ad-Empty (control) virus, and exposed to chemotherapeutic challenge. In the RB-deficient cells (H1734 and H2172), RB overexpression (compared to the control infected cells) increases the G1 population after exposure to chemotherapeutic agents thereby restoring G1 checkpoint activity. (B) MTT assays were utilized to determine cellular viability of Ad-RB infected cells versus Ad-Empty (control) infection after chemotherapeutic exposure. Return of RB function via adenoviral delivery leads to decreased chemosensitivity in RB-deficient cells (H1734 and H2172) to varying degrees depending on the agent and the cell line. All values represent the average of three independent experiments and error bars represent one standard deviation. CDDP = cisplatin; VP-16 = etoposide; 5-FU = 5-fluorouracil.
After RB restoration, cellular viability after chemotherapeutic treatment was assessed using the MTT assay (Fig. 4B). The difference in viability attributable to RB reconstitution (compared to Ad-Empty control) was determined in the setting of chemotherapeutic exposure. RB overexpression in the RB-deficient NSCLC cells yielded a moderate decrease in chemosensitivity (represented by increased viability) compared to the empty vector control, although the extent of chemoresistance depended on the cell line and the chemotherapeutic agent. For example, the H1734 cells are relatively resistant to cisplatin compared to H2172 cells (Fig. 2B). When RB is reconstituted, this difference is sustained (Fig. 4B). Notably, the H1734 cells harbor intact p53 (RB−/p53+) while p53 is absent in the H2172 line (RB−/p53−). Thus, other determinants of chemoresponsiveness, such as the p53 pathway, certainly contribute to regulation of the chemotherapeutic effect and may account for variation depending on the cell line and the therapeutic agent. However, RB overexpression in the RB-proficient line resulted in no alteration in chemosensitivity. Hence, RB activity is able to alter chemosensitivity in NSCLC lines to varying degrees, even in the genetic background of diverse mutations present in NSCLC lines.
Discussion
Mutation or functional inactivation of RB is frequent in human malignancies including lung cancer [3–8, 20, 31]. A critical role of RB is checkpoint control, which includes arrest mechanisms invoked after genotoxic insults such as chemotherapeutic exposure. While chemotherapy has been employed for decades in the treatment of metastatic lung cancer, recent clinical studies have defined an important role for chemotherapy in all but the earliest stage of NSCLC [32, 33]. Thus, in order to improve the design of efficacious treatment modalities, it is imperative to define the mechanism by which deregulation of RB checkpoint control in NSCLC modifies responsiveness to chemotherapy. In this study we restored RB expression in RB-deficient NSCLC cells (Fig. 3). Re-introduction of RB restored the regulation of RB/E2F target gene products (Fig. 3A). RB overexpression, particularly in the RB-deficient cells, also led to a G1 arrest (Fig. 3B). We then determined the effect of RB re-introduction on checkpoint responses to chemotherapeutic treatments. Re-introduction of RB activity in RB-deficient NSCLC cells caused an increase in G1 accumulation upon chemotherapeutic treatment (Fig. 4A). We also assessed cellular viability after exposure to chemotherapeutics. Restoration of RB activity in RB-deficient cells conferred increased chemoresistance (Fig 4B). Together these results demonstrate that return of RB activity in RB-deficient NSCLC lines establishes regulated expression of RB/E2F target genes and confers a G1 arrest. Furthermore, return of RB function enhances the G1 checkpoint response to chemotherapeutics and decreases chemosensitivity.
Prior studies of the role of RB in chemotherapeutic response have lacked an ideal model system. One approach has been to focus on one cancer type and to correlate RB status among different lines with chemoresponsiveness [25]. This strategy is limited by the diverse genetic background present in human tumors, thus precluding the attribution of findings specifically to the activity of RB. Our group and others have therefore used the approach of specifically manipulating RB in an otherwise consistent genetic background. For example, mice harboring defined genetic loss of RB have been engineered. Since homozygous germline deletion of RB causes embryonic lethality, such studies have been limited to cells obtained early in embryonic development, such as murine embryo fibroblasts (MEFs) [22, 23, 34]. This system was utilized to compare Rb+/+ to Rb−/− MEFs and showed that cell cycle arrest in G1/S invoked after genotoxic insults are RB-dependent. A confounding issue proposed for this model was that other pocket proteins, namely p107 and p130, possess some functional redundancy with RB and might compensate for chronic RB loss. Thus, our group took the approach of acute removal of RB activity using a Cre/LoxP system to target disruption of Rb [26, 27]. In murine adult fibroblasts, chemotherapy-induced cell cycle inhibition was determined to be RB-dependent even with acute RB loss. In this study, we show that RB status alone in NSCLC lines does not determine the cell cycle response to chemotherapeutic exposure; however, re-introduction of RB into the RB-deficient cells increases G1 accumulation in response to chemotherapeutic exposure and decreases chemosensitivity
While the central role of RB activity on cell cycle progression and its activity as a transcriptional co-repressor are well-accepted, the known genetic targets of RB repression were limited. Therefore, Markey and colleagues used an unbiased approach of RNA microarray analysis comparing cells of identical genetic background, only differing in RB status [24]. Exploiting a tetracycline-inducible constitutively active RB, a panel of target genes (e.g. TS, cyclin A, cyclin E) exhibited RB-dependence. Here we have demonstrated that reintroduction of RB into RB-deficient NSCLC cells restores the regulation of RB/E2F target genes. Furthermore, loss of RB in fibroblasts compromises the checkpoint response to DNA damaging agents with distinct primary targets (ionizing radiation, topoisomerase I inhibitors, alkylating agents), as well as antimetabolites (5-FU, methotrexate, hydroxyurea) [26, 27, 35, 36]. In RB-deficient NSCLC cells, similar agents induce checkpoint responses that are reconstituted by RB replacement.
In contrast to murine fibroblast models, human tumors possess a multiplicity of genetic anomalies. Indeed, in the evolution of human tumors, loss of RB activity is usually not the initiating event, but occurs in conjunction with other genetic lesions such as loss of p53 function [37]. Likewise, human NSCLC typically possesses multiple mutations [3]. Thus, specific manipulation of RB in the genetic background of a human neoplasm can offer a system that more accurately models the clinical setting in which the effect of RB on checkpoint control is relevant. Our approach to replace RB in RB-deficient lines has been used by others [25, 38, 39]. Yet, it is possible that overexpressed RB after adenoviral delivery may not respond to the complex regulation to which the endogenous protein is normally subjected. In fact, this possibility is somewhat borne out by the demonstration of further downregulation of RB target genes in the H1299 (RB-proficient) cells. However, this also suggests that the targets of RB activity are appropriately responsive to the overexpressed RB, indicating an intact control mechanism. A comparable effect is also seen with an increased G1 population when RB was overexpressed in the H1299 cells. Thus, RB replacement by adenoviral induction may not exactly replicate reintroduction of physiological RB level; however, it models a mechanism of altering checkpoints specific to RB re-introduction.
In conclusion, this study demonstrates that RB activity affects chemoresponsiveness in NSCLC cells. Despite a variety of checkpoints evoked by chemotherapeutics in NSCLC lines, RB restoration caused G1 phase accumulation, thus demonstrating that downstream effector mechanisms of RB activity are sufficiently intact to achieve a consistent cell cycle arrest. Additionally, reconstitution of RB activity restores the regulation of RB/E2F target genes, alters checkpoint responses to chemotherapeutics, and leads to decreased chemosensitivity. Knowledge of RB-dependent chemoresponsiveness may ultimately contribute to individualized targeting of lung cancer therapy based on molecular characterization of human tumors.
Acknowledgments
MFR is supported by an American Cancer Society Institutional Grant (#92-026-09). ESK is supported by an NIH grant (CA106471). Susanne Wells provided valuable assistance with the preparation of adenoviral constructs. We are grateful to members of the Knudsen laboratories for technical, administrative, and logistical support. We are particularly indebted to C. Mayhew and A. K. McClendon for critical review of the manuscript.
Footnotes
Presented at the 38th Annual Meeting of the Association for Academic Surgery in Houston, TX, November 11-13, 2004.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carney DN. Lung Cancer -- Time to Move on from Chemotherapy. N Engl J Med. 2002;346:126. doi: 10.1056/NEJM200201103460211. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Thomas A, Murray T, Thun M. Cancer Statistics. CA Cancer J Clin. 2002;52:23. doi: 10.3322/canjclin.52.1.23. [DOI] [PubMed] [Google Scholar]
- 3.Meyerson M, Franklin WA, Kelley MJ. Molecular classification and molecular genetics of human lung cancers. Semin Oncol. 2004;31:4. doi: 10.1053/j.seminoncol.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 4.Cagle PT, el-Naggar AK, Xu HJ, Hu SX, Benedict WF. Differential retinoblastoma protein expression in neuroendocrine tumors of the lung. Potential diagnostic implications. Am J Pathol. 1997;150:393. [PMC free article] [PubMed] [Google Scholar]
- 5.Dosaka-Akita H, Hu SX, Fujino M, et al. Altered retinoblastoma protein expression in nonsmall cell lung cancer: its synergistic effects with altered ras and p53 protein status on prognosis. Cancer. 1997;79:1329. [PubMed] [Google Scholar]
- 6.Zochbauer-Muller S, Fong KM, Virmani AK, Geradts J, Gazdar AF, Minna JD. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001;61:249. [PubMed] [Google Scholar]
- 7.Marchetti A, Buttitta F, Pellegrini S, et al. Alterations of P16 (MTS1) in node-positive non-small cell lung carcinomas. J Pathol. 1997;181:178. doi: 10.1002/(SICI)1096-9896(199702)181:2<178::AID-PATH741>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 8.Shapiro GI, Park JE, Edwards CD, et al. Multiple mechanisms of p16INK4A inactivation in non-small cell lung cancer cell lines. Cancer Res. 1995;55:6200. [PubMed] [Google Scholar]
- 9.Harbour JW, Dean DC. Rb function in cell-cycle regulation and apoptosis. Nat Cell Biol. 2000;2:E65. doi: 10.1038/35008695. [DOI] [PubMed] [Google Scholar]
- 10.Wang JY, Knudsen ES, Welch PJ. The retinoblastoma tumor suppressor protein. Adv Cancer Res. 1994;64:25. doi: 10.1016/s0065-230x(08)60834-9. [DOI] [PubMed] [Google Scholar]
- 11.Zhang HS, Postigo AA, Dean DC. Active transcriptional repression by the Rb-E2F complex mediates G1 arrest triggered by p16INK4a, TGFbeta, and contact inhibition. Cell. 1999;97:53. doi: 10.1016/s0092-8674(00)80714-x. [DOI] [PubMed] [Google Scholar]
- 12.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 13.Strobeck MW, Fribourg AF, Puga A, Knudsen ES. Restoration of retinoblastoma mediated signaling to Cdk2 results in cell cycle arrest. Oncogene. 2000;19:1857. doi: 10.1038/sj.onc.1203510. [DOI] [PubMed] [Google Scholar]
- 14.Zhang HS, Gavin M, Dahiya A, et al. Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell. 2000;101:79. doi: 10.1016/S0092-8674(00)80625-X. [DOI] [PubMed] [Google Scholar]
- 15.Ewen ME, Sluss HK, Sherr CJ, Matsushime H, Kato J, Livingston DM. Functional interactions of the retinoblastoma protein with mammalian D- type cyclins. Cell. 1993;73:487. doi: 10.1016/0092-8674(93)90136-e. [DOI] [PubMed] [Google Scholar]
- 16.Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993;7:331. doi: 10.1101/gad.7.3.331. [DOI] [PubMed] [Google Scholar]
- 17.Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859. doi: 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- 18.Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- 19.Nyberg KA, Michelson RJ, Putnam CW, Weinert TA. Toward maintaining the genome: DNA damage and replication checkpoints. Annu Rev Genet. 2002;36:617. doi: 10.1146/annurev.genet.36.060402.113540. [DOI] [PubMed] [Google Scholar]
- 20.Bartkova J, Lukas J, Bartek J. Aberrations of the G1- and G1/S-regulating genes in human cancer. Prog Cell Cycle Res. 1997;3:211. doi: 10.1007/978-1-4615-5371-7_16. [DOI] [PubMed] [Google Scholar]
- 21.Flatt PM, Pietenpol JA. Mechanisms of cell-cycle checkpoints: at the crossroads of carcinogenesis and drug discovery. Drug Metab Rev. 2000;32:283. doi: 10.1081/dmr-100102335. [DOI] [PubMed] [Google Scholar]
- 22.Harrington EA, Bruce JL, Harlow E, Dyson N. pRB plays an essential role in cell cycle arrest induced by DNA damage. Proc Natl Acad Sci U S A. 1998;95:11945. doi: 10.1073/pnas.95.20.11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knudsen KE, Booth D, Naderi S, et al. RB-dependent S-phase response to DNA damage. Mol Cell Biol. 2000;20:7751. doi: 10.1128/mcb.20.20.7751-7763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markey MP, Angus SP, Strobeck MW, et al. Unbiased Analysis of RB-mediated Transcriptional Repression Identifies Novel Targets and Distinctions from E2F Action. Cancer Res. 2002;62:6587. [PubMed] [Google Scholar]
- 25.Li W, Fan J, Hochhauser D, et al. Lack of functional retinoblastoma protein mediates increased resistance to antimetabolites in human sarcoma cell lines. Proc Natl Acad Sci U S A. 1995;92:10436. doi: 10.1073/pnas.92.22.10436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bosco EE, Mayhew CN, Hennigan RF, Sage J, Jacks T, Knudsen ES. RB signaling prevents replication-dependent DNA double-strand breaks following genotoxic insult. Nucleic Acids Res. 2004;32:25. doi: 10.1093/nar/gkg919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mayhew CN, Perkin LM, Zhang X, Sage J, Jacks T, Knudsen ES. Discrete signaling pathways participate in RB-dependent responses to chemotherapeutic agents. Oncogene. 2004;23:4107. doi: 10.1038/sj.onc.1207503. [DOI] [PubMed] [Google Scholar]
- 28.Knudsen ES, Wang JY. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J Biol Chem. 1996;271:8313. doi: 10.1074/jbc.271.14.8313. [DOI] [PubMed] [Google Scholar]
- 29.Knudsen ES, Buckmaster C, Chen TT, Feramisco JR, Wang JY. Inhibition of DNA synthesis by RB: effects on G1/S transition and S- phase progression. Genes Dev. 1998;12:2278. doi: 10.1101/gad.12.15.2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimizu E, Coxon A, Otterson GA, et al. RB protein status and clinical correlation from 171 cell lines representing lung cancer, extrapulmonary small cell carcinoma, and mesothelioma. Oncogene. 1994;9:2441. [PubMed] [Google Scholar]
- 31.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 32.The International Adjuvant Lung Cancer Trial Collaborative Group. Cisplatin-Based Adjuvant Chemotherapy in Patients with Completely Resected Non-Small-Cell Lung Cancer. N Engl J Med. 2004;350:351. doi: 10.1056/NEJMoa031644. [DOI] [PubMed] [Google Scholar]
- 33.Winton T, Livingston R, Johnson D, et al. Vinorelbine plus cisplatin vs. observation in resected non-small-cell lung cancer. N Engl J Med. 2005;352:2589. doi: 10.1056/NEJMoa043623. [DOI] [PubMed] [Google Scholar]
- 34.Almasan A, Yin Y, Kelly RE, et al. Deficiency of retinoblastoma protein leads to inappropriate S-phase entry, activation of E2F-responsive genes, and apoptosis. Proc Natl Acad Sci U S A. 1995;92:5436. doi: 10.1073/pnas.92.12.5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayhew CN, Bosco EE, Solomon DA, Knudsen ES, Angus SP. Analysis of RB Action in DNA Damage Checkpoint Response. Methods Mol Biol. 2004;281:3. doi: 10.1385/1-59259-811-0:003. [DOI] [PubMed] [Google Scholar]
- 36.Bosco EE, Knudsen ES. Differential role of RB in response to UV and IR damage. Nucleic Acids Res. 2005;33:1581. doi: 10.1093/nar/gki283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shackney SE, Shankey TV. Common patterns of genetic evolution in human solid tumors. Cytometry. 1997;29:1. [PubMed] [Google Scholar]
- 38.Schnier JB, Nishi K, Goodrich DW, Bradbury EM. G1 arrest and down-regulation of cyclin E/cyclin-dependent kinase 2 by the protein kinase inhibitor staurosporine are dependent on the retinoblastoma protein in the bladder carcinoma cell line 5637. Proc Natl Acad Sci U S A. 1996;93:5941. doi: 10.1073/pnas.93.12.5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shinohara H, Zhou J, Yoshikawa K, et al. Retinoblastoma protein-initiated cellular growth arrest overcomes the ability of cotransfected wild-type p53 to induce apoptosis. Br J Cancer. 2000;83:1039. doi: 10.1054/bjoc.2000.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]