

Abstract
Background: Novel molecular therapies for metastatic breast cancer (MBC) are necessary to improve the dismal prognosis of this condition. Imatinib mesylate (Gleevec®) inhibits several protein tyrosine kinases, including platelet-derived growth factor receptor (PDGFR) and c-kit, which are preferentially expressed in tumor cells. We tested the activity of imatinib mesylate in MBC with overexpression of PDGFR or c-kit. Additionally, we sought to determine the biological correlates and immunomodulatory effects.
Patients and methods: Thirteen patients were treated with Imatinib administered orally at 400 mg p.o. b.i.d. (800 mg/day), until disease progression. All patients demonstrated PDGFR-β overexpression and none showed c-kit expression.
Results: No objective responses were observed among the 13 patients treated in an intention-to-treat analysis. All patients experienced disease progression, with a median time to progression of 1.2 months. Twelve patients have died, and the median overall survival was 7.7 months. No patient had a serious adverse event. Imatinib therapy had no effect on the plasma levels of the angiogenesis-related cytokines, vascular endothelial growth factor, PDGF, b-fibroblast growth factor, and E-selectin. Immune studies showed imatinib inhibits interferon-γ production by TCR-activated CD4+ T cells.
Conclusion: Imatinib as a single agent has no clinical activity in PDGFR-overexpressing MBC and has potential immunosuppressive effects.
Keywords: c-kit expression, imatinib, immune-suppression, metastatic breast cancer
introduction
Metastatic breast cancer (MBC) is an incurable condition; current medical treatments have provided appropriate palliation but are unable to eradicate this disease [1, 2]. It is hoped that expanded knowledge of the biological bases of breast carcinogenesis and the mechanisms of cancer progression will lead to the development of more directed and effective treatment of this disease. The use of tyrosine kinase inhibitors (TKIs) may represent one such attempt at targeted ‘translational’ research.
Among these inhibitors, imatinib (STI571, Gleevec®) is the first successful, rationally developed, receptor-targeted agent for chronic myelogenous leukemia (CML) [3]. Imatinib inhibits the constitutively active Bcr–Abl tyrosine kinase protein encoded by the fusion gene generated by the Philadelphia chromosome translocation, which is responsible for the pathogenesis of the disease [3]. Interestingly, recent in vitro studies have suggested a possible negative immunomodulatory effect of imatinib therapy that is likely related to the drug's effect on T-cell-specific kinases [4–6]. Imatinib also inhibits c-kit and platelet-derived growth factor receptor (PDGFR) kinases, with affinities similar to those described for the Bcr–Abl kinases [7, 8] C-kit encodes for KIT (CD117), a 145- to 160-kDa transmembrane receptor tyrosine kinase that plays an important role in the development of gastrointestinal stromal tumors, small-cell lung cancer, melanoma, and breast cancer [9–12].
PDGFR expression has been demonstrated in malignant breast tissue and surrounding stromal cells, including pericytes that support blood vessels [13, 14]. In preclinical studies, imatinib has shown antitumor activity in a breast carcinoma model, particularly in osteolytic bone metastases [15, 16]. Because breast cancer has been shown to variably express PDGFR and c-kit, we investigated the clinical activity of imatinib in women with MBC that expressed c-kit or PDGFR-β or both. Additionally, we sought to determine the biological correlates [17–19] and immunomodulatory effects associated with the administration of imatinib in women with breast cancer [4–6].
patients and methods
patient population
A prospective, open-label phase II study of imatinib for MBC was conducted at The University of Texas M. D. Anderson Cancer Center from September 2002 to February 2003. Eligible patients included those with measurable MBC, who were ≥18 years of age, had normal organ and marrow functions, had a score of ≤2 on the Eastern Cooperative Oncology Group performance status scale or a Karnofsky index of >60%, had received at least one and not more than two prior chemotherapy regimens for metastatic disease, had received treatment with both an anthracycline and a taxane either as adjuvant or for advanced disease, and had a life expectancy of >12 weeks. Moreover, patients were required to have a prescreening assessment for c-kit (CD117) and PDGFR-β expression by the metastatic lesion as only patients with demonstrable expression of c-kit and/or PDGFR-β were considered for enrollment and treatment. Patients were excluded from the study if they had brain metastasis (or other symptomatic evidence of central nervous system disease) or if bone metastasis was the only disease site that could be evaluated. The Cancer Therapy Evaluation Program of the National Cancer Institute (CTEP/NCI) and M. D. Anderson's Institutional Review Board approved the protocol.
study design
Patients received imatinib mesylate [supplied by Novartis Pharmaceutical Corporation (Cambridge, MA) through CTEP/NCI] at a dose of 400 mg by mouth b.i.d. (800 mg/day) taken with a meal. Patients were treated continuously on a 4-week cycle. Treatment was discontinued for progression or severe toxicity. Dose reductions were permitted for patients with intolerable non-hematologic grade 2 toxicity or any grade 3 or 4 toxicity. If imatinib dose reduction was required, doses were reduced in 100-mg increments. Recurrent toxicity of similar severity resulted in another dose reduction, but patients who required more than two dose reductions or who had any delay of ≥2 weeks in scheduled therapy as a result of toxicity were withdrawn from the study. All patients were required to have absolute neutrophil counts >1500/μl and platelet counts of >75 000/μl in order to receive treatment on day 1 of each cycle of therapy.
Patients were reevaluated for response with standard imaging studies (computed tomography scans) every 8 weeks. In addition to a baseline scan, confirmatory scans were obtained 4 weeks following initial documentation of objective response. Definitions of response and disease progression were according to Response Evaluation Criteria in Solid Tumors (RECIST) [20].
pathology studies
Immunohistiochemical staining was carried out on 4-μm sections cut from a representative paraffin block specimen of the invasive breast carcinoma. Immunostaining for c-kit and PDGFR-β was carried out in a DAKO autostainer with the LSAB-2 peroxidase kit (DAKO Corporation, Carpinteria, CA). Tissue sections were subjected to antigen retrieval using citrate buffer (pH 6.0) before immunostaining. Primary antibodies were used against c-kit (DAKO cytoautomation, dilution of 1 : 400) and PDGFR-β (Santa Cruz biotechnology Inc., dilution of 1 : 50). For phosphorylated PDGFR-β, we carried out epitope retrieval using EDTA solution (pH 8.0) and stained the tissue sections with antibodies against phosphorylated PDGFR-β (Cell Signaling, Danvers, MA, 01923), dilution of 150, using the Biocare detection system. The chromogen utilized for detection of immunostaining was diaminobenzidine. Positivity for the markers was recognized as granular brown cytoplasmic staining. The extent of immunostaining was expressed as the percentage of positively stained tumor cells.
laboratory correlates
Peripheral blood samples from patients were collected at baseline, at 2–4 weeks after therapy and beyond 5 weeks after imatinib therapy. Whole blood was used for determining lymphocyte immunophenotypes by flow cytometry. Moreover, peripheral blood mononuclear cells (PBMCs) and plasma were separated from the whole blood for the detection of cytokine synthesis by activated T-cells and circulating biomarkers of angiogenesis, respectively.
measurement of biomarkers of angiogenesis
Plasma was analyzed in batches for interleukin (IL)-8, PDGF, tumor necrosis factor-alpha (TNF-α), transforming growth factor-beta, E-selectin, vascular endothelial growth factor (VEGF), and basic fibroblast growth factor (b-FGF), using commercially available ELISA kits (R&D Systems, Minneapolis, MN) and the assays were run according to the manufacturer's instructions. Samples were run in duplicate, and the amount of each analyte was interpolated from a standard curve constructed with known concentrations of the particular analyte.
cell surface immunophenotypes of T cells
Total peripheral blood leukocyte counts and immunophenotypes of peripheral blood lymphocytes were determined pre- and postimatinib therapy. Percentages of total T cells (CD3+), helper T (CD4+), and suppressor/cytotoxic T (CD8+) cells were determined by flow cytometry. Absolute numbers of CD3+, CD4+, and CD8+ T cells were calculated to determine the effect of imatinib therapy.
intracellular cytokine synthesis by TCR-activated T-cells
The effect of imatinib therapy on T-cell function was studied in patients and the results compared with those of normal blood donors who were employees or graduate students at the M. D. Anderson Cancer Center. Normal donors of matched age were recruited by word of mouth, provided consent for a phlebotomy, and received a monetary compensation for their blood. PBMCs were activated with immobilized anti-CD3 antibody to measure cytokine syntheses by CD4+ T and CD8+ T cells, as we previously described [6].
statistical analysis
The primary objective of this study was to determine the efficacy of imatinib in MBC that demonstrated expression of CD117 (c-kit) and/or PDGFR as measured by objective tumor response. Secondary objectives included evaluating progression-free survival (PFS) of patients and the toxicity and tolerability of imatinib and defining plasma, tissue, and imaging surrogate end points of activity. In addition, we carried out correlative studies to identify potential surrogate markers of response.
The primary clinical end point was achievement of an objective tumor response (complete and partial responses) as determined by the RECIST [20]. To be of interest for further study, imatinib would have to produce a response rate of at least 30% and would have not been considered of interest for further study if the response rate was <10%. Based on Simon's optimal two-stage design [21] with false-positive and false-negative error rates of 10%, a maximum of 35 patients were to be accrued with one interim analysis planned after the first 12 patients were evaluated for response. The study would be continued to the second stage if at least two patients achieved an objective response. If the trial continued to the maximum of 35 patients, at least six responses would be required for imatinib to be considered of interest for further study.
PFS was measured from the start of treatment to the date of disease progression. Overall survival (OS) was measured from the start of treatment to the date of death from any cause or to the date of last follow-up. Estimates of time to progression and of OS were calculated using the Kaplan–Meier method with 95% confidence intervals (CIs).
Statistical differences in median levels of plasma cytokines, median percentages of leukocyte immunophenotypes, and median percentages of TCR-activated CD4+ T and CD8+ T cells that synthesized cytokines before and after initiation of therapy with imatinib were determined by the Mann–Whitney test. Evaluations were done at baseline, after 2–4 weeks and after 4 weeks. All specimens were compared with normal controls.
All analyses were carried out using SAS version 9.1 for Windows (SAS Institute, Cary, NC), S-Plus version 7 (Insightful Corp.)
results
patient characteristics
The characteristics of the 13 patients enrolled in this trial are presented in Table 1. All patients demonstrated PDGFR-β overexpression and none showed c-kit expression (Figure 1 A and B). Eight patients (61%) had visceral metastasis and seven patients (54%) had already received chemotherapy for metastatic disease.
Table 1.
Patients characteristics
| Characteristics | n (%) |
| Sex | |
| Female | 12 |
| Male | 1 |
| Performance status (ECOG) | |
| 0–1 | 12 |
| 2 | 1 |
| Age, years | |
| Median (range) | 52 (38–63) |
| Histology | |
| Ductal carcinoma | 11 |
| Lobular carcinoma | 2 |
| Site of metastasis | |
| Visceral | 4 |
| Bone/soft tissue | 3 |
| Both | 4 |
| Prior therapy for metastasis | |
| None | 6 |
| Chemotherapy | 7 |
| Hormonal therapy | 4 |
ECOG, Eastern Cooperative Oncology Group.
Figure 1.
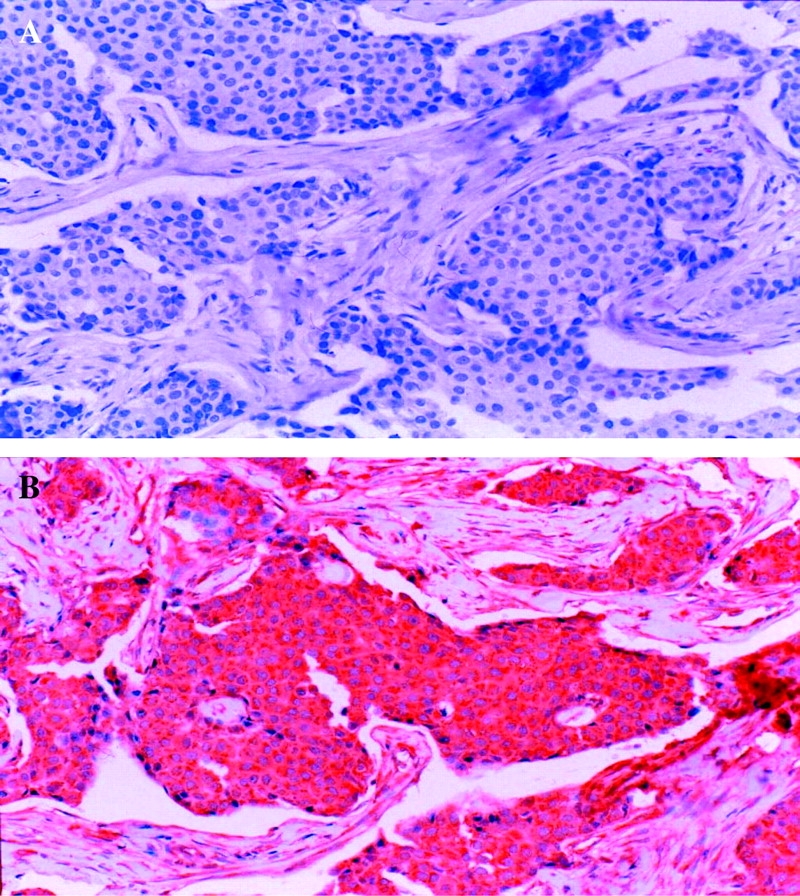
(A) A representative section of invasive ductal carcinoma immunostained for c-kit. Note that the tumor cells are entirely negative for c-kit. (B) A representative section of invasive ductal carcinoma immunostained for platelet-derived growth factor receptor (PDGF)β. The tumor cells show diffuse and strong cytoplasmic positivity for PDGFβ.
response and survival
No objective responses were included in the intention-to treat analysis among the 13 patients treated. One of the patients discontinued treatment at home after a few doses and was initially considered not assessable. This allowed for an additional patient to be enrolled as for initial design. The subsequent information obtained on that patient suggested an early progression of disease and therefore 13 patients were included in the final intention-to-treat analysis. All patients experienced disease progression, with a median time to progression of 1.2 months (Figure 2). One patient with lobular carcinoma (hormone receptor positive) had biopsy-confirmed liver disease and was stable for >335 days. At 6 months, all but one patient had experienced a disease progression (PFS at 6 months = 8%, 95% CI = 1%, 51%). Twelve patients had died, and the median OS was 7.7 months (Figure 3). OS at 6 months was 62% (95% CI = 40%, 95%), and at 12 months it was 38% (95% CI = 19%, 77%).
Figure 2.

Kaplan–Meier representation of time to progression for patients with metastatic breast cancer treated with imatinib mesylate.
Figure 3.

Kaplan–Meier representation of overall survival in the intention-to-treat analysis for patients with metastatic breast cancer treated with imatinib mesylate.
toxicity
The toxicity profile of patients who received imatinib consisted mainly of general symptoms and gastrointestinal and hematologic toxicity (Table 2). Most adverse events were mild to moderate in intensity. The most severe events requiring dose reduction included vomiting and edema (Table 2).
Table 2.
Toxicity/adverse event with imatinib
| Any grade | Grade III/IV | |
| No. of patients (%) | No. of patients (%) | |
| Nausea | 10 | 1 |
| Diarrhea | 7 | 1 |
| Vomiting | 6 | 4 |
| Constipation | 5 | 1 |
| Edema | 5 | 1 |
| Stomatitis | 3 | 0 |
| Granulocytopenia | 3 | 3 |
| Infection without | ||
| Myalgia | 5 | 1 |
| Rash/desquamation | 3 | 0 |
| Neutropenia | 2 | 0 |
| Thrombocytopenia | 1 | 0 |
| Anemia | 1 | 0 |
| Arthralgia | 1 | 2 |
| Dyspnea | 1 | 1 |
correlative studies
serum cytokines.
Imatinib therapy had no effect on the plasma levels of the angiogenesis-related cytokines, particularly VEGF, PDGF, b-FGF, and E-selectin (Figure 4 A–D). There were no significant differences in the median levels of IL-8 and TNF-α before and after therapy with imatinib.
Figure 4.

Plasma levels of (A) vascular endothelial growth factor, (B) platelet-derived growth factor, (C) basis fibroblast growth factor, and (D) E-selectin in metastatic breast cancer patients treated with imatinib mesylate.
cell surface immunophenotypes of T cells.
Imatinib had no effect on the percentage and absolute number of peripheral blood lymphocytes or on the percentages of CD3+ pan-T cells, CD4+ T, and CD8+ T cells. However, there was a noticeable, but not statistically significant, decline in the absolute number of total CD3+ pan-T cells by 2–4 weeks after therapy (1003/μl versus 839/μl), which reached a statistically significant decline after 4 weeks of therapy with imatinib (1003/μl versus 736/μl; P = 0.035) (Table 3).
Table 3.
Effect of imatinib therapy on peripheral blood T-lymphocyte subsets
| Parameter | Median (range) relative to initiation of imatinib therapy |
||
| Pretreatment | 2–4 after | >4 weeks after | |
| White blood cell per microliter | 4.9 (3.1, 6.7) | 4.2 (2.9, 6.3) | 3.7 (1.7, 9.6) |
| % Lymphocytes | 24.0 (6.0, 41.0) | 22.0 (5.0, 69.0) | 20.0 (7.0, 53.0) |
| % CD3+ T | 69.9 (59.4, 81.2) | 71.9 (47.5, 84.1) | 70.1 (56.6, 86.2) |
| % CD4+ T | 45.6 (20.0, 58.4) | 46.5 (35.4, 57.7) | 51.1 (29.1, 59.1) |
| % CD8+ T | 22.2 (12.1, 39.7) | 22.8 (12.0, 27.1) | 20.9 (13.0, 32.5) |
| Lymph per microliter | 1372 (868, 1876) | 1176 (812, 1764) | 1022 (476, 2688) |
| CD3+ T per microliter | 1003 (580, 1460) | 839 (591, 1233) | 736a (410, 1784) |
| CD4+ T per microliter | 560 (252, 1064) | 602 (348, 849) | 5.2 (1.0, 18.3) |
| CD8+ T per microliter | 298 (136, 744) | 241 (198, 369) | 235 (134, 482) |
Pretreatment versus >4 weeks after imatinib therapy, P = 0.035.
T-cell activation by anti-CD3 through the TCR.
The ability of CD4+ T and CD8+ T cells to synthesize IL-2, interferon (IFN)-γ, TNF-α, or IL-10 in response to activation through the TCR was determined for 29 samples obtained from 13 patients. The breakdown of the samples consisted of 11 samples obtained at from patients at baseline, eight samples collected after 2–4 weeks of therapy, and 10 samples after >4 weeks of therapy with imatinib. After >4 weeks of therapy with imatinib, PBMC cultures had a decrease in the median percentage of TCR-activated CD4+ T cells, and not CD8+ T cells, that synthesized IFN-γ, which evolved into a statistically significant decrease in the median percentage of activated CD4+ T cells that synthesized IFN-γ (8.0% versus 3.6%; P = 0.032). Therapy with imatinib had no effect on the percentages of CD4+ T and CD8+ T cells that synthesized IL-2, TNF-α, or IL-10 following activation through the TCR with anti-CD3 antibody (Table 4).
Table 4.
Effect of imatinib therapy on cytokine syntheses by peripheral blood CD4+ and CD8+ T cells activated through the T-cell receptor
| Cell type | Cytokine synthesized | Median percentages (range) of TCR-activated T-cell subsets synthesizing cytokines relative to start of therapy |
||
| Pretreatment | 2–4 weeks after | >4 weeks after | ||
| CD8+ T | IL-2 | 4.4 (0.0, 11.7) | 3.0 (0.3, 7.9) | 3.0 (1.4, 13.0) |
| IFN-γ | 9.2 (1.1, 19.4) | 6.9 (3.1, 14.8) | 8.7 (2.3, 15.1) | |
| TNF-α | 9.7 (1.4, 17.1) | 6.6 (1.2, 18.0) | 5.2 (1.0, 18.3) | |
| IL-10 | 2.5 (0.3, 6.2) | 5.6 (1.5, 15.8) | 6.9 (2.2, 10.2) | |
| CD4+ T | IL-2 | 7.0 (0.1, 10.7) | 3.7 (1.3, 8) | 7.3 (1.7, 11.0) |
| IFN-γ | 8.0 (1.9, 16.2) | 5.2 (3.2, 12.6) | 3.6a (0.56, 15.8) | |
| TNF-α | 12.2 (0.4, 17.8) | 9.0 (2.1, 19.8) | 11.7 (3.2, 22.1) | |
| IL-10 | 3.3 (0.1, 5.0) | 6.7 (1.6, 31.1) | 4.2 (1.6, 23.4) | |
Pretreatment versus >4 weeks after imatinib therapy, P = 0.032.
discussion
The use of molecularly targeted therapies with mAbs or orally bioavailable, low-molecular weight TKIs has demonstrated the capacity of these therapies to improve the outcome of patients with both primary and MBC [22, 23]. The molecular ‘promiscuity’ of imatinib has expanded its therapeutic role in different tumor types supported through expression or persistent activation of c-kit and/or PDGFR tyrosine kinases [7, 14, 24–26]. Imatinib demonstrated extremely effective in gastrointestinal stromal tumors in which ∼90% of tumor show c-kit abnormally express or mutated [27]. In invasive breast cancer, c-kit is rarely expressed while PDGFR is quite commonly overexpressed both in tumor and stromal cells, albeit at different levels [9, 10, 28–31]. These receptors and their downstream effectors trigger a cascade that regulates cell proliferation, differentiation, and survival [32]. In preclinical settings, the growth inhibitory effect of imatinib is related to a dose-dependent decrease of activation of PDGFR-β and Akt [33].
In this study, we demonstrated that imatinib as a single agent lacks clinical activity in patients with MBC selected for tissue expression of either PDGFR-β (c-kit was not expressed). A few other clinical trials have tested imatinib either as a single agent or in combination regimens for MBC. Modi et al. [34] reported similar results with the same imatinib schedule as we used in 16 unselected patients; in their study, only one patient was c-kit positive and four were PDGFR positive. Toxicity in that study, mainly gastrointestinal, was significant in a heavily pretreated study group [34]. In our series, one patient with extensive liver involvement experienced a long stabilization (335 days) of disease. Unfortunately, a liver biopsy obtained at disease progression was impossible to repeat, making any speculation as to the reason for the stabilization useless.
In breast cancer, c-kit expression is found infrequently (1%–13%) but varies across the different tumor subtypes [28, 29]. The majority of c-kit-positive breast tumors appear to belong to the basal-like breast cancer subtype [35, 36]. By mRNA expression, c-kit is one of the best basal-specific markers; however, by immunohistochemistry, many of the tumors that showed c-kit mRNA were not positive for c-kit protein expression [29]. C-kit-positive metastatic breast carcinoma accounts for <5% of breast carcinomas and appears to belong to the basal-like subtype of breast cancer. Mutational analysis of epidermal growth factor and c-kit receptor genes in this disease has been recently presented [37]. Expression of c-kit was detectable in ∼25% of the tumors, but mutations predictive of responsiveness to imatinib were absent. PDGFRs are pivotal in peritumoral vasculature, stroma, and bone [13, 24, 25, 38, 39]. In another study, signaling through PDGF-β receptors was shown to increase interstitial fluid pressure and therefore to affect tumor cells' chemosensitivity [14]. Inhibition of this pathway by imatinib can enhance drug delivery to the tumor [40]. In vitro evidence from another study suggested that inhibition of the PDGF pathway would decrease the concentration of VEGF and other proangiogenic molecules [41]. The clinical significance of plasma levels of angiogenic factors like VEGF, b-FGF, PDGF, and E-selectin is presently unknown. In our study, we did not observe any statistically significant changes in the plasma levels of these angiogenesis-related molecules with imatinib therapy even though others have reported the ability of imatinib to decrease these factors.
Conflicting in vitro and in vivo results have been published on the negative immune-modulating effect of imatinib mesylate; this effect is likely related to a direct or indirect effect of the drug on T-cell-specific kinases [4–6, 42–44]. In preclinical tumor models, stimulation and proliferation assays showed these kinases to be inhibited, and a reduction in delayed hypersensitivity was demonstrated [5, 42]. On the other hand, whereas a positive effect of imatinib on bone marrow-derived antigen-presenting cells' restoring the responsiveness of tolerant T-cells from tumor-bearing hosts has been reported [44], we found imatinib to inhibit the synthesis of IFN-γ by TCR-activated CD4+ T cells. This was not unexpected as we previously found imatinib to interfere with the signaling of TCR-activated CD4+ T cells [6].
Retrospective analyses of patients with CML receiving imatinib demonstrated a relative low incidence of reactivation of herpes zoster infections [45]. In the current study, alteration in T-cell number and function as demonstrated by a decrease in the absolute number of CD3+ T cells and by TCR-activated CD4+ T cells synthesizing IFN-γ were observed. These data are consistent with other reports in hematological malignancies [4, 6] even though the consequences of immunological manifestations in patients with solid tumors are presently unknown. However, according to another study, no significant increase in adverse infectious events was reported at a 5-year follow-up in CML patients treated with imatinib [46].
In conclusion, our results indicate that targeting PDGFR with imatinib mesylate monotherapy in MBC is ineffective. Future studies should consider a careful selection of patients with c-kit overexpression (possibly basal-like breast cancers) and concomitant administration of chemotherapy because it appears unlikely to demonstrate single-agent activity. Nevertheless, our findings regarding immune functions should be taken into account in cases of concomitant administration of imatinib with chemotherapy in previously treated patients with advanced disease [40].
funding
NCI (N01-CM-17003) and NCI/CTEP Translational Research Initiative.
References
- 1.Ellis MHD, Lippman ME. Treatment of metastatic disease. In: Harris J LM, Morrow M, et al., editors. Diseases of the Breast. 2nd edition. Philadelphia: Lippincott-Raven; 2000. pp. 749–799. [Google Scholar]
- 2.Greenberg PA, Hortobagyi GN, Smith TL, et al. Long-term follow-up of patients with complete remission following combination chemotherapy for metastatic breast cancer. J Clin Oncol. 1996;14:2197–2205. doi: 10.1200/JCO.1996.14.8.2197. [DOI] [PubMed] [Google Scholar]
- 3.O'Dwyer ME, Druker BJ. Chronic myelogenous leukaemia—new therapeutic principles. J Intern Med. 2001;250:3–9. doi: 10.1046/j.1365-2796.2001.00823.x. [DOI] [PubMed] [Google Scholar]
- 4.Appel S, Boehmler AM, Grunebach F, et al. Imatinib mesylate affects the development and function of dendritic cells generated from CD34+ peripheral blood progenitor cells. Blood. 2004;103:538–544. doi: 10.1182/blood-2003-03-0975. [DOI] [PubMed] [Google Scholar]
- 5.Dietz AB, Souan L, Knutson GJ, et al. Imatinib mesylate inhibits T-cell proliferation in vitro and delayed-type hypersensitivity in vivo. Blood. 2004;104:1094–1099. doi: 10.1182/blood-2003-12-4266. [DOI] [PubMed] [Google Scholar]
- 6.Gao H, Lee BN, Talpaz M, et al. Imatinib mesylate suppresses cytokine synthesis by activated CD4 T cells of patients with chronic myelogenous leukemia. Leukemia. 2005;19:1905–1911. doi: 10.1038/sj.leu.2403933. [DOI] [PubMed] [Google Scholar]
- 7.Heinrich MC, Blanke CD, Druker BJ, Corless CL. Inhibition of KIT tyrosine kinase activity: a novel molecular approach to the treatment of KIT-positive malignancies. J Clin Oncol. 2002;20:1692–1703. doi: 10.1200/JCO.2002.20.6.1692. [DOI] [PubMed] [Google Scholar]
- 8.Wang WL, Healy ME, Sattler M, et al. Growth inhibition and modulation of kinase pathways of small cell lung cancer cell lines by the novel tyrosine kinase inhibitor STI 571. Oncogene. 2000;19:3521–3528. doi: 10.1038/sj.onc.1203698. [DOI] [PubMed] [Google Scholar]
- 9.Chui X, Egami H, Yamashita J, et al. Immunohistochemical expression of the c-kit proto-oncogene product in human malignant and non-malignant breast tissues. Br J Cancer. 1996;73:1233–1236. doi: 10.1038/bjc.1996.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DiPaola RS, Kuczynski WI, Onodera K, et al. Evidence for a functional kit receptor in melanoma, breast, and lung carcinoma cells. Cancer Gene Ther. 1997;4:176–182. [PubMed] [Google Scholar]
- 11.Lammie A, Drobnjak M, Gerald W, et al. Expression of c-kit and kit ligand proteins in normal human tissues. J Histochem Cytochem. 1994;42:1417–1425. doi: 10.1177/42.11.7523489. [DOI] [PubMed] [Google Scholar]
- 12.Tsuura Y, Hiraki H, Watanabe K, et al. Preferential localization of c-kit product in tissue mast cells, basal cells of skin, epithelial cells of breast, small cell lung carcinoma and seminoma/dysgerminoma in human: immunohistochemical study on formalin-fixed, paraffin-embedded tissues. Virchows Arch. 1994;424:135–141. doi: 10.1007/BF00193492. [DOI] [PubMed] [Google Scholar]
- 13.Bhardwaj B, Klassen J, Cossette N, et al. Localization of platelet-derived growth factor beta receptor expression in the periepithelial stroma of human breast carcinoma. Clin Cancer Res. 1996;2:773–782. [PubMed] [Google Scholar]
- 14.Ostman A. PDGF receptors-mediators of autocrine tumor growth and regulators of tumor vasculature and stroma. Cytokine Growth Factor Rev. 2004;15:275–286. doi: 10.1016/j.cytogfr.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Roussidis AE, Mitropoulou TN, Theocharis AD, et al. STI571 as a potent inhibitor of growth and invasiveness of human epithelial breast cancer cells. Anticancer Res. 2004;24:1445–1447. [PubMed] [Google Scholar]
- 16.Lev DC, Kim SJ, Onn A, et al. Inhibition of platelet-derived growth factor receptor signaling restricts the growth of human breast cancer in the bone of nude mice. Clin Cancer Res. 2005;11:306–314. [PubMed] [Google Scholar]
- 17.Benoy IH, Salgado R, Van Dam P, et al. Increased serum interleukin-8 in patients with early and metastatic breast cancer correlates with early dissemination and survival. Clin Cancer Res. 2004;10:7157–7162. doi: 10.1158/1078-0432.CCR-04-0812. [DOI] [PubMed] [Google Scholar]
- 18.Sheen-Chen SM, Chen WJ, Eng HL, Chou FF. Serum concentration of tumor necrosis factor in patients with breast cancer. Breast Cancer Res Treat. 1997;43:211–215. doi: 10.1023/a:1005736712307. [DOI] [PubMed] [Google Scholar]
- 19.Salven P, Manpaa H, Orpana A, et al. Serum vascular endothelial growth factor is often elevated in disseminated cancer. Clin Cancer Res. 1997;3:647–651. [PubMed] [Google Scholar]
- 20.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 21.Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials. 1989;10:1–10. doi: 10.1016/0197-2456(89)90015-9. [DOI] [PubMed] [Google Scholar]
- 22.Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733–2743. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 23.Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 24.Pietras K, Sjoblom T, Rubin K, et al. PDGF receptors as cancer drug targets. Cancer Cell. 2003;3:439–443. doi: 10.1016/s1535-6108(03)00089-8. [DOI] [PubMed] [Google Scholar]
- 25.Roussidis AE, Theocharis AD, Tzanakakis GN, Karamanos NK. The importance of c-Kit and PDGF receptors as potential targets for molecular therapy in breast cancer. Curr Med Chem. 2007;14:735–743. doi: 10.2174/092986707780090963. [DOI] [PubMed] [Google Scholar]
- 26.Went PT, Dirnhofer S, Bundi M, et al. Prevalence of KIT expression in human tumors. J Clin Oncol. 2004;22:4514–4522. doi: 10.1200/JCO.2004.10.125. [DOI] [PubMed] [Google Scholar]
- 27.Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626–632. doi: 10.1200/JCO.2007.13.4452. [DOI] [PubMed] [Google Scholar]
- 28.Sihto H, Sarlomo-Rikala M, Tynninen O, et al. KIT and platelet-derived growth factor receptor alpha tyrosine kinase gene mutations and KIT amplifications in human solid tumors. J Clin Oncol. 2005;23:49–57. doi: 10.1200/JCO.2005.02.093. [DOI] [PubMed] [Google Scholar]
- 29.Simon R, Panussis S, Maurer R, et al. KIT (CD117)-positive breast cancers are infrequent and lack KIT gene mutations. Clin Cancer Res. 2004;10:178–183. doi: 10.1158/1078-0432.ccr-0597-3. [DOI] [PubMed] [Google Scholar]
- 30.Ulivi P, Zoli W, Medri L, et al. c-kit and SCF expression in normal and tumor breast tissue. Breast Cancer Res Treat. 2004;83:33–42. doi: 10.1023/B:BREA.0000010694.35023.9e. [DOI] [PubMed] [Google Scholar]
- 31.Yared MA, Middleton LP, Meric F, et al. Expression of c-kit proto-oncogene product in breast tissue. Breast J. 2004;10:323–327. doi: 10.1111/j.1075-122X.2004.21351.x. [DOI] [PubMed] [Google Scholar]
- 32.Dibb NJ, Dilworth SM, Mol CD. Switching on kinases: oncogenic activation of BRAF and the PDGFR family. Nat Rev Cancer. 2004;4:718–727. doi: 10.1038/nrc1434. [DOI] [PubMed] [Google Scholar]
- 33.Mundhenke C, Weigel MT, Meinhold-Heerlein I, et al. Imatinib mesylate inhibits the growth of breast cancer cells mediated by PDGF-receptor-β and Akt inactivation. Breast Cancer Rest Treat. 2006;100(Suppl 1) (Abstr 4118) [Google Scholar]
- 34.Modi S, Seidman AD, Dickler M, et al. A phase II trial of imatinib mesylate monotherapy in patients with metastatic breast cancer. Breast Cancer Res Treat. 2005;90:157–163. doi: 10.1007/s10549-004-3974-0. [DOI] [PubMed] [Google Scholar]
- 35.Nielsen TO, Hsu FD, Jensen K, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10:5367–5374. doi: 10.1158/1078-0432.CCR-04-0220. [DOI] [PubMed] [Google Scholar]
- 36.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gilbert JA, Goetz MP, Giordano KF, et al. Mutational analysis of the epidermal growth factor receptor and stem cell factor receptor genes in metaplastic breast carcinoma. Breast Cancer Rest Treat. 2006;100(Suppl 1) (Abstr 3025) [Google Scholar]
- 38.Carvalho I, Milanezi F, Martins A, et al. Overexpression of platelet-derived growth factor receptor alpha in breast cancer is associated with tumour progression. Breast Cancer Res. 2005;7:R788–R795. doi: 10.1186/bcr1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hotte SJ, Winquist EW, Lamont E, et al. Imatinib mesylate in patients with adenoid cystic cancers of the salivary glands expressing c-kit: a Princess Margaret Hospital phase II consortium study. J Clin Oncol. 2005;23:585–590. doi: 10.1200/JCO.2005.06.125. [DOI] [PubMed] [Google Scholar]
- 40.Yardley DA, Barton JH, Liggett W, et al. Preliminary results from a phase II pilot of imatinib mesylate with weekly docetaxel in metastatic breast cancer. Breast Cancer Rest Treat. 2006;100(Suppl 1) (Abstr 6068) [Google Scholar]
- 41.Legros L, Bourcier C, Jacquel A, et al. Imatinib mesylate (STI571) decreases the vascular endothelial growth factor plasma concentration in patients with chronic myeloid leukemia. Blood. 2004;104:495–501. doi: 10.1182/blood-2003-08-2695. [DOI] [PubMed] [Google Scholar]
- 42.Cwynarski K, Laylor R, Macchiarulo E, et al. Imatinib inhibits the activation and proliferation of normal T lymphocytes in vitro. Leukemia. 2004;18:1332–1339. doi: 10.1038/sj.leu.2403401. [DOI] [PubMed] [Google Scholar]
- 43.Reuben JM, Lee BN, Johnson H, et al. Restoration of Th1 cytokine synthesis by T cells of patients with chronic myelogenous leukemia in cytogenetic and hematologic remission with interferon-alpha. Clin Cancer Res. 2000;6:1671–1677. [PubMed] [Google Scholar]
- 44.Wang H, Cheng F, Cuenca A, et al. Imatinib mesylate (STI-571) enhances antigen-presenting cell function and overcomes tumor-induced CD4+ T-cell tolerance. Blood. 2005;105:1135–1143. doi: 10.1182/blood-2004-01-0027. [DOI] [PubMed] [Google Scholar]
- 45.Mattiuzzi GN, Cortes JE, Talpaz M, et al. Development of Varicella-Zoster virus infection in patients with chronic myelogenous leukemia treated with imatinib mesylate. Clin Cancer Res. 2003;9:976–980. [PubMed] [Google Scholar]
- 46.Druker BJ, Guilhot F, O'Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]