

Abstract
Reduction of nitrite to nitric oxide during ischemia protects the heart against injury from ischemia/reperfusion. However the optimal dose of nitrite and the mechanisms underlying nitrite-induced cardioprotection are not known. We determined the ability of nitrite and nitrate to confer protection against myocardial infarction in two rat models of ischemia/reperfusion injury and the role of xanthine oxidoreductase, NADPH oxidase, nitric oxide synthase and KATP channels in mediating nitrite-induced cardioprotection. In vivo and in vitro rat models of myocardial ischemia/reperfusion injury were used to cause infarction. Hearts (n=6/group) were treated with nitrite or nitrate for 15 min prior to 30 min regional ischemia and 180 min reperfusion. Xanthine oxidoreductase activity was measured after 15 min aerobic perfusion and 30 min ischemia. Nitrite reduced myocardial necrosis and decline in ventricular function following ischemia/reperfusion in the intact and isolated rat heart in a dose or concentration-dependent manner with an optimal dose of 4 mg/kg in vivo and concentration of 10 μM in vitro. Nitrate had no effect on protection. Reduction in infarction by nitrite was abolished by inhibition of flavoprotein reductases and the molybdenum site of xanthine oxidoreductase, and was associated with an increase in activity of xanthine dehydrogenase and xanthine oxidase during ischemia. Inhibition of nitric oxide synthase had no effect on nitrite-induced cardioprotection. Inhibition of NADPH oxidase and KATP channels abolished nitrite-induced cardioprotection. Nitrite but not nitrate protects against infarction by a mechanism involving xanthine oxidoreductase, NADPH oxidase and KATP channels.
Keywords: nitrite, xanthine oxidoreductase, NADPH oxidase, KATP channels, infarction
INTRODUCTION
Until recently the nitrite anion (NO2−) has been considered to be an oxidation product of nitric oxide, useful as an index of the catalytic activity of nitric oxide synthase [1]. Approximately 80–90% of plasma nitrite is derived from endothelial nitric oxidase synthase derived nitric oxide [2]. Recently it has been shown that plasma cerulloplasmin has a nitric oxide oxidase activity, and may play a role in the conversion of nitric oxide to nitrite in vivo [3]. The nitrite anion, which is present between 0.2 and 10 μM in the blood and tissues [4, 5], may represent a storage form of nitric oxide that is made available under conditions of oxygen deprivation to maintain cell survival. Cardioprotective effects of nitrite have recently been demonstrated. Reduction of nitrite to nitric oxide during ischemia protects the heart against injury from ischemia-reperfusion [6, 7]. The underlying mechanisms by which nitrite is reduced to nitric oxide have not yet been fully defined. Of the candidates for nitrite reduction in vivo, deoxyhemoglobin, deoxymyoglobin and xanthine oxidoreductase have been shown to have an oxygen-sensitive nitrite-reductase activity [8–10]. Data suggest that deoxyhemoglobin plays a role in nitrite-dependent control of vascular tone during hypoxia [11], while xanthine oxidoreductase has been implicated in the cardioprotective effects of nitrite under conditions of oxygen deprivation [7]. In mammals, xanthine oxidoreductase exists in two interconvertible forms, xanthine dehydrogenase (EC 1.1.1.204), which predominates in vivo, and xanthine oxidase (EC 1.1.3.22). The extent to which nitrite alters the activity of xanthine dehydrogenase and xanthine oxidase during myocardial ischemia is not known.
In this study we have selected in vitro and in vivo models of regional myocardial ischemia and reperfusion to determine the ability of nitrite to confer acute cardioprotection in both the presence and absence of hemoglobin, The objectives of our study were to determine 1) the ability of nitrite and nitrate to confer protection against injury from regional ischemia-reperfusion in vivo and in vitro and 2) to assess the contribution of xanthine oxidoreductase, nitric oxide synthase, NADPH oxidase and the sarcolemmal and mitochondrial ATP-sensitive potassium (KATP) channels to nitrite-induced cardioprotection.
MATERIALS AND METHODS
Male Sprague Dawley rats at 8 weeks of age used in this study received humane care in compliance with the “Guide for the Care and Use of Laboratory Animals” formulated by the National Research Council, 1996.
Nitrite, nitrate and cardioprotection studies in vivo
An in vivo anesthetized rat model was used for these experiments with the general surgical protocol and determination of infarct size described previously [12]. For infarct size studies, rats underwent 30 minutes of regional ischemia followed by 2 hours of reperfusion. Rats were treated with KATP channel blockers as needed.
Nitrite, nitrate and cardioprotection studies in vitro
Isolated rat hearts were perfused in a retrograde manner with bicarbonate buffer at constant perfusion pressure (98 mmHg) with a balloon inflated in the left ventricle. End-diastolic pressure was set to 5 mmHg and developed pressure was recorded during steady-state conditions [13]. Hearts were allowed to beat spontaneously. Hearts were kept in temperature-controlled chambers to maintain myocardial temperature at 37°C during periods of perfusion and ischemia. Hearts were subjected to 30 minutes of regional no-flow ischemia and 3 hours of reperfusion. Inhibitors of xanthine oxidoreductase, nitric oxide synthase, NADPH oxidase and scavengers of nitric oxide were added to the coronary perfusate as needed.
Xanthine oxidoreductase studies/analysis
The frozen free wall of the left ventricle from perfused rat hearts was powdered in a prechilled mortar and pestle with liquid nitrogen. Ice-cold homogenization buffer (0.25 M sucrose, 10 mM DTT, 0.2 mM PMSE, 0.1 mM EDTA, and 50 mM K+-phosphate, pH 7.4) was added to the powdered tissue (200mg/ml) and homogenized in a Wheaton 903475 homogenizer. The homogenate was centrifuged at a of speed 40,000×g at 4 °C for 30 min and xanthine dehydrogenase and xanthine oxidase activities were measured in the supernatant according to the method of Beckman et al [14]. Data (means ± SE) are shown as mIUs of xanthine oxidase/xanthine dehydrogenase per gram protein.
Statistical analysis
Data reported are mean ± SD. Statistical analysis was performed by use of repeated measures ANOVA with the Greenhouse-Geisser adjustment used to correct for the inflated risk of a Type I error [15]. If significant, the Mann-Whitney test was used as a second step to identify which groups were significantly different [15]. Significance was set at P < 0.05.
RESULTS
Nitrite, nitrate and cardioprotection in vivo
We determined the dose-response relationship for nitrite to confer protection against injury from myocardial infarction using an in vivo model of regional myocardial ischemia and reperfusion. Rats were treated with an I.V. bolus of nitrite (0.04, 0.4, 1.0, 4.0, 7.0 or 10.0 mg/kg) 15 minutes prior to 30 minutes occlusion of the left anterior descending coronary artery and 2 hours of reperfusion. The effects of increasing nitrite dosage on hemodynamic parameters in the in vivo rat model of ischemia reperfusion injury are shown in Table 1. Heart rate, mean arterial pressure, and rate-pressure product showed significant differences in baseline heart rate for the vehicle-treated group and the experimental groups when nitrite was administered 15 minutes prior to ischemia. Rate pressure product was better maintained in nitrite treated animals both 15 minutes and 3 hours after ischemia as compared to vehicle-treated controls.
Table 1.
Hemodynamic values for nitrite acute dose-response studies in vivo.
| BASELINE |
15min ISCHEMIA |
3hr REPERFUSION |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Heart rate, beats/min |
MAP, mmHg |
RPP mmHg /sec |
Heart rate, beats/min |
MAP, mmHg |
RPP mmHg /sec |
Heart rate, beats /min |
MAP, mmHg |
RPP mmHg /sec |
|
| Drug-free control | 378±15 | 124±6 | 42±4 | 360±14 | 94±11 | 29±5 | 370±14 | 66±9 | 20±4 |
| Nitrite (0.04mg/kg) | 390±12 | 124±5 | 49±3 | 390±12 | 97±5 | 38±3 | 373±7 | 67±3 | 25±3 |
| Nitrite (0.4 mg/kg) | 380±12 | 119±4 | 45±13 | 377±11 | 103±5 | 38±11 | 353±13 | 70±11 | 25±8 |
| Nitrite (1.0 mg/kg) | 395±6 | 136±9 | 48±4 | 380±8 | 105±9 | 40±9 | 333±10 | 94±8 | 31±8* |
| Nitrite (4.0 mg/kg) | 368±12 | 123±5 | 44±3 | 365±13 | 97±9 | 36±4 | 335±12 | 74±3 | 25±3 |
| Nitrite (7.0 mg/kg) | 368±10 | 130±7 | 47±6 | 375±12 | 101±6 | 38±3 | 358±11 | 78±8 | 27±3 |
| Nitrite (10.0 mg/kg) | 370±10 | 122±4 | 45±2 | 370±13 | 98±8 | 37±6 | 353±9 | 66±6 | 23±2 |
Data are mean ± SE, n = 6/group,
= P<0.05, nitrite plus ischemia vs ischemia alone. MAP = mean arterial pressure, RPP = rate pressure product.
Figure 1A shows the effect of nitrite dose on infarct size. The area of the left ventricle at risk for myocardial infarction was equivalent in all experimental groups (data not shown). Increased resistance to myocardial ischemia was determined by a reduction in infarct size. The dose-dependent effect of nitrite on infarct size was biphasic, with protection being afforded up to a dose of 4 mg/kg which was lost at higher doses. The dose of nitrite that afforded maximum reduction in infarct (4.0 mg/kg) resulted in an infarct size of 42±3% of the area at risk which corresponds to a 32% reduction in infarct size over controls. Equivalent doses of nitrate (0.04, 0.4, 1.0, 4.0, 7.0 or 10.0 mg/kg I.V.) had no effect on infarct size (Figure 1A).
Figure 1.
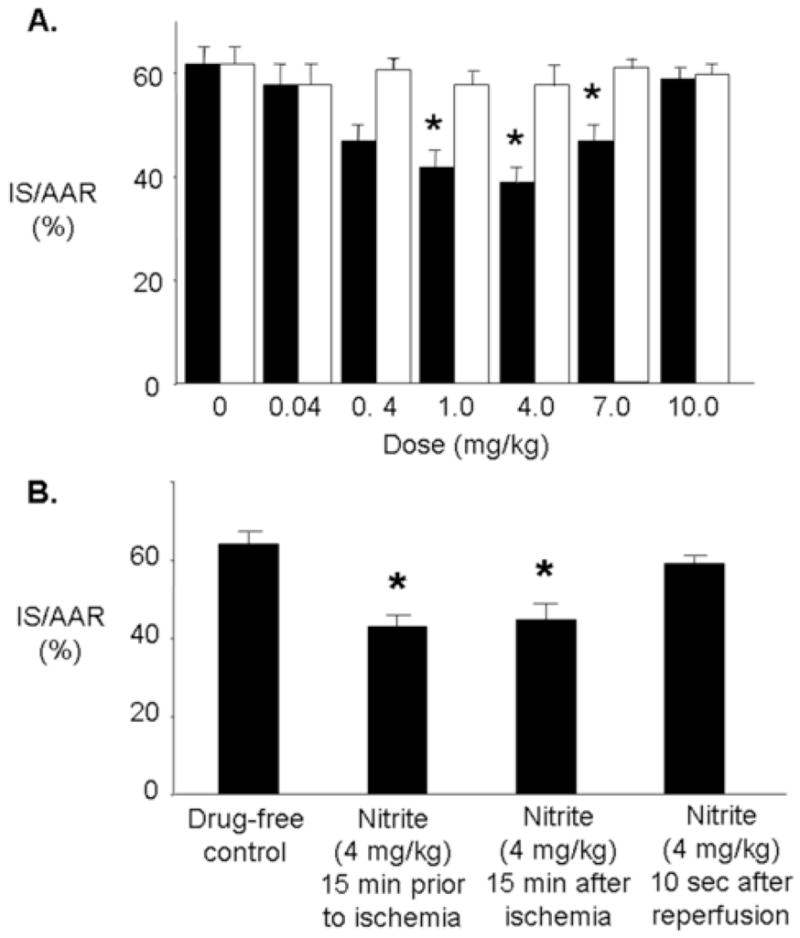
Nitrite-induced cardioprotection in vivo. (A.) Dose-response study. Rats were treated intravenously with either saline or increasing doses of nitrite (■) or nitrate (□) administered as an IV bolus at 15 minutes prior to ischemia, followed by 30 minutes regional ischemia and 120 minutes reperfusion. Infarct size was determined at the end of 120 minutes of reperfusion. (B.) Phase of action of nitrite. Infarct size was determined after 30 minutes of regional ischemia and 120 minutes of reperfusion. Data are mean ± SD, n=6/group. IS, infarct size * = p<0.05, nitrite vs. drug-free control.
Timing of nitrite administration
Clinically, patients generally receive medical treatment for acute myocardial infarction after the onset of symptoms. Thus, we determined whether nitrite is able to protect the heart against injury after the onset of ischemia or when administered at the time of reperfusion. Rats were treated with a bolus of nitrite (4 mg/kg I.V.) 15 minutes prior to ischemia, 15 minutes after the onset of ischemia or 10 seconds after the onset of reperfusion. Nitrite was able to reduce infarct size when administered before and during ischemia but not immediately at the onset of reperfusion (Figure 1B).
Nitrite, nitrate and cardioprotection in vitro
Recent studies have suggested hemoglobin can act as a nitrite reductase under conditions of oxygen deprivation such as that which occurs during ischemia [11]. To examine if cardiac tissue contains an intrinsic ability to realize the protective effects of nitrite, we used a buffer perfused isolated heart model of regional ischemia and reperfusion where hemoglobin is absent. Rat hearts were isolated and perfused with nitrite (1.0, 10.0, 30.0, 100.0 or 300 μM) for 15 minutes prior to 30 minutes of regional ischemia and 3 hours of reperfusion. Nitrite (10 μM) had no effect on coronary flow rate prior to ischemia, but increased left ventricular developed pressure (LVDP) from 138±6 mmHg to 149±7 mmHg and decreased heart rate from 238±23 beats/minute to 208±14 beats/minute (Table 2). Increased cardioprotection was measured by a reduction in infarct size and an increase in recovery of developed pressure. In a similar way to the in vivo response, nitrite exhibited a biphasic concentration-dependent effect on post-ischemic infarct size (Figure 2A). The protection afforded by lower concentrations of nitrite was lost at the highest concentration. The presence of 10 μM nitrite resulted in an infarct size of 23±5% which corresponds to a 47% reduction in infarct size over control hearts. Nitrite also increased the recovery of LVDP following ischemia and reperfusion, reaching significance between 10 and 100 μM nitrite; an effect that was again lost at the highest nitrite concentration (Figure 2B). Recovery of heart rate and coronary flow was unaffected in hearts treated with 10 μM nitrite compared with untreated hearts. Nitrate (10 μM) had no effect on infarct size or on post-ischemic recovery of LVDP (Figure 2A and B).
Table 2.
Hemodynamic values for nitrite concentration-response studies in vitro.
| PRE DRUG |
POST DRUG |
REPERFUSION |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Groups | Heart rate (beats/ min) |
Coronary flow rate (ml/min/g) |
Left ventricle developed pressure (mmHg) |
Heart rate (beats/ min) |
Coronary flow rate (ml/min/g) |
Left ventricle developed pressure (mmHg) |
Heart rate (beats/ min) |
Coronary flow rate (ml/min/g) |
Left ventricle developed pressure (mmHg) |
| Drug-free control | 274±13 | 6±1 | 124±4 | --- | --- | ---(70) | 193±23 | 3±1 | 73±5 |
| Nitrite (1.0μM) | 308±15 | 6±1 | 135±5 | 287±16 | 6±1 | 136±4 | 235±22 | 4±1 | 85±7 |
| Nitrite (10.0μM) | 238±23 | 5±1 | 138±6 | 208±14 | 5±1 | 149±7 | 155±17 | 3±1 | 100±7* |
| Nitrite (30.0μM) | 253±12 | 5±1 | 126±6 | 235±13 | 5±1 | 134±7 | 175±16 | 3±1 | 83±7* |
| Nitrite (100μM) | 248±13 | 5±1 | 128±6 | 230±12 | 5±1 | 136±7 | 169±12 | 3±1 | 85±6* |
| Nitrite (300μM) | 256±19 | 5±1 | 121±7 | 224±24 | 5±1 | 132±6 | 186±21 | 3±1 | 71±6 |
Data are mean ± SE, n = 6/group,
= P<0.05, Nitrite vs. drug-free control.
Figure 2.
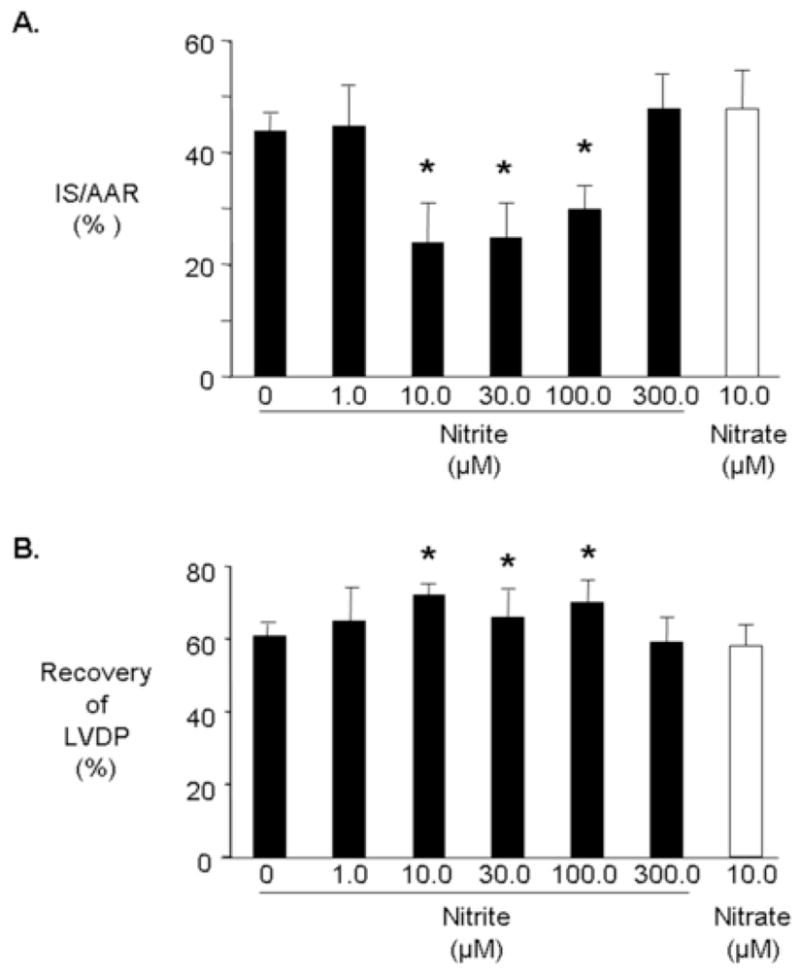
Nitrite and cardioprotection in vitro. Rats were perfused for 15 minutes with nitrite (■) or nitrate (□) prior to 30 minutes regional ischemia and 180 minutes reperfusion. Infarct size was determined at the end of 180 minutes of reperfusion (A). Percent infarction after treatment with nitrite or nitrate (B). Recovery of left ventricular developed pressure (LVDP). Data are mean ± SD, n=6/group. * = p<0.05, nitrite vs. drug-free control.
Role of xanthine oxidoreductase in nitrite-induced cardioprotection
It has been previously demonstrated that xanthine oxidoreductase can reduce nitrite at the molybdenum site [16]. To determine the role of xanthine oxidoreductase in mediating nitrite-induced cardioprotection hearts were perfused with oxypurinol, an inhibitor of the molybdenum site of xanthine oxidoreductase, or DPI, a non-specific flavoprotein inhibitor, prior to ischemia. Oxypurinol (20 μM) abolished the cardioprotective effect of nitrite (10 μM) but had no effect when used alone (Figure 3A and B). DPI also inhibited nitrite-mediated cardioprotection suggesting a role for both the molybdenum site of xanthine oxidoreductase and a flavoprotein (Figure 3A and B). These data implicate a major role for xanthine oxidoreductase in mediating nitrite reduction.
Figure 3.
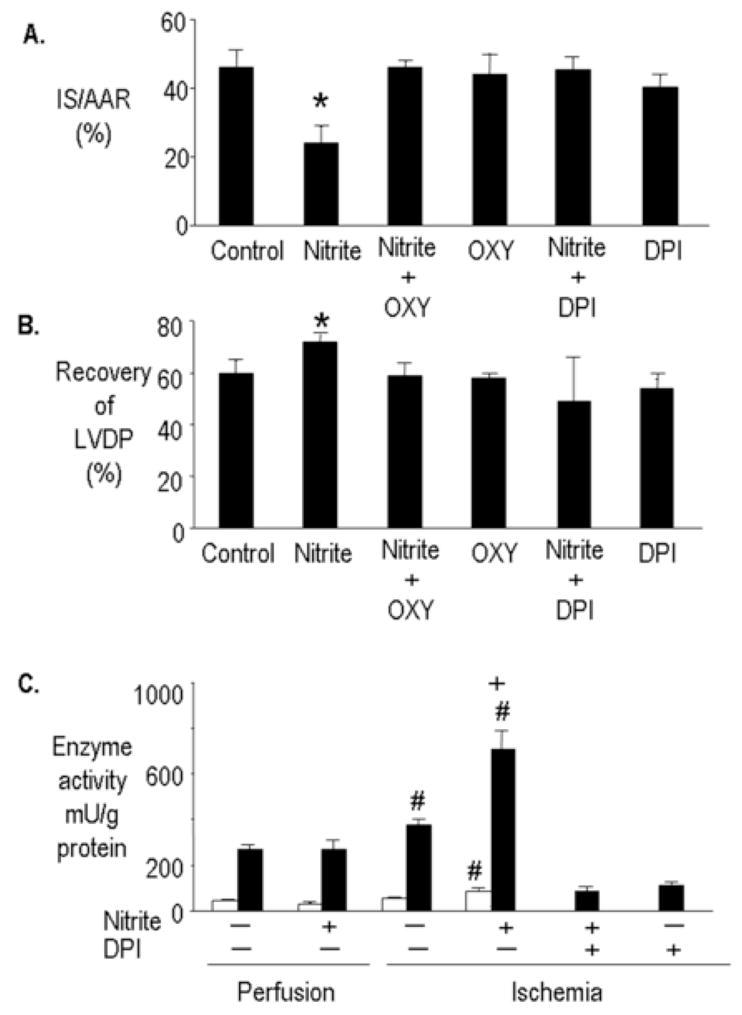
Inhibition of molybdenum and FAD site in xanthine oxidoreductase prevents nitrite-induced cardioprotection. Rat hearts were perfused with and without nitrite (10 μM) for 15 minutes prior to 30 minutes regional ischemia and 180 minutes of reperfusion. Oxypurinol (OXY) (10 μM) or DPI (2 μM) were present in the coronary perfusate for 30 minutes prior to ischemia (A). Percent infarction (B). Recovery of left ventricular developed pressure (LVDP). Data are mean ± SD, n=6/group. * = p<0.05, vs. control. (C) Ischemia is required for nitrite to increase xanthine oxidase and xanthine dehydrogenase activity. Rat hearts were perfused with nitrite (10 μM) for 15 minutes and subjected to 30 minutes regional ischemia with and without DPI (2μM). Xanthine oxidase (□) and xanthine dehydrogenase (■) activity was then determined. Data are mean ± SD, n=6/group. # = p<0.05, vs. perfusion. + = p<0.05 ischemia plus nitrite, vs. ischemia.
Nitrite can also act as a suicide substrate for XOR, inactivating the enzyme to the desulfo form [17]. To examine if nitrite affected xanthine oxidoreductase activity, the activity of both the dehydrogenase and oxidase activities of xanthine oxidoreductase were determined before and after ischemia. Aerobic perfusion of hearts with nitrite (10 μM) had no effect on xanthine oxidase or xanthine dehydrogenase activity. Interestingly, ischemia alone (without nitrite) increased the activity of xanthine dehydrogenase but not xanthine oxidase, whereas ischemia plus nitrite increased the activity of xanthine oxidase and almost doubled xanthine dehydrogenase activity. DPI (2 μM) inhibited both xanthine oxidase and xanthine dehydrogenase activity in hearts subjected to ischemia with or without nitrite (Figure 3C).
Role of nitric oxide synthase in nitrite-induced cardioprotection
To determine the role of nitric oxide synthase in mediating nitrite-induced cardioprotection hearts were treated with L-NMA (100 μM), a general nitric oxide synthase inhibitor, prior to ischemia. L-NMA did not prevent nitrite-induced cardioprotection (Figure 4). L-NMA alone had no effect on cardioprotection.
Figure 4.
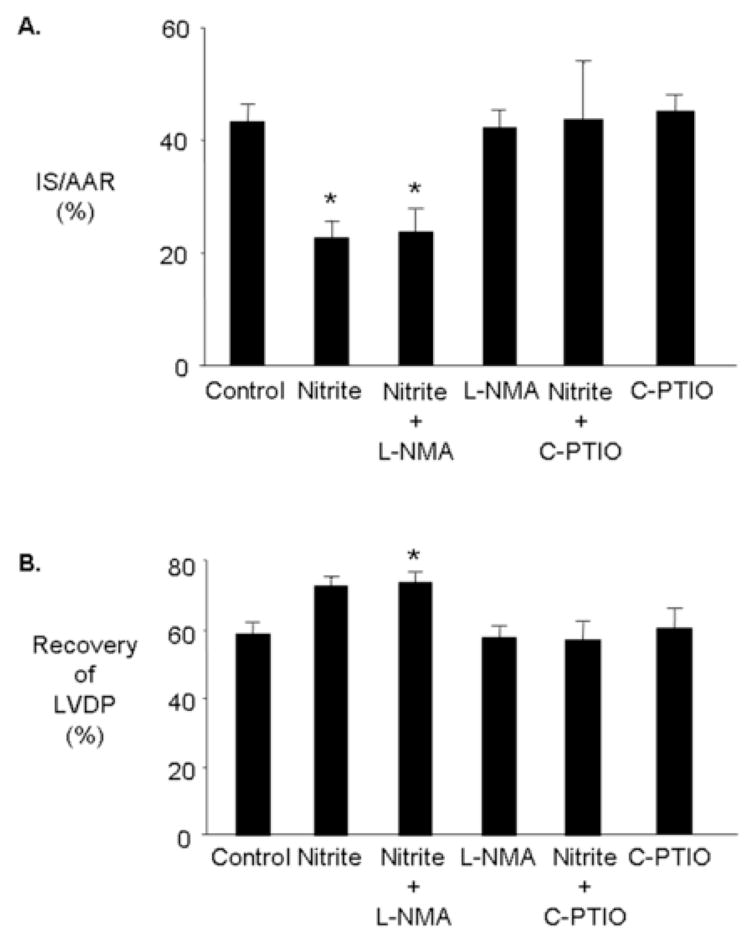
Mechanisms underlying nitrite-induced cardioprotection. Rat hearts were perfused with and without nitrite (10 μM) and either L-NMA (100 μM) or CPTIO (100 μM) for 15 minutes prior to 30 minutes regional ischemia and 180 minutes of reperfusion. (A) Percent infarction (B) Recovery of left ventricular developed pressure (LVDP). Data are mean ± SD, n=6/group. * = p<0.05, vs. control.
Role of nitric oxide in mediating nitrite-induced cardioprotection
To determine whether the heart generates nitric oxide from nitrite to confer cardioprotection hearts were treated with the nitric oxide scavenger, C-PTIO. C-PTIO (100 μM) abolished nitrite-induced cardioprotection. C-PTIO alone had no effect on cardioprotection (Figure 4).
Role of NADPH oxidase in nitrite-induced cardioprotection in vitro
To determine a possible role for NADPH oxidase in mediating nitrite-induced cardioprotection hearts were treated with apocynin (30 μM), an inhibitor of NADPH oxidase. Apocynin abolished nitrite-induced cardioprotection. Apocynin alone had no effect on cardioprotection (Figure 5).
Figure 5.
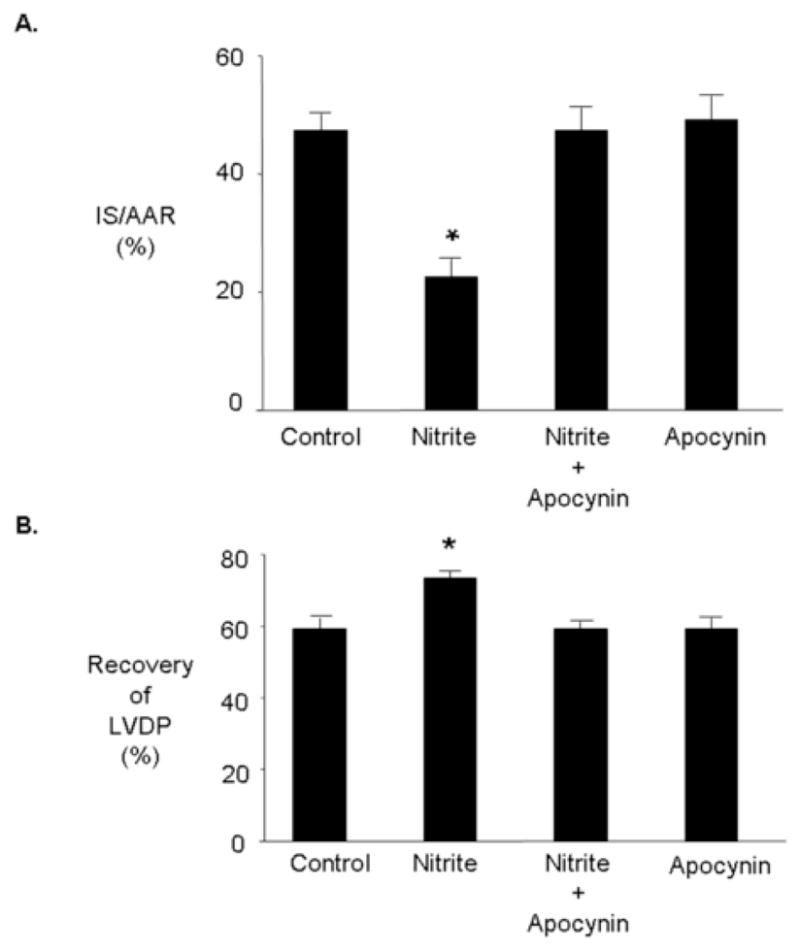
Nitrite-induced cardioprotection is mediated by NADPH oxidase. Rat hearts were perfused with and without nitrite (10 μM) for 15 minutes prior to 30 minutes regional ischemia and 180 minutes of reperfusion. Apocynin (30 μM) was present in the coronary perfusate for 30 minutes prior to ischemia. (A) Percent infarction (B) Recovery of left ventricular developed pressure (LVDP). Data are mean ± SD, n=6/group. * = p<0.05, vs. control.
Role of KATP channels in nitrite-induced cardioprotection in vitro
KATP channels, highly expressed in myocardial sarcolemma and thought to be expressed in myocardial mitochondria, have been found to serve as triggers and mediators of cardioprotection [18–20]. To investigate a role for KATP channels in mediating nitrite-induced cardioprotection, rats were treated with and without nitrite (4 mg/kg) 15 minutes prior to 30 minutes regional myocardial ischemia and 120 minutes reperfusion. The mitochondrial KATP channel blocker 5-HD (10 mg/kg) or sarcolemmal KATP channel blocker HMR 1098 (3 mg/kg) were administered either 20 minutes prior to ischemia or 5 minutes prior to reperfusion. Prior administration of 5-HD or HMR 1098 before ischemia abolished infarct size reduction with nitrite. 5-HD and HMR 1098 given alone before ischemia had no effect on infarct size (Figure 6A). Administration of 5-HD but not HMR 1098 prior to reperfusion abolished infarct size reduction produced by nitrite (Figure 6B). Administration of 5-HD and HMR 1098 alone prior to reperfusion had no effect on infarct size.
Figure 6.
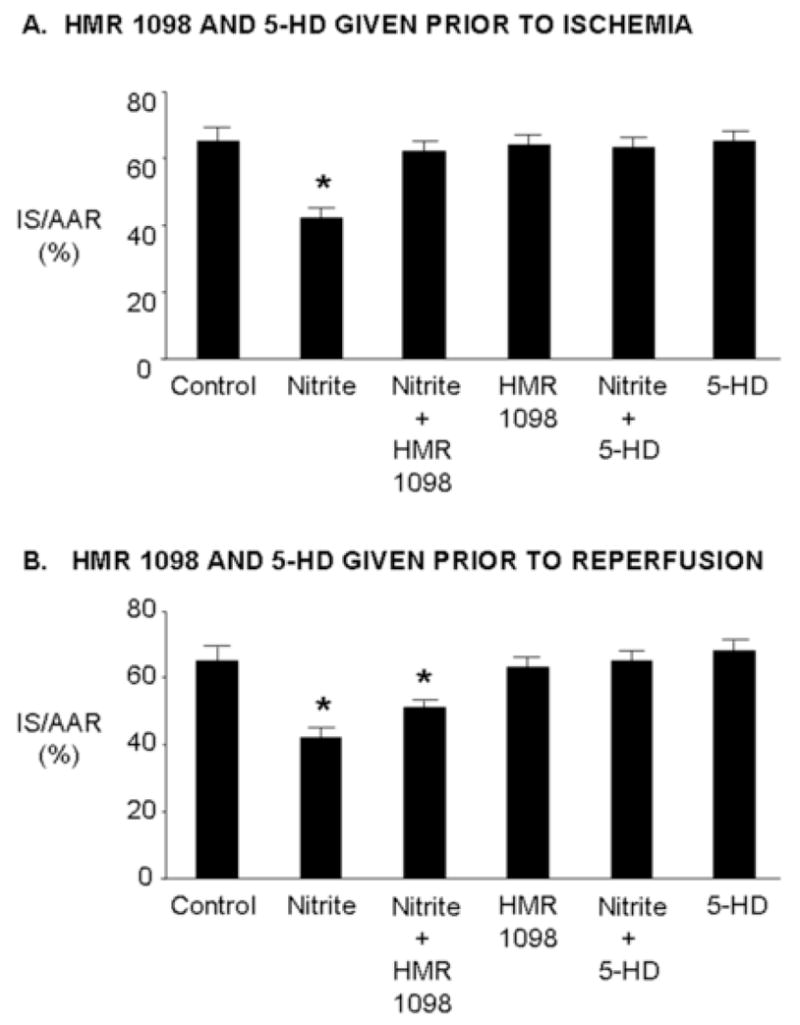
Nitrite-induced cardioprotection is mediated by KATP channels. Rats were treated with and without nitrite (4 mg/kg) 15 minutes prior to 30 minutes regional ischemia and 180 minutes of reperfusion. HMR 1098 (3 mg/kg) or 5-HD (10 mg/kg) were administered either 20 minutes prior to ischemia or 5 minutes prior to reperfusion. Data are mean ± SD, n=6/group. * = p<0.05, vs. control.
DISCUSSION
The present data indicate that nitrite protects the heart against injury from infarction and decreased post-ischemic ventricular function in both in vivo and in vitro models of ischemia/reperfusion. This effect of nitrite is dose- and concentration-dependent with optimal protection occurring at 4 mg/kg I.V. in vivo and 10 μM in vitro. Nitrite conferred cardioprotection over the range of 10–100 μM in vitro. One hundred micromolar nitrite also vasodilates aortic ring preparations [21], with subsequent studies showing the effect is mediated by soluble guanylyl cyclase [22, 23]. These observations suggested nitrite is being reduced to nitric oxide which elicits protection. Our studies on cardioprotection by nitrite are in strong agreement with the studies of Webb et al [7] and Duranski et al [6] and in addition describe novel findings on the mechanisms underlying nitrite-induced cardioprotection.
We calculated the nitrite concentration in the plasma at the time the heart becomes ischemic 15 minutes after nitrite administration. Previous pharmacokinetic studies of nitrite in the rat [24] predict a plasma concentration of 30 μM 15 minutes after intravenous administration at the optimal dose of 4 mg/kg. Thirty micromolar nitrite conferred cardioprotection in our in vitro studies. Thus the optimal dose of 4 mg/kg for the in vivo studies is within the range of concentrations (10–100 μM) for nitrite that confer protection in the in vitro studies.
Nitric oxide is an important defense against injury from myocardial ischemia/reperfusion. Generation of nitric oxide from L-arginine by nitric oxide synthase requires molecular oxygen as an essential substrate. Under conditions of oxygen deprivation, as encountered during ischemia, oxygen levels are rapidly depleted so that nitric oxide synthase activity will be inhibited. Generation of nitric oxide from the reduction of nitrite under ischemic conditions may represent an important mechanism to maintain nitric oxide production for cell survival and function until aerobic conditions are restored, following reperfusion, so that nitric oxide synthase can resume production of nitric oxide. In control studies, nitrate administered in an identical protocol did not confer cardioprotection in both in vivo and in vitro models of myocardial ischemia/reperfusion indicating that the protective effect is specific to nitrite.
Xanthine oxidoreductase is a molybdoflavin enzyme present in the highest concentrations in gut, liver and breast milk as well as in plasma from patients with inflammation [25]. Xanthine dehydrogenase is responsible for the conversion of xanthine to uric acid. Xanthine dehydrogenase activity can be converted to xanthine oxidase, under reducing conditions present during ischemia, and it is the oxidase form of the enzyme that is responsible for superoxide production. The activity of xanthine dehydrogenase in the rat hearts we studied accounts for approximately 80% of total enzymatic activity with the balance contributed by xanthine oxidase. This distribution in catalytic activity between the two forms of the enzyme is in agreement with previous studies on xanthine oxidoreductase activity [26, 27]. The protective effects of nitrite against infarction and decreased post-ischemic ventricular dysfunction were abolished by oxypurinol, an inhibitor of the molybdo-pterin sites of xanthine oxidoreductase. This site is responsible for the binding and oxidation of purine substrates and has also been shown, in in vitro studies to be the site of nitrite reduction [28]. Surprisingly, the effect was also inhibited by DPI, an inhibitor of flavoprotein reductases, which will block electron flow from the molybdenum site to electron acceptors (oxygen or NAD+). It has been shown previously that DPI is unable to inhibit nitrite reduction by xanthine oxidoreductase using xanthine as a substrate, but could inhibit NADH-driven nitrite reduction [28]. However, DPI is a relatively non-specific inhibitor and it is possible that other flavoprotein reductase enzymes are contributing to nitric oxide formation. We show that apocynin, an inhibitor of the NADPH oxidase, a flavoprotein reductase [29], abolishes nitrite-induced cardioprotection. This novel finding may explain why DPI abolishes the protective effect of nitrite and identifies a component of another mechanism by which nitrite may confer its salubrious actions. Although the reduction of nitrite by NADPH oxidase has not previously been observed, the reduction of nitrite by ferrous heme proteins, such as hemoglobin, is well established and may be mechanistically similar [30].
Our study suggests that nitrite can act as a substrate for both xanthine dehydrogenase and xanthine oxidase during ischemia to produce nitric oxide. Harrison has also shown that nitric oxide formation results from reduction of nitrites, both inorganic [16, 31] and organic [32], catalyzed by xanthine oxidoreductase. Activity of xanthine oxidase and xanthine dehydrogenase is increased by nitrite during ischemic conditions, where oxygen and pH are reduced, but not during aerobic conditions. Increased xanthine oxidoreductase activity as a consequence of ischemia has been reported previously [33–36]. Alternately, ischemia may repair the inactive desulfo form of the enzyme to the fully active form as previously suggested [25].
The spin trap carboxy-PTIO abolished nitrite-induced cardioprotection indicating that nitric oxide is responsible for protection against post-ischemic ventricular dysfunction and necrosis. Inhibition of the enzyme nitric oxide synthase with L-NMA did not abolish cardioprotection by nitrite, suggesting nitrite-induced cardioprotection occurs by a nitric oxide synthase-independent mechanism. In addition it has been shown that NOS itself may function as a nitrite reductase in the absence of oxygen [37, 38], but the lack of effect of L-NMA rules out this possibility in the isolated cardiac preparation used in these studies.
The signaling pathway by which nitrite protects against injury from ischemia/reperfusion has not been established. We show here that nitrite-mediated protection is mediated in part by the activation of KATP channels. Currently there are two KATP channels thought to be present in the cardiac myocyte, one in the sarcolemma (sarc KATP channel) and one in the inner mitochondrial membrane (mito KATP channel). Our data show that the sarcolemmal KATP channel appears to act as trigger and the mitochondrial KATP channel as an effector of nitrite-induced cardioprotection. There is evidence to suggest that opening either channel may be important in producing cardioprotection, although the bulk of evidence suggests that it is the mito KATP channel that is responsible for mediating the protective effect of ischemic preconditioning [39, 40]. The role of additional components in the signaling pathway by which the heart is protected by nitrite against injury remains unknown.
Furthermore, we found that a single treatment with nitrite before ischemia reduces cardiac necrosis and increases the recovery of ventricular function following reperfusion. The cardioprotective effect of nitrite is observed immediately after treatment suggesting that the production of new protein is not necessary to manifest its cardioprotective effect. Nitrite also confers protection against injury when given after the onset of ischemia, i.e. after the onset of symptoms. In patients experiencing symptoms of a myocardial infarction, or those who are about to undergo cardiac surgery, nitrite could be administered acutely to decrease injury to the heart from ischemia/reperfusion. Thus, nitrite may represent an important and potent agent for immediately conferring cardioprotection. Nitrite is well tolerated and does not require an elaborate drug delivery system as is needed for several gene-based therapies to confer cardioprotection.
Sodium nitrite, a naturally occurring chemical and common meat preservative, is only used medically to treat cyanide poisoning. Our study suggests this chemical can be used to protect and preserve organs such as the heart in the setting of myocardial infarction and preservation of the heart for transplantation. In support of this notion, nitrite is undergoing clinical trials to increase nitric oxide production in patients with coronary artery disease [41] and in preconditioning mediated tolerance to ischemia [42]. These clinical trials will determine whether dietary nitrite and nitrates can produce nitric oxide to dilate peripheral blood vessels and whether protection by nitrite is equivalent to preconditioning in the setting of ischemia/reperfusion in the human forearm.
We conclude that nitrite, a simple inorganic anion, is reduced by xanthine oxidoreductase to nitric oxide to increase resistance of the heart to injury from ischemia/reperfusion. The cardioprotective effects of nitrite are also mediated by NADPH oxidase and KATP channels suggesting nitrite acts by triggering multiple pathways. These observations contribute to a re-evaluation of how we view the mechanisms and biological effects of nitrite.
Acknowledgments
This work was supported in part by grants HL54075 (JEB), GM55792 (NH) and HL08361 (GJG) from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shi Y, Hutchins W, Ogawa H, Chang C-C, Pritchard K, Jr, Zhang C, et al. Increased Resistance to Myocardial Ischemia in the Brown Norway vs. Dahl S Rat: Role of Nitric Oxide Synthase and Hsp90. J Mol Cell Cardiol. 2005;38:625–35. doi: 10.1016/j.yjmcc.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Kleinbongard P, Dejam A, Lauer T, Rassaf T, Schindler A, Picker O, et al. Plasma nitrite reflects constitutive nitric oxide synthase activity in mammals. Free Radic Biol Med. 2003;35:790–6. doi: 10.1016/s0891-5849(03)00406-4. [DOI] [PubMed] [Google Scholar]
- 3.Shiva S, Wang X, Ringwood LA, Xu X, Yuditskaya S, Annavajjhala V, et al. Ceruloplasmin is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat Chem Biol. 2006;2:486–93. doi: 10.1038/nchembio813. [DOI] [PubMed] [Google Scholar]
- 4.Pelletier MM, Kleinbongard P, Ringwood L, Hito R, Hunter CJ, Schechter AN, et al. The measurement of blood and plasma nitrite by chemiluminescence: pitfalls and solutions. Free Radic Biol Med. 2006;41:541–8. doi: 10.1016/j.freeradbiomed.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez J, Maloney RE, Rassaf T, Bryan NS, Feelisch M. Chemical nature of nitric oxide storage forms in rat vascular tissue. Proc Natl Acad Sci USA. 2003;100:336–41. doi: 10.1073/pnas.0234600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langston W, et al. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest. 2005;115:1232–40. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Webb A, Bond R, McLean P, Uppal R, Benjamin N, Ahluwalia A. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc Natl Acad Sci USA. 2004;101:13683–8. doi: 10.1073/pnas.0402927101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang KT, Keszler A, Patel N, Patel RP, Gladwin MT, Kim-Shapiro DB, et al. The reaction between nitrite and deoxyhemoglobin. Reassessment of reaction kinetics and stoichiometry. J Biol Chem. 2005;280:31126–31. doi: 10.1074/jbc.M501496200. [DOI] [PubMed] [Google Scholar]
- 9.Huang Z, Shiva S, Kim-Shapiro DB, Patel RP, Ringwood LA, Irby CE, et al. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest. 2005;115:2099–107. doi: 10.1172/JCI24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, Samouilov A, Liu X, Zweier JL. Characterization of the effects of oxygen on xanthine oxidase-mediated nitric oxide formation. J Biol Chem. 2004;279:16939–46. doi: 10.1074/jbc.M314336200. [DOI] [PubMed] [Google Scholar]
- 11.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498–505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 12.Gross ER, Peart JN, Hsu AK, Grover GJ, Gross GJ. K(ATP) opener-induced delayed cardioprotection: involvement of sarcolemmal and mitochondrial K(ATP) channels, free radicals and MEK1/2. J Mol Cell Cardiol. 2003;35:985–92. doi: 10.1016/s0022-2828(03)00183-4. [DOI] [PubMed] [Google Scholar]
- 13.Cabigas BP, Su J, Hutchins W, Shi Y, Schaefer RB, Recinos RF, et al. Hyperoxic and hyperbaric-induced cardioprotection: role of nitric oxide synthase 3. Cardiovasc Res. 2006;72:143–51. doi: 10.1016/j.cardiores.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 14.Beckman JS, Parks DA, Pearson JD, Marshall PA, Freeman BA. A sensitive fluorometric assay for measuring xanthine dehydrogenase and oxidase in tissues. Free Radic Biol Med. 1989;6:607–15. doi: 10.1016/0891-5849(89)90068-3. [DOI] [PubMed] [Google Scholar]
- 15.Baker JE, Holman P, Gross GJ. Preconditioning in immature rabbit hearts: role of KATP channels. Circulation. 1999;99:1249–54. doi: 10.1161/01.cir.99.9.1249. [DOI] [PubMed] [Google Scholar]
- 16.Godber BL, Doel JJ, Sapkota GP, Blake DR, Stevens CR, Eisenthal R, et al. Reduction of nitrite to nitric oxide catalyzed by xanthine oxidoreductase. J Biol Chem. 2000;275:7757–63. doi: 10.1074/jbc.275.11.7757. [DOI] [PubMed] [Google Scholar]
- 17.Godber BL, Doel JJ, Goult TA, Eisenthal R, Harrison R. Suicide inactivation of xanthine oxidoreductase during reduction of inorganic nitrite to nitric oxide. Biochem J. 2001;358:325–33. doi: 10.1042/0264-6021:3580325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cole WC, McPherson CD, Sontag D. ATP-regulated K+ channels protect the myocardium against ischemia/reperfusion damage. Circ Res. 1991;69:571–81. doi: 10.1161/01.res.69.3.571. [DOI] [PubMed] [Google Scholar]
- 19.Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, DiAlonzo AJ, et al. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Potential mechanism of cardioprotection. Circ Res. 1997;81:1072–82. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- 20.Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–8. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- 21.Furchgott RF, Bhadrakom S. Reactions of strips of rabbit aorta to epinephrine, isopropylarterenol, sodium nitrite and other drugs. J Pharmacol Exp Ther. 1953;108:129–43. [PubMed] [Google Scholar]
- 22.Ignarro LJ, Lippton H, Edwards JC, Baricos WH, Hyman AL, Kadowitz PJ, et al. Mechanism of vascular smooth muscle relaxation by organic nitrates, nitrites, nitroprusside and nitric oxide: evidence for the involvement of S-nitrosothiols as active intermediates. J Pharmacol Exp Ther. 1981;218:739–49. [PubMed] [Google Scholar]
- 23.Kimura H, Mittal CK, Murad F. Activation of guanylate cyclase from rat liver and other tissues by sodium azide. J Biol Chem. 1975;250:8016–22. [PubMed] [Google Scholar]
- 24.Kohn MC, Melnick RL, Ye F, Portier CJ. Pharmacokinetics of sodium nitrite-induced methemoglobinemia in the rat. Drug Metab Dispos. 2002;30:676–83. doi: 10.1124/dmd.30.6.676. [DOI] [PubMed] [Google Scholar]
- 25.Harrison R. Structure and function of xanthine oxidoreductase: where are we now? Free Radic Biol Med. 2002;33:774–97. doi: 10.1016/s0891-5849(02)00956-5. [DOI] [PubMed] [Google Scholar]
- 26.Frederiks WM, Bosch KS. The proportion of xanthine oxidase activity of total xanthine oxidoreductase activity in situ remains constant in rat liver under various (patho)physiological conditions. Hepatology. 1996;24:1179–84. doi: 10.1002/hep.510240533. [DOI] [PubMed] [Google Scholar]
- 27.Parks DA, Williams TK, Beckman JS. Conversion of xanthine dehydrogenase to oxidase in ischemic rat intestine: a reevaluation. Am J Physiol. 1988;254:G768–74. doi: 10.1152/ajpgi.1988.254.5.G768. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Cui H, Liu X, Zweier JL. Xanthine oxidase catalyzes anaerobic transformation of organic nitrates to nitric oxide and nitrosothiols: characterization of this mechanism and the link between organic nitrate and guanylyl cyclase activation. J Biol Chem. 2005;280:16594–600. doi: 10.1074/jbc.M411905200. [DOI] [PubMed] [Google Scholar]
- 29.Isabelle M, Vergeade A, Moritz F, Dautreaux B, Henry JP, Lallemand F, et al. NADPH oxidase inhibition prevents cocaine-induced up-regulation of xanthine oxidoreductase and cardiac dysfunction. J Mol Cell Cardiol. 2007;42:326–32. doi: 10.1016/j.yjmcc.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 30.Doyle MP, Pickering RA, DeWeert TM, Hoekstra JW, Pater D. Kinetics and mechanism of the oxidation of human deoxyhemoglobin by nitrites. J Biol Chem. 1981;256:12393–8. [PubMed] [Google Scholar]
- 31.Godber BL, Doel JJ, Durgan J, Eisenthal R, Harrison R. A new route to peroxynitrite: a role for xanthine oxidoreductase. FEBS Lett. 2000;475:93–6. doi: 10.1016/s0014-5793(00)01639-2. [DOI] [PubMed] [Google Scholar]
- 32.Doel JJ, Godber BL, Goult TA, Eisenthal R, Harrison R. Reduction of organic nitrites to nitric oxide catalyzed by xanthine oxidase: possible role in metabolism of nitrovasodilators. Biochem Biophys Res Commun. 2000;270:880–5. doi: 10.1006/bbrc.2000.2534. [DOI] [PubMed] [Google Scholar]
- 33.Brown JM, Terada LS, Grosso MA, Whitmann GJ, Velasco SE, Patt A, et al. Xanthine oxidase produces hydrogen peroxide which contributes to reperfusion injury of ischemic, isolated, perfused rat hearts. J Clin Invest. 1988;81:1297–301. doi: 10.1172/JCI113448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferdinandy P, Panas D, Schulz R. Peroxynitrite contributes to spontaneous loss of cardiac efficiency in isolated working rat hearts. Am J Physiol. 1999;276:H1861–7. doi: 10.1152/ajpheart.1999.276.6.H1861. [DOI] [PubMed] [Google Scholar]
- 35.Weinbroum A, Nielsen VG, Tan S, Gelman S, Matalon S, Skinner KA, et al. Liver ischemia-reperfusion increases pulmonary permeability in rat: role of circulating xanthine oxidase. Am J Physiol. 1995;268:G988–96. doi: 10.1152/ajpgi.1995.268.6.G988. [DOI] [PubMed] [Google Scholar]
- 36.Zweier JL, Broderick R, Kuppusamy P, Thompson-Gorman S, Lutty GA. Determination of the mechanism of free radical generation in human aortic endothelial cells exposed to anoxia and reoxygenation. J Biol Chem. 1994;269:24156–62. [PubMed] [Google Scholar]
- 37.Vanin AF, Bevers LM, Slama-Schwok A, van Faassen EE. Nitric oxide synthase reduces nitrite to NO under anoxia. Cell Mol Life Sci. 2007;64:96–103. doi: 10.1007/s00018-006-6374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Z, Naughton D, Winyard PG, Benjamin N, Blake DR, Symons MC. Generation of nitric oxide by a nitrite reductase activity of xanthine oxidase: a potential pathway for nitric oxide formation in the absence of nitric oxide synthase activity. Biochem Biophys Res Commun. 1998;249:767–72. doi: 10.1006/bbrc.1998.9226. [DOI] [PubMed] [Google Scholar]
- 39.Hanley PJ, Daut J. K(ATP) channels and preconditioning: a re-examination of the role of mitochondrial K(ATP) channels and an overview of alternative mechanisms. J Mol Cell Cardiol. 2005;39:17–50. doi: 10.1016/j.yjmcc.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 40.Gross GJ, Fryer RM. Sarcolemmal versus mitochondrial ATP-sensitive K+ channels and myocardial preconditioning. Circ Res. 1999;84:973–9. doi: 10.1161/01.res.84.9.973. [DOI] [PubMed] [Google Scholar]
- 41.NIH. Dietary Nitrate and Nitrite to Increase Nitric Oxide in Patients with Coronary Artery Disease. National Heart, Lung, and Blood Institute National Institutes of Health Clinical Center (CC) . 2006 [cited Available from: http://www.clinicaltrials.gov/ct/show/nct00069654.
- 42.NIH. The role of nitrite in preconditioning mediated tolerance to ischemic stress. National Institutes of Health Clinical Center . 2006 Sept [cited Available from: http://www.clinicaltrials.gov/ct/show/NCT00250185.