

Abstract
Loss of brain melanocortin receptors (Mc3rKO and Mc4rKO) causes increased adiposity and exacerbates diet-induced obesity (DIO). Little is known about how Mc3r or Mc4r genotype, diet, and obesity affect insulin sensitivity. Insulin resistance, assessed by insulin and glucose tolerance tests, Ser307 phosphorylation of insulin receptor substrate 1, and activation of protein kinase B, was examined in control and DIO wild-type (WT), Mc3rKO and Mc4rKO C57BL/6J mice. Mc4rKO mice were hyperphagic and had increased metabolic efficiency (weight gain per kilojoule consumed) relative to WT; both parameters increased further on high-fat diet. Obesity of Mc3rKO was more dependent on fat intake, involving increased metabolic efficiency. Fat mass of DIO Mc3rKO and Mc4rKO was similar, although Mc4rKO gained weight more rapidly. Mc4rKO develop hepatic insulin resistance and severe hepatic steatosis with obesity, independent of diet. DIO caused further deterioration of insulin action in Mc4rKO of either sex and, in male Mc3rKO, compared with controls, associated with increased fasting insulin, severe glucose intolerance, and reduced insulin signaling in muscle and adipose tissue. DIO female Mc3rKO exhibited very modest perturbations in glucose metabolism and insulin sensitivity. Consistent with previous data suggesting impaired fat oxidation, both Mc3rKO and Mc4rKO had reduced muscle oxidative metabolism, a risk factor for weight gain and insulin resistance. Energy expenditure was, however, increased in Mc4rKO compared with Mc3rKO and controls, perhaps due to hyperphagia and metabolic costs associated with rapid growth. In summary, DIO affects insulin sensitivity more severely in Mc4rKO compared with Mc3rKO, perhaps due to a more positive energy balance.
Ventromedial hypothalamic lesions cause hyperphagia, obesity, and hyperinsulinemia, indicating that hypothalamic neurons are critical for energy homeostasis (1–3). The best-characterized hypothalamic neurons involved in energy homeostasis express proopiomelanocortin (POMC), a propeptide processed into α-, β-, and γ-MSH; agouti-related peptide (AgRP), or melanocortin receptors (Mc3r and Mc4r) (2, 4). POMC and AgRP fibers project from the arcuate nucleus of the hypothalamus to hypothalamic and extra-hypothalamic centers involved in energy homeostasis (2, 5). These neurons release agonists (MSH) or an Mc3r/Mc4r antagonist (AgRP), respectively, in response to orexigenic and anorectic inputs from sources in the periphery and brain (2, 5). A small population of POMC-positive neurons in the nucleus tractus solitarius in the brain stem also regulates feeding behavior (6).
The prevailing model of the regulation of energy homeostasis by melanocortins proposes that increased POMC neuronal activity inhibits food intake and increases energy expenditure (2, 3). Conversely, stimulation of AgRP neurons has the opposite effect by inhibiting MSH action and, through the release of the inhibitory neurotransmitter γ-aminobutyric acid, reducing the activity of POMC neurons (2, 7). Neuropeptide Y (NPY) release from AgRP neurons may also inhibit POMC neuronal activity (8). In humans, Pomc or Mc4r gene mutations are associated with childhood hyperphagia and obesity (9). Mutations reducing efficacy of Mc3r activation by MSH may also cause childhood obesity (10). The phenotypes of transgenic mutant mice are generally consistent with the prevailing model, with loss of POMC synthesis (11, 12) or melanocortin receptor function (13–16) resulting in obesity, and ablation of AgRP neurons in adults results in hypophagia and weight loss (17–19).
Mc3r and Mc4r knockout (Mc3rKO and Mc4rKO) mice have been used to investigate the role of each receptor in regulating energy homeostasis. Comparison of single and double knockouts suggests that Mc3r and Mc4r function independently to regulate energy homeostasis (13–15). Analysis of conditional Mc4r mutant mice, where expression is limited to neurons, suggests that most, if not all, of Mc4r regulation of energy balance occurs in the brain (16). The acute effects of nonselective melanocortin receptor agonists on feeding behavior and energy expenditure require Mc4r (13, 15, 16, 20). Mc4r are required for stimulation of sympathetic nervous activity (21, 22) and thyroid function (23) by nonselective melanocortin receptor agonists. Stimulation of sympathetic nervous activity and the thyroid are mechanisms by which Mc4r activation could increase energy expenditure. Inhibition of food intake by the satiety factor cholecystokinin involves stimulation of POMC neurons in the brain stem and also requires Mc4r (6).
The regulation of energy homeostasis by the Mc3r has not been extensively studied. Mc3rKO initially exhibited a very modest obesity syndrome, involving modest increases in fat mass (FM) and reduced lean mass without hyperphagia and with a normal anorectic response to melanocortin agonists (13, 15). However, a recent study indicated that Mc3r and Mc4r may be of approximately equal importance in preventing weight gain on moderately high-fat chow and demonstrated that Mc3rKO also exhibit aberrant regulation of feeding behavior by leptin (22). Moreover, male Mc3rKO backcrossed onto the C57BL/6J (B6) background exhibit a modest hyperphagia on high-fat diets (24). Intracerebroventricular administration of AgRP increases food intake of Mc4rKO, suggesting that inhibition of Mc3r may increase food intake (25). Other factors that may contribute to increased adiposity of Mc3rKO include low fatty acid oxidation (FAO), indicated by a high respiratory exchange ratio, and reduced physical activity (13, 15, 24).
Diet and genetic background profoundly affect obesity and diabetes (“diabesity”) in mice (26, 27). Studies using mice on mixed genetic backgrounds and various diets indicate that diet-induced obesity (DIO) is more severe in Mc3rKO, Mc4rKO, Pomc−/−, or Pomc+/− mice (11, 15, 28). However, very little is known about how genotype and diet affect insulin sensitivity of melanocortin mutant mice at the tissue level. Central administration of nonselective melanocortin receptor agonists or antagonist in rats improves, or worsens, insulin sensitivity independently of changes in adiposity, which is thought to primarily involve Mc4r (29).
The current study further investigated the effects of Mc3r and Mc4r deficiency on DIO, metabolism, and insulin resistance of B6 mice. Insulin receptor signaling was examined in liver, muscle, and retroperitoneal adipose tissue of knockout and wild-type (WT) B6 mice fed low- or high-fat diets. Insulin resistance was assessed by measuring the phosphorylation of insulin receptor substrate 1 (IRS1) on Ser307, which inhibits association of IRS1 with the insulin receptor tyrosine kinase (30). Activation of protein kinase B (PKB), which is a target of phosphatidyl-3 kinase (PI3K) and involved in the regulation of glucose uptake (31), and phosphorylation of downstream targets of PKB [Forkhead Box O1 (FoxO1) and glycogen synthase kinase-3 (GSK3)], were also measured in liver, skeletal muscle (quadriceps), and retroperitoneal adipose tissue to assess insulin responsiveness. The original hypothesis, based on analysis of Mc3rKO and Mc4rKO on outbred backgrounds (13–15, 20, 28), was that diabesity would be most severe in Mc4rKO and mild in Mc3rKO. However, our data suggest a more complex interaction between Mc3r and Mc4r deficiency and dietary fat on the development of obesity and insulin resistance in B6 mice.
Materials and Methods
Experimental animals
The Pennington Biomedical Research Center Institutional Animal Care and Use Committee approved all experiments. Mc3rKO, Mc4rKO, and WT B6 littermates were obtained from mating heterozygote parents, as described previously (13, 32). Mice were housed on a 12-h light, 12-h dark period (lights on 0600–1800 h). Adiposity (percent body fat) was estimated by measuring FM and fat-free mass (FFM) by nuclear magnetic resonance (NMR) (Bruker Mice Minispec NMR Analyzer; Bruker Optics Inc., Billerica, MA), validated against dual x-ray absorptiometry and standard chemical composition analysis (33). Low-fat (Research Diets no. 12450B), moderately high-fat (no. 12451), and high-fat (no. 12492) diet, with 10, 45, or 60% kJ from fat, respectively, were purchased from Research Diets, Inc (New Brunswick, NJ).
Food intake and energy balance
Food intake (kilojoules per day) and gain in body mass energy (kilojoules per day) were measured over 8 wk (black line in Fig. 1, A and B) using mice housed in wire-mesh caging; spillage was collected in trays beneath the cage floor and subtracted from the reduction of food weight in the feeder to provide an accurate measure of actual intake. Mice were placed in the wire mesh caging at 7–8 wk of age and allowed 10 d to acclimate to the new conditions. Gain in body mass energy was calculated using NMR measurements of FM and FFM, using a value of 37.7 kJ/g FM and 4.2 kJ/g FFM (assumes 25% of fat-free mass is protein, with the remainder being water). For estimating basal daily energy expenditure in a weight-stable state (bEEest), the energy cost of depositing FM and protein was estimated using a value of 55.3 (FM) and 9.2 (FFM) kJ/g (34). Metabolic efficiency (ME%) is the gain in body mass energy as a percentage of total energy intake (35).
Fig. 1.
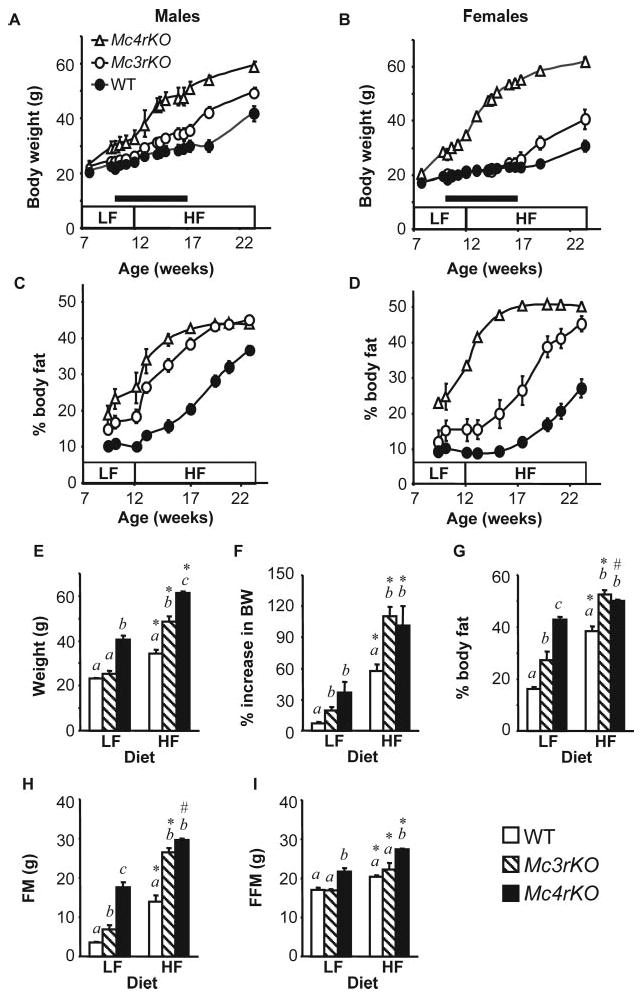
Genotype-specific effects of dietary fat content on weight gain of Mc3rKO and Mc4rKO. Data shown are for individually housed male (A and C) and female (B and D) mice that were fed low-fat diet (LF, 10% kJ from fat) from weaning to 12 wk, followed by high-fat diet (HF, 60% kJ from fat) for 12 wk. Growth curves show body mass (A and B) and adiposity (percent body fat) (C and D) of male Mc4rKO (n = 4), Mc3rKO (n = 4), and WT (n = 8) and female Mc4rKO (n = 6), Mc3rKO (n = 3), and WT (n = 11) mice. Introduction of the high-fat diet accelerated weight gain and adiposity irrespective of sex and genotype. The black bars in A and B indicate the period when mice were housed in wire-mesh-bottom cages to allow for collection of spillage for accurate measurements of food intake. E–I, Comparison of body mass measurements from 6-month-old females (n = 8–9 per group for WT; n = 5 per group for Mc3rKO and Mc4rKO) group housed and fed either low-fat or high-fat diet for 12 wk. Increasing dietary fat content had equal effects on body mass (E), the increase in weight over 12 wk as a percentage of baseline or starting weight (F), and FM (H) of Mc3rKO relative to Mc4rKO. G, On the low-fat diet, adiposity of Mc3rKO was intermediate between lean WT and obese Mc4rKO. Adiposity of Mc3rKO and Mc4rKO were increased to the same extent, relative to WT controls, on the high-fat diet. I, FFM of Mc4rKO was significantly increased relative to WT and Mc3rKO, irrespective of diet. Two-way ANOVA (independent variables were diet and genotype) indicated significant effects of diet and genotype for all parameters. Significant effect of diet within genotype are indicated as follows: *, P < 0.001; #, P < 0.05. Significant effects of genotype within diet are indicated using letters; groups not sharing the same letter (a, b, or c) are significantly different (P < 0.05).
Energy expenditure, fatty acid oxidation, and mitochondrial function
Total and resting energy expenditure (TEEcal and REE cal) were also measured by indirect calorimetry, using a 16-chamber Oxymax system (Columbus Instruments, Columbus, OH), as previously described (24, 36). Ambulatory activity in the x and z axes were evaluated using an OPTO-M3 sensor system (Columbus Instruments) (24).
Calorimetry data collected over several days provides a more accurate assessment of energy expenditure, compared with short-term measurements (i.e. 3–4 h). Calorimetry data were collected over a 7- to 10-d period, with chambers disassembled for maintenance every 3 d. Energy expenditure of mice housed in this system is stable over a 14-d period (Refs. 24 and 36 and data not shown). Some of the data presented are from studies published previously (24, 32, 36). Nonresting energy expenditure (NREEcal), due to ambulation and the thermic effects of feeding (37), was calculated by subtracting REEcal from TEEcal. Energy expenditure data are presented as kilojoules per animal (kilojoules per day), adjusted for body weight (kilojoules per gram body weight per day), or fat-free mass determined using NMR (kilojoules per gram FFM per day). REEcal from TEEcal were also analyzed by linear regression against body weight and FFM (37).
FAO was assessed by measuring production of CO2 and acid-soluble metabolites after incubation of fresh liver and muscle extracts with [14C]palmitate (38, 39). Citrate synthase activity was measured as previously described (40).
Histology and serum assays
Hematoxylin and eosin staining was performed on 15-μm sections of liver tissue collected in 10% paraformaldehyde, dehydrated, and mounted in paraffin. Hepatic lipid and triglyceride content was measured as previously described (32). Total plasma cholesterol and triglycerides were measured after an overnight fast using a Beckman Synchron CX7. Insulin ELISAs were performed in duplicate using kits from Crystal Chem (Downer's Grove, IL).
Gene expression
Total RNA from liver was isolated using TRI Reagent (Molecular Research Centre Inc., Cincinnati, OH) and reverse transcribed using Superscript III RT system (Invitrogen, Carlsbad, CA). Oligonucleotide primers (Integrated DNA Technologies, Coralville, IA) were designed using Primer Express 2.0 software (Applied Biosystems, Foster City, CA). Quantitation of target gene mRNA using cyclophilin B as a reference was performed in 384-well plates using SYBR Green, or TaqMan Universal PCR Master Mix (Applied Biosystems) and an ABI PRISM 7900 HT Sequence Detection System (Applied Biosystems). Sequences of primers and probes used for expression analyses were as follows: cyclophilin B sense 5′-ggtggagagcaccaagacaga-3′ and antisense 5′-gccggagtcgacaatgatg-3′; apolipoprotein A IV (Apoa4) sense 5′-tggtgcccttt-gtcgtacag-3′, antisense 5′-catcatgcggtcacgtaggt-3′, and probe 5′-tgagtgggcatctagccaaggaaactga-3′; stearoyl CoA desaturase 1 (Scd1) sense 5′-caacaccatggcgttcca-3′ and antisense 5′-ggtgggcgcggtgat-3′. 6′-Carboxyfluorescein (FAM)-labeled probes were synthesized by Applied Biosystems (Foster City, CA).
Insulin sensitivity and insulin receptor signaling
Whole-body insulin sensitivity and glucose tolerance were estimated using insulin tolerance tests (ITT) (1 U/kg Humulin; Eli Lilly, Indianapolis, IN) or ip glucose tolerance test (IPGTT) (1 g/kg glucose), as previously described (32). Blood glucose was measured using a Glucometer Elite XL (Bayer Corp., Elkhart, IN).
Insulin receptor signaling was analyzed using muscle, liver, and retroperitoneal adipose tissue extracts from mice killed 10 min after receiving a single ip injection of 1 U/kg insulin or saline after overnight fast. Tissues were homogenized in ice-cold buffer [20 mm Tris (pH 7.5), 150 mm NaCl, 1 mm EDTA, 1 mm EDTA, 1% Triton X-100, 2.5 mm Na pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, and 1 μg/ml leupeptin], and 20 μg protein was diluted in sample buffer [50 mm Tris HCl (pH 6.7), 4% wt/vol glycerol, 4% SDS, 1% 2-mercaptoethanol, and 0.02 mg/ml bromphenol blue] and separated on a 10% SDS-polyacrylamide gel. Protein was transferred to Immobilon-P polyvinylidene difluoride membranes in Towbin-transfer buffer (25 mm Tris, 192 mm glycine, 20% methanol, and 0.01% SDS).
Membranes were incubated with antibodies for phospho-PKB, phospho-FoxO1, or phospho-GSK3 α/β or antibodies against total PKB, FoxO1, or GSK3 α/β (Cell Signaling Technology, Beverly, MA). Antigen-antibody-peroxidase complexes were detected using enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ). Phosphorylation of IRS1 on Ser307 was determined by immunoprecipitation using a polyclonal antibody against the C terminus, followed by blotting using a polyclonal antibody against Ser307 of IRS1 (Cell Signaling Technology).
PKB kinase activity was measured using a nonradioactive assay kit (Cell Signaling Technology). PKB was immunoprecipitated from 200 μg lysate using a mouse monoclonal antibody (1G1). PKB-bound beads were then incubated at 37 C for 30 min with ATP (10 mm) and 1 μg of a fusion protein containing the phosphorylation sites of GSK-3 α/β (Ser21 for GSK-3α and Ser9 for GSK-3β). The GSK-3 fusion protein was solubilized in Laemmli sample buffer and separated on 10% SDS-PAGE gels, and GSK-3 phosphorylation was detected using an antibody recognizing both Ser21 and Ser9 of GSK-3α and -β. Signal on autoradiograms was quantified by densitometry using QuantityOne (Bio-Rad, Hercules, CA). Data are presented as arbitrary units, relative to the mean of insulin-treated WT mice. Phosphospecific antibody data were also normalized for total protein.
Statistics
Data presented are mean ± se. Sigmastat software (SPSS Inc., Chicago, IL) was used for one-way and two-way ANOVA. The strength of association between energy expenditure and body mass was assessed using the cor and lm functions in R (R Development Core Team, http://cran.r-project.org/). Tukey or Student-Newman-Keuls post hoc tests were performed for data exhibiting homogeneous or nonhomogeneous variance, respectively. Significance was assumed for P values < 0.05.
Results
Effect of diet-genotype interactions on obesity and energy balance
Weight gain and adiposity
The effect of a high-fat diet (60% kJ from fat) on weight gain and adiposity of male and female Mc3rKO (n = 4 males and 3 females), Mc4rKO (n = 4 males and 6 females), and WT mice (n = 8 males and 11 females) is shown in Fig. 1. The increase in weight gain associated with the high-fat diet was significantly affected by gender and genotype (Fig. 1, A–D, and Table 1). In descending order, the increase gain in body mass energy per week on the high-fat diet, and the net increase on the high-fat diet compared with the low-fat diet [Δ (high-fat − low-fat)] was greatest for male and female Mc4rKO (15.2 or 19.3 kJ/d, an increase of 8.6–9.3 kJ/d over low-fat), followed by male Mc3rKO (11.3 kJ/d, + 9.0 kJ), followed by female Mc3rKO and male WT (4.4 or 5.1 kJ/d, + 3.9 or + 3.6 kJ), whereas female WT mice were most resistant to DIO (1.5 kJ/d, + 1.1 kJ).
TABLE 1.
Effect of dietary fat content on energy balance of male and female WT, Mc3rKO, and Mc4rKO mice
| Genotype | Low-fat diet | High-fat diet | Δ (High-fat − low-fat) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| WT | Mc3rKO | Mc4rKO | WT | Mc3rKO | Mc4rKO | WT | Mc3rKO | Mc4rKO | |
| Gain in body mass energy (kJ/d) | |||||||||
| Male | 0.5 ± 0.2a | 2.3 ± 0.7a | 6.6 ± 1.8b | 4.4 ± 0.6a# | 11.3 ± 1.1 b* | 15.2 ± 1.4c* | 3.9 ± 0.6a | 9.0 ± 0.7b | 8.6 ± 2.9b |
| Female | 0.4 ± 0.2a | 1.7 ± 0.4a | 10.0 ± 1.1b | 1.5 ± 0.5a | 5.1 ± 1.3b # | 19.3 ± 0.6c* | 1.1 ± 5a | 3.6 ± 1.4a* | 9.3 ± 1.4b |
| Energy intake (kJ/d) | |||||||||
| Male | 58.6 ± 2.3a | 57.0 ± 2.1a | 70.6 ± 2.2b | 62.4 ± 2.8a | 63.9 ± 1.3a# | 84.6 ± 2.4b* | 3.8 ± 1.6a | 6.9 ± 1.9a | 14.0 ± 4.0b |
| Female | 54.3 ± 1.6a | 53.5 ± 2.5a | 78.0 ± 0.5b | 58.2 ± 1.8a* | 55.1 ± 2.6a | 90.8 ± 1.7b* | 3.9 ± 1.4a | 1.6 ± 0.9a | 12.8 ± 1.9b |
| bEEest (kJ/d) | |||||||||
| Male | 57.6 ± 2.3 | 53.5 ± 1.6 | 60.7 ± 2.1 | 55.7 ± 2.8a | 47.1 ± 1.1a # | 61.8 ± 2.0a,b | -1.9 ± 1.1 | −6.4 ± 2.5 | + 1.1 ± 3.9 |
| Female | 53.4 ± 1.5a | 50.9 ± 2.5a | 63.0 ± 1.6b | 55.8 ± 1.7a | 47.3 ± 0.9b | 62.0 ± 1.6c | + 2.4 ± 1.3 | −3.6 ± 2.0 | −0.9 ± 3.1 |
| ME% | |||||||||
| Male | 0.9 ± 0.3a | 4.0 ± 1.1a | 9.2 ± 2.4b | 7.1 ± 0.9a* | 17.6 ± 1.4b* | 17.9 ± 1.5b* | 6.2 ± 1.1a | 13.7 ± 1.0b | 8.7 ± 3.7a,b |
| Female | 0.7 ± 0.4a | 3.1 ± 0.8a | 12.9 ± 1.4b | 2.5 ± 0.8a | 9.1 ± 1.9b # | 21.3 ± 0.7c* | 1.7 ± 0.7a* | 6.0 ± 2.1a,b* | 8.4 ± 1.9b |
Data are from the mice whose growth curves are shown in Fig. 1, A–D, and represent weekly means collected during 3 wk on a low-fat and 5 wk on a high-fat diet. bEEest represents the proportion of kilojoules consumed less the metabolic cost of fat and protein deposition and is an estimate of the kilojoules available for non-growth-related basal metabolism and physical activity. The effect of genotype and diet on each of the parameters shown was analyzed using two-way ANOVA with repeated measures (low-fat and high-fat diet). Values not sharing a superscript letter (a, b, and c) within rows and within diets are statistically different from each other at P < 0.05; for the effects of diet within sex and within genotype:
P < 0.05;
P < 0.01.
The Δ (high-fat − low-fat) data were analyzed using two-way ANOVA with sex and genotype as independent variables. Values not sharing superscripts within rows (i.e. genotype) are statistically different from each other at P < 0.05. *, Significant effect of sex (within column) at P < 0.05.
Weight gain and adiposity of 6-month-old female Mc3rKO, Mc4rKO, and WT mice (n = 4–6 per group) was also compared after 3 months on either a low-fat or high-fat diet (Fig. 1, E–I). Significantly increased body mass of Mc3rKO relative to WT was observed only on the high-fat diet (Fig. 1E), although Mc3rKO fed a low-fat diet exhibited a modest increase in adiposity. The high-fat diet resulted in a doubling of body mass of Mc3rKO and Mc4rKO (Fig. 1F). On the high-fat diet, adiposity and FM of female Mc3rKO were not significantly different from Mc4rKO (Fig. 1, D, G, and H). Data shown in Fig. 1 and Table 1 are representative of several experiments. Similar results were also observed using a moderately high-fat diet (45% kJ from fat) (data not shown and Refs. 24, 32, and 36).
Food intake and metabolic efficiency
Mc4rKO were hyperphagic compared with Mc3rKO and WT mice, irrespective of sex and diet (Table 1 and Fig. 2, A and B). The high-fat diet further increased energy intake of male and female Mc4rKO [diet effect within genotype for males and females, P < 0.01; comparison of Δ (high-fat − low-fat) intake between WT and Mc4rKO, P < 0.01]. Diet effects on energy intake of WT and Mc3rKO were gender specific (Table 1). Male Mc3rKO increased energy consumption on the high-fat diet by +6.9 kJ/d, compared with the low-fat diet (effect of diet, P < 0.05). There was a modest, and not statistically significant, increase in kilojoules intake of male WT on a high-fat diet (+3.8 kJ/d). Diet did not significantly affect daily kilojoules consumed of female Mc3rKO (+ 1.6 kJ/d). However, female WT mice exhibited a small increase comparable to that observed in males that was statistically significant (+3.9 kJ/d, P < 0.01) (Table 1).
Fig. 2.
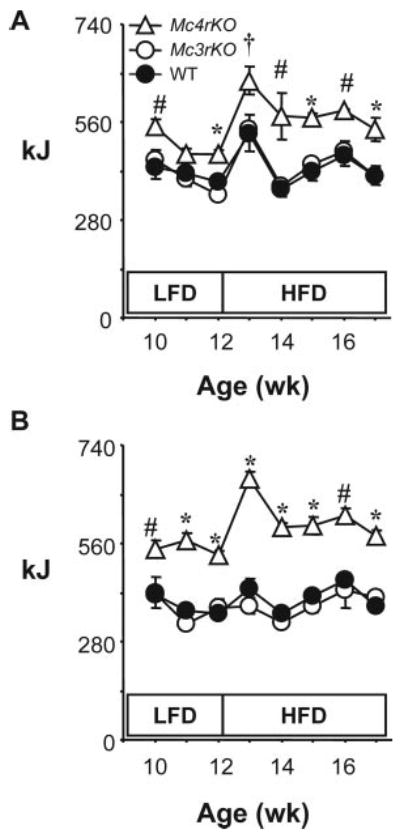
Genotype-specific effects of dietary fat content on food intake of Mc3rKO and Mc4rKO. Weekly food intake (kJ) for male (A) and female (B) WT, Mc3rKO, and Mc4rKO. The legend for this figure is the same as that for Fig. 1, A–D. Food intake was measured during the period indicated by a black bar in Fig. 1, A and B. For each week, significant differences (P < 0.05) are indicated as follows: *, Mc4rKO vs. Mc3rKO, WT; #, Mc4rKO vs. WT; †, Mc4rKO vs. Mc3rKO. Results of two-way ANOVA with repeated-measures analysis examining the effects of diet and genotype for mean weekly intake on low-fat diet (LFD, 10% kJ from fat) and high-fat diet (HFD, 60% kj from fat) are reported in Table 1. Food intake in kilojoules of Mc4rKO, but not Mc3rKO, is increased compared with WT controls, irrespective of diet. Male and female Mc4rKO also increased kilojoules of food consumed when exposed to the high-fat diet. Male WT and Mc3rKO exhibited a transient hyperphagia on the high-fat diet, whereas for female WT and Mc3rKO, kilojoules consumed was not affected by diet.
With the exception of female WT, the high-fat diet significantly increased ME% (Table 1). ME% of Mc4rKO of either sex was always higher compared with WT. For Mc3rKO, ME% was significantly increased compared with WT only on the high-fat diet, with the high-fat diet having a greater impact in males (Table 1).
Energy expenditure
For this study, bEEest is assumed to be energy available for basal metabolism and physical activity once the metabolic cost of protein and fat deposition is taken into account. The bEEest was lower in Mc3rKO compared with WT and Mc4rKO, with the high-fat diet increasing the difference (Table 1). For female Mc3rKO, bEEest was 5% lower compared with WT on the low-fat diet, the difference increasing to 15% on the high-fat diet (P < 0.05 on high-fat diet). A similar difference was observed in male Mc3rKO, with a strong tendency (P = 0.058) for bEEest of male Mc3rKO to be lower compared with WT. Moreover, the decline in bEEest of male Mc3rKO from 53.5 to 47.1 kJ/d was statistically significant (P < 0.05). The bEEest of male Mc4rKO was not statistically different from WT irrespective of diet. The bEEest of female Mc4rKO was modestly increased and was also not affected by diet. These data suggest that energy available for physical activity and basal metabolism, once metabolic costs of fat deposition have been subtracted from the energy budget, is significantly reduced in Mc3rKO relative to WT and Mc4rKO.
The efficiency of deposition of fat and protein is estimated at 70 and 50%, respectively (34). It is worth noting that the metabolic costs estimated for fat and protein deposition were high for Mc4rKO of either sex (8–10 kJ/d on the high-fat diet) relative to WT controls (1.0–2.5 kJ/d). Mc3rKO on the high-fat diet also exhibited a significantly higher value compared with WT, particularly in males (5.5 kJ/d). Metabolic costs of fat deposition in mice rapidly gaining weight, such as Mc4rKO (Fig. 1, A and B), may therefore represent a significant proportion of the energy expenditure budget.
Indirect calorimetry data from a large cohort of mice, whose body mass data are shown in Fig. 3A, also indicated that Mc4rKO of either sex had increased TEEcal, due to increases in REEcal and NREEcal relative to WT (Fig. 3B). There was an intermediate increase in TEEcal, REEcal, and NREEcal of male, but not female, Mc3rKO. The increase in TEEcal of Mc4rKO appeared most severe for females. TEEcal adjusted for FFM or body mass of female Mc4rKO was still increased relative to WT and Mc3rKO. However, in males, energy expenditure was not significantly different between genotype when the data were adjusted for FFM or body mass.
Fig. 3.
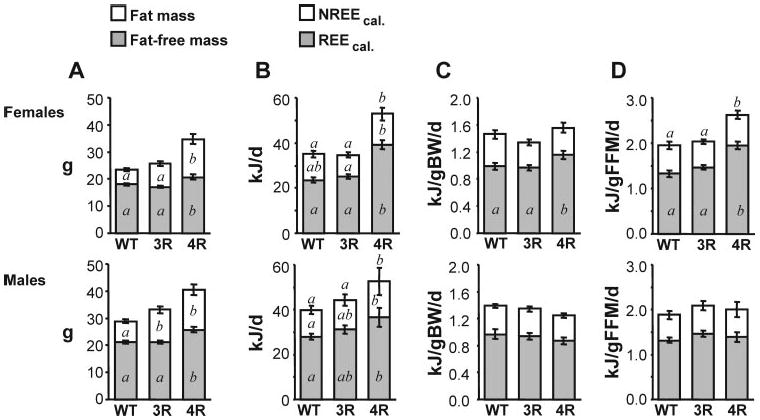
Energy expenditure of male and female Mc3rKO, Mc4rKO, and WT mice determined by indirect calorimetry. Energy expenditure (kJ/d) was determined using indirect calorimetry for WT females (n = 21) and males (n = 29), Mc3rKO females (n = 22) and males (n = 23), and Mc4rKO females (n = 16) and males (n = 10) fed a moderately high-fat diet (45% kJ from fat) for 6–8 wk. FM and FFM (A) were determined using NMR. Note that there was a strong tendency for increased FM of female Mc3rKO compared with WT (P = 0.055). REEcal, NREEcal, and TEEcal are presented per animal (B) and adjusted for body weight (C) or FFM (D). Significant differences (P < 0.05) between groups are indicated using letters; groups not sharing the same letter are significantly different. TEEcal in kilojoules per day was significantly increased in Mc4rKO, irrespective of sex. Female Mc4rKO also exhibited an increase in TEEcal and REEcal adjusted for FFM and for REEcal adjusted for body weight. TEEcal and REEcal of female Mc3rKO was normal when expressed per animal and when adjusted for body weight and FFM. REEcal and NREEcal of Mc3rKO exhibited an intermediate increase, compared with a large increase observed Mc4rKO. In males, energy expenditure parameters adjusted for body weight or FFM were not significantly different from WT.
Multiple regression analysis of the combined groups indicated a positive association of TEEcal and REEcal with FM (R = 0.58 and 0.60), FFM (R = 0.57 and 0.53), and total body mass (R = 0.62) (Fig. 4, A and B, and data not shown). Linear regression with genotype indicated that the association between body mass and either TEEcal or REEcal of Mc4rKO was significantly higher relative to the regression line for Mc3rKO (Fig. 4, A and B). No association was evident in WT mice, perhaps due to the narrow range of body weights. These data suggest that metabolic rate of Mc4rKO is higher compared with Mc3rKO as a function of body mass.
Fig. 4.
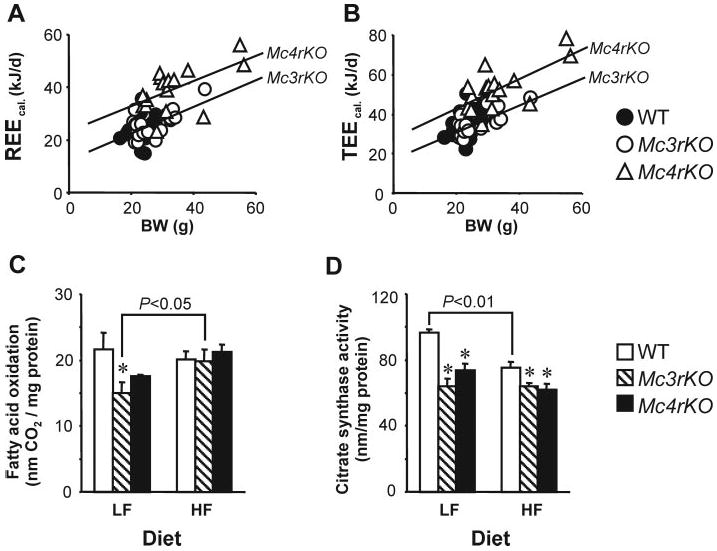
Regression analysis of energy expenditure data and muscle oxidative metabolism of female Mc3rKO, Mc4rKO, and WT mice. Regression analysis of REEcal and TEEcal of female mice suggest reduced energy expenditure of Mc3rKO, relative to Mc4rKO, independently of differences in body weight (A and B). The differences in energy expenditure of female Mc3rKO and Mc4rKO, determined using calorimetry, did not correlate with skeletal muscle oxidative capacity. FAO (C) was significantly reduced in Mc3rKO fed the low-fat (LF) diet but not the moderately high-fat (HF) diet. Citrate synthase activity in skeletal muscle of Mc3rKO and Mc4rKO was lower compared with WT controls, irrespective of diet (D). Body mass data for mice shown in C and D are shown in Fig. 1, E–I. *, P < 0.05 compared with WT.
FAO and mitochondrial function in red muscle
FAO ([14C]palmitate oxidation) and citrate synthase activity were measured in red oxidative muscle isolated from the gastrocnemius of female WT, Mc3rKO, and Mc4rKO fed a low-fat or high-fat diet (Fig. 4, C and D). Body mass data for this cohort were shown in Fig. 1, E–I. On the low-fat diet, FAO was significantly reduced in Mc3rKO compared with WT, with a tendency for a reduction in Mc4rKO (Fig. 4C). On the high-fat diet, FAO was not significantly different between genotype groups. This was due to FAO in the melanocortin receptor knockout being higher on the high-fat diet, a change that was statistically significant for Mc3rKO (P < 0.05). Citrate synthase activity was significantly reduced in Mc3rKO and Mc4rKO compared with WT mice irrespective of diet (Fig. 4 D). The high-fat diet also significantly reduced citrate synthase activity in female WT mice (P < 0.01) but had no effect on citrate synthase activity of either Mc3rKO or Mc4rKO.
Spontaneous locomotor behavior
Male and female Mc3rKO (n = 5) and Mc4rKO (n = 4 females and 3 males) mice fed a high-fat diet were hypoactive compared with WT (n = 5) (least squares mean for total beam breaks per day was 50,286 ± 4,457 for WT; 29,334 ± 4,457 for Mc3rKO; and 29,220 ± 5,382 for Mc4rKO; WT vs. Mc3rKO and Mc4rKO, P < 0.05).
Effect of diet-genotype interactions on insulin resistance
Fasting insulin and glucose
Fasting insulin and glucose were measured in male and female Mc3rKO, Mc4rKO, and WT mice at 6 months of age after 3 months on diets with variable fat content (10, 45, or 60% kJ from fat) (Fig. 5, A–D). In males, Mc3r and Mc4r deficiency exacerbated fasting hyperinsulinemia associated with consumption of the 60% high-fat diet (Fig. 5, A and C, and data not shown). For males on the 45% fat diet, a significant increase in fasting insulin was observed only in Mc4rKO. In females, Mc4rKO exhibited a more marked response to both the 45% and 60% high-fat diet, with Mc3rKO having insulin levels comparable to WT irrespective of diet.
Fig. 5.
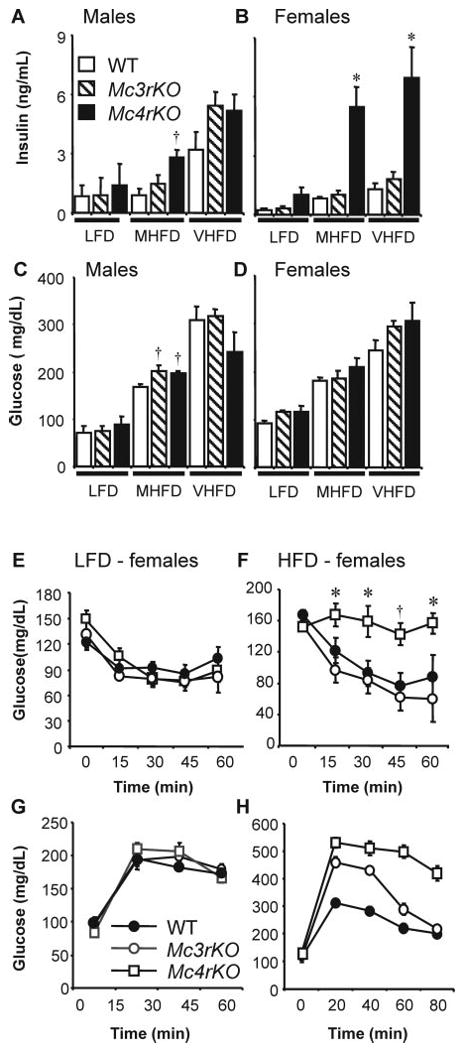
Effects of genotype and diet on fasting insulin and glucose and on ITT and IPGTT data. Mc4rKO are hyperinsulinemic and are glucose intolerant and insulin resistant compared with Mc3rKO and WT mice on a high-fat diet. A–D, Fasting serum insulin (A and B) and blood glucose (C and D) of female and male Mc4rKO, Mc3rKO, and WT mice fed a low-fat diet (LFD, 10% kJ from fat), moderately high-fat diet (MHFD, 45% kJ from fat), or very high-fat diet (VHFD, 60% kJ from fat). *, P < 0.05 compared with WT and Mc3rKO; †, P < 0.05 compared with WT. E–H, ITT (E and F) and IPGTT (G and H) result of obese female Mc4rKO, Mc3rKO, and WT mice fed either a low-fat diet (LFD, n = 3–5 per group) (E and G) or moderately high-fat diet (HFD, n = 5–11) (F and H).*, P < 0.05 vs. Mc3rKO; †, P < 0.05 vs. WT and Mc3rKO. For IPGTT data (H), there was no significant difference between groups at 0 min (i.e. baseline fasting glucose); between 20 and 60 min, all genotypes were significantly different (P < 0.05); at 80 min, glucose levels were significantly (P < 0.01) higher in Mc4rKO but had returned to WT levels in Mc3rKO.
ITT and GTT results
Fasting insulin data suggest that Mc4rKO are more sensitive, relative to Mc3rKO, to the effects of dietary fat on insulin resistance. In particular, female Mc3rKO on high-fat diets appeared to develop a syndrome of obesity not associated with severe insulin resistance. Further investigation of insulin sensitivity used mice fed a moderately high-fat diet (45% kJ from fat) to avoid type 2 diabetes (fasting blood glucose > 200 mg/dl) observed on the high-fat diet (60% kJ from fat) (Fig. 5, C and D).
ITT and GTT results from 6-month-old WT, Mc3rKO, and Mc4rKO on the low-fat diet were not significantly different (Fig. 5, E and G). The moderately high-fat diet caused reduced glucose tolerance that was particularly severe for Mc4rKO (Fig. 5H). The glucose-lowering action of insulin was also significantly compromised in Mc4rKO, compared with Mc3rKO and WT mice, on the moderately high-fat diet (Fig. 5F).
Effect of genotype and diet on insulin receptor signaling
Phosphorylation of PKB, a downstream mediator of PI3K action, is involved in mediating intracellular effects of insulin in target organs, including stimulating glucose uptake (31). Phosphorylation of PKB at Ser473 and Thr308 is an indicator of kinase activation (41–43). In Mc4rKO, insulin-stimulated PKB activity was significantly reduced compared with both WT and Mc3rKO irrespective of diet (Fig. 6, A and B). Phosphorylation of two downstream targets of PKB, GSK3β and FoxO1, was also reduced in liver of Mc4rKO compared with both WT and Mc3rKO.
Fig. 6.
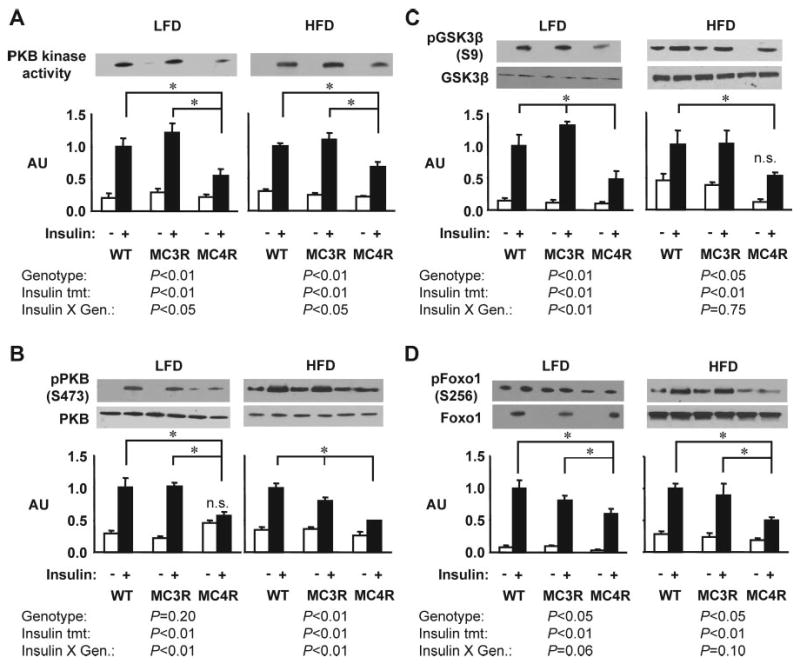
Insulin signaling in liver is impaired in female Mc4rKO relative to WT and Mc3rKO, irrespective of diet. Representative Western blots and quantified data (n = 3 per group) are shown for basal (white bar) or insulin-stimulated (black bar) PKB kinase activity (A), phosphorylation of PKB on Ser473 (B), phosphorylation of GSK3β on Ser9 (C), or phosphorylation of FoxO1 on Ser256 (D). Quantified data were analyzed within diet using two-way ANOVA (independent variables were genotype and insulin treatment). Phosphospecific antibody data (B–D) was normalized for total protein content. For most outcomes measured, insulin-stimulated activity was reduced in liver of Mc4rKO (MC4R), irrespective of diet, compared with Mc3rKO (MC3R) and WT mice. Significant differences between groups are indicated by lines: *, P < 0.05. For PKB activity assessed by kinase assay (A) or serine phosphorylation (B), two-way ANOVA indicated that the degree of insulin stimulation was dependent on genotype. The effects of insulin treatment were statistically significant (P < 0.05), unless indicated otherwise (n.s., not significant). AU, Arbitrary units; LFD, low-fat diet (10% kJ from fat); HFD, high-fat diet (45% kJ from fat).
Insulin stimulation of PKB activity in muscle and adipose was not significantly affected by genotype in mice fed the low-fat diet (Fig. 7, A and B, and Fig. 8, A and B). Phosphorylation of GSK3β in muscle, and FoxO1 in adipose tissue, was also not affected by genotype in mice on the low-fat diet. However, on the moderately high-fat diet, there was a marked deterioration of insulin-stimulated PKB activity in Mc4rKO in skeletal muscle (Fig. 7, A and B) and modest reduction of PKB activity in adipose (Fig. 8, A and B). This correlated with a significant reduction in the phosphorylation of GSK3β in muscle, and FoxO1 in adipose tissue, after insulin treatment (Figs. 7C and 8C).
Fig. 7.
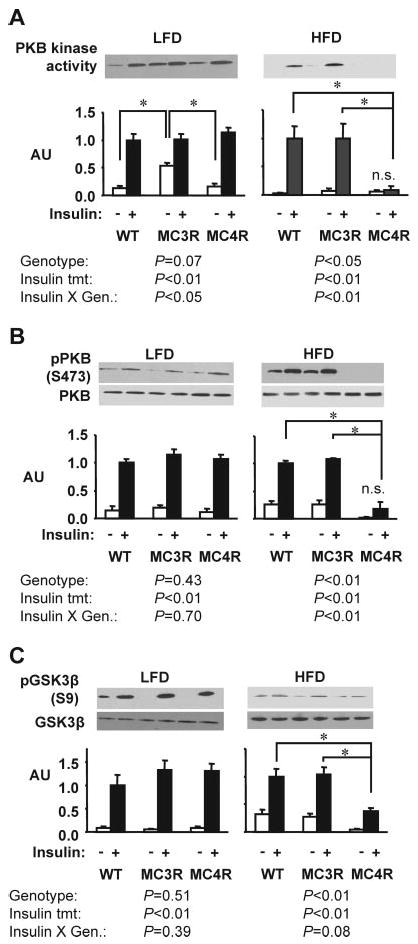
High-fat diet is associated with a marked reduction in insulin receptor signaling in skeletal muscle of female Mc4rKO compared with Mc3rKO and WT mice. Representative Western blots, with quantified data (n = 3 per group), are shown for basal and insulin-stimulated PKB kinase activity (A), phosphorylation of PKB on Ser473 (B), or phosphorylation of GSK3β on Ser9 (C). A significant effect of genotype was observed only for Mc4rKO (MC4R) fed the high-fat diet, resulting in the loss of a significant effect of insulin on PKB kinase activity, phosphorylation of PKB, and phosphorylation of GSK3β. Significant differences between groups are indicated by lines: *, P < 0.05. Unless indicated otherwise (n.s., not significant), treatment effects of insulin on kinase activity and phosphorylation were statistically significant (P < 0.05). AU, Arbitrary units; MC3R, Mc3rKO mice; LFD, Low-fat diet (10% kJ from fat); HFD, high-fat diet (45% kJ from fat).
Fig. 8.
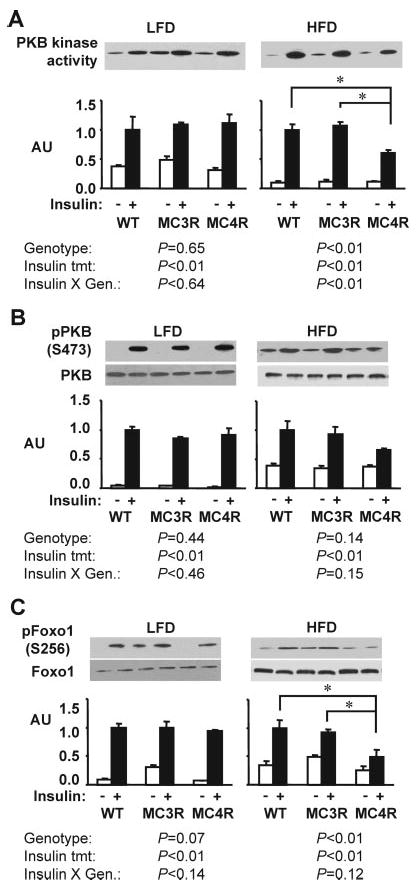
High-fat diet is associated with reduced insulin receptor signaling in adipose of Mc4rKO compared with Mc3rKO and WT mice. Representative Western blots, with quantified data (n = 3 per group), are shown for basal and insulin-stimulated PKB kinase activity (A), phosphorylation of PKB on Ser473 (B), or phosphorylation of FoxO1 on Ser256 (C). A significant effect of genotype on insulin-stimulated PKB activity and phosphorylation of FoxO1 was observed only for Mc4rKO (MC4R) fed the high-fat diet. Significant differences between groups are indicated by lines: *, P < 0.05. Unless indicated otherwise (n.s., not significant), treatment effects of insulin were statistically significant (P < 0.05). AU, Arbitrary units; MC3R, Mc3rKO mice; LFD, low-fat diet (10% kJ from fat); HFD, high-fat diet (45% kJ from fat).
Ser307 phosphorylation of IRS1 reduces the efficiency of association with the insulin receptor tyrosine kinase, reducing insulin activation of PI3K and MAPK cascades (30). Insulin resistance of knockout and WT mice fed the moderately high-fat diet was further compared by measuring Ser307 phosphorylation of IRS1 in liver, muscle, and adipose (Fig. 9). Ser307 phosphorylation of IRS1 was significantly increased in liver and muscle of insulin-treated Mc4rKO compared with WT (Fig. 9, A and B). In muscle, there was a strong tendency (P = 0.06) for Ser307 phosphorylation of IRS1 to be significantly increased in Mc4rKO compared with Mc3rKO. A significant increase in Ser307 phosphorylation of IRS1 was also observed in liver of Mc3rKO although not as severe as that observed in Mc4rKO. Genotype did not significantly affect Ser307 phosphorylation of IRS1 in adipose tissue (Fig. 9C).
Fig. 9.
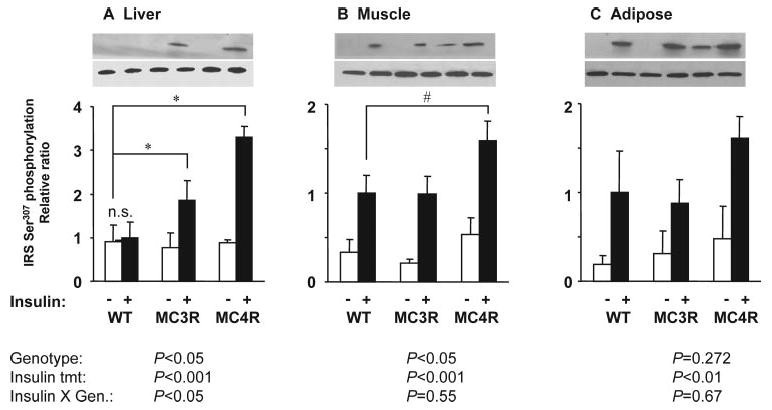
Increased Ser307 phosphorylation of IRS1 in liver and skeletal muscle of Mc4rKO compared with WT fed a moderately high-fat diet. Representative Western blots, with quantified data (n = 3 per group), are shown for basal and insulin-stimulated IRS1 phosphorylation on Ser307 in liver (A), skeletal muscle (B), and retroperitoneal adipose tissue (C). These samples were from the high-fat diet groups shown in Figs. 6–8. A significant effect of genotype on insulin-stimulated IRS1 Ser307 phosphorylation was observed for Mc4rKO (MC4R) and Mc3rKO (MC3R) compared with WT in liver, and for Mc4rKO compared with WT in skeletal muscle. Significant difference between groups is indicated by lines: *, P < 0.05. Unless indicated otherwise (n.s., not significant), treatment effects of insulin were statistically significant (P < 0.05). Mice were maintained on moderately high-fat diet (45% kJ from fat).
Liver metabolism
Severe insulin resistance associated with leptin deficiency is associated with hepatomegaly, steatosis, and abnormal hepatic expression of genes involved in lipid metabolism (41, 44, 45). Liver weight and lipid content and hepatic ApoA4 and Scd1 mRNA expression were measured in male and female mice after 3 months on the 60% high-fat or 45% moderately high-fat diet (Fig. 10 and data not shown). Compared with WT and Mc3rKO, Mc4rKO were hepatomegalic and had more severe steatosis (Fig. 10, A and C). Analysis of liver weight as a function of adiposity indicated a positive linear association in Mc4rKO, with no effect of increasing adiposity in WT or Mc3rKO (Fig. 10B). Measurement of hepatic lipid content indicated mild steatosis in Mc3rKO compared with Mc4rKO (lipid content in mg/g: WT, 39.1 ± 6.4; Mc3rKO, 103.5 ± 19.7; and Mc4rKO, 184.9 ± 18.6; all P < 0.05). In females, the expression of ApoA4 and Scd1 mRNA was significantly increased in Mc4rKO compared with WT, with normal expression in Mc3rKO. In males, Mc3rKO exhibited an intermediate increase in ApoA4 and Scd1 mRNA compared with Mc4rKO (Fig. 10, D and E).
Fig. 10.
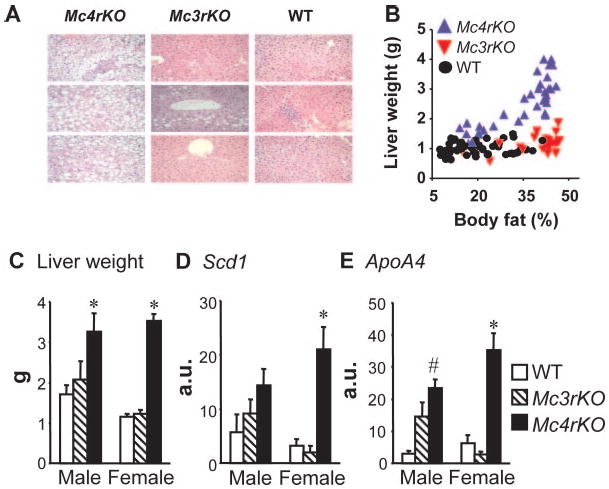
Hepatomegaly and liver gene expression data suggest hepatic insulin resistance is most severe for Mc4rKO. Mc4rKO demonstrate marked hepatic steatosis and hepatomegaly compared with Mc3rKO and WT. A, Hematoxylin and eosin-stained cross-sections of liver tissue from female Mc4rKO, Mc3rKO, and WT mice fed a high-fat diet. B, Plot of liver weight vs. adiposity from several studies where mice were fed low- or high-fat diets. C, Mean liver weight of female Mc4rKO, Mc3rKO, and WT mice fed a moderately high-fat diet. Mc4rKO also exhibited generally increased lipogenic gene expression compared with both Mc3rKO and WT mice (n = 5–6 per group). D and E, Expression of stearoyl-CoA desaturase 1 (Scd1) (D), and apolipoprotein A4 (Apoa4) (E) in the liver of WT, Mc3rKO, and Mc4rKO fed the high-fat diet. Data are expressed as arbitrary units (a.u.). *, P < 0.05 vs. WT and Mc3rKO; #, P < 0.05 vs. WT mice, within gender.
Discussion
Melanocortins have an essential role in regulating energy homeostasis in mice and humans, acting through two receptors that are expressed in regions of the central nervous system involved in regulating energy homeostasis (2, 9). These experiments demonstrate the complexity, and importance, of the interaction between the two melanocortin receptors and dietary fat in the development of obesity and insulin resistance. A strength of these experiments is that both knockout strains have been backcrossed onto C57BL/6J, removing the confounding effect of genetic background on obesity and diabetes. Overall, these results suggest that the Mc4r has a critical role in preventing hepatic insulin resistance, with the high-fat diet associated with a broader insulin resistance.
The results from mice fed a low-fat diet are consistent with previous results demonstrating a modest obese phenotype of Mc3rKO compared with Mc4rKO. On a high-fat diet, the phenotype of the two knockout strains is far less distinct. Surprisingly, DIO male Mc3rKO mice are very similar to male Mc4rKO in terms of weight gain, metabolic efficiency, and fasting hyperinsulinemia. Additional experiments comparing the insulin-resistant phenotype of DIO male Mc3rKO and Mc4rKO, and their response to compounds stimulating melanocortin receptor activity, are currently in progress. The interaction of the Mc3r with dietary fat is gender specific. The less rapid weight gain of female Mc3rKO on a high-fat diet may be a factor in the modest perturbations of glucose homeostasis compared with Mc4rKO.
DIO of Mc3rKO and Mc4rKO
Reduced MSH synthesis (11, 12), or loss of MSH action through Mc3r (15) or Mc4r (28, 32), is associated with weight gain and an exacerbation of DIO. The results of these experiments confirm these findings and demonstrate that obesity associated with Mc3r deficiency is more dependent on dietary fat. On a low-fat diet, Mc3rKO exhibit a modest increase in FM, and normal body weight, which was previously reported (13, 15). On a high-fat diet, FM of Mc3rKO is comparable to that of Mc4rKO. Mc4rKO also exhibit a marked response to a high-fat diet, consistent with previous results using outbred mice and standard or moderately high-fat chows (22, 28). However, Mc4rKO exhibit a robust obesity that is independent of dietary fat content.
Food intake is clearly a factor in the different sensitivity of Mc3rKO and Mc4rKO to a high-fat diet. Previous data indicate that hyperphagia contributes to the obese phenotype of Mc4rKO on a low-fat diet (14, 46, 47). In this study, on the low-fat diet, Mc4rKO were massively hyperphagic compared with Mc3rKO and WT, a difference further compounded by the high-fat diet. As previously reported, hyperphagia was not a factor in the obese phenotype of Mc3rKO on a low-fat diet (13, 15). However, on the high-fat diet, male Mc3rKO were modestly hyperphagic. We have previously observed a modest hyperphagia, restricted to the lights-on period, in male Mc3rKO after introduction of a high-fat diet (24). Loss of Mc3r in the ventromedial hypothalamus could be involved in obesity of Mc3rKO. Lesions in the ventromedial hypothalamus of rats are associated with obesity (48, 49), at least partially due to hyperphagia during the lights-on period (49).
Although activation of Mc3r does not appear to inhibit feeding behavior (20, 50, 51), these are not the first data to indicate that inhibition or loss of Mc3r modestly increases food intake. Intracerebroventricular infusion of the Mc3r/Mc4r antagonist AgRP modestly increases food intake in Mc4rKO (23, 25). Loss of Mc3r also attenuates the inhibition of food intake by leptin (22). However, female Mc3rKO were not hyperphagic on the high-fat diet, which may be a factor in the less rapid gain of body mass energy observed in female Mc3rKO (5.1 kJ/d) compared with male Mc3rKO (11.3 kJ/d). The regulation of feeding behavior by Mc3r thus appears to be gender specific, having a modest role in males to prevent hyperphagia on a high-fat diet.
Energy metabolism of Mc3rKO and Mc4rKO
Obesity results from an imbalance of energy intake and energy expenditure. Mc3r and Mc4r are expressed in regions of the hypothalamus and hind brain involved in the regulation of energy expenditure (5, 52). However, examination of the response of knockout mice to nonselective melanocortin receptor agonists suggest a primary role for the Mc4r in the acute regulation of energy expenditure (16, 20–22, 47). The Mc4r has a crucial role in the regulation of pathways known to regulate energy expenditure, including the hypothalamo-pituitary-thyroid axis (23), sympathetic nervous activity (21), and stimulation of brown adipose tissue thermogenesis (22, 47), with no compensatory role for the Mc3r. Diet-induced thermogenesis, an adaptive metabolic response to hyperphagia that is dependent on β-adrenergic receptor signaling (53), requires a functional Mc4r but is normal in Mc3r-deficient mice (13, 28).
Despite a lack of experimental evidence supporting a role for the Mc3r in regulating energy expenditure, the results from these experiments and previous studies (13, 15) demonstrating obesity of Mc3rKO without hyperphagia strongly suggests an important role in regulating peripheral metabolism and energy balance. Indeed, the comparable increase in metabolic efficiency of male Mc3rKO and Mc4rKO fed a high-fat diet suggests an equal role for the receptors in promoting a thrifty phenotype. It has, however, been difficult to demonstrate a reduction in energy expenditure of Mc3rKO (13, 15) or Mc4rKO (20, 32, 47). The problems associated with interpreting energy expenditure data from lean and obese subjects of any species have been discussed by other investigators (37, 54). This study measured energy expenditure using two methods, first using food intake and estimates of gain in body mass energy (34) and second by indirect calorimetry. Mitochondrial activity in skeletal muscle was also measured to obtain another estimate of potential metabolic rate.
The energy expenditure data from the current study suggest that energy expenditure of Mc4rKO is significantly increased compared with WT, with no significant difference in Mc3rKO. The seemingly paradoxical finding that energy expenditure was highest in the strain with the fastest rate of fat deposition may be explained by two observations. The metabolic costs of digestion will be higher for Mc4rKO consuming an extra 21–36 kJ/d compared with Mc3rKO and WT. In addition, the metabolic cost of fat and protein deposition is estimated to be increased for Mc4rKO. This variable was estimated at 3–5 kJ/d for Mc4rKO on the low-fat diet, increasing to 7–10 kJ/d on the high-fat diet, or 5- to 10-fold greater than that estimated for WT mice. Such a large metabolic cost of weight gain in Mc4rKO is likely transient and may not occur once a new set-point in body weight has been achieved. Clearly, taking into account the growth rate of Mc4rKO and other models where obesity is associated with rapid gains in body mass is important when analyzing and interpreting energy expenditure data. The metabolic cost of fat deposition was also significant for male Mc3rKO (5.5 kJ/d) but not for female Mc3rKO depositing FM at a slower rate.
Preferential nutrient partitioning to adipose remains an attractive hypothesis explaining the obese phenotype in Mc3rKO. Indeed, removing the metabolic costs of fat deposition from the daily energy budget reveals a significant reduction in the amount of energy available for other aspects of basal metabolism of Mc3rKO compared with WT and Mc4rKO. Preferential nutrient partitioning to fat is also probably a factor in Mc4rKO (47). However, for a reason that is not clear, but may involve the metabolic cost of digestion, a comparable reduction in bEEest was not observed for Mc4rKO.
The mechanisms involved in the nutrient partitioning defect of Mc3rKO and Mc4rKO are not known. However, on the B6 background, both knockout mice have a high respiratory exchange ratio, indicating reduced oxidation of fatty acids that is associated with a positive imbalance of fat intake (24, 32). In this study, skeletal muscle oxidative metabolism, as determined by oxidation of [14C]palmitate and citrate synthase activity, was reduced in both Mc3rKO and Mc4rKO when compared with controls. Citrate synthase activity has been suggested to be an indicator of mitochondrial content and function (40, 55). Reduced capacity of FAO in muscle may therefore be a factor contributing to the high respiratory exchange ratio of Mc3rKO and Mc4rKO. The mechanisms by which Mc3r and Mc4r regulate oxidative metabolism in muscle are not clear at this time. Both receptors are expressed in the hypothalamus, which has been shown to be an important site for the regulation of FAO in muscle by leptin, likely through the regulation of the autonomic nervous system (56).
Insulin resistance of Mc3rKO and Mc4rKO is largely secondary to obesity
The melanocortin system is thought to be a promising target for the treatment of obesity and insulin resistance. Treatment of DIO C57BL/6J with nonselective melanocortin agonists reduces body weight and improves insulin sensitivity (57, 58). Moreover, infusion of SHU9119 intracerebroventricularly in rats is associated with the development of hepatic insulin resistance (29). Mc4rKO exhibit marked hyperinsulinemia and hyperglycemia, which can be attenuated but not completely reversed by pair-feeding with WT controls (14, 47). Type 2 diabetes and glucose intolerance are also observed in 6- to 8-month-old viable yellow (Avy) and lethal yellow (Ay/a) mice, which ectopically express an antagonist of the Mc1r and Mc4r (59). In contrast, Mc3rKO exhibit a very mild insulin-resistance phenotype (13, 15). Taken together, these observations suggest that the Mc4r has a more important role in preventing insulin resistance.
A goal of these experiments was to compare insulin action in age- and weight-matched Mc3rKO and Mc4rKO. The results of this study, and those of a previous investigation (32), suggest the development of a comparatively mild insulin resistance of Mc4rKO fed a low-fat diet. Obese Mc4rKO fed a low-fat diet exhibit a modest increase in fasting insulin and a deterioration of hepatic insulin sensitivity. This result is surprising, when the diabetic phenotype of viable yellow (Avy) and lethal yellow (Ay/a) mice is taken into consideration (59). It is tempting to speculate that ectopically expressed agouti may affect insulin sensitivity or β-cell function independently of Mc4r, as recently demonstrated for adipocyte metabolism (60, 61). However, carefully controlled studies using Mc4rKO, Ay/a and Avy mice on the same B6 background are required to test this hypothesis.
The current study extended previous investigations to include insulin receptor signaling in tissues of female knockout and WT mice, which exhibited the clearest distinction between genotype in terms of fasting hyperinsulinemia, glucose tolerance, and response to insulin treatment. The stimulation of PKB activity and the phosphorylation of targets of PKB, FoxO1, and GSK3β after insulin treatment was reduced in liver of 6-month-old female Mc4rKO. Insulin stimulation of PKB kinase activity or Ser473 phosphorylation is normal in liver, and also in muscle, of 6-wk-old Mc4rKO (Sutton, G. M., and A. A. Butler, unpublished data).
The development of severe insulin resistance in skeletal muscle of Mc4rKO, associated with a marked decline in the stimulation of PKB activity and GSK3β phosphorylation by insulin, was observed only on the high-fat diet. This was associated with a marked deterioration of glucose tolerance and insulin-stimulated glucose disposal, consistent with the importance of muscle as a site of glucose disposal. The mechanisms involved in the development of severe insulin resistance in Mc4rKO on a high-fat diet were not determined in this study. However, several factors could be involved in the development of severe muscle insulin resistance of Mc4rKO fed a high-fat diet, including but not limited to the intramyocellular accumulation of fatty acids (62) and inflammation (63). At the level of neuroendocrine systems, the progressive development of hypothalamic leptin resistance might also be involved in the deterioration of insulin action in Mc4rKO, independently of diet (36, 44, 50). Alternatively, leptin-independent regulation of Mc4r activity may be able to compensate for hypothalamic leptin resistance in WT mice, preventing severe hepatic insulin resistance and steatosis.
Reduced mitochondrial function has also been proposed as a risk factor for type 2 diabetes (64). Interestingly, reduced mitochondrial function does not appear to explain the increased sensitivity of Mc4rKO to muscle insulin resistance induced by a high-fat diet. Although female Mc3rKO and Mc4rKO exhibiting a comparable reduction in citrate synthase activity, insulin stimulation of PKB activity was severely compromised in Mc4rKO. These data may suggest that other factors, such as hyperphagia and high-fat diet, may interact with reduced mitochondrial function to severely impair insulin signaling in muscle.
C57BL/6J mice are a commonly used model of DIO (65) and rapidly develop insulin resistance in liver, muscle, and brown and white adipose tissue on a high-fat diet (66). The interaction of diet with melanocortin receptor genotype thus represents an additive effect, over and above what is occurring in WT controls. It may also be premature to state, based on the data presented here, that the stimulation of glucose uptake in muscle and adipose are not lower, relative to controls, in Mc4rKO fed a low-fat diet. However, if present, any deficits are likely to be modest and were undetectable using glucose clearance after insulin or glucose injections.
Gender has a marked effect on the insulin-resistant state of DIO Mc3rKO. Irrespective of the high-fat diet used (45 or 60% kJ from fat), DIO female Mc3rKO exhibit modest perturbations of glucose metabolism. DIO female Mc3rKO exhibited a modest glucose intolerance compared with DIO WT, perhaps related to the development of mild hepatic insulin resistance indicated by the increase in Ser307 phosphorylation of IRS1. A deterioration of pancreatic β-cell function may also be a factor in glucose intolerance observed in DIO mice, irrespective of genotype, although this was not examined in these experiments. However, the fact that glucose intolerance of female DIO Mc3rKO is very mild, with glucose returning to normal levels within 90 min, is not consistent with a severe decompensation of β-cell function.
Male Mc3rKO on a very-high-fat diet may also develop severe insulin resistance, with fasting hyperinsulinemia not significantly different from Mc4rKO. Compared with female Mc4rKO with similar body fat content, female Mc3rKO fed high-fat diets exhibited only a very modest deterioration of glucose clearance. The mechanisms explaining the gender difference in DIO insulin resistance of Mc3rKO are not known. One possible factor may be the mild perturbations of energy balance causing weight gain, and the absence of hyperphagia, in female Mc3rKO.
To summarize, these data suggest that reduced energy expenditure is likely an important factor contributing to obesity of Mc3rKO. A very modest hyperphagia may also contribute to DIO of male Mc3rKO relative to WT mice. Because energy expenditure of Mc4rKO is increased relative to WT controls, hyperphagia coupled with nutrient partitioning to adipose is likely a primary mechanism driving obesity. Reduced capacity for FAO in red muscle may also be a factor contributing to the lower energy expenditure of Mc3rKO and may also be involved in low whole-body fat oxidation of Mc3rKO and Mc4rKO. The metabolic costs associated with digestion and fat and protein deposition may also account for the higher energy expenditure of Mc4rKO compared with Mc3rKO. Finally, Mc3rKO on the B6 background may be a useful model for investigating the development of insulin resistance in the obese state.
Acknowledgments
We thank Drs. Leslie Kozak, Randall Mynatt, Jennifer Rood, and the staff of the Department of Comparative Biology at the Pennington Biomedical Research Center for advice, comments, and assistance with the preparation of this manuscript. Dr. Dennis Huszar (Millennium Pharmaceuticals) and Dr. Roger Cone (Vollum Institute) provided knockout mice used for these studies.
This work was supported by grants from the Health Excellence Fund of the Louisiana State University Board of Regents (to A.B.), Metabolife Settlement Fund (to A.B.), the American Diabetes Association (to A.B. and M.W.H.), National Institutes of Health (DK068330 to A.B.), and the Pennington Biomedical Research Foundation (to A.B. and M.W.H.).
Abbreviations
- AgRP
Agouti-related peptide
- bEEest
basal daily energy expenditure in a weight-stable state
- DIO
diet-induced obesity
- FAO
fatty acid oxidation
- FFM
fat-fee mass
- FM
fat mass
- FoxO1
forkhead box O1
- GSK3
glycogen synthase kinase-3
- IPGTT
ip glucose tolerance test
- IRS1
insulin receptor substrate 1
- ITT
insulin-tolerance test
- Mc3r
melanocortin-3 receptor
- Mc4r
melanocortin-4 receptor
- ME%
metabolic efficiency
- NMR
nuclear magnetic resonance
- NPY
neuropeptide Y
- NREEcal
nonresting energy expenditure
- PI3K
phosphatidyl-3 kinase
- PKB
protein kinase B
- POMC
proopiomelanocortin
- REEcal
resting energy expenditure
- TEEcal
total energy expenditure
- WT
wild type
Footnotes
Author disclosure statement: G.S., J.T., M.W., R.M., N.M., M.B., E.M., and A.B. have nothing to declare.
References
- 1.Bray GA, York DA. The MONA LISA hypothesis in the time of leptin. Recent Prog Horm Res. 1998;53:95–117. [PubMed] [Google Scholar]
- 2.Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- 3.Barsh GS, Schwartz MW. Genetic approaches to studying energy balance: perception and integration. Nat Rev Genet. 2002;3:589–600. doi: 10.1038/nrg862. [DOI] [PubMed] [Google Scholar]
- 4.Zigman JM, Elmquist JK. From anorexia to obesity: the yin and yang of body weight control. Endocrinology. 2003;144:3749–3756. doi: 10.1210/en.2003-0241. [DOI] [PubMed] [Google Scholar]
- 5.Elmquist JK, Coppari R, Balthasar N, Ichinose M, Lowell BB. Identifying hypothalamic pathways controlling food intake, body weight, and glucose homeostasis. J Comp Neurol. 2005;493:63–71. doi: 10.1002/cne.20786. [DOI] [PubMed] [Google Scholar]
- 6.Fan W, Ellacott KL, Halatchev IG, Takahashi K, Yu P, Cone RD. Cholecystokinin-mediated suppression of feeding involves the brainstem melanocortin system. Nat Neurosci. 2004;7:335–336. doi: 10.1038/nn1214. [DOI] [PubMed] [Google Scholar]
- 7.Jobst EE, Enriori PJ, Cowley MA. The electrophysiology of feeding circuits. Trends Endocrinol Metab. 2004;15:488–499. doi: 10.1016/j.tem.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Roseberry AG, Liu H, Jackson AC, Cai X, Friedman JM. Neuropeptide Y-mediated inhibition of proopiomelanocortin neurons in the arcuate nucleus shows enhanced desensitization in ob/ob mice. Neuron. 2004;41:711–722. doi: 10.1016/s0896-6273(04)00074-1. [DOI] [PubMed] [Google Scholar]
- 9.Coll AP, Farooqi IS, Challis BG, Yeo GS, O'Rahilly S. Proopiomelanocortin and energy balance: insights from human and murine genetics. J Clin Endocrinol Metab. 2004;89:2557–2562. doi: 10.1210/jc.2004-0428. [DOI] [PubMed] [Google Scholar]
- 10.Feng N, Young SF, Aguilera G, Puricelli E, Adler-Wailes DC, Sebring NG, Yanovski JA. Co-occurrence of two partially inactivating polymorphisms of MC3R is associated with pediatric-onset obesity. Diabetes. 2005;54:2663–2667. doi: 10.2337/diabetes.54.9.2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Challis BG, Coll AP, Yeo GS, Pinnock SB, Dickson SL, Thresher RR, Dixon J, Zahn D, Rochford JJ, White A, Oliver RL, Millington G, Aparicio SA, Colledge WH, Russ AP, Carlton MB, O'Rahilly S. Mice lacking pro-opiomelanocortin are sensitive to high-fat feeding but respond normally to the acute anorectic effects of peptide-YY(3–36) Proc Natl Acad Sci USA. 2004;101:4695–4700. doi: 10.1073/pnas.0306931101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med. 1999;5:1066–1070. doi: 10.1038/12506. [DOI] [PubMed] [Google Scholar]
- 13.Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141:3518–3521. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- 14.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 15.Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van der Ploeg LH. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet. 2000;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- 16.Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 17.Bewick GA, Gardiner JV, Dhillo WS, Kent AS, White NE, Webster Z, Ghatei MA, Bloom SR. Post-embryonic ablation of AgRP neurons in mice leads to a lean, hypophagic phenotype. FASEB J. 2005;19:1680–1682. doi: 10.1096/fj.04-3434fje. [DOI] [PubMed] [Google Scholar]
- 18.Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, Barsh GS, Horvath TL, Bruning JC. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci. 2005;8:1289–1291. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- 19.Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–685. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- 20.Chen AS, Metzger JM, Trumbauer ME, Guan XM, Yu H, Frazier EG, Marsh DJ, Forrest MJ, Gopal-Truter S, Fisher J, Camacho RE, Strack AM, Mellin TN, MacIntyre DE, Chen HY, Van der Ploeg LH. Role of the melanocortin-4 receptor in metabolic rate and food intake in mice. Transgenic Res. 2000;9:145–154. doi: 10.1023/a:1008983615045. [DOI] [PubMed] [Google Scholar]
- 21.Rahmouni K, Haynes WG, Morgan DA, Mark AL. Role of melanocortin-4 receptors in mediating renal sympathoactivation to leptin and insulin. J Neurosci. 2003;23:5998–6004. doi: 10.1523/JNEUROSCI.23-14-05998.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Kilroy GE, Henagan TM, Prpic-Uhing V, Richards WG, Bannon AW, Mynatt RL, Gettys TW. Targeted deletion of melanocortin receptor subtypes 3 and 4, but not CART, alters nutrient partitioning and compromises behavioral and metabolic responses to leptin. FASEB J. 2005;19:1482–1491. doi: 10.1096/fj.05-3851com. [DOI] [PubMed] [Google Scholar]
- 23.Fekete C, Marks DL, Sarkar S, Emerson CH, Rand WM, Cone RD, Lechan RM. Effect of agouti-related protein in regulation of the hypothalamic-pituitary-thyroid axis in the melanocortin 4 receptor knockout mouse. Endocrinology. 2004;145:4816–4821. doi: 10.1210/en.2004-0476. [DOI] [PubMed] [Google Scholar]
- 24.Butler AA. The melanocortin system and energy balance. Peptides. 2006;27:301–309. doi: 10.1016/j.peptides.2005.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marsh DJ, Miura GI, Yagaloff KA, Schwartz MW, Barsh GS, Palmiter RD. Effects of neuropeptide Y deficiency on hypothalamic agouti-related protein expression and responsiveness to melanocortin analogues. Brain Res. 1999;848:66–77. doi: 10.1016/s0006-8993(99)01962-9. [DOI] [PubMed] [Google Scholar]
- 26.Coleman DL, Hummel KP. The influence of genetic background on the expression of the obese (Ob) gene in the mouse. Diabetologia. 1973;9:287–293. doi: 10.1007/BF01221856. [DOI] [PubMed] [Google Scholar]
- 27.Leiter EH. Mice with targeted gene disruptions or gene insertions for diabetes research: problems, pitfalls, and potential solutions. Diabetologia. 2002;45:296–308. doi: 10.1007/s00125-001-0743-z. [DOI] [PubMed] [Google Scholar]
- 28.Butler AA, Marks DL, Fan W, Kuhn CM, Bartolome M, Cone RD. Melanocortin-4 receptor is required for acute homeostatic responses to increased dietary fat. Nat Neurosci. 2001;4:605–611. doi: 10.1038/88423. [DOI] [PubMed] [Google Scholar]
- 29.Obici S, Feng Z, Tan J, Liu L, Karkanias G, Rossetti L. Central melanocortin receptors regulate insulin action. J Clin Invest. 2001;108:1079–1085. doi: 10.1172/JCI12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem. 2002;277:1531–1537. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- 31.Welsh GI, Hers I, Berwick DC, Dell G, Wherlock M, Birkin R, Leney S, Tavare JM. Role of protein kinase B in insulin-regulated glucose uptake. Biochem Soc Trans. 2005;33:346–349. doi: 10.1042/BST0330346. [DOI] [PubMed] [Google Scholar]
- 32.Albarado DC, McClaine J, Stephens JM, Mynatt RL, Ye J, Bannon AW, Richards WG, Butler AA. Impaired coordination of nutrient intake and substrate oxidation in melanocortin-4 receptor knockout mice. Endocrinology. 2004;145:243–252. doi: 10.1210/en.2003-0452. [DOI] [PubMed] [Google Scholar]
- 33.Tinsley FC, Taicher GZ, Heiman ML. Evaluation of a quantitative magnetic resonance method for mouse whole body composition analysis. Obes Res. 2004;12:150–160. doi: 10.1038/oby.2004.20. [DOI] [PubMed] [Google Scholar]
- 34.Pullar JD, Webster AJ. The energy cost of fat and protein deposition in the rat. Br J Nutr. 1977;37:355–363. doi: 10.1079/bjn19770039. [DOI] [PubMed] [Google Scholar]
- 35.Rothwell NJ, Stock MJ. A role for brown adipose tissue in diet-induced thermogenesis. Nature. 1979;281:31–35. doi: 10.1038/281031a0. [DOI] [PubMed] [Google Scholar]
- 36.Trevaskis JL, Butler AA. Double leptin (Lepob) and melanocortin-4 receptor (Mc4r) gene mutations have an additive effect on fat mass, and are associated with reduced effects of leptin on weight loss and food intake. Endocrinology. 2005;146:4257–4265. doi: 10.1210/en.2005-0492. [DOI] [PubMed] [Google Scholar]
- 37.Ravussin E, Bogardus C. Relationship of genetics, age, and physical fitness to daily energy expenditure and fuel utilization. Am J Clin Nutr. 1989;49:968–975. doi: 10.1093/ajcn/49.5.968. [DOI] [PubMed] [Google Scholar]
- 38.Hulver MW, Berggren JR, Carper MJ, Miyazaki M, Ntambi JM, Hoffman EP, Thyfault JP, Stevens R, Dohm GL, Houmard JA, Muoio DM. Elevated stearoyl-CoA desaturase-1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metab. 2005;2:251–261. doi: 10.1016/j.cmet.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hulver MW, Berggren JR, Cortright RN, Dudek RW, Thompson RP, Pories WJ, MacDonald KG, Cline GW, Shulman GI, Dohm GL, Houmard JA. Skeletal muscle lipid metabolism with obesity. Am J Physiol Endocrinol Metab. 2003;284:E741–E747. doi: 10.1152/ajpendo.00514.2002. [DOI] [PubMed] [Google Scholar]
- 40.Sparks LM, Xie H, Koza RA, Mynatt R, Hulver MW, Bray GA, Smith SR. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes. 2005;54:1926–1933. doi: 10.2337/diabetes.54.7.1926. [DOI] [PubMed] [Google Scholar]
- 41.Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000;6:77–86. [PubMed] [Google Scholar]
- 42.Shimomura I, Bashmakov Y, Horton JD. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J Biol Chem. 1999;274:30028–30032. doi: 10.1074/jbc.274.42.30028. [DOI] [PubMed] [Google Scholar]
- 43.Yahagi N, Shimano H, Hasty AH, Matsuzaka T, Ide T, Yoshikawa T, Amemiya-Kudo M, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Nagai R, Ishibashi S, Yamada N. Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. J Biol Chem. 2002;277:19353–19357. doi: 10.1074/jbc.M201584200. [DOI] [PubMed] [Google Scholar]
- 44.Asilmaz E, Cohen P, Miyazaki M, Dobrzyn P, Ueki K, Fayzikhodjaeva G, Soukas AA, Kahn CR, Ntambi JM, Socci ND, Friedman JM. Site and mechanism of leptin action in a rodent form of congenital lipodystrophy. J Clin Invest. 2004;113:414–424. doi: 10.1172/JCI19511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cohen P, Miyazaki M, Socci ND, Hagge-Greenberg A, Liedtke W, Soukas AA, Sharma R, Hudgins LC, Ntambi JM, Friedman JM. Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science. 2002;297:240–243. doi: 10.1126/science.1071527. [DOI] [PubMed] [Google Scholar]
- 46.Weide K, Christ N, Moar KM, Arens J, Hinney A, Mercer JG, Eiden S, Schmidt I. Hyperphagia, not hypometabolism, causes early onset obesity in melanocortin-4 receptor knockout mice. Physiol Genomics. 2003;13:47–56. doi: 10.1152/physiolgenomics.00129.2002. [DOI] [PubMed] [Google Scholar]
- 47.Ste Marie L, Miura GI, Marsh DJ, Yagaloff K, Palmiter RD. A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proc Natl Acad Sci USA. 2000;97:12339–12344. doi: 10.1073/pnas.220409497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi S, Sparks R, Clay M, Dallman MF. Rats with hypothalamic obesity are insensitive to central leptin injections. Endocrinology. 1999;140:4426–4433. doi: 10.1210/endo.140.10.7064. [DOI] [PubMed] [Google Scholar]
- 49.Choi S, Wong LS, Yamat C, Dallman MF. Hypothalamic ventromedial nuclei amplify circadian rhythms: do they contain a food-entrained endogenous oscillator? J Neurosci. 1998;18:3843–3852. doi: 10.1523/JNEUROSCI.18-10-03843.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marsh DJ, Hollopeter G, Huszar D, Laufer R, Yagaloff KA, Fisher SL, Burn P, Palmiter RD. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat Genet. 1999;21:119–122. doi: 10.1038/5070. [DOI] [PubMed] [Google Scholar]
- 51.Abbott CR, Rossi M, Kim M, AlAhmed SH, Taylor GM, Ghatei MA, Smith DM, Bloom SR. Investigation of the melanocyte stimulating hormones on food intake. Lack of evidence to support a role for the melanocortin-3-receptor. Brain Res. 2000;869:203–210. doi: 10.1016/s0006-8993(00)02386-6. [DOI] [PubMed] [Google Scholar]
- 52.Roselli-Rehfuss L, Mountjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro JB, Entwistle ML, Simerly RB, Cone RD. Identification of a receptor for γ-melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proc Natl Acad Sci USA. 1993;90:8856–8860. doi: 10.1073/pnas.90.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bachman ES, Dhillon H, Zhang CY, Cinti S, Bianco AC, Kobilka BK, Lowell BB. β-AR signaling required for diet-induced thermogenesis and obesity resistance. Science. 2002;297:843–845. doi: 10.1126/science.1073160. [DOI] [PubMed] [Google Scholar]
- 54.Himms-Hagen J. On raising energy expenditure in ob/ob mice. Science. 1997;276:1132–1133. doi: 10.1126/science.276.5315.1132. [DOI] [PubMed] [Google Scholar]
- 55.Rasmussen UF, Rasmussen HN. Human skeletal muscle mitochondrial capacity. Acta Physiol Scand. 2000;168:473–480. doi: 10.1046/j.1365-201x.2000.00699.x. [DOI] [PubMed] [Google Scholar]
- 56.Minokoshi Y, Kim YB, Peroni OD, Fryer LGD, Muller C, Carling D, Kahn BB. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–343. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 57.Bluher S, Ziotopoulou M, Bullen JW, Jr, Moschos SJ, Ungsunan L, Kokkotou E, Maratos-Flier E, Mantzoros CS. Responsiveness to peripherally administered melanocortins in lean and obese mice. Diabetes. 2004;53:82–90. doi: 10.2337/diabetes.53.1.82. [DOI] [PubMed] [Google Scholar]
- 58.Pierroz DD, Ziotopoulou M, Ungsunan L, Moschos S, Flier JS, Mantzoros CS. Effects of acute and chronic administration of the melanocortin agonist MTII in mice with diet-induced obesity. Diabetes. 2002;51:1337–1345. doi: 10.2337/diabetes.51.5.1337. [DOI] [PubMed] [Google Scholar]
- 59.Wolff GL, Roberts DW, Mountjoy KG. Physiological consequences of ectopic agouti gene expression: the yellow obese mouse syndrome. Physiol Genomics. 1999;1:151–163. doi: 10.1152/physiolgenomics.1999.1.3.151. [DOI] [PubMed] [Google Scholar]
- 60.Smith SR, Gawronska-Kozak B, Janderova L, Nguyen T, Murrell A, Stephens JM, Mynatt RL. Agouti expression in human adipose tissue: functional consequences and increased expression in type 2 diabetes. Diabetes. 2003;52:2914–2922. doi: 10.2337/diabetes.52.12.2914. [DOI] [PubMed] [Google Scholar]
- 61.Mynatt RL, Stephens JM. Agouti regulates adipocyte transcription factors. Am J Physiol Cell Physiol. 2001;280:C954–C961. doi: 10.1152/ajpcell.2001.280.4.C954. [DOI] [PubMed] [Google Scholar]
- 62.Savage DB, Petersen KF, Shulman GI. Mechanisms of insulin resistance in humans and possible links with inflammation. Hypertension. 2005;45:828–833. doi: 10.1161/01.HYP.0000163475.04421.e4. [DOI] [PubMed] [Google Scholar]
- 63.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 65.Collins S, Martin TL, Surwit RS, Robidoux J. Genetic vulnerability to diet-induced obesity in the C57BL/6J mouse: physiological and molecular characteristics. Physiol Behav. 2004;81:243–248. doi: 10.1016/j.physbeh.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 66.Park SY, Cho YR, Kim HJ, Higashimori T, Danton C, Lee MK, Dey A, Rothermel B, Kim YB, Kalinowski A, Russell KS, Kim JK. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes. 2005;54:3530–3540. doi: 10.2337/diabetes.54.12.3530. [DOI] [PubMed] [Google Scholar]