

Summary
The biotin repressor is an allosterically regulated site-specific DNA binding protein. Binding of the small ligand, bio-5'-AMP, activates repressor dimerization, which is a prerequisite to DNA binding. Multiple disorder-to-order transitions, some of which are known to be important for the functional allosteric response, occur in the vicinity of the ligand binding site concomitant with effector binding to the repressor monomer. In this work the extent to which these local changes are coupled to additional changes in the structure/dynamics of the repressor was investigated using Hydrogen-Deuterium exchange coupled to Mass Spectrometry. Measurements were performed on the apo-protein and on complexes of the protein bound to four different effectors that elicit a range of thermodynamic responses in the repressor. Global exchange measurements indicate that binding of any effector to the intact protein is accompanied by protection from exchange. Mass spectrometric analysis of pepsin-cleavage products generated from the exchanged complexes reveals that the protection is distributed throughout the protein. Furthermore, the magnitude of the level of protection in each peptide from H-D exchange correlates with the magnitude of the functional allosteric response elicited by a ligand. These results indicate that local structural changes in the binding site that occur concomitant with effector binding nucleate global dampening of dynamics. Moreover, the magnitude of dampening of repressor dynamics tracks with magnitude of the functional response to effector binding.
Keywords: Allostery, HDX-MS, MALDI-ToF MS, biotin repressor
Introduction
Thermodynamic linkage or cooperativity is ubiquitous in biology and is integral to regulation of metabolism, gene transcription, and signal transduction. Despite its widespread nature, the physical-chemical basis of cooperativity is not yet understood. Classic work focused on modeling cooperative behavior evidenced in binding isotherms and kinetic analysis using the two-state allosteric MWC (1) or the stepwise Koshland Nemethy and Filmer (2) formalisms. These types of analyses provide low-resolution, mechanical views of cooperativity. However, elucidation of the mechanism of cooperativity requires determination of how a signal that impinges on one region of a biological macromolecule or complex is transmitted throughout the system. Recent development of experimental tools including dynamic methods in NMR spectroscopy, hydrogen-deuterium exchange detected by either mass spectrometry or NMR spectroscopy and computational tools have provided insight into structural and dynamic changes that accompany energy transduction in biological macromolecules (3). Results of these studies have prompted development of models of signaling the extremes of which are those that posit defined pathways of allosteric communication (3,4) and global models that support distributed signaling that occurs through changes in the dynamics of the cooperative macromolecule (5).
The biotin repressor: An allosteric site-specific DNA binding protein
The Escherichia coli biotin repressor, BirA is an allosteric site-specific DNA binding protein. BirA, carries out two biological functions including catalysis of biotin linkage to a biotin-dependent carboxylase and binding to the operator sequence (bioO) of the biotin biosynthetic operon (6,7). The active BirA species in both functions is bound to bio-5'-AMP, which is synthesized from substrates biotin and ATP (8,9). The adenylated biotin serves both as an intermediate in the biotin transfer reaction and as a corepressor in assembly of the BirA.bioO transcription repression complex. This assembly occurs through coupled dimerization and DNA binding (Figure 1A) and bio-5'-AMP enhances transcription repression complex assembly by selectively driving the dimerization step (10,11). Thus, elucidation of the mechanism of allosteric communication in this system requires determination of the structural and or dynamic changes accompanying effector binding to the repressor monomer that are responsible for the enhanced dimerization energetics.
Figure 1.
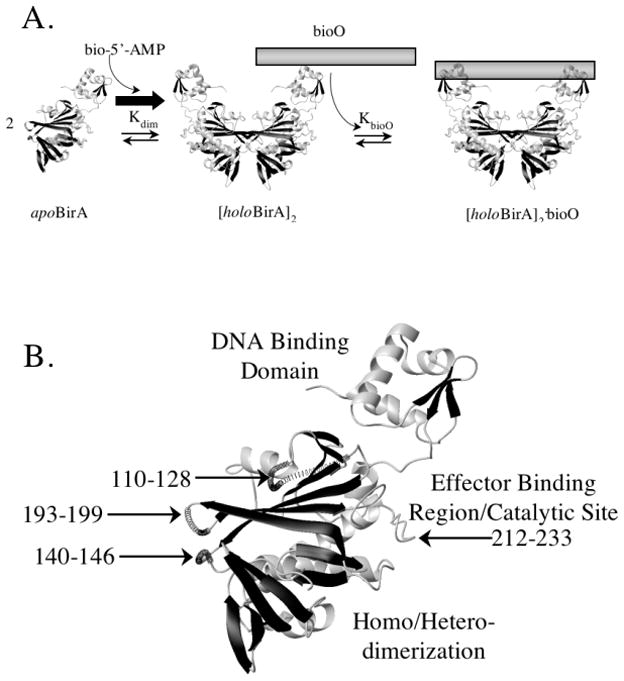
A. Assembly of the Biotin Operon Repression Complex occurs by coupled dimerization and DNA binding. Binding of the effector, bio-5'-AMP, induces repressor dimerization, which is a prerequisite to site-specific DNA binding to the biotin operator sequence. B. Model of the apoBirA structure with the positions of the four partially disordered loops highlighted. The models were created in MolMol (35) using PDB files 1hxd and 2ewn as input.
Structures of the apoBirA monomer and dimeric complexes of the protein bound to biotin and btnOH-AMP, an analogue of bio-5'-AMP, reveal several-ligand induced changes in the repressor monomer (12–14). Functional studies indicate that some of the changes in the vicinity of the allosteric effector binding site are important for the allosteric response. The high-resolution structure of apoBirA reveals that the ligand binding site/active site is characterized by four loops that are partially disordered in the unliganded protein (Figure 1B). One of these loops, the biotin binding loop or BBL composed of residues 110–128, is folded over biotin in the BirA.biotin and BirA.btnOH-AMP structures. This loop as well as two of the other partially disordered loops composed of residues 140–146 and 193–199 form part of the protein-protein interface in both liganded dimers (13,15). Thus, the disorder-to-order transition in the BBL that accompanies ligand binding is important for the ligand-linked dimerization. A fourth loop, the adenylate binding loop or ABL, composed of residues 212–233, folds around the adenine base in the adenylate bound repressor. Consistent with the structural data, solution measurements of subtilisin-mediated proteolytic digestion of the repressor revealed that corepressor binding leads to protection of this loop from digestion (16). Inspection of the adenylate-bound structure determined by x-ray crystallography reveals that loop folding around the adenine base is accompanied by formation of a hydrophobic core involving side chains of ABL residues V114, V119 and W223. Both corepressor-induced loop folding and the allosteric response are compromised by replacement of any of these residues with alanine (17). Thus, the allosteric response requires local folding of the ABL around the adenylate moiety of the corepressor. However, since the ABL is distal to the BirA surface that directly participates in dimerization, it is likely that ligand-induced folding of the ABL is coupled to other structural and/or dynamic changes that are significant for the allosteric response.
The magnitude of the energetic response to ligand binding in the biotin repressor is tunable. Four biotin analogs have been subjected to analysis with respect to effects of their binding on energetics of both repressor dimerization and total assembly of the repressor:operator complex (18). As shown in Figure 1, total assembly refers to combined dimerization and site-specific DNA binding of the dimer. In order to determine the magnitude of the coupling free energy associated with each ligand, the free energies of dimerization and total assembly were compared for the unliganded repressor and the repressor bound to each of the four ligands (Figure 2). Results of these studies allowed classification of two ligands, biotin and biotinoyl-sulfamoyl adenylate as weak effectors and biotinol-5'-AMP and the physiological effector, bio-5'-AMP, as strong. Thus, the presence of the adenine moiety, which should serve as a nucleus for folding of the adenylate binding loop, is not sufficient to elicit the maximum enhancement of dimerization energetics. A notable feature of the thermodynamic data is that for the two weak effectors the coupling free energy measured for dimerization is not equal to that measured for the total assembly process. Moreover, this coupling free energy was more favorable for total assembly than for dimerization; suggesting the occurrence of ligand-linked changes in the monomer that promote DNA binding but not, necessarily, dimerization.
Figure 2.
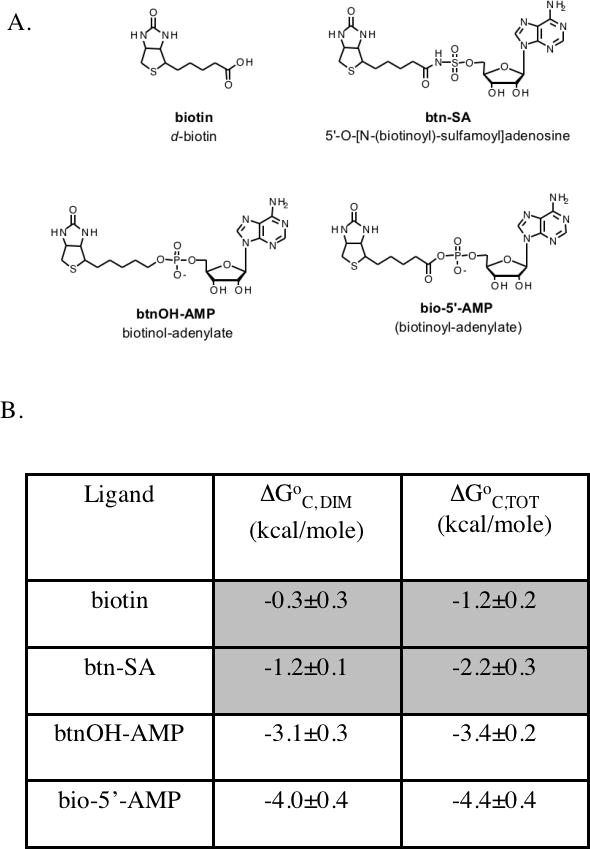
A. Chemical structure of four ligands that activate biotin repressor dimerization to different extents. The figures were created in ChemDraw. B. Coupling free energies associated with binding of the effectors to the biotin repressor. The coupling energies associated with dimerization, ΔG°C, DIM, and total assembly of the repression complex, dimerization plus DNA binding of the dimer, ΔG°C, TOT, are shown.
As discussed above, elucidation of the molecular basis of allosteric activation of BirA requires determination of the structural and dynamic changes that occur in the repressor monomer in response to effector binding. The linkage between effector binding to the monomer and dimerization renders use of either x-ray crystallography or NMR spectroscopy inappropriate for addressing this question in this particular system. Indeed, even the complex of BirA bound to the weakest effector, biotin, crystallizes as a dimer, presumably because of the high effective concentration associated with crystallization (13). NMR spectroscopy is problematic for the same reason. Hydrogen-deuterium exchange coupled to mass spectrometric (HDX-MS) analysis provides an alternative approach to determining ligand-linked structural and/or dynamic changes in the BirA monomer (19,20). This method has proven powerful in localizing changes in the levels and rates of peptide backbone H-D exchange in proteins as they form complexes and it has been successfully applied to analyzing the allosteric response in several protein systems (21–24). Its major advantage over other techniques for analyzing the allosteric response in BirA is that the exchange can be performed at concentrations at which even the liganded forms of BirA are predominantly monomeric.
The availability of four allosteric effectors that yield a range of functional responses in BirA provides an opportunity to investigate structural/dynamic correlates to the allosteric response. In this work partial proteolysis and H-D exchange coupled to mass spectrometry are combined to investigate ligand-linked changes in the BirA monomer. Results of partial proteolytic measurements reveal no correlation between the rate of cleavage of the ABL and the magnitude of the allosteric response. Measurement of global H-D exchange on the unliganded and liganded intact BirA using ESI-ToF MS indicate that in all cases ligand binding results in protection of the protein backbone from H-D exchange. MALDI-ToF MS analysis of pepsin digestion products of repressor exchanged in the absence and presence of saturating ligand was performed to localize the regions that changed in their levels of exchange at long time frames. These measurements reveal that ligand binding results, for the most part, in protection of local regions from exchange. Moreover, the protection extends from the N-terminal DNA binding domain to the C-terminal domain of the repressor. Furthermore, the extent of protection, in terms of number of backbone amide hydrogens exchanged, is greatest for the physiological effector, bio-5'-AMP. These results support a distributive model for transmission of the allosteric response in BirA in which local folding in the ligand binding site triggers changes in the dynamics of the entire protein.
Results
In the adenylate-bound structure of BirA the adenylate binding loop is folded around the adenine ring of the ligand and this folding contributes to the allosteric response. The relationship between the loop conformation, as probed by subtilisin-mediated partial proteolysis, and the allosteric response associated with binding of each of the four effectors was investigated. Initial cleavage of the repressor with subtilisin occurs at the peptide bond linking amino acid residues 217 and 218. The pseudo-first order rate of BirA cleavage by subtilisin has previously been shown to be moderately decreased upon biotin binding and decreased to a greater extent upon binding of bio-5'-AMP (16). These rates were measured for apoBirA and its complexes with biotin, btnSA, btnOH-AMP and bio-5'-AMP. In each case the ligand concentration was sufficiently high to saturate the protein and the repressor concentration was sufficiently low to avoid significant contribution of dimer to the total species population. Results of the measurements, shown in Table 1, indicate that biotin binding results in no decrease in the cleavage rate. While previous studies indicated a 2-fold decrease in the rate with biotin binding, they are consistent with the current results in indicating a small change in the rate. The three other ligands, which induce a range of allosteric responses, confer greater protection from proteolytic digestion than does biotin. However, the rates of cleavage of the repressor bound to all three adenylated ligands are similar in magnitude.
Table 1.
Pseudo-first order rates of cleavage of the adenylate binding loop of apoBirA and its complexes with effector ligands.
| Ligand | rate (s−1)a |
|---|---|
| None | 0.019±0.002 |
| biotin | 0.021±0.001 |
| btnSA | 0.0050±0.0004 |
| btnOH-AMP | 0.0042±0.0001 |
| bio-5'-AMP | 0.0038±0.0004 |
The reported values represent the average of at least three independent measurements of the cleavage rate and errors are the 68% confidence limits.
Global H-D exchange reveals that binding of all effectors results in protection from H-D exchange
In order to determine if any differences exist at a global level, ligand-induced changes in the rate of hydrogen-deuterium exchange were first measured for intact BirA. These measurements were performed on the apo-protein and each of the liganded species. In all cases the exchange was performed at sufficiently low total protein concentration to ensure minimal contribution of the liganded dimer to the total species population. The experiments were initiated by dilution of the protein, either alone or combined with saturating concentration of the appropriate ligand, into deuterated buffer. Samples were removed at specific time intervals, quenched into cold low pH buffer, rapidly desalted, and immediately subjected to ESI-ToF mass spectrometry. Results of these measurements, which are shown in Figure 3, indicate that, relative to the aporepressor, all liganded species are protected from H-D exchange. At 30 minutes after initiating the exchange reaction approximately 40 fewer backbone hydrogens in the liganded species are exchanged relative to the aporepressor. The time courses were subjected to nonlinear least squares analysis and found, consistent with previous studies (19), to be well-described by a triple exponential model. While the rates and amplitudes of the three phases were not, in all cases, well resolved, they can, in agreement with other studies of H-D exchange, be categorized as fast, medium and slow.
Figure 3.

Time course of global exchange of deuterium into apoBirA and its complexes with the four effectors shows protection from exchange. ApoBirA, □ BirA.biotin;x, BirA.btnSA, ▲, BirA.btnOH-AMP; ■, BirA.bio-5'-AMP; ◆. The continuous lines are the best-fit curves obtained from nonlinear least squares analysis of the data using a triple exponential model with Prism GraphPad.
Localization of differences in H-D exchange
While the global exchange measurements indicate that binding of each of the four effectors leads to protection of the backbone amides from H-D exchange, these measurements cannot reveal specific locations in the BirA primary structure of this reduced exchange. In order to localize the changes in exchange, mass spectrometry was performed on products of pepsin digestion of the exchanged samples. Prior to performing measurements on exchanged samples the proteolysis was optimized to maximize coverage of the protein sequence and the peptide digestion products were subjected to MS/MS analysis in order to identify their primary sequences.
The method of choice for MS analysis of the pepsin digest products of H-D exchange in this system was MALDI-ToF and a spectrum of the unliganded, unexchanged protein subjected to pepsin digestion is shown in Figure 4A. The protein was digested using pepsin immobilized on agarose beads for 30 sec. This short digestion time was employed in order to minimize back-exchange in analysis of the exchanged protein. The identities of the peptides in the MALDI-ToF spectrum were determined using MS/MS analysis on a Q-ToF mass spectrometer with the goal of maximizing identification of the peptides observed in the MALDI-ToF spectrum. The maximum coverage that could be reproducibly obtained was approximately 65% of the protein sequence. While these peptides represented segments of the entire primary sequence of BirA (Figure 4B), the coverage in the amino and carboxy terminal segments of the sequence was better than that obtained for the region that forms the central domain in the 3-dimensional structure.
Figure 4.


A. MALDI-ToF spectrum of products of pepsin digestion of unexchanged apo-BirA. The spectrum was acquired as indicated in Materials and Methods. B. Sequences of peptic fragments of BirA that could both be resolved in MALDI-ToF measurements and identified by MS/MS analysis using the Q-ToF instrument.
The hydrogen-deuterium exchange measurements to localize regions of difference in exchange were performed in steady-state mode. The global exchange measurements indicate that after 30 minutes of exchange the mass of BirA, either in its apo or liganded states exhibits a very small increase with time, consistent with leveling off of the in-exchange. The locations of differences in deuterium content in the repressor sequence were, therefore, determined after 60 minutes of exchange. The protein, either apoBirA or its complexes with the four small ligands, was subjected to exchange that was rapidly quenched by lowering the pH and temperature of the solution. After a 30 second digestion with immobilized pepsin the proteolytic fragments were subjected to MALDI-ToF MS analysis. Given the very short time required to achieve sufficient proteolytic digestion of the protein, the total time from quenching of the exchange to mass spectrometric analysis was approximately 4 minutes, a design that helped to minimize back-exchange. Examples of exchange profiles for two of the peptides obtained for the apo-repressor and all four liganded species are shown in Figure 5. Inspection of the figure reveals that while peptide 1–10 undergoes significant exchange, there is no difference between the unliganded and liganded forms of the protein. By contrast, the peptide corresponding to residues 85–101 also undergoes significant exchange. However, relative to the unliganded protein, this region is protected in all liganded forms of the repressor. Moreover, the extent of protection differs for each of the ligands.
Figure 5.
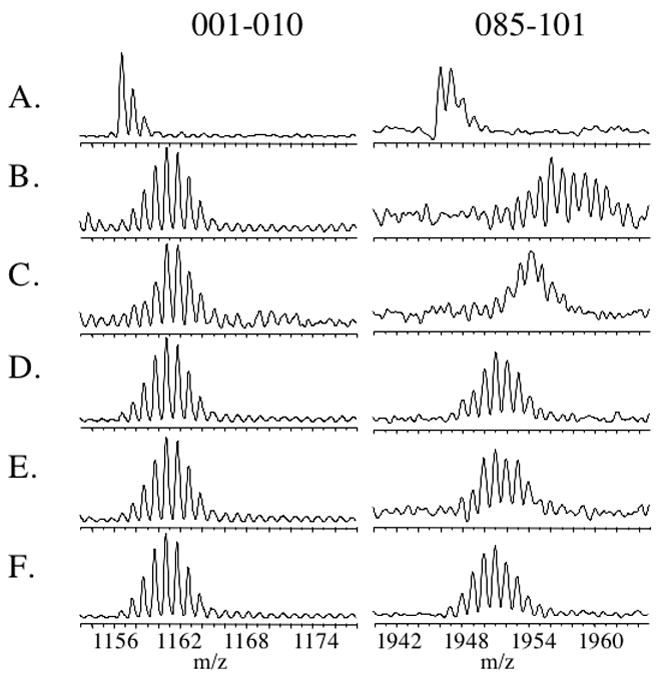
Mass envelopes obtained for two peptic fragments of BirA, 1–10 and 88–101. A. ApoBirA no H-D exchange, B-F: 60 minutes of deuterium in-exchange in the following conditions: B: No ligand, C: biotin, D: btnSA, E: btnOH-AMP, F: bio-5'-AMP. Masses of each peptide were obtained from the centroids of the mass envelope for each.
Deuteration levels of the pepsin digestion products were determined from the average mass of each peptides, which were obtained from the centroids of the mass envelopes for each peptide. Repeated measurements indicated that deuteration levels could be reproducibly obtained in independent experiments to less than 0.5 deuterons. Thus, in our analysis differences greater than 0.5 deuterons are considered significant. The total number of deuterons associated with each peptide was corrected for both the fact that exchange was performed in 90% vol/vol D2O and for back exchange. The back-exchange control experiments were performed by first subjecting apoBirA to pepsin digestion and then incubating the resulting peptide products in deuterated buffer at high temperature for 90 minutes to effect complete exchange. This variation on the back-exchange control measurements was used because extensive precipitation occurred during the long incubations required to completely exchange all backbone amides into the intact protein under native conditions (25). The resulting exchanged peptides were treated identically to the pepsin digestion products obtained from protein that had been subjected to H-D exchange. Results of these control measurements revealed that the extent of back exchange varied for the different pepsin digestion products. Therefore, in correcting for back exchange the number of deuterons exchanged into each peptide was corrected using the appropriate measured back exchange value for that specific peptide. The numbers of deuterons associated with peptides for the unliganded protein and the protein complexed to each ligand are shown in Table 2. Inspection of the values reveals one technical difficulty associated with the experiments. In a few cases, for example peptide 085–101, more deuteriums than theoretically possible are associated with the peptide in one of the species, the unliganded protein. However, the signal to noise ratio for this particular peptide (see Figure 5, line B.) rendered determination of the centroid difficult. This poor signal to noise ratio stems from the necessity of working with relatively low protein concentrations to ensure that all liganded species are predominantly monomeric. Fortunately, this problem was encountered for only a few peptides.
Table 2.
Number of deuterons in each peptide after 60 minutes of exchange
| #Deuterium | ||||||
|---|---|---|---|---|---|---|
| Peptide | MaxD | -ligand | biotin | btnSA | btnOH-AMP | bio-5'-AMP |
| 001–010 | 8 | 8.0 | 8.9 | 7.6 | 8.0 | 7.4 |
| 014–050 | 36 | 28.3 | 29.8 | 27.8 | 26.9 | 25.7 |
| 051–066 | 12 | 12.0 | 11.8 | 9.4 | 9.0 | 8.4 |
| 085–101 | 15 | 17.0 | 10.8 | 6.4 | 6.6 | 5.7 |
| 095–101 | 6 | 5.6 | 6.2 | 4.9 | 5.4 | 5.1 |
| 136–146 | 9 | 7.3 | 7.7 | 6.8 | 6.6 | 5.7 |
| 136–148 | 11 | 9.0 | 9.7 | 7.4 | 8.0 | 6.9 |
| 137–146 | 8 | 6.9 | 7.3 | 6.9 | 6.7 | 6.0 |
| 178–188 | 10 | 5.7 | 3.7 | 3.1 | 2.8 | 2.2 |
| 180–188 | 8 | 4.0 | 1.6 | 1.2 | 1.0 | 0.8 |
| 201–209 | 8 | 8.4 | 10.5 | 8.4 | 9.3 | 8.4 |
| 242–248 | 6 | −0.2 | −0.4 | 0.0 | 0.0 | 0.0 |
| 243–252 | 9 | 1.3 | 1.1 | 0.7 | 0.7 | 0.4 |
| 253–265 | 11 | 8.4 | 9.9 | 9.1 | 9.1 | 8.8 |
| 266–278 | 11 | 6.9 | 6.9 | 6.4 | 6.6 | 5.6 |
| 271–278 | 6 | 1.6 | 0.9 | 1.3 | 1.3 | 1.3 |
| 272–278 | 5 | 1.3 | 0.8 | 0.8 | 0.8 | 0.8 |
| 279–298 | 19 | 9.5 | 10.0 | 9.2 | 9.5 | 8.3 |
| 279–299 | 20 | 9.8 | 9.8 | 8.9 | 9.3 | 8.1 |
| 287–299 | 12 | 6.9 | 4.3 | 3.8 | 4.5 | 3.8 |
| 300–313 | 12 | 10.5 | 13.1 | 11.4 | 11.8 | 9.8 |
| 300–316 | 15 | 13.5 | 16.1 | 14.5 | 14.1 | 12.9 |
| 300–321 | 20 | 15.6 | 19.1 | 17.3 | 17.4 | 15.3 |
The differences in exchange between the ligand-free and ligand-bound states of BirA, which are shown Table 3, are relevant in analyzing the allosteric response using H-D exchange. As indicated above, a conservative estimate of the reproducibility with which amounts of exchange could be measured in our hands is ±0.5 deuteron. Thus, only differences greater than 0.5 in magnitude, which are highlighted in grey, were considered significant. In figure 6 the locations of these differences in H-D exchange are mapped onto the 3-dimensional structure of BirA. The ligand dependence of deuteration levels of each peptide highlighted on the structure are shown in the bar graphs using the same color code. Consistent with the results of global exchange, binding of any ligand affords a net protection of the backbone from exchange. However, the magnitude of the protection observed for a particular peptide differs for each ligand. Moreover, in all cases of protection from exchange the strongest effector, bio-5'-AMP, provides the greatest protection from exchange. Even for peptides for which ligand binding results in enhanced exchange, the amount of exchange is less in those derived from the bio-5'-AMP-bound protein.
Table 3.
Differences in deuterium levels for peptides derived from liganded BirA relative to the unliganded protein.
| ΔDeuterium | ||||
|---|---|---|---|---|
| Peptide | biotin | btnSA | btnOH | bioAMP |
| 001–010 | +0.9 | −0.4 | 0.0 | −0.7 |
| 014–050 | +1.5 | −0.5 | −1.4 | −2.6 |
| 051–066 | −0.2 | −2.5 | −2.9 | −3.5 |
| 085–101 | −6.2 | −10.6 | −10.4 | −11.3 |
| 095–101 | +0.5 | −0.8 | −0.3 | −0.5 |
| 136–146 | +0.5 | −0.5 | −0.7 | −1.6 |
| 136–148 | +0.8 | −1.5 | −1.0 | −2.1 |
| 137–146 | +0.4 | 0.0 | −0.2 | −0.9 |
| 178–188 | −2.0 | −2.6 | −2.8 | −3.5 |
| 180–188 | −2.4 | −2.8 | −3.0 | −3.2 |
| 201–209 | +2.1 | 0.0 | +0.9 | 0.0 |
| 242–248 | −0.2 | +0.2 | +0.2 | +0.2 |
| 243–252 | −0.2 | −0.7 | −0.7 | −0.9 |
| 253–265 | +1.5 | +0.7 | +0.7 | +0.4 |
| 266–278 | 0.0 | −0.5 | −0.3 | −1.3 |
| 271–278 | −0.7 | −0.4 | −0.4 | −0.4 |
| 272–278 | −0.5 | −0.5 | −0.5 | −0.5 |
| 279–298 | +0.5 | −0.3 | 0.0 | −1.3 |
| 279–299 | +0.0 | −0.9 | −0.5 | −1.7 |
| 287–299 | −2.6 | −3.1 | −2.4 | −3.1 |
| 300–313 | +2.6 | +0.9 | +1.3 | −0.7 |
| 300–316 | +2.5 | +1.0 | +0.6 | −0.6 |
| 300–321 | +3.5 | +1.7 | +1.8 | −0.3 |
Values were calculated by subtracting the value found for the peptide in the liganded protein from that found for that in the unliganded. Results for the shaded peptides are shown in Fig. 6.
Figure 6.
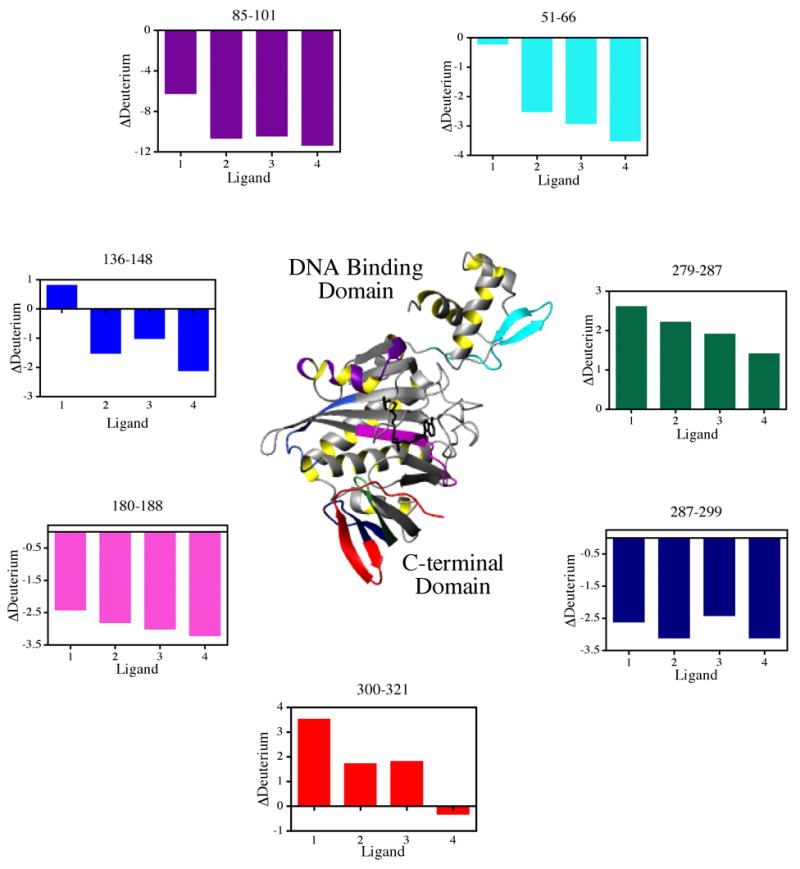
Summary of results of hydrogen-deuterium exchange mapped on the 3-dimensional structure of BirA. The color-coded bar graphs show the difference in deuterium content relative to that measured for the unliganded repressor for each of the relevant peptides. The model was created in MolMol (35) using the input PDB file 2ewn.
The regions of protection from H-D exchange extend throughout the 3-dimensional structure of BirA. Again, because of the limited coverage of the central domain in the peptide identification, few peptides in this region could be assessed for changes in exchange. Those that exhibited changes show protection and are in the central β-sheet over which the ligand binds. The level of protection in the 180–188 peptide is significantly greater for the bio-5'-AMP bound protein. Peptide 136–148, which includes a region of the protein, loop 140–146, that is directly involved in dimerization, becomes protected upon binding of any of the adenylate derivatives. However, the exchange is slightly enhanced upon biotin binding. Binding of the adenylated ligands results in protection of peptide segments in the N-terminal domain from exchange. These include the entire wHTH motif. By contrast, no protection in this region is afforded by biotin binding. The region of the protein that lies close to the interface of the N-terminal and central domains, peptide 85–101, exhibits the largest magnitude of protection in the entire protein. However, for biotin the protection is roughly half of what is measured for any of the other effector ligands. Finally, the C-terminal domain exhibits increased exchange for segments 279–287 and 300–321 (for three ligands), and decreased exchange in segment 287–299.
Discussion
Allosteric activation of the biotin repressor by binding of bio-5'-AMP effects a 1000-fold increase in the equilibrium association constant governing dimerization. Although x-ray crystallographic structures have provided snapshots of the unliganded apo-monomer and two liganded dimers (12–14) elucidation of the mechanism of allosteric activation requires determination of the structural and/or dynamic changes that occur in the repressor monomer in response to effector binding. The availability of four effectors that elicit a range of functional responses in BirA (18) provides an opportunity to investigate the relationship of the thermodynamic effect of a small ligand to the structural and dynamic changes induced upon it’s binding. Results of the combined partial proteolysis and HDX-MS measurements reveal that both local folding and global dampening of dynamics are significant for the allosteric response in BirA.
Local folding of partially disordered loops in BirA is insufficient to elicit the maximal allosteric response. One feature of the apoBirA structure is the four partially disordered loops consisting of residues 110–128, 140–146, 193–199 and 212–223 (12). The first three loops are folded in both the biotin-bound and adenylate-bound repressor structures and form part of the dimerization interface (13,14). The 110–128 loop or biotin binding loop (BBL) organizes over the biotin moiety in both structures and is, therefore, presumably folded in the liganded monomer. Although studies of BirA mutants indicate that the BBL is important for dimerization (15), thermodynamic measurements reveal that the biotin-induced folding of the BBL does not contribute significantly to allosteric activation or repressor dimerization (Figure 2) (10). By contrast, folding of the fourth loop or ABL around the adenine moiety of the adenylate contributes significantly to the functional allosteric response. Replacements of single amino acid residues in the loop both disrupt folding and cause losses in free energy of allosteric activation ranging from 1.0 to 1.5 kcal/mole (17). In this work proteolysis measurements were performed on complexes of BirA bound to the four allosteric activators in order to determine the extent of loop folding in the complexes. Previous results of combined binding measurements using ITC and kinetic measurements of subtilisin-mediated proteolysis indicate that the degree of protection from proteolysis agrees with the thermodynamic measure of loop-induced folding (ΔCp°) (17). The protection, as judged by the rates of subtilisin-mediated proteolysis, are the same for btnSA, btnOH-AMP and bio-5'-AMP, consistent with similar extents of folding in all three complexes. By contrast, the coupling free energies at the levels of dimerization are −1.0, −3.0 and −4.0 kcal/mole, respectively.
Hydrogen-deuterium exchange coupled to mass spectrometric detection was performed to investigate the response of the entire protein to effector binding. Results of global measurements of time courses of deuterium exchange into the protein reveal that binding of any of the four ligands results in net protection of the protein backbone amides from exchange. In order to localize the regions of protection pepsin digestion products of the exchanged protein were analyzed by mass spectrometry to determine the average deuterium content. This analysis is typically performed using either electrospray ionization or MALDI techniques (20,26). Based on trials performed on BirA pepsin digestion products using the two techniques with the available instruments, the MALDI method provided better coverage of the protein sequence. The MALDI-ToF measurements were performed using a modification of previously published methods (26). However, the ease with which BirA is cleaved by pepsin allowed for a very short time between quenching of the exchange and mass spectrometric analysis. On average there was a 3–4 minute interval between the quench and spectrum acquisition. The speed with which the analysis was performed minimized the levels of back exchange to 9 to 50% with an average value of 33%. One of the challenges associated with analysis of this particular system by HDX-MS is that in order to avoid significant contributions from the liganded dimer to the species population exchange measurements were performed at a relatively low total protein concentration of 2 μM. This low concentration compromised the signal to noise ratio in the mass spectra and, in some cases, rendered challenging the determination of average deuteration levels of peptides from the mass envelopes. In localizing differences in H-D exchange to specific peptide segments it is customary to measure the changes in rates of exchange (27). However, the concentration constraints in the biotin repressor system made such kinetic measurements challenging. Consequently, in this work only differences in the steady-state levels of deuterium incorporation into pepsin digestion products were measured. Although this strategy precluded quantifying ligand-induced changes in the stability of specific regions of the protein (27), it does allow mapping of differences in solvent access to the protein backbone.
Mapping of ligand-induced changes in hydrogen-deuterium exchange indicates that segments of the protein sequence throughout the repressor three-dimensional structure become, for the most part, protected from H-D exchange when bound to the ligand. Even the very weak effector, biotin, elicits net protection. The majority of these differences in H-D exchange observed for the apo- and liganded BirA species likely reflect changes in dynamics. This conclusion is based on the known structures of the protein. The major structural differences between the apo-monomer and liganded monomer derived from the dimer structure are localized to the four partially disordered loops observed in the structure of the apo-repressor (12–14). Overlay of the apo-BirA and BirA.biotin monomer structures reveals root-mean-squared discrepancies between α-carbon atoms for the entire protein of 1.26 Å. Thus, no dramatic changes in the structures of regions outside of the loops occur upon ligand binding.
Several peptide segments of the repressor monomer that directly participate in dimerization exhibit ligand-induced changes in H-D exchange. In the btnOH-AMP dimer regions of both the central and C-terminal domains form the intersubunit interface. The interface is, in part, composed of an extended β-sheet formed by side-by-side antiparallel alignment of the central β-sheets of the two individual monomers. The strand from each monomer that forms the intermolecular contact in this extended sheet contains residues 182–192 (13,14). H-D exchange measurements indicate that the peptide containing residues 180–188 is protected from exchange upon binding of any of the four effectors and that the magnitude of protection correlates with the magnitude of the functional response to ligand binding. In addition, peptide 136–148, which contains loop 140–146 that directly participates in dimerization (13–15), becomes protected from exchange upon binding of btnSA, btnOH-AMP or bio-5'-AMP. By contrast, biotin binding slightly enhances exchange in this region. HD exchange in the central segment of the C-terminal domain, peptide 287–299, also decreases upon binding of all four ligands. Segment 293–295 forms part of the dimerization interface in the structure of repressor bound to btnOH-AMP (14). These changes in H-D exchange in segments that form part of the dimerization interface indicate that, even in the repressor monomer, effector binding induces rigidity that may facilitate dimerization.
Several regions of the repressor protein that are distal to both the dimerization interface and the ligand binding site are altered in their H-D exchange levels in response to binding effectors. For example, the peptide segment that undergoes the greatest ligand-associated protection from H-D exchange extends from residue 85 to 101. Based on the values obtained for exchange into peptide 95–101 (see Table 3), most of the protection in peptide 85–101 is associated with residues 85–94. While this segment abuts the β-sheet that forms part of the dimerization interface, it is distal to the edge of the sheet that forms part of the interface. Differences in exchange are also observed in the N-terminal domain, which forms the DNA binding domain. Finally, even the far C-terminus of the repressor structure exhibits ligand-linked changes in H-D exchange. In this case binding of the three ligands biotin, btnSA or btnOH-AMP is associated with increases in exchange. In general ligand binding to BirA results in decreases in H-D exchange for the majority of the protein sequence that could be analyzed. These results are consistent with ligand-induced global tightening of the protein structure.
The global tightening of the repressor structure upon allosteric effector binding is inconsistent with previous interpretation of results of thermodynamic measurements. Isothermal titration calorimetry measurements of binding of the four allosteric ligands to the BirA monomer indicate a correlation between the magnitude of the allosteric response and the ligand binding thermodynamics(28). Weak effectors bind with large favorable enthalpies that are opposed by unfavorable entropies and strong effector binding is characterized by more modest favorable enthalpy and modest favorable entropy. These data were originally interpreted as indicating that in binding of the strong effectors some of the favorable enthalpy associated with bond formation between ligand and protein is used to drive an enthalpically unfavorable structural transition in the protein. If one focuses on the protein alone, the results of the HDX measurements, which indicate a global tightening of the protein structure, the extent of which correlates with effector strength, do not support this interpretation of the thermodynamic data. Indeed, the increased tightening observed with the strong effectors should be accompanied by an enthalpic gain and an entropic loss, neither of which are observed in the thermodynamic data. One possibility is that the differences in the thermodynamic patterns of effector binding reflect solvent reorganization that would not be detectable in the HDX measurements.
The combined structural, thermodynamic and hydrogen-deuterium exchange measurements suggest a two-stage model for transducing the signal for effector binding throughout the biotin repressor structure. Disorder-to-order transitions in loops localized to the binding site initiate the signaling process. Other allosteric processes have been shown to utilize analogous localized ordering (29). The H-D exchange measurements presented in this work indicate that the signal is propagated from the binding site to effect global changes in repressor dynamics. Moreover, the magnitude of the dampening of dynamics tracks with the magnitude of the functional change in the repressor induced by an allosteric activator. In mechanical models of allostery signal transmission occurs through a pathway(s) in which alterations in bonding interactions propagate from the effector binding site to the site of functional change in a macromolecule. Recent studies indicate that allosteric signaling can also be entropically based (5,30,31). The effector-induced changes hydrogen-deuterium exchange in BirA indicate that this system provides another example of transmission of an allosteric response through alterations in protein fluctuations.
Materials & Methods
Chemicals and Biochemicals
Deuterium oxide (D2O) 99.9%, Ultra grade trisodium citrate and disodium succinate, d-biotin 99%, trifluoroacetic acid (TFA) 99%, and immobilized pepsin-agarose from porcine gastric mucosa were purchased from Sigma. HPLC grade acetonitrile (ACN) was obtained from ThermoFisher Scientific. All other chemicals used in preparation of buffers were at least reagent or analytical grade. E. coli biotin repressor, BirA, was overexpressed and purified as previously described (18). Biotinoyl-5-adenosine monophosphate, bio-5-AMP, was synthesized and purified as described by Abbott and Beckett (8,32). The analogs of bio-5'-AMP, biotinol adenylate, btnOH-AMP and 5-O-[N-(biotinoyl sulfamoyl]-adenosine, btn-SA were purchased from RNA-Tech (Leuven, Belgium) or synthesized as previously reported (18).
Subtilisin-mediated Proteolysis Measurements
The pseudo-first order rates of subtilisin-catalyzed cleavage of BirA, apo- and ligand-bound, were measured. Solutions of either BirA alone or with excess ligand were first prepared in standard buffer (10 mM Tris-HCl (pH 7.50 ± 0.02 at 20.0 ± 0.1°C), 200 mM KCl, 2.5 mM MgCl2) and equilibrated for 30 minutes at 20°C. Subtilisin was freshly diluted in Standard Buffer and added to the BirA solution to obtain a final weight ratio of repressor:subtilisin of 33 to 50:1 and incubation was continued at 20°C. At 15 minute time intervals 10 μL aliquots were removed from the reaction and proteolysis was stopped by additional of a 1 μL aliquot of 100 mM PMSF freshly prepared in absolute ethanol. A 6 μL volume of Laemmli sample buffer was added to each sample and the products were separated by electrophoresis in a 15% SDS-polyacrylamide gel. Protein bands were visualized by staining with Coomassie Brilliant Blue and the amount of intact BirA for each time point in the proteolytic digestion was quantified by scanning the gel using a Molecular Dynamics Laser Scanning Personal Densitometer (GE Healthcare). Control experiments have shown that high concentrations of the ligands do not affect the catalytic activity of subtilisin (16). The rates of subtilisin-catalyzed cleavage of BirA were obtained by relating the time-dependent decrease in the amount of intact BirA to a pseudo-first order process. The apparent rates of proteolysis were estimated from linear least-squares analysis using the following equation:
| (1) |
where [BirAOD]t and [BirAOD]t=0 are the integrated optical densities for the bands corresponding to intact BirA at time t and the zero time point, respectively, t is time in minutes and k is the rate or slope of the line.
Global H/D Exchange Study on BirA with Small Ligands
Time courses of deuterium in-exchange to intact BirA in the absence and presence of ligands were performed following the methods outlined by Hoofnagle et al. with the following changes (19). The biotin repressor protein was dialyzed extensively against Standard Buffer (10 mM Tris pH 7.50 ± 0.02 at 20.0 ± 0.1°C, 200 mM KCl, and 2.5mM MgCl2) and its concentration was determined spectrophotometrically using a molar extinction coefficient of 47510 M−1cm−1 at 280 nm (33). Solutions of BirA, both with and without the ligands biotin, bio-5'-AMP, btnOH-AMP, and btn-SA were prepared in Standard Buffer to give concentrations of 20 μM protein and 30 μM ligand. All solutions were equilibrated for 30 minutes at 20°C.
Individual deuterium in-exchange reactions were produced by diluting 2 μL of the protein solution into a final volume of 20 μL with 20°C Standard Buffer made in D2O: 10mM Tris pD 7.50 ± 0.02 at 20.0 ± 0.1°C, 200 mM KCl, and 2.5 mM MgCl2 to yield final concentrations of 2μM protein and, in relevant samples, 3μM ligand. These protein and ligand concentrations are sufficiently high to ensure saturation of the protein with ligand. Time points from 5 to 60 minutes in-exchange were taken as 20 μL aliquots at appropriate intervals from a master reaction of 100 μL, prepared by combining 10 μL of the original protein solution, with or without ligand, with 90 μL D2O Standard Buffer. All reactions were treated identically by quenching with 18 μL of 0°C 25 mM sodium citrate, 25 mM sodium succinate, pH 2.4 resulting in a final pH of 2.5, chilled on a dry ice/salt/ice bath for 10 seconds, desalted using an equilibrated 0°C Zip Tip C18 pipette tip (Millipore) and eluted with 0°C ACN/0.1% TFA (7:3 v/v) in mQ H2O. Samples of unexchanged and exchanged protein were directly infused in an ESI-TOF mass spectrometer to determine molecular weight.
Local H/D Exchange Measurements
Reactions for hydrogen-deuterium exchange followed by pepsin cleavage were performed according to the methods described by Mandell et al. with the following changes (26). The Standard Buffer used contained 10 mM Tris pH 7.50 ± 0.02 at 20.0 ± 0.1°C, 200 mM KCl, and 2.5 mM MgCl2. Solutions of BirA, either as an apo (or unliganded) control, or with the addition of the ligands biotin, bio-5-AMP, btnOH-AMP, or btn-SA at a molar ratio of 1:1.5 were equilibrated in Standard Buffer for 30 minutes at 20°C. Hydrogen-deuterium exchange was initiated by the dilution of 2 μL of each of these solutions into a total volume of 20 μL with Standard Buffer prepared in D2O: 10 mM Tris pD 7.50 ± 0.02 at 20.0 ± 0.1°C, 200 mM KCl, and 2.5 mM MgCl2 to give 2μM BirA +/- 3μM ligand. A control reaction was similarly diluted into Standard Buffer prepared in H2O and treated identically. In-exchange was allowed to proceed for 60 minutes at 20°C. Immediately prior to use, agarose-immobilized pepsin for each reaction was exchanged twice with 500 μL of 0°C mQ H2O and twice with 500 μL of 0°C 0.1% TFA in mQ H2O and resuspended to 25 mg/mL w/v in 0°C 0.1% TFA in mQ H2O. The pepsin-agarose in a 100 μL aliquot, containing 95 units of pepsin, was pelleted at 3000 rpm at −2°C for 30 seconds in an Eppendorf tube and excess liquid removed by careful pipetting.
Following in-exchange, each reaction was chilled for 2 minutes on ice, quenched by the addition of 80 μL of 0°C 0.1% TFA in mQ H2O, resulting in a final pH of 2.5, mixed, and transferred to the drained pepsin-agarose aliquot on ice. Digestion reactions were briefly mixed and allowed to incubate at 0°C for 30 seconds before being separated from the immobilized pepsin using a pre-chilled Ultrafree MC centrifuge filter device (Millipore) and centrifugation for 30 seconds at 3000 rpm at −2°C. The liquid flow through was desalted on ice using a 0°C pre-equilibrated ZipTip C18 (Millipore) pipette tip and peptides were eluted from the tip with 1μL of 0°C pH 2.5 10 mg/mL α-cyano-4-hydroxycinnamic acid matrix solution in ACN/0.1% TFA (7:3 v/v). Eluant was directly spotted onto the MALDI target plate, prechilled on a −20°C freezer bag under vacuum in a dessicator. The vacuum was broken only to allow for spotting then immediately reapplied and the spots typically dried in less than 60 seconds. Thereafter, the target was quickly transferred to the adjacent mass spectrometer to avoid condensation of water on the plate. The average total time from quenching of the H/D exchange to transfer of the plate to the mass spectrometer was 4:03±0:15 minutes.
Back-exchange Controls
Measurements of the fully exchanged controls were obtained following the method described by Resing and Ahn (25). Reactions were set up identically to the unliganded, unexchanged control reactions described above with the exception that digestion preceded the in-exchange in deuterated buffer. Following digestion of 2μM BirA at 0°C pH 2.5 for 30 seconds with pepsin, the immobilized pepsin was removed using a chilled Ultrafree MC centrifuge filter device. The resulting peptides were desalted with a Zip Tip C18 pipette tip and eluted from the tip with 10μL of ACN/0.1% TFA (7:3 v/v) into a 200μL thin wall thermal cycler tube. Peptides were lyophilized and resuspended in 20μL of 10mM MOPS pD 8.00 ± 0.02, 200mM KCl, 2.5mM MgCl2, heated 90 minutes at 90°C in a PCR Sprint thermal cycler (Thermo) and chilled on ice. The resulting 100% deuterium–exchanged peptides were desalted with a Zip Tip C18 pipette tip at 0°C, eluted with 1μL of 0°C pH 2.5 10 mg/mL α-cyano-4-hydroxycinnamic acid matrix solution in ACN/0.1% TFA (7:3 v/v) and spotted directly onto the chilled MALDI target plate. Spectra were acquired as described below.
Mass Spectrometry
Samples subjected to global H/D exchange were analyzed using an AccuTOF-CS ESI-TOF mass spectrometer (JEOL, Tokyo, Japan). Samples were introduced into the mass spectrometer by direct infusion and the instrument was externally calibrated with polypropylene glycol. Mass spectra were recorded at m/z 250–2500.
Local H/D exchange studies were performed using an Axima-CFR Plus MALDI-TOF mass spectrometer (Shimadzu Biotech, Columbia, MD). The instrument was equipped with a nitrogen laser (λ=337 nm) and operated on reflectron mode. The matrix of choice was α-cyano-4-hydroxycinnamic acid from Sigma (St. Louis, MO) and the sample for external calibration was a PPG Standard from Applied Biosystems (Foster City, CA). MALDI mass spectra were recorded at m/z 400–4400 and 500 profiles were averaged for every spectrum in the HDX studies.
Identities of peptic peptides in the local HDX study were confirmed by MS/MS using a PE Sciex API QSTAR Pulsar i Q-TOF mass spectrometer (Concord, ON) and an in-house version of Mascot software (Matrix Science). Samples were introduced through an LC Packings Ultimate nanoflow LC system (Abberdaan, the Netherlands) equipped with a C18 PepMap100 column (75 μm, 15 cm, 3 μm, 100 Å) and connected to a nano ionspray source (Protana, Odense, Denmark) or by direct infusion into the ion spray source (PE Sciex).
Data Analysis
In analysis of the global exchange data the molecular weight of the protein was obtained from deconvolution of the spectrum obtained for each time point using the MagTran 1.0 software (34). The data were plotted as molecular weight versus time and subjected to nonlinear least squares analysis using a triple exponential model (GraphPad Prism).
Spectra obtained from MALDI-ToF analysis of peptide digestion products of the protein that had been subjected to hydrogen-deuterium exchange provide isotope peak envelopes for the relevant peptides. The centroids of these envelopes, which were directly determined using a tool in the Kratos Kompact MALDI software for Axima Instruments 2.3.4., provided the masses of the peptides. These values were subjected to two corrections. The first correction accounted for the fact that exchange was performed in buffer that was 90%, not 100%, D2O. The second correction, which took into account the back exchange that occurred between quenching of the exchange and acquisition of the mass spectra, was performed using the following equation:
| (2) |
where D is the corrected deuterium level in the peptide, m is the experimentally determined mass of the peptide obtained as the centroid of the mass envelope, m0% is a centroid mass of the undeuterated peptide, m100% is the experimentally determined centroid mass of the fully deuterated peptide obtained as described above., and N is a total number of exchangeable amide hydrogens in the peptide fragment (20). For the peptides in this work subjected to analysis for HD exchange back exchange values ranged from 9–50%.
Acknowledgments
Supported by NIH Grant GM46511 to DB, NIH Grant GM21248 to CCF and a grant from the Helsingin Sanomat 100th Anniversary Foundation to OL. Funds from NIH Grant S10RR19341 were used to purchase the JEOL ESI-ToF Mass Spectrometer.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Monod J, Wyman J, Changeux JP. On the Nature of Allosteric Transitions: A Plausible Model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 2.Koshland DE, Jr, Nemethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 3.Suel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 4.Yang J, Garrod SM, Deal MS, Anand GS, Woods V, Jr, Taylor S. Allosteric network of cAMP-dependent protein kinase revealed by mutation of Tyr204 in the P+1 loop. J Mol Biol. 2005;346:191–201. doi: 10.1016/j.jmb.2004.11.030. [DOI] [PubMed] [Google Scholar]
- 5.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barker DF, Campbell AM. The birA gene of Escherichia coli encodes a biotin holoenzyme synthetase. J Mol Biol. 1981;146:451–467. doi: 10.1016/0022-2836(81)90042-5. [DOI] [PubMed] [Google Scholar]
- 7.Barker DF, Campbell AM. Genetic and biochemical characterization of the birA gene and its product: evidence for a direct role of biotin holoenzyme synthetase in repression of the biotin operon in Escherichia coli. J Mol Biol. 1981;146:469–492. doi: 10.1016/0022-2836(81)90043-7. [DOI] [PubMed] [Google Scholar]
- 8.Lane MD, Rominger KL, Young DL, Lynen F. The enzymatic synthesis of holotranscarboxylase from apotranscarboxylase and (+)-biotin. J Biol Chem. 1964;239:2865–2871. [PubMed] [Google Scholar]
- 9.Prakash O, Eisenberg MA. Biotinyl 5'-adenylate: corepressor role in the regulation of the biotin genes of Escherichia coli K-12. Proc Natl Acad Sci U S A. 1979;76:5592–5595. doi: 10.1073/pnas.76.11.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Streaker ED, Gupta A, Beckett D. The biotin repressor: thermodynamic coupling of corepressor binding, protein assembly, and sequence-specific DNA binding. Biochemistry. 2002;41:14263–14271. doi: 10.1021/bi0203839. [DOI] [PubMed] [Google Scholar]
- 11.Streaker ED, Beckett D. Coupling of protein assembly and DNA binding: biotin repressor dimerization precedes biotin operator binding. J Mol Biol. 2003;325:937–948. doi: 10.1016/s0022-2836(02)01308-6. [DOI] [PubMed] [Google Scholar]
- 12.Wilson KP, Shewchuk LM, Brennan RG, Otsuka AJ, Matthews BW. Escherichia coli biotin holoenzyme synthetase/bio repressor crystal structure delineates the biotin- and DNA-binding domains. Proc Natl Acad Sci U S A. 1992;89:9257–9261. doi: 10.1073/pnas.89.19.9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weaver LH, Kwon K, Beckett D, Matthews BW. Corepressor-induced organization and assembly of the biotin repressor: a model for allosteric activation of a transcriptional regulator. Proc Natl Acad Sci U S A. 2001;98:6045–6050. doi: 10.1073/pnas.111128198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wood ZA, Weaver LH, Brown PH, Beckett D, Matthews BW. Co-repressor induced order and biotin repressor dimerization: a case for divergent followed by convergent evolution. J Mol Biol. 2006;357:509–523. doi: 10.1016/j.jmb.2005.12.066. [DOI] [PubMed] [Google Scholar]
- 15.Kwon K, Streaker ED, Ruparelia S, Beckett D. Multiple disordered loops function in corepressor-induced dimerization of the biotin repressor. J Mol Biol. 2000;304:821–833. doi: 10.1006/jmbi.2000.4249. [DOI] [PubMed] [Google Scholar]
- 16.Xu Y, Nenortas E, Beckett D. Evidence for distinct ligand-bound conformational states of the multifunctional Escherichia coli repressor of biotin biosynthesis. Biochemistry. 1995;34:16624–16631. doi: 10.1021/bi00051a010. [DOI] [PubMed] [Google Scholar]
- 17.Naganathan S, Beckett D. Nucleation of an allosteric response via ligand-induced loop folding. J Mol Biol. 2007;373:96–111. doi: 10.1016/j.jmb.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown P, Cronan JE, Grotli M, Beckett D. The biotin repressor: Modulation of allostery by corepressor analogs. J Mol Biol. 2004;337:857–869. doi: 10.1016/j.jmb.2004.01.041. [DOI] [PubMed] [Google Scholar]
- 19.Hoofnagle AN, Resing KA, Ahn NG. Practical methods for deuterium exchange/mass spectrometry. Methods Mol Biol. 2004;250:283–298. doi: 10.1385/1-59259-671-1:283. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emrick MA, Lee T, Starkey PJ, Mumby MC, Resing KA, Ahn NG. The gatekeeper residue controls autoactivation of ERK2 via a pathway of intramolecular connectivity. Proc Natl Acad Sci U S A. 2006;103:18101–18106. doi: 10.1073/pnas.0608849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koeppe JR, Komives EA. Amide H/2H exchange reveals a mechanism of thrombin activation. Biochemistry. 2006;45:7724–7732. doi: 10.1021/bi060405h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brock M, Fan F, Mei FC, Li S, Gessner C, Woods V, Jr, Cheng X. Conformational analysis of Epac activation using amide hydrogen/deuterium exchange mass spectrometry. J Biol Chem. 2007;282:32256–32263. doi: 10.1074/jbc.M706231200. [DOI] [PubMed] [Google Scholar]
- 24.Englander JJ, Del Mar C, Li W, Englander SW, Kim JS, Stranz DD, Hamuro Y, Woods VL., Jr Protein structure change studied by hydrogen-deuterium exchange, functional labeling, and mass spectrometry. Proc Natl Acad Sci U S A. 2003;100:7057–7062. doi: 10.1073/pnas.1232301100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Resing KA, Ahn NG. Deuterium exchange mass spectrometry as a probe of protein kinase activation. Analysis of wild-type and constitutively active mutants of MAP kinase kinase-1. Biochemistry. 1998;37:463–475. doi: 10.1021/bi971750x. [DOI] [PubMed] [Google Scholar]
- 26.Mandell JG, Falick AM, Komives EA. Measurement of amide hydrogen exchange by MALDI-TOF mass spectrometry. Anal Chem. 1998;70:3987–3995. doi: 10.1021/ac980553g. [DOI] [PubMed] [Google Scholar]
- 27.Englander SW. Hydrogen exchange and mass spectrometry: A historical perspective. J Am Soc Mass Spectrom. 2006;17:1481–1489. doi: 10.1016/j.jasms.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown PH, Beckett D. Use of binding enthalpy to drive an allosteric transition. Biochemistry. 2005;44:3112–3121. doi: 10.1021/bi047792k. [DOI] [PubMed] [Google Scholar]
- 29.Keramisanou D, Biris N, Gelis I, Sianidis G, Karamanou S, Economou A, Kalodimos CG. Disorder-order folding transitions underlie catalysis in the helicase motor of SecA. Nat Struct Mol Biol. 2006;13:594–602. doi: 10.1038/nsmb1108. [DOI] [PubMed] [Google Scholar]
- 30.Hilser VJ, Thompson EB. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci U S A. 2007;104:8311–8315. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsai CJ, Del Sol A, Nussinov R. Allostery: Absence of a Change in Shape Does Not Imply that Allostery Is Not at Play. J Mol Biol. 2008 doi: 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abbott J, Beckett D. Cooperative binding of the Escherichia coli repressor of biotin biosynthesis to the biotin operator sequence. Biochemistry. 1993;32:9649–9656. doi: 10.1021/bi00088a017. [DOI] [PubMed] [Google Scholar]
- 33.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Z. MAG-TRAN. Amgen, Inc.; Thousand Oaks, CA: [Google Scholar]
- 35.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]