

Abstract
Mitochondrial damage is implicated in the progression of cardiac disease. Considerable evidence suggests that proinflammatory cytokines induce oxidative stress and contribute to cardiac dysfunction. This study was conducted to determine whether a TNF-induced increase in superoxide ( ) contributes to mitochondrial damage in the left ventricle (LV) by impairing respiratory complex I activity. We employed an electron paramagnetic resonance (EPR) method to measure and oxygen consumption in mitochondrial respiratory complexes, using an oxygen label. Adult male Sprague–Dawley rats were divided into four groups: control, TNF treatment (ip), TNF+ apocynin (APO; 200 μmol/kg bw, orally), and TNF+ Tempol (Temp; 300 μmol/kg bw, orally). TNF was injected daily for 5 days. Rats were sacrificed, LV tissue was collected, and mitochondria were isolated for EPR studies. Total LV ROS production was significantly higher in TNF animals than in controls; APO or Temp treatment ameliorated TNF-induced LV ROS production. Total mitochondrial ROS production was significantly higher in the TNF and TNF+ APO groups than in the control and TNF+ Temp groups. These findings suggest that TNF alters the cellular redox state, reduces the expression of four complex I subunits by increasing mitochondrial production and depleting ATP synthesis, and decreases oxygen consumption, thereby resulting in mitochondrial damage and leading to LV dysfunction.
Keywords: TNF, Free radicals, Oxidative stress, Respiratory complex I, Mitochondrial damage, Oxygen spin label
Neurohumoral mechanisms play important roles in the pathophysiology of cardiovascular disease. Current treatments aimed at blocking neurohormones such as angiotensin have considerably reduced mortality and morbidity; however, the progressive clinical course of heart disease emphasizes the need for innovative approaches to therapy. A growing body of evidence indicates that, along with neurohormones, proinflammatory cytokines (PICs) contribute to the progression of heart disease [1–3]. The PICs, including tumor necrosis factor (TNF), interleukin-1β (IL-1β), and IL-6, can induce oxidative stress and contribute to the pathophysiology of cardiovascular disease [4–7]. TNF is the most studied cytokine; it increases with the severity of heart disease and is of prognostic significance. TNF stimulates reactive oxygen species (ROS) in mitochondria by altering membrane permeability and by inhibiting the electron transport chain (ETC), thereby causing mitochondrial damage [8,9]. However, at the mitochondrial level, the potential implications of chronic TNF infusion in the progression of left ventricular (LV) dysfunction are not known.
Proper myocardial function depends on the energy produced by mitochondria, which is primarily generated by oxidative phosphorylation and fatty acid β-oxidation [10–12]. Damage to mitochondria results in the inability to generate energy in the form of ATP [13,14]. Overproduction of ROS also occurs as a result of this damage and contributes to cardiac dysfunction [15–17]. Complexes I and III of the mitochondrial ETC are potent sources of cellular superoxide; deficiencies in the ETC result in increased mitochondrial superoxide production, which is a major cause of cellular damage [18–20]. Complex I defects are some of the most frequent causes of ETC disorders. Mitochondrial complex I contains 46 distinct subunits; the number of subunits directly involved in electron transport is unknown. We explored the effects of TNF on mitochondrial complex I superoxide production in the LV and examined four specific protein subunits of complex I, the 17-, 20-, 30-, and 39-kDa subunits, which are known participants in oxidative phosphorylation and ATP production [21,22]. We also examined alterations in the protein levels of these subunits in response to TNF infusion. We used a gain-of-function strategy by blocking NAD(P)H oxidase using apocynin (APO) or by scavenging superoxide using Tempol (Temp).
We used electron paramagnetic resonance (EPR), the most sensitive and definitive method for quantification of oxygen consumption in mitochondria. EPR is superior to other free radical detection methods in that it allows for the direct measurement of specific free radicals using specific spin probes [23]. Under pathological conditions, increased oxidative stress itself can alter oxygen levels; this might affect mitochondrial oxygen consumption. To circumvent this problem, we developed an EPR method for measuring superoxide and oxygen consumption in mitochondrial respiratory complexes, using the oxygen label NOX-13.1-OS. By using a gas controller and newly synthesized nontoxic spin label, we were able to set up a physiological oxygen concentration of 20 mm Hg [24] and to follow the consumption of oxygen during detection of ROS. The merit of this method is that it allows us to measure superoxide production, complex activity, and oxygen consumption in parallel using the same incubation medium, temperature, and substrate concentration in each mitochondrial preparation.
Materials and methods
Chemicals and drugs
The spin probes 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH) and 1-hydroxy-4-phosphono-oxy-2,2,6, 6-tetramethylpiperidine (PPH), the metal chelators defferoxamine (DF) and diethyldithiocarbamate (DETC), Krebs–Hepes buffer (KHB), and the oxygen label NOX-13.1-OS were obtained from Noxygen Science Transfer and Diagnostics (Elzach, Germany). Recombinant rat TNF was obtained from Biosource (Camarillo, CA, USA). Bovine erythrocyte superoxide dismutase (SOD), polyethylene glycol-conjugated superoxide dismutase (PEG-SOD), APO, and Temp were obtained from Sigma–Aldrich (St. Louis, MO, USA). All other chemicals and reagents used were of analytical grade and were purchased from Sigma–Aldrich unless otherwise specified.
Animals
Studies were conducted using adult male Sprague–Dawley rats (n= 8 in each group), weighing 325–350 g, obtained from Harlan (Indianapolis, IN, USA). Animals were housed in temperature-(23 ± 2°C) and light-controlled (12 h light/dark cycle) animal quarters; standard rat chow and water were provided ad libitum. All animal experimental protocols were approved by the Louisiana State University Institutional Animal Care and Use Committee.
Experimental protocol
Rats were treated for 5 days with TNF (40 μg/kg body wt (bw), intraperitoneally (ip)), TNF+ APO (200 μmol/kg bw, orally), TNF+ Temp (300 μmol/kg bw, orally), or vehicle. APO-only and Temp-only groups served as positive controls. Body weights were measured at baseline and on day 6 (study end). Tail plethysmography was performed daily as previously described [9]. On day 6, rats were injected with heparin (100 U/25 mg bw, ip) to prevent excessive blood clotting. Rats were then sacrificed, and LV were separated from hearts for mitochondrial isolation and tissue processing for EPR studies.
Echocardiography
Echocardiography was performed as previously described [25]. The Tei index (an indicator of diastolic dysfunction) was determined from Doppler recordings of LV inflow and outflow as described previously [26].
Isolation of LV mitochondria
LV mitochondria were isolated by differential centrifugation of heart homogenates as described previously [9]. Mitochondrial purity was assessed by transmission electron microscopy.
Electron spin resonance studies
Total LV ROS and production as well as mitochondrial total ROS and H2O2 production were measured by EPR using the spin probes CMH and PPH [27], respectively. Detection of ROS and oxygen concentrations was conducted under the following EPR settings: center field g= 2.002, field sweep 50 G, microwave power 20 mW, modulation amplitude 1.90 G, conversion time 10.24 ms, time constant 81.92 ms.
Preparation of LV tissue for EPR studies
Small portions (15–20 mg) of LV tissue from each animal were minced and placed into 4 wells of a 24-well plate with 20 mM KHB containing 25 μM DF and 5 μM DETC. Tissue pieces were then washed twice with the same buffer to remove any trace contamination and incubated at 37°C with specific spin probes for 30 min. The incubation of tissue was terminated by placing the plate on ice. All tissue EPR experiments measuring the concentration of oxidized CM• and PP• were conducted at 20°C in disposable capillary tubes as previously described [27].
Total LV tissue ROS production
Tissue pieces were incubated at 37°C with CMH (200 μM) for 30 min. Aliquots of the incubated probe medium were then taken into 50-μl glass capillary tubes (Noxygen Science Transfer and Diagnostics) for determination of total LV ROS production.
LV tissue production
Superoxide production was measured as previously described [27]. Briefly, tissue pieces were incubated at 37°C with PEG-SOD (50 U/ml) for 30 min, then the spin probe CMH (200 μM) was added for another 30-min incubation period. Aliquots of the incubated probe medium were then taken into 50-μl glass capillary tubes for determination of total LV superoxide production. Preincubation of tissue with PEG-SOD allows competitive inhibition of CMH by intracellular and extracellular released . Because it is cell permeative, PEG-SOD can competitively inhibit the interaction in both the intracellular and the extracellular spaces, thus allowing accurate measurement of tissue production. For tissue production, the values obtained from incubation with PEG-SOD and CMH were subtracted from the values obtained from incubation with CMH only. For determination of LV production, the above-mentioned EPR settings were used.
Total mitochondrial ROS production
LV mitochondria from each rat (7–10 μg protein) were mixed with CMH. After addition of CMH, aliquots of the mitochondria were taken into 50-μl glass capillary tubes [11]. Total mitochondrial ROS production was detected using EPR under the following settings: center field g= 2.002, field sweep 9.000 G, microwave power 20 mW, modulation amplitude 1.90 G, conversion time 10.24 ms, time constant 81.92 ms, receiver gain 3.17×103. For the detection of ROS production, we used the Time Scan mode with the averaging of EPR amplitude every 10 scans over 10 min. Total mitochondrial ROS production experiments were performed at 37°C under 20 mm Hg of oxygen partial pressure. The setup of the oxygen concentration in KHB buffer was performed using the Gas-Controller NOX-E.4-GC (Noxygen Science Transfer and Diagnostics GmbH).
Mitochondrial and hydrogen peroxide (H2O2) production
Mitochondrial and H2O2 production was measured using PPH as the spin probe [11,28]. Aliquots of LV mitochondria (approximately 7–10 μg protein) were probed with PPH (500 μM) alone or PPH and SOD (50 U/ml) for quantification of production. Catalase (50 U/ml) was added to measure H2O2 formation. PPH allows the detection of extracellular and extramitochondrial production of [28]. PPH reacts with to produce a stable PP nitroxide radical, which can be detected with EPR [11]. The PPH probe is highly sensitive (1 nM) and requires a very small amount of mitochondria. Further, PPH is non-membrane-permeative, which allows the measurement of mitochondrial superoxide without the interference of oxidation by mitochondrial oxidases [11]. After adequate mixing, 50 μl of mitochondria was taken into 50-μl glass capillary tubes. Mitochondrial production and H2O2 production were determined by EPR under the same settings as were used for the measurement of total mitochondrial ROS.
Measurements of MnSOD activity
MnSOD activity was measured in isolated mitochondria using a commercially available kit (Dojindo Molecular Technologies, Kumamoto, Japan). Protein concentration was determined according to the Bradford method using bovine serum albumin as the standard. All assays were run in triplicate and averaged to obtain a mean value per sample.
Enzymatic and respiratory activity of mitochondrial complexes I–IV
We have developed a method for simultaneous detection of mitochondrial oxygen consumption, using a newly synthesized oxygen spin label (NOX-13.1-OS), and detection of ROS using the spin probe CMH or PPH. Aliquots of mitochondria were mixed with oxygenated KHB (20 mm Hg–pO2) containing 1 mM EGTA. Then, the oxygen spin label NOX-13.1-OS (5 μM), CMH (200 μM), and one of the following substrates were added to the mitochondrial suspension: 20 mM glutamate (complex I), 5 mM succinate (complex II), 5 mM pyruvate (complex III), or 15 μM cytochrome c (complex IV). After adequate mixing, the samples were taken into capillary tubes [29] and the enzymatic as well respiratory activity of each complex ( ) and oxygen consumption were measured using EPR. The increase in EPR amplitude from peak 1, 2, or 4 (g= 2.0129, 2.0036, or 1.9943; see Fig. 3) corresponds with the production of ROS ( ), and the decrease in EPR amplitude of peak 3 (g= 2.0011, Fig. 3) corresponds with consumption of oxygen. Enzymatic/respiratory activity of each mitochondrial complex was quantified by EPR under the same settings described above.
Fig. 3.

(A) Total ROS production in LV mitochondria. EPR spectra and their graphic interpretations are shown. TNF administration significantly increased total ROS production in LV mitochondria. Note the similarities between the TNFand the TNF+ APO groups. Temp significantly decreased mitochondrial ROS. All values are presented as means ± SEM (*p< 0.05, control vs TNF; $p< 0.05, TNF vs TNF+ Temp; @p< 0.05, TNF+ APO vs TNF+ Temp). (B) Superoxide production in LV mitochondria. Significantly higher superoxide production was noted in the TNF group compared to the control and TNF+ Temp groups. In the graphical representation, all values are presented as means ± SEM (*p< 0.05, control vs TNF; $p< 0.05, TNF vs TNF+ Temp; @p< 0.05, TNF+ APO vs TNF+ Temp). (C) Hydrogen peroxide production in LV mitochondria. Significantly lower rates of hydrogen peroxide production were found in the TNF+ Temp and control groups compared to the TNF and TNF+ APO groups. All values are presented as means ± SEM (*p< 0.05, control vs TNF; $p< 0.05 TNF vs TNF+ Temp; @p< 0.05, TNF+ APO vs TNF+ Temp).
Enzymatic activity of mitochondrial complex I under inhibitory and stimulatory conditions
Mitochondrial complexes I and III have major roles in both total ROS and production. We chose mitochondrial complex I for studies examining the effects of inhibition and stimulation on enzyme activity. The experiment was conducted in four parts. First, basal levels of enzyme activity were measured. Second, enzyme activity was initiated using 5 mM glutamate, and complex I enzyme activity was then measured. Third, enzyme activity was initiated with glutamate and then inhibited with 100 pmol rotenone (ETC blocker)/mg mitochondria, and complex I enzyme activity was then measured. Last, enzyme activity was initiated with glutamate and inhibited with rotenone. SOD (50 U/ml) was then added to quench production, and measurements of complex I enzyme activity were performed.
Western blot analysis
LV mitochondrial complex I subunit protein content was determined by Western blot. The immunoreactive proteins (anti-17-,-20-, -30-, and-39-kDa antibodies; 1:1000 dilutions) were visualized with rabbit HRP-conjugated anti-mouse IgG (1:10,000 dilution). The band intensities were quantified using a Bio-Rad ChemiDoc imaging system and normalized to voltage dependent anion channel (VDAC). Densitometry analysis was performed using NIH ImageJ software.
Assessment of ATP levels
ATP levels of LV mitochondria were measured using a luminometric assay (Apo SENSOR assay kit; BioVision, Mountain View, CA, USA).
Statistical analyses
Data were analyzed by ANOVA, followed by Bonferroni’s multiple comparison tests. p≤0.05 was considered significant.
Results
Echocardiography
Blood pressure measurements were obtained for all experimental animals during the 5-day treatment period. Treatment with TNF and other drugs did not have any effect on blood pressure parameters. Echocardiographic analysis revealed significantly decreased fractional shortening (FS%) and significantly higher Tei index in the TNF and TNF+ APO groups compared to controls and to TNF+ TEMP animals (p< 0.05 for all; Table 1). The changes produced by APO were not statistically significant compared with the control group.
Table 1.
Echocardiographic analysis of study groups
| Parameter | Control | TNF | TNF+ APO | TNF+ Temp |
|---|---|---|---|---|
| IVSD (mm) | 1.32 ± 0.04 | 1.71 ± 0.04* | 1.56 ± 0.03 | 1.42 ± 0.02‡ |
| IVSS (mm) | 2.69 ± 0.13 | 2.67 ± 0.14 | 2.68 ± 0.05 | 2.56 ± 0.03 |
| LVD (mm) | 6.51 ± 0.07 | 7.72 ± 0.11* | 7.34 ± 0.10 | 6.85 ± 0.04‡,§ |
| LVS (mm) | 2.92 ± 0.16 | 4.67 ± 0.06* | 4.12 ± 0.01† | 3.37 ± 0.08‡,§ |
| PWD(mm) | 1.41 ± 0.02 | 1.71 ± 0.03* | 1.50 ± 0.03† | 1.45 ± 0.01‡ |
| PWS (mm) | 2.53 ± 0.09 | 2.58 ± 0.09 | 2.44 ± 0.14 | 2.54 ± 0.14 |
| FS% | 55.19 ± 2.42 | 34.20 ± 1.31* | 40.86 ± 0.76† | 50.79 ± 1.12‡ |
| HR | 352.25 ± 13.70 | 342.20 ± 14.73 | 350.40 ± 15.3 | 357.0 ± 14.8 |
| Tei | 0.37 ± 0.02 | 0.51 ± 0.02* | 0.49 ± 0.04 | 0.38 ± 0.02‡,§ |
Echocardiographic analysis revealed significantly greater left ventricular diastolic (LVD) and systolic (LVS) dimensions in TNF-treated animals. TNF treatment also decreased fractional shortening (FS%) and increased Tei index. Cotreatment with Tempol prevented these changes. IVSD, intraventricular septal thickness at end diastole; IVSS, intraventricular septal thickness at end systole; PWD, posterior wall thickness at end diastole; PWS, posterior wall thickness at end systole; HR, heart rate. Values are expressed as means ± SEM.
p< 0.05, control vs TNF.
p< 0.05, TNF vs TNF+ APO.
p< 0.05, TNF vs TNF+ Temp.
p< 0.05, TNF+ APO vs TNF+ Temp.
Total LV ROS
Total ROS production in the TNF-treated group was significantly higher than that in the control group (p< 0.05; Fig. 1A). When the TNF+ APO and TNF+ Temp groups were compared to the TNF-treated group, significantly lower ROS production rates were observed (p< 0.05 for all). The lower total tissue ROS production rates in the TNF+ APO and TNF+ Temp groups support the assertion that APO and Temp are both capable of attenuating oxidative stress.
Fig. 1.

(A) Total ROS production in LV tissue. EPR spectra and their graphic interpretations are given. TNF administration significantly increased total ROS production in LV tissue. APO or Temp significantly decreased total LV ROS production. All values are presented as means ± SEM (*p< 0.05, control vs TNF; #p< 0.05, TNF vs TNF+ APO; *$p< 0.05 TNF vs TNF+ Temp). (B) Superoxide production in LV tissue. EPR spectra and their graphic interpretations are given. TNF administration significantly increased total superoxide production in LV tissue. APO or Temp significantly decreased total tissue superoxide production. All values are presented as means ± SEM (*p< 0.05, control vs TNF; #p< 0.05, TNF vs TNF+ APO; $p< 0.05, TNF vs TNF+ Temp).
LV levels
Left ventricular levels in the TNF-treated group were significantly higher than those in the control group (p< 0.05; Fig. 1B). When the TNF+ APO and TNF+ Temp groups were compared to the TNF-treated group, significantly decreased production rates were observed (p< 0.05).
Purity of mitochondria
Mitochondrial purity was verified with transmission electron microscopy (Fig. 2).
Fig. 2.
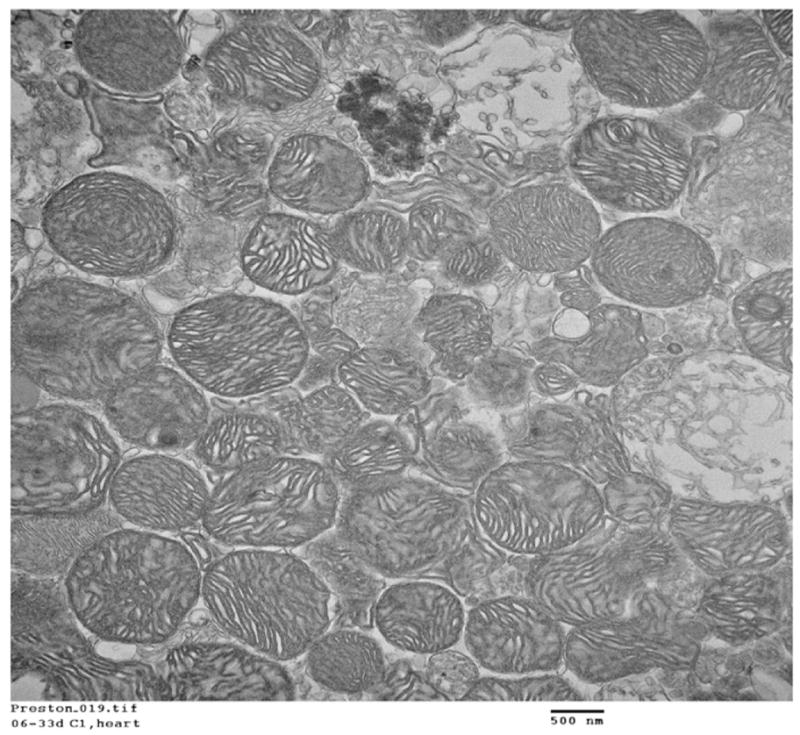
Mitochondrial purity as determined with transmission electron microscopy.
Mitochondrial total ROS production
Mitochondrial total ROS production was significantly higher in both the TNF and the TNF+ APO groups than in the control and TNF+ Temp groups (p< 0.05 for all; Fig. 3A). No significant differences were observed between the TNF+ APO and the TNF-treated groups. Interestingly, mitochondrial total ROS production in the TNF+ Temp group was significantly lower than in the TNF+ APO group (p< 0.05). These results clearly suggest that APO attenuates oxidative stress only at the cytosolic level, and not at the mitochondrial level, whereas Temp preserves redox status at both levels.
Mitochondrial production
Mitochondrial levels were significantly higher in the TNF and TNF+ APO groups compared to the control and TNF+ Temp groups (p < 0.05; Fig. 3B). No significant differences in mitochondrial levels were found between the control and the TNF+ Temp groups or between the TNF and the TNF+ APO groups.
Mitochondrial H2O2 levels
H2O2 production was significantly higher in the TNF group than in the control and TNF+ Temp groups (p< 0.05; Fig. 3C). No significant differences in complex I H2O2 production were found among other groups.
Mitochondrial complex enzyme activity
Significant decreases in the activities of mitochondrial complexes I, II, III, and IV were observed in the TNF and TNF+ APO groups compared to the control group (p< 0.05 for all; Table 2A and Fig. 4). No significant differences in complex I, II, III, or IV enzyme activity were noted between the TNF+ Temp and the control groups. Significant decreases in complex II and III activity were also noted in the TNF+ APO group compared to the TNF+ Temp group (p< 0.05 for both).
Table 2.
| Table 2A. Mitochondrial respiratory complexes I–IV | ||||
|---|---|---|---|---|
| Parameter (nM/mg protein/s) | Control | TNF | TNF+ APO | TNF+ Temp |
| Complex I activity | 11.90 ± 1.21 | 3.91 ± 0.65* | 4.96 ± 0.46 | 8.51 ± 0.99‡,§ |
| Complex II activity | 17.70 ± 0.90 | 9.34 ± 0.5* | 10.49 ± 0.96 | 15.17 ± 0.94‡,§ |
| Complex III activity | 22.61 ± 1.08 | 12.68 ± 1.69* | 12.37 ± 0.74 | 18.25 ± 1.97‡,§ |
| Complex IV activity | 22.05 ± 0.7 | 16.74 ± 0.98* | 16.50 ± 1.8 | 20.81 ± 0.85 |
| Table 2B. Stimulatory effect of glutamate on mitochondrial respiratory complex I–enzyme activity | ||||
| Parameter (nM/mg protein/s) | Control | TNF | TNF+ APO | TNF+ Temp |
|
| ||||
| Basal | 8.23 ± 0.21 | 3.21 ± 0.22s* | 3.14 ± 0.21† | 7.12 ± 0.4‡,§ |
| Glutamate | 12.67 ± 0.75 | 5.02 ± 0.08* | 6.37 ± 0.11 | 12.15 ± 0.28‡,§ |
| Glutamate+ rotenone | 15.55 ± 0.73 | 8.79 ± 0.49* | 11.69 ± 1.44 | 17.53 ± 1.49‡,§ |
| Glutamate+ rotenone+ SOD | 24.90 ± 2.70 | 13.77 ± 0.54* | 20.10 ± 0.99 | 30.32 ± 1.32‡,§ |
| Table 2C. Mitochondrial respiratory complex I–superoxide production | ||||
| Parameter (nM/mg protein/s) | Control | TNF | TNF+ APO | TNF+ Temp |
|
| ||||
| Basal | 5.24 ± 0.19 | 8.16 ± 0.17* | 6.72 ± 0.06 | 4.37± 0.16‡,§ |
| Glutamate | 3.72 ± 0.08 | 7.63 ± 0.12* | 7.47 ± 0.11 | 4.03 ± 0.22‡,§ |
| Glutamate+ rotenone | 5.73 ± 0.20 | 11.59 ± 0.36* | 8.73 ± 0.39 | 4.46 ± 0.29‡,§ |
| Glutamate+ rotenone+ SOD | 3.71 ± 0.18 | 7.13 ± 0.12* | 7.27 ± 0.21 | 4.19 ± 0.20‡,§ |
Enzyme activity of mitochondrial respiratory complexes I–IV. Mitochondria were probed with CMH and one of the following substrates was added: 20 mM glutamate (complex I), 5 mM succinate (complex II), 5 mM pyruvate (complex III), or 15 μM cytochrome c (complex IV). Values are expressed as means ± SEM.
Mitochondrial complex I activity under inhibitory and stimulatory conditions. Values are expressed as means ± SEM.
p< 0.05, control vs TNF.
p< 0.05, TNF vs TNF+ APO.
p< 0.05, TNF vs TNF+ Temp.
p< 0.05, TNF+ APO vs TNF+ Temp.
Mitochondrial complex I superoxide production. Values are expressed as means ± SEM.
Fig. 4.

Complex I enzyme activities (total ROS production) in LV mitochondria. Note the significant decreases in the activity levels of complex I in the TNF and TNF+ APO groups and the restoration of activity in the TNF+ Temp group. All values are presented as means ± SEM (*p< 0.05, control vs TNF; #p< 0.05, TNF vs TNF+ APO).
Stimulatory effect of glutamate on mitochondrial complex I enzyme activity
A decrease in basal complex I enzyme activity was seen in the TNF group compared to the TNF+ Temp group (p< 0.05; Table 2B). Significant decreases in glutamate-stimulated complex I activity were noted in the TNF and TNF+ APO groups compared to the control and TNF+ Temp groups (p< 0.05). When enzyme activity was assessed in the presence of glutamate and rotenone, decreases in activity were seen in the TNF and TNF+ APO groups compared to the control group, although these differences were not significant.
In the presence of glutamate+ rotenone+ SOD, a significant decrease in complex I enzyme activity was seen in the TNF group compared with the control group (p< 0.05). A significant increase in complex I enzyme activity was also seen in the TNF+ Temp group compared to the TNF and TNF+ APO groups (p< 0.05).
Stimulatory effect of glutamate on mitochondrial complex I production
A significant increase in basal complex I production was seen in the TNF group compared to the control and TNF+ Temp groups (p< 0.05 for all; Table 2C). The TNF+ APO group had significantly higher levels of than the TNF+ TEMP group (p< 0.05).
Glutamate stimulation significantly increased production in the TNF and TNF+ APO groups compared to the control group (p< 0.05). Conversely, significantly lower levels were seen in the TNF+ TEMP group compared to the TNF group (p< 0.05). In the presence of glutamate and rotenone, a significant increase in production was seen in the TNF group compared to the control or TNF+ Temp group (p < 0.05 for both). In mitochondrial preparations containing glutamate+ rotenone+ SOD, significant increases in levels were seen in the TNF and TNF+ APO groups compared with the control and TNF+ Temp groups (p < 0.05 for all).
Mitochondrial MnSOD activity
Figure 5A shows MnSOD activities from control and treatment groups. MnSOD activity in heart mitochondria was significantly decreased (3.32 ± 0.16 units/mg protein) in the TNF group compared with the control group (5.55 ± 1.0 units/mg protein) and the TNF+ Tempol group (4.37 ± 0.18 units/mg protein). Treatment of TNF-induced rats with Tempol restored MnSOD activity, suggesting that Tempol treatment prevented the decrease in MnSOD activity.
Fig. 5.

(A) MnSOD and (B) oxygen consumption in LV mitochondria. MnSOD/oxygen consumption was significantly lower in the TNF and TNF+ APO groups compared to the control and TNF+ Temp groups. All values are presented as means ± SEM (*p< 0.05, control vs TNF; #p< 0.05, TNF vs TNF+ APO; $p < 0.05, TNF vs TNF+ Temp; @p< 0.05, TNF+ APO vs TNF+ Temp).
Respiratory activity of mitochondrial complex I
Significantly lower levels of complex I respiratory activity were noted in the TNF group in comparison with the control and TNF+ TEMP groups (p< 0.05; Fig. 5B). No significant differences in oxygen consumption were found among any of the other groups.
Western blotting
To determine the effect of TNF on the mitochondrial ETC, we systematically examined the protein levels of complex I subunits using Western blotting. Significant decreases in the expression of mitochondrial complex I 17-, 20-, 30-, and 39-kDa subunits were observed in the TNF and TNF+ APO groups compared to the control and TNF+ TEMP groups (p< 0.05 for all; Figs. 6A and 6B). No significant differences in protein expression of these complex I subunits were noted between the TNF+ Temp and the control group.
Fig. 6.

(A) Effect of TNF on expression of mitochondrial respiratory complex I subunits. Significant decreases in the expression of mitochondrial complex I 17-, 20-, 30-, and 39-kDa subunits were observed in the TNF and TNF+ APO groups compared to the control and TNF+ Temp groups. (B) Densitometry values are represented as relative intensities in mean arbitrary units calculated from the Western blots shown in (A). All values are presented as means ± SEM (*p< 0.05, control vs TNF; #p< 0.05, TNF vs TNF+ APO; $p< 0.05, TNF vs TNF+ Temp; @p < 0.05, TNF+ APO vs TNF+ Temp).
ATP levels and calculated ADP/ATP ratios
Significant decreases in ATP levels were found in the TNF and TNF+ APO groups compared to the control and TNF+ Temp groups (p< 0.05). No significant differences in ATP levels were found among other groups (Fig. 7A). Similar trends were seen in ADP/ATP ratios (Fig. 7B). These results suggest the presence of mitochondrial damage in the TNF and TNF+ APO groups and reaffirm the inability of APO to penetrate the mitochondrial membrane to provide antioxidant protection.
Fig. 7.

(A) ATP levels and (B) ATP/ADP ratio in LV mitochondria. TNF administration significantly decreased ATP levels in the TNF and TNF+ APO groups compared to the control and TNF+ Temp groups. All values are presented as means ± SEM (*p< 0.05, control vs TNF; #p< 0.05, TNF vs TNF+ APO; $p< 0.05, TNF vs TNF+ Temp; @p< 0.05, TNF+ APO vs TNF+ Temp).
Discussion
In this study, we investigated the effects of TNF on mitochondrial respiratory complexes and we observed that complex I enzymatic activity and protein levels of the complex I 17-, 20-, 30-, and 39-kDa subunits were significantly reduced with TNF administration, and that the resultant impairment of electron transfer led to the deleterious production of superoxide in the intracellular space; these results explain the LV dysfunction we saw in this study. The three aims of our study were: (1) to determine if ROS produced by mitochondrial complex I would affect tissue and mitochondrial functionality, (2) to identify the subunits of complex I affected during TNF infusion, and (3) to evaluate the mechanism of superoxide production by the ETC as it relates to decreased complex I activity and alterations in both oxygen consumption and ATP synthesis.
We have previously demonstrated that TNF alters the cellular redox state, thus suggesting a role for ROS in cardiac dysfunction [9]. The present study demonstrates that, in the LV of hearts that exhibited decreased FS% and increased LV end-systolic diameter and volume, mitochondrial ROS and superoxide production rates are increased, thereby affecting the ETC. Enhanced generation of ROS, and of in particular (Fig. 1), was observed in TNF-treated animals, indicating the toxic effect of TNF on the antioxidant defense system of LV tissue. This response was further substantiated by increased mitochondrial ROS production (especially ) and complex activity in the LV and a decrease in MnSOD activity.
Because the mitochondrial electron transport chain is the major intracellular producer of and H2O2, we focused on these free radicals in mitochondria. Mitochondrial and H2O2 can be produced by complexes I, II, and III of the ETC and released primarily toward the mitochondrial matrix [11,30]. To determine the stimulatory site of TNF action responsible for the increased production of , we incubated isolated LV mitochondria with various ETC substrates and measured the activities of each mitochondrial complex. Here, we show that TNF reacts with mitochondrial membranes and significantly inhibits the activities of complexes I, II, III, and IV, but with less effect upon complex IV (Table 2A); this decline of individual mitochondrial enzymes involved in energy metabolism could decrease the overall activity of the ETC in the LV. Impairments in the LV ETC, in addition to their obvious impairment of ATP production, may lead to an altered flow of electrons into the intracellular space and a subsequent generation of oxidative damage due to free radicals; this process may result in LV dysfunction. To our surprise, we found that Tempol, a scavenger that permeates biological membranes [31], prevented increases in mitochondrial and H2O2 and preserved activities of both complex I and the antioxidant enzyme MnSOD. Complex I, II, III, and IV activities remained reduced with APO treatment, demonstrating that APO is beneficial only at the cytosolic level; these results are consistent with data from our previous study [9]. It is possible that TNF decreases MnSOD protein levels, thereby causing a functional decline in the respiratory chain and increased ROS generation, coupled with an inability to efficiently scavenge free radicals, which may perpetuate mitochondrial dysfunction. The decreased activities of the mitochondrial respiratory complexes (CI, CII, CIII and CIV) and the decreased MnSOD content all suggest mitochondrial damage in the TNF group. The involvement of mitochondrial electron transport in TNF-induced ROS generation has been demonstrated in various cell types [32–34] and in isolated mitochondria from hepatocytes. These results are consistent with those obtained from skin fibroblasts [35] and cardiac myocytes [32].
We chose to focus on complex I in this study because it is the initial complex in the ETC and is known to be a major site of production [36–38]. We analyzed basal enzyme activity under physiological oxygen tension and after adding glutamate, glutamate+ rotenone, and finally glutamate+ rotenone+ SOD. We found a significant decrease in complex I activity and an increase in complex I levels in TNF and TNF+ APO rats. When glutamate was added to initiate the complex I reaction, enzyme activity levels rose and levels decreased in both control and TNF+ Temp rats. Enzyme activity remained low and levels remained increased in TNF and TNF+ APO rats. The same trend was observed in the presence of glutamate+ rotenone or glutamate+ rotenone+ SOD. The decreases in activities of the TNF and TNF+ APO groups are likely the result of TNF-induced mitochondrial damage. Further, we selected four subunits of complex I (17-, 20-, 30-, and 39-kDa) that have known involvement in oxidative phosphorylation, ATP production, and cellular respiration [39,40]. The decreases in complex I activity, oxygen consumption, MnSOD activity, and ATP production seen in the TNF and TNF+ APO groups are also the likely result of TNF-induced mitochondrial damage. The decreased protein expression levels of each of the four subunits further suggest that TNF negatively affects mitochondrial ETC activity. APO requires myeloperoxidase for its activation [42] and myeloperoxidase is absent from the mitochondrial fraction; thus, the effects of the coadministration of TNF and APO on complex I activity, ATP production, and subunit protein expression were similar to those of TNF administration alone, indicating that APO merely acts as a free radical scavenger system. These results suggest that TNF, along with losses of mitochondrial ETC and antioxidant enzymes, generates more radicals and that Tempol prevents the loss of these enzymes, in part, by acting on complex I and its subunits in the ETC. These observations are in accordance with previous studies in which inhibition of one or more ETC enzymes led to the increased levels of radicals [35,41].
TNF generates overproduction of ROS, particularly superoxide, which can induce opening of the mitochondrial membrane. As a consequence, the outer membrane is disrupted, thereby causing decreased expression of mitochondrial permeability transition pore proteins and release of cytochrome c into the cytosol. Further, TNF present in the cytosol may penetrate the damaged mitochondrial membrane through an unknown mechanism, causing increased production of mitochondrial superoxide and a decrease in MnSOD activity. Mitochondrial superoxide could then induce further production of cytosolic superoxide by NAD(P)H oxidase, thereby initiating a ROS-induced ROS mechanism. These possible scenarios may explain how oxidative stress damages mitochondria and impedes the flow of electrons within the respiratory chain and how electron flow may be further decreased by TNF, which can cause release of cytochrome c from mitochondria. Owing to the imbalance between a high electron input and a restricted outflow, electrons may accumulate within complex I and react with oxygen to form the superoxide anion radical and thereby cause increased mitochondrial production and decrease the expression of individual complex I subunits.
The myocardium is a tissue that depends almost entirely on energy production for its proper function. Mitochondria are the major energy producers in cardiomyocytes, and therefore, the implications of alterations in this energy production must be considered. In our study, TNF treatment resulted in significant damage to the mitochondrial membrane and was accompanied by a decrease in ATP production. Decreased ATP production was also associated with a worsening of cardiac function as exhibited by a reduction in fractional shortening and an increase in the Tei index (an indicator of diastolic dysfunction) in the TNF-treated group. Interestingly, APO-treated rats exhibited the same degree of mitochondrial damage and reduced ATP production in the mitochondria. These animals also had reduced fractional shortening and increased Tei index, similar to the values seen in the TNF-treated group. In contrast, treatment with Tempol, a membrane-permeative superoxide scavenger, reduced mitochondrial damage, restored ATP production rates, and normalized fractional shortening and Tei index. These findings clearly suggest that, in the myocardium, mitochondrial energetic/ATP production rates are critical in maintaining the proper functionality of the organ and that disruption of mitochondrial energy production results in worsened cardiac function. These data corroborate the previous finding that overexpression of TNF in the heart, or high levels of TNF in the circulation, contributes to cardiac dysfunction.
In summary, TNF acts on ETC complexes I, II, III, and IV and can significantly inhibit their activities; here we show the effects of TNF on complex I in particular. This study addresses the mechanism of decreased complex I activity in LV mitochondria after TNF infusion. Our results provide compelling evidence that chronic TNF administration reduces the expression of four individual complex I subunits by increasing mitochondrial levels and depleting ATP levels and ATP synthesis, as well as oxygen consumption, thereby resulting in mitochondrial damage and leading to LV dysfunction.
Acknowledgments
These studies were supported by a grant from the National Heart, Lung, and Blood Institute (RO1 HL080544-01) to Dr. Joseph Francis. We are grateful to Dr. Philip J. Ebenezer for his technical assistance and to Sherry Ring.
References
- 1.Sekiguchi K, Li X, Coker M, Flesch M, Barger PM, Sivasubramanian N, Mann DL. Cross-regulation between the renin–angiotensin system and inflammatory mediators in cardiac hypertrophy and failure. Cardiovasc Res. 2004;63:433–442. doi: 10.1016/j.cardiores.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Kang YM, Ma Y, Elks C, Zheng JP, Yang ZM, Francis J. Cross-talk between cytokines and renin–angiotensin in hypothalamic paraventricular nucleus in heart failure: role of nuclear factor-κB. Cardiovasc Res. 2008;79:671–678. doi: 10.1093/cvr/cvn119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Francis J, Zhang ZH, Weiss RM, Felder RB. Neural regulation of the proinflammatory cytokine response to acute myocardial infarction. Am J Physiol Heart Circ Physiol. 2004;287:H791–797. doi: 10.1152/ajpheart.00099.2004. [DOI] [PubMed] [Google Scholar]
- 4.Pignatelli P, Cangemi R, Celestini A, Carnevale R, Polimeni L, Martini A, Ferro D, Loffredo L, Violi F. Tumour necrosis factor α upregulates platelet CD40 L in patients with heart failure. Cardiovasc Res. 2008;78:515–522. doi: 10.1093/cvr/cvn040. [DOI] [PubMed] [Google Scholar]
- 5.Yu Y, Kang YM, Zhang ZH, Wei SG, Chu Y, Weiss RM, Felder RB. Increased cyclooxygenase-2 expression in hypothalamic paraventricular nucleus in rats with heart failure: role of nuclear factor kappaB. Hypertension. 2007;49:511–518. doi: 10.1161/01.HYP.0000257356.20527.c5. [DOI] [PubMed] [Google Scholar]
- 6.Vidali M, Hietala J, Occhino G, Ivaldi A, Sutti S, Niemela O, Albano E. Immune responses against oxidative stress-derived antigens are associated with increased circulating tumor necrosis factor-α in heavy drinkers. Free Radic Biol Med. 2008;45:306–311. doi: 10.1016/j.freeradbiomed.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 7.Vasquez-Vivar J, Whitsett J, Ionova I, Konorev E, Zielonka J, Kalyanaraman B, Shi Y, Pieper GM. Cytokines and lipopolysaccharides induce inducible nitric oxide synthase but not enzyme activity in adult rat cardiomyocytes. Free Radic Biol Med. 2008;45:994–1001. doi: 10.1016/j.freeradbiomed.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moe GW, Marin-Garcia J, Konig A, Goldenthal M, Lu X, Feng Q. In vivo TNF-alpha inhibition ameliorates cardiac mitochondrial dysfunction, oxidative stress, and apoptosis in experimental heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H1813–1820. doi: 10.1152/ajpheart.00036.2004. [DOI] [PubMed] [Google Scholar]
- 9.Mariappan N, Soorappan RN, Haque M, Sriramula S, Francis J. TNF-α-induced mitochondrial oxidative stress and cardiac dysfunction: restoration by superoxide dismutase mimetic Tempol. Am J Physiol Heart Circ Physiol. 2007;293:H2726–2737. doi: 10.1152/ajpheart.00376.2007. [DOI] [PubMed] [Google Scholar]
- 10.Fink B, Laude K, McCann L, Doughan A, Harrison DG, Dikalov S. Detection of intracellular superoxide formation in endothelial cells and intact tissues using dihydroethidium and an HPLC-based assay. Am J Physiol Cell Physiol. 2004;287:C895–902. doi: 10.1152/ajpcell.00028.2004. [DOI] [PubMed] [Google Scholar]
- 11.Panov A, Dikalov S, Shalbuyeva N, Taylor G, Sherer T, Greenamyre JT. Rotenone model of Parkinson disease: multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J Biol Chem. 2005;280:42026–42035. doi: 10.1074/jbc.M508628200. [DOI] [PubMed] [Google Scholar]
- 12.Choksi KB, Nuss JE, Deford JH, Papaconstantinou J. Age-related alterations in oxidatively damaged proteins of mouse skeletal muscle mitochondrial electron transport chain complexes. Free Radic Biol Med. 2008;45:826–838. doi: 10.1016/j.freeradbiomed.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stanley WC, Hoppel CL. Mitochondrial dysfunction in heart failure: potential for therapeutic interventions? . Cardiovasc Res. 2000;45:805–806. doi: 10.1016/s0008-6363(99)00419-8. [DOI] [PubMed] [Google Scholar]
- 14.Lopez-Campistrous A, Hao L, Xiang W, Ton D, Semchuk P, Sander J, Ellison MJ, Fernandez-Patron C. Mitochondrial dysfunction in the hypertensive rat brain: respiratory complexes exhibit assembly defects in hypertension. Hypertension. 2008;51:412–419. doi: 10.1161/HYPERTENSIONAHA.107.102285. [DOI] [PubMed] [Google Scholar]
- 15.Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 2007;49:241–248. doi: 10.1161/01.HYP.0000254415.31362.a7. [DOI] [PubMed] [Google Scholar]
- 16.Tsutsui H, Ide T, Kinugawa S. Mitochondrial oxidative stress, DNA damage, and heart failure. Antioxid Redox Signal. 2006;8:1737–1744. doi: 10.1089/ars.2006.8.1737. [DOI] [PubMed] [Google Scholar]
- 17.Dabkowski ER, Williamson CL, Hollander JM. Mitochondria-specific transgenic overexpression of phospholipid hydroperoxide glutathione peroxidase (GPx4) attenuates ischemia/reperfusion-associated cardiac dysfunction. Free Radic Biol Med. 2008;45:855–865. doi: 10.1016/j.freeradbiomed.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 18.Li YL, Gao L, Zucker IH, Schultz HD. NADPH oxidase-derived superoxide anion mediates angiotensin II-enhanced carotid body chemoreceptor sensitivity in heart failure rabbits. Cardiovasc Res. 2007;75:546–554. doi: 10.1016/j.cardiores.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones DP. Disruption of mitochondrial redox circuitry in oxidative stress. Chem Biol Interact. 2006;163:38–53. doi: 10.1016/j.cbi.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 20.Xiong Y, Liu X, Lee CP, Chua BH, Ho YS. Attenuation of doxorubicin-induced contractile and mitochondrial dysfunction in mouse heart by cellular glutathione peroxidase. Free Radic Biol Med. 2006;41:46–55. doi: 10.1016/j.freeradbiomed.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 21.Papa S, Scacco S, Sardanelli AM, Vergari R, Papa F, Budde S, van den Heuvel L, Smeitink J. Mutation in the NDUFS4 gene of complex I abolishes cAMP-dependent activation of the complex in a child with fatal neurological syndrome. FEBS Lett. 2001;489:259–262. doi: 10.1016/s0014-5793(00)02334-6. [DOI] [PubMed] [Google Scholar]
- 22.Scacco S, Petruzzella V, Budde S, Vergari R, Tamborra R, Panelli D, van den Heuvel LP, Smeitink JA, Papa S. Pathological mutations of the human NDUFS4 gene of the 18-kDa (AQDQ) subunit of complex I affect the expression of the protein and the assembly and function of the complex. J Biol Chem. 2003;278:44161–44167. doi: 10.1074/jbc.M307615200. [DOI] [PubMed] [Google Scholar]
- 23.Shi H, Timmins G, Monske M, Burdick A, Kalyanaraman B, Liu Y, Clement JL, Burchiel S, Liu KJ. Evaluation of spin trapping agents and trapping conditions for detection of cell-generated reactive oxygen species. Arch Biochem Biophys. 2005;437:59–68. doi: 10.1016/j.abb.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez C, Sanz-Alfayate G, Agapito MT, Gomez-Nino A, Rocher A, Obeso A. Significance of ROS in oxygen sensing in cell systems with sensitivity to physiological hypoxia. Respir Physiol Neurobiol. 2002;132:17–41. doi: 10.1016/s1569-9048(02)00047-2. [DOI] [PubMed] [Google Scholar]
- 25.Francis J, Wei SG, Weiss RM, Felder RB. Brain angiotensin-converting enzyme activity and autonomic regulation in heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H2138–2146. doi: 10.1152/ajpheart.00112.2004. [DOI] [PubMed] [Google Scholar]
- 26.Guggilam A, Haque M, Kerut EK, McIlwain E, Lucchesi P, Seghal I, Francis J. TNF-alpha blockade decreases oxidative stress in the paraventricular nucleus and attenuates sympathoexcitation in heart failure rats. Am J Physiol Heart Circ Physiol. 2007;293:H599–609. doi: 10.1152/ajpheart.00286.2007. [DOI] [PubMed] [Google Scholar]
- 27.Dikalov SI, Li W, Mehranpour P, Wang SS, Zafari AM. Production of extracellular superoxide by human lymphoblast cell lines: comparison of electron spin resonance techniques and cytochrome C reduction assay. Biochem Pharmacol. 2007;73:972–980. doi: 10.1016/j.bcp.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dikalov SI, Dikalova AE, Mason RP. Noninvasive diagnostic tool for inflammation-induced oxidative stress using electron spin resonance spectroscopy and an extracellular cyclic hydroxylamine. Arch Biochem Biophys. 2002;402:218–226. doi: 10.1016/S0003-9861(02)00064-4. [DOI] [PubMed] [Google Scholar]
- 29.Panov A, Dikalov S, Shalbuyeva N, Hemendinger R, Greenamyre JT, Rosenfeld J. Species-and tissue-specific relationships between mitochondrial permeability transition and generation of ROS in brain and liver mitochondria of rats and mice. Am J Physiol Cell Physiol. 2007;292:C708–718. doi: 10.1152/ajpcell.00202.2006. [DOI] [PubMed] [Google Scholar]
- 30.Yan LJ, Rajasekaran NS, Sathyanarayanan S, Benjamin IJ. Mouse HSF1 disruption perturbs redox state and increases mitochondrial oxidative stress in kidney. Antioxid Redox Signaling. 2005;7:465–471. doi: 10.1089/ars.2005.7.465. [DOI] [PubMed] [Google Scholar]
- 31.Perez MJ, Cederbaum AI. Spin trapping agents (Tempol and POBN) protect HepG2 cells overexpressing CYP2E1 against arachidonic acid toxicity. Free Radic Biol Med. 2001;30:734–746. doi: 10.1016/s0891-5849(01)00461-0. [DOI] [PubMed] [Google Scholar]
- 32.Suematsu N, Tsutsui H, Wen J, Kang D, Ikeuchi M, Ide T, Hayashidani S, Shiomi T, Kubota T, Hamasaki N, Takeshita A. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation. 2003;107:1418–1423. doi: 10.1161/01.cir.0000055318.09997.1f. [DOI] [PubMed] [Google Scholar]
- 33.Li J, Li Q, Xie C, Zhou H, Wang Y, Zhang N, Shao H, Chan SC, Peng X, Lin SC, Han J. Beta-actin is required for mitochondria clustering and ROS generation in TNF-induced, caspase-independent cell death. J Cell Sci. 2004;117:4673–4680. doi: 10.1242/jcs.01339. [DOI] [PubMed] [Google Scholar]
- 34.Takano H, Zou Y, Hasegawa H, Akazawa H, Nagai T, Komuro I. Oxidative stress-induced signal transduction pathways in cardiac myocytes: involvement of ROS in heart diseases. Antioxid Redox Signal. 2003;5:789–794. doi: 10.1089/152308603770380098. [DOI] [PubMed] [Google Scholar]
- 35.Pitkanen S, Robinson BH. Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of superoxide dismutase. J Clin Invest. 1996;98:345–351. doi: 10.1172/JCI118798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cocheme HM, Murphy MP. Complex I is the major site of mitochondrial superoxide production by paraquat. J Biol Chem. 2008;283:1786–1798. doi: 10.1074/jbc.M708597200. [DOI] [PubMed] [Google Scholar]
- 37.Matsuzaki S, Szweda LI. Inhibition of complex I by Ca2+reduces electron transport activity and the rate of superoxide anion production in cardiac submitochondrial particles. Biochemistry. 2007;46:1350–1357. doi: 10.1021/bi0617916. [DOI] [PubMed] [Google Scholar]
- 38.Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Abe Y. Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension. 2005;45:438–444. doi: 10.1161/01.HYP.0000157169.27818.ae. [DOI] [PubMed] [Google Scholar]
- 39.Triepels RH, Hanson BJ, van den Heuvel LP, Sundell L, Marusich MF, Smeitink JA, Capaldi RA. Human complex I defects can be resolved by monoclonal antibody analysis into distinct subunit assembly patterns. J Biol Chem. 2001;276:8892–8897. doi: 10.1074/jbc.M009903200. [DOI] [PubMed] [Google Scholar]
- 40.Ghafourifar P, Sen CK. Mitochondrial nitric oxide synthase. Front Biosci. 2007;12:1072–1078. doi: 10.2741/2127. [DOI] [PubMed] [Google Scholar]
- 41.Sharma P, Mongan PD. Ascorbate reduces superoxide production and improves mitochondrial respiratory chain function in human –fibroblasts with electron transport chain deficiencies. Mitochondrion. 2001;1:191–198. doi: 10.1016/s1567-7249(01)00016-2. [DOI] [PubMed] [Google Scholar]
- 42.Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HHHW, Busse R, Schroder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]